

Summary
Recent studies provide evidence of correlations of DNA methylation and expression of protein‐coding genes with human aging. The relations of microRNA expression with age and age‐related clinical outcomes have not been characterized thoroughly. We explored associations of age with whole‐blood microRNA expression in 5221 adults and identified 127 microRNAs that were differentially expressed by age at P < 3.3 × 10−4 (Bonferroni‐corrected). Most microRNAs were underexpressed in older individuals. Integrative analysis of microRNA and mRNA expression revealed changes in age‐associated mRNA expression possibly driven by age‐associated microRNAs in pathways that involve RNA processing, translation, and immune function. We fitted a linear model to predict ‘microRNA age’ that incorporated expression levels of 80 microRNAs. MicroRNA age correlated modestly with predicted age from DNA methylation (r = 0.3) and mRNA expression (r = 0.2), suggesting that microRNA age may complement mRNA and epigenetic age prediction models. We used the difference between microRNA age and chronological age as a biomarker of accelerated aging (Δage) and found that Δage was associated with all‐cause mortality (hazards ratio 1.1 per year difference, P = 4.2 × 10−5 adjusted for sex and chronological age). Additionally, Δage was associated with coronary heart disease, hypertension, blood pressure, and glucose levels. In conclusion, we constructed a microRNA age prediction model based on whole‐blood microRNA expression profiling. Age‐associated microRNAs and their targets have potential utility to detect accelerated aging and to predict risks for age‐related diseases.
Keywords: aging, cardiometabolic traits, methylation, microRNA, mortality, mRNA
Introduction
Human aging is a complex process that has been linked to dysregulation of numerous cellular and molecular processes, including shortened telomere length (Harley et al., 1990), altered DNA damage response (Moskalev et al., 2013), loss of protein homeostasis (Tomaru et al., 2012), cellular senescence (Childs et al., 2015), and mitochondrial dysfunction (Green et al., 2011). Those cellular and molecular processes can lead to a variety of diseases including cancer, cardiovascular disease, and neurological disease, as well as an increased risk of mortality (Fontana et al., 2010; López‐Otín et al., 2013).
Recent studies have revealed that human aging can be characterized by changing patterns of DNA methylation (Hannum et al., 2013; Horvath, 2013) and expression of protein‐coding genes (Peters et al., 2015). A growing body of research suggests that aging is associated with changes in DNA methylation both genome‐wide and at specific C‐G dinucleotide (CpG) loci. Two recent studies developed age predictors based on the methylation state of CpGs in whole blood and other tissues (Hannum et al., 2013; Horvath, 2013). The resultant DNA methylation‐based predicted age (i.e., DNAm age) was associated with chronological age in several independent studies. The difference between DNAm age and chronological age (i.e., DNAm Δage) has been proposed as an index of accelerated aging and was reported to be associated with all‐cause mortality and several coronary heart disease risk factors (Marioni et al., 2015; Christiansen et al., 2016; Horvath et al., 2016).
At the mRNA level, a recent meta‐analysis of whole‐blood gene expression in ~15 000 individuals identified 1497 mRNAs that are differentially expressed in relation to age (Peters et al., 2015). An age predictor based on mRNA expression (i.e., mRNA age) highlighted genes involved in mitochondrial, metabolic, and immune function‐related pathways as key components of aging processes. The difference between mRNA age and chronological age (i.e., mRNA Δage) correlated with many metabolic risk factors including blood pressure, total cholesterol levels, fasting glucose, and body mass index (BMI) (Peters et al., 2015).
MicroRNAs (miRNAs) are a class of small noncoding RNAs that downregulate protein‐coding genes by either cleaving target mRNAs or suppressing translation of mRNAs into proteins (Lee & Ambros, 2001; Lee et al., 2004; Cordes & Srivastava, 2009). Research in a Caenorhabditis elegans model system revealed changes in miRNA expression in relation to lifespan and longevity (Boehm & Slack, 2005; Ibáñez‐Ventoso et al., 2006; Pincus et al., 2011). In humans, highly specific miRNA expression patterns are correlated with many age‐related diseases including cardiovascular disease (Small & Olson, 2011; Huan et al., 2015c) and cancer (Lu et al., 2005; Hayes et al., 2014). Recent studies have examined differentially expressed miRNAs in relation to age in whole blood (ElSharawy et al., 2012), peripheral blood mononuclear cells (PBMC) (Noren Hooten et al., 2010), and serum (Noren Hooten et al., 2013; Zhang et al., 2014). These studies, however, were based on small sample sizes, limiting the power to investigate age‐related changes in miRNA expression. We hypothesized, a priori, that it would be possible to create a miRNA signature of age that is predictive of chronological age and that age prediction based on miRNA expression is biologically meaningful and can be used as a biomarker of risk for age‐related outcomes including all‐cause mortality.
In a previous study, we measured miRNA expression in whole blood from more than 5000 Framingham Heart Study (FHS) participants. We investigated the heritability of miRNA expression and performed a genome‐wide association study (GWAS) of miRNA expression to identify miR‐eQTLs (Huan et al., 2015b). Our results showed that miRNAs are under strong genetic control. In the present study, we further investigated whole‐blood miRNA expression in relation to chronological age in FHS participants. Fig. S1 shows the overall study design. We identified 127 miRNAs that were differentially expressed in relation to chronological age, and performed internal validation by splitting the samples 1:1 into two independent sample sets. An integrative miRNA–mRNA coexpression analysis and miRNA target prediction revealed many age‐related pathways underlying age‐associated molecular changes. We also defined and evaluated an age predictor based on miRNA expression levels (i.e., miRNA age). Our results indicate that the difference between miRNA age and chronological age (i.e., miRNA Δage) is associated with multiple age‐related clinical traits including all‐cause mortality, coronary heart disease (CHD), hypertension, blood pressure, and glucose levels. In addition, we compared miRNA age with DNAm age and mRNA age in FHS participants.
Results
Study population
Table 1 shows the characteristics of the 5221 FHS participants in this study (2295 participants from the FHS offspring cohort and 2926 participants from the FHS third‐generation cohort). At the time of measured sample collection for miRNA isolation, the FHS Offspring cohort was on average 20 years older than the Third Generation cohort (mean age 66 vs. 46 years). As expected, given the age differences between cohorts, the Offspring cohort had a higher prevalence of cardiovascular disease risk factors. In addition, during 6 years of follow‐up, incident deaths occurred more commonly in the Offspring cohort (257 vs. 12).
Table 1.
Framingham heart study offspring and third generation cohort study participant characteristics
| Phenotypes/Covariates | Offspring cohort N = 2295 | Third generation cohort N = 2926 |
|---|---|---|
| Male (%) | 44 | 46 |
| Age (years), mean (SD) | 66 (9) | 46 (9) |
| Body mass index (kg m−2), mean (SD) | 28.3 (5.4) | 28.0 (5.9) |
| Systolic blood pressure (mm Hg), mean (SD) | 129 (17) | 116 (14) |
| Diastolic blood pressure (mm Hg), mean (SD) | 73 (10) | 74 (9) |
| Serum glucose (mg dL−1), mean (SD) | 107 (23) | 96 (18) |
| High‐density lipoprotein (mg dL−1), mean (SD) | 59 (18) | 60 (18) |
| Total cholesterol (mg dL−1), mean (SD) | 186 (37) | 187 (35) |
| Triglycerides (mg dL−1), mean (SD) | 118 (70) | 113 (79) |
| Current smokers, n (%) | 147 (6) | 341 (12) |
| Hypertension, n (%) | 1449 (63) | 719 (25) |
| Hypertension, no hypertensive treatment, n (%) | 209 (9) | 170 (6) |
| Prevalent diabetes mellitus, n (%) | 317 (14) | 156 (5) |
| Prevalent diabetes mellitus, no diabetes treatment, n (%) | 108 (5) | 56 (2) |
| Prevalent coronary heart disease, n (%) | 219 (10) | 31 (1) |
| Deaths, n (%) | 257 (11) | 12 (0) |
| Hypertensive treatment, n (%) | 1196 (52) | 544 (19) |
| Diabetes treatment, n (%) | 209 (9) | 103 (4) |
| Lipids treatment, n (%) | 951 (41) | 453 (15) |
We split the 5221 FHS samples 1:1 by pedigrees into independent discovery and replication sets (see Methods). The mean age was 54.9 in the discovery set and 55.4 in the replication set (P = 0.21). Other clinical factors did not differ between the discovery and replication sets.
Identification of differentially expressed miRNAs in relation to chronological age
We identified 127 miRNAs that were differentially expressed in relation to chronological age at P < 3.3 × 10−4 (Bonferroni‐corrected, 0.05/150). The incremental proportion of interindividual difference in age explained by the 127 differentially expressed miRNAs ranged from partial r 2 of 0.002–0.15. Table 2 provides results for the top 25 miRNAs, and Table S1 provides the full list. Of the 127 age‐associated miRNAs, 103 (81%) miRNAs were negatively correlated with age and 24 (19%) miRNAs were positively correlated (Fig. 1). Age‐related miRNA expression studies in whole blood (ElSharawy et al., 2012), PBMCs (Noren Hooten et al., 2010), and serum (Noren Hooten et al., 2013; Zhang et al., 2014) have similarly reported that the majority of age‐related miRNAs show decreased expression in older individuals.
Table 2.
Top 25 differentially expressed miRNAs in relation to chronological age
| miRNA | Estimated Betaa | SE | R‐squared | P‐Value |
|---|---|---|---|---|
| miR‐99b‐5p | 0.07 | 0.002 | 0.15 | 1.16E‐286 |
| miR‐130b‐5p | 0.06 | 0.002 | 0.11 | 3.04E‐227 |
| miR‐505‐5p | 0.06 | 0.002 | 0.12 | 3.04E‐226 |
| miR‐425‐3p | 0.08 | 0.002 | 0.10 | 8.58E‐203 |
| miR‐144‐5p | 0.11 | 0.004 | 0.09 | 1.55E‐165 |
| miR‐182‐5p | 0.10 | 0.003 | 0.08 | 7.29E‐157 |
| miR‐1275 | 0.08 | 0.003 | 0.08 | 5.40E‐149 |
| miR‐601 | 0.09 | 0.003 | 0.08 | 4.48E‐146 |
| miR‐206 | 0.07 | 0.003 | 0.08 | 4.48E‐139 |
| miR‐30a‐5p | 0.03 | 0.001 | 0.08 | 9.07E‐132 |
| miR‐218‐5p | 0.07 | 0.003 | 0.07 | 4.94E‐130 |
| miR‐30d‐5p | 0.04 | 0.002 | 0.07 | 9.10E‐127 |
| miR‐502‐3p | 0.05 | 0.002 | 0.06 | 5.28E‐116 |
| miR‐28‐3p | −0.08 | 0.003 | 0.06 | 7.96E‐114 |
| miR‐197‐3p | 0.04 | 0.002 | 0.06 | 3.69E‐111 |
| miR‐320b | 0.04 | 0.002 | 0.07 | 4.77E‐110 |
| miR‐576‐3p | 0.06 | 0.002 | 0.06 | 2.58E‐105 |
| miR‐181a‐5p | 0.06 | 0.003 | 0.05 | 2.61E‐87 |
| miR‐18a‐5p | 0.05 | 0.003 | 0.05 | 2.93E‐86 |
| miR‐223‐5p | 0.05 | 0.002 | 0.05 | 6.86E‐86 |
| miR‐339‐5p | 0.04 | 0.002 | 0.05 | 1.65E‐85 |
| miR‐24‐3p | 0.03 | 0.001 | 0.05 | 2.08E‐84 |
| miR‐22‐3p | −0.06 | 0.003 | 0.05 | 2.14E‐84 |
| miR‐345‐5p | 0.04 | 0.002 | 0.05 | 1.66E‐82 |
| miR‐302c‐3p | 0.08 | 0.004 | 0.05 | 2.83E‐82 |
Higher Ct values indicate lower miRNA expression levels. Therefore, positive beta values indicate negative associations between miRNA expression and age.
Figure 1.
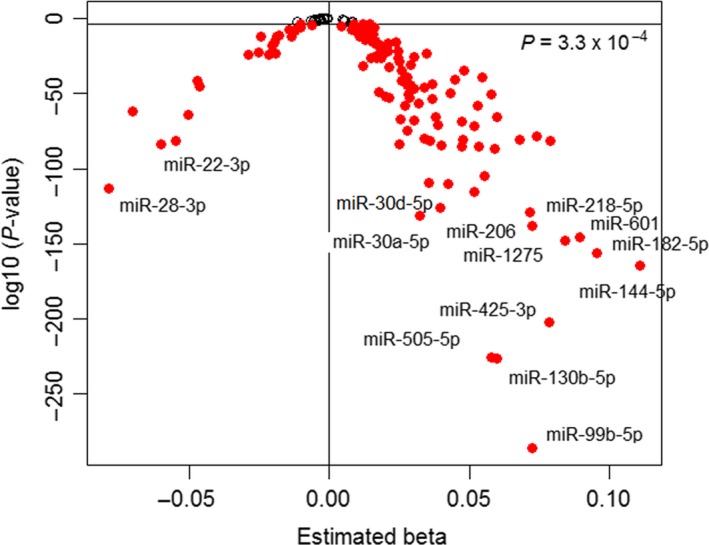
Volcano plot of differentially expressed miRNAs in relation to chronological age. Higher Ct values indicate lower miRNA expression levels. Therefore, positive beta values indicate negative associations between miRNA expression and age.
The age associations of miRNAs in the discovery and replication sets were highly consistent with Pearson's correlations (r) of beta values (effect size) of > 0.99 and of log10‐transformed P values of 0.97 (Fig. S2). Ninety‐one percentage of differentially expressed miRNAs in relation to age in the discovery set (at P < 3.3 × 10−4) replicated in the replication set (at P < 3.3 × 10−4). The validation results suggest that age‐associated miRNA expression signatures are robust and highly replicable.
In conducting a sensitivity analysis, we identified differentially expressed miRNAs in relation to age separately in the FHS Offspring cohort and FHS Third Generation cohort (TablesS2and S3). At P < 3.3 × 10−4, there were 32 age‐associated miRNAs identified in the FHS Offspring cohort and 57 in the larger FHS Third Generation cohort. A total of 23 miRNAs overlapped between cohorts. The Pearson correlation of beta values for all 150 measured miRNAs between cohorts was 0.67 (Fig. S3). These results suggest that age‐associated miRNA expression differs slightly across age groups.
miRNA age prediction
We used elastic net regression (Friedman et al., 2010) to select miRNAs for building an age prediction model using 10‐fold cross‐validation. To ensure unbiased validation, the age prediction model was trained only in the discovery sample set. Table S4> reports the results of the prediction model that included 80 miRNAs. Seventy‐two of the 80 miRNAs in the prediction model showed differential expression in relation to age at P < 3.3 × 10−4.
The correlation between miRNA predicted age (miRNA age) and chronological age was significant in the discovery (Pearson's correlation r = 0.70; P < 1 × 10−320) and replication sets (r = 0.65; P = 2.7 × 10−311) (Fig. 2). We hypothesized that the difference between miRNA age and chronological age (miRNA Δage) is an index of ‘biological’ aging, with a positive Δage indicating accelerated aging, and a negative Δage reflecting slower aging in comparison with chronological age. miRNA Δage was not associated with miRNA age (Fig. S4).
Figure 2.
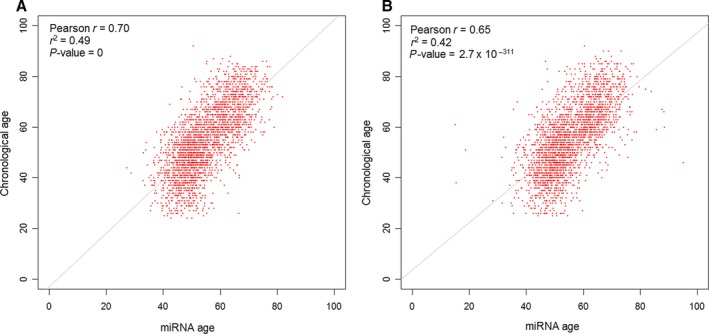
miRNA age vs. chronological age. (A) In the discovery set; (B) in the replication set.
In conducting a sensitivity analysis to account for cohort effects, we used the same method to train a miRNA age prediction model in the FHS Offspring and Third Generation cohorts separately (Fig. S5). When the two cohorts were used as separate discovery‐replication sets, the correlation between miRNA age and chronological age was stronger in the Offspring cohort (Pearson's correlation r = 0.50 and P = 1.2 × 10−73) than in FHS Third Generation cohort (Pearson's correlation r = 0.36 and P = 2.7 × 10−45). The different correlation between miRNA age and chronological age in cohort‐specific analysis could be explained by differences in mean age, age range, and unmeasured technical factors.
Heritability of miRNA Δage
The estimate of heritability of miRNA Δage () was 0.38. To determine whether any specific genetic variants correlate with miRNA Δage, we performed a genome‐wide association study of miRNA Δage using all available FHS samples (see Fig. S6 for the Manhattan plot). There were no SNPs associated with miRNA Δage at P < 5 × 10−8.
We previously reported that miRNA expression levels are highly heritable, and identified 5269 miRNA expression‐associated SNPs for 76 miRNAs within +/‐ 1 MB of the associated SNP (i.e., cis‐miR‐eSNPs). We further explored whether miRNA Δage is linked to cis‐miR‐eSNPs. No cis‐miR‐eSNPs were associated with miRNA Δage at P < 1 × 10−5.
Influence of blood cell types on miRNA age
We compared the differentially expressed miRNAs in relation to age both with and without adjustment of blood cell types. As shown in Fig. S7, the Pearson correlation coefficients of beta values with vs. without adjustment for blood cell types were > 0.99, and for log10‐transformed P values, it was > 0.99. In addition, 122 of the 127 age‐associated miRNAs (96%) remained significant after adjusting for blood cell types, indicating that adjusting for blood cell type proportions had little effect on our results.
In conducting a sensitivity analysis adjusting for white blood cell counts, the miRNA age predictor exhibited a weaker but still high correlation with chronological age (r = 0.61) in the replication set. Cell type effects explained a proportion of age variability, which may have resulted in the weaker correlation observed between miRNA age and chronological age after adjusting for cell types.
miRNA Δage is predictive of all‐cause mortality and correlated with many metabolic traits
The association of miRNA Δage with all‐cause mortality was tested in FHS Offspring participants (there were too few deaths in the Third Generation cohort for meaningful analysis). We found that miRNA Δage was associated with mortality with a hazards ratio (HR) of 1.10 (95% CI 1.05–1.14; P = 4.2 × 10−5) per year of miRNA Δage after adjustment for chronological age and sex. Kaplan–Meier survival curves for miRNA Δage tertiles are presented in Fig. 3. The plot illustrates higher mortality rates for those with higher Δage. Sensitivity analyses were performed to control for additional potential confounders (cigarette smoking, HDL cholesterol, total cholesterol, triglycerides, systolic and diastolic blood pressure, fasting blood glucose, BMI, lipid treatment, diabetes treatment, hypertension treatment, prevalent cancer, prevalent CHD, and prevalent diabetes). The fully adjusted HR was 1.15 (95% CI 1.08–1.22; P = 2.5 × 10−5).
Figure 3.
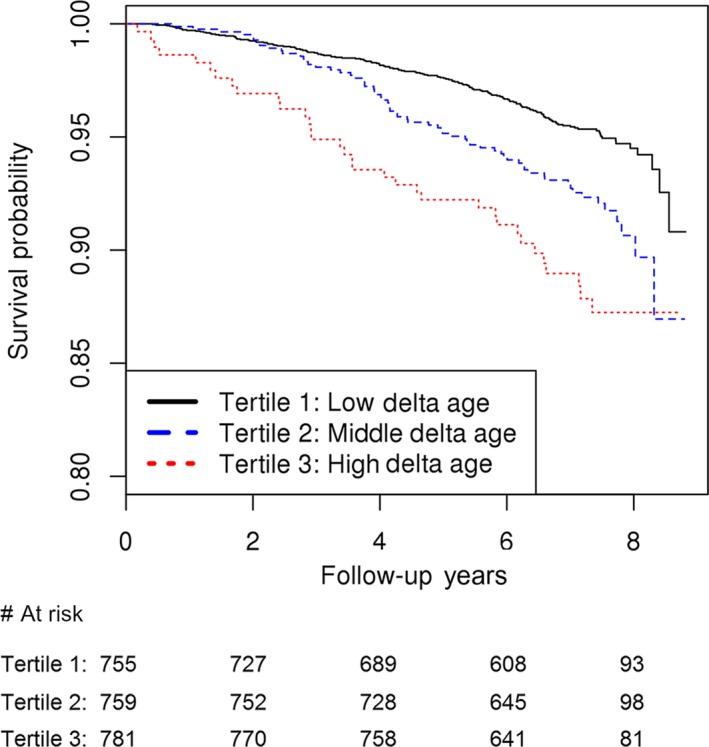
Survival probability by tertiles of miRNA Δage.
The association of miRNA Δage with prevalent CHD was tested in all available individuals (a total of 250 prevalent CHD cases). The associations of miRNA Δage with prevalent diabetes and hypertension were tested in individuals who were not receiving medications to treat diabetes and hypertension (including 164 diabetes cases and 379 hypertension cases), respectively. We found miRNA Δage to be positively associated with CHD (P = 3.8 × 10−17) and hypertension (P = 0.002), after adjustment for chronological age, sex, and BMI (Table 3).
Table 3.
Associations of miRNA Δage with prevalent CHD, diabetes, hypertension, and cardiometabolic traits
| Trait/Disease | Chronological age | miRNA Δage | ||||||
|---|---|---|---|---|---|---|---|---|
| Estimated Beta | SE | T‐Value | P‐Value | Estimated Beta | SE | T‐Value | P‐Valuea | |
| CHD | 15.28 | 0.84 | 18.20 | 8.72E‐72 | 5.42 | 0.64 | 8.45 | 3.76E‐17 |
| Hypertension | 9.45 | 0.64 | 14.84 | 2.76E‐48 | 1.62 | 0.53 | 3.05 | 2.34E‐03 |
| Diabetes | 8.42 | 1.05 | 8.02 | 1.28E‐15 | 1.01 | 0.77 | 1.30 | 1.94E‐01 |
| Total cholesterol | 0.82 | 0.05 | 15.84 | 3.81E‐54 | −0.05 | 0.07 | −0.82 | 4.14E‐01 |
| High‐density lipoprotein | 0.07 | 0.02 | 2.73 | 6.34E‐03 | 0.06 | 0.03 | 2.00 | 4.61E‐02 |
| Triglycerides | 0.26 | 0.10 | 2.51 | 1.21E‐02 | −0.18 | 0.13 | −1.36 | 1.73E‐01 |
| Systolic blood pressure | 0.55 | 0.02 | 26.27 | 5.09E‐136 | 0.10 | 0.03 | 3.78 | 1.58E‐04 |
| Diastolic blood pressure | 0.08 | 0.01 | 6.00 | 2.24E‐09 | 0.06 | 0.02 | 3.65 | 2.67E‐04 |
| Glucose | 0.31 | 0.02 | 15.81 | 5.78E‐54 | 0.07 | 0.02 | 2.86 | 4.30E‐03 |
| Body mass index | 0.004 | 0.01 | 0.52 | 6.01E‐01 | −0.002 | 0.01 | −0.24 | 8.11E‐01 |
The P‐value threshold for significance for miRNA Δage vs. traits analyses (P < 0.005) was determined by the Bonferroni method (0.05/10). Significant P‐values are shown in boldface.
We also tested the associations of miRNA Δage with multiple cardiometabolic traits using all available FHS participants who were not receiving medications to treat hypertension, dyslipidemia, or diabetes (N = 2993). These analyses revealed miRNA Δage to be positively associated with systolic blood pressure, diastolic blood pressure, and fasting glucose levels at P < 0.005 (Bonferroni‐corrected, 0.05/10 tests) after adjusting for age, sex, and BMI (Table 3).
miRNA–mRNA coexpression, miRNA targets, and pathway analyses
To better understand how miRNAs might contribute to aging processes, we further tested whether expression levels of age‐associated mRNA transcripts are mirrored by differential expression of age‐associated miRNAs. For this purpose, we analyzed miRNA–mRNA coexpression in the same set of FHS individuals (n = 5012) in whom miRNA and mRNA expression data were both available. For the 127 age‐associated miRNAs, the coexpression analysis identified 4682 mRNAs that were highly coexpressed with the 123 age‐related miRNAs at FDR < 0.05, including 24 310 miRNA–mRNA coexpression pairs. Of the 4682 mRNAs that were coexpressed with age‐associated miRNAs, 551 mRNAs were previously reported to be age‐associated (Peters et al., 2015); the total number of reported age‐associated mRNAs in that study was 1497. Comparison of the two ratios (4682/17 318 measured mRNAs and 551/1497) yielded P < 1 × 10−16 (by the hypergeometric test), suggesting that age‐associated differences in miRNA expression are indicative of age‐associated changes in their coexpressed mRNAs. Gene ontology enrichment analysis (Table S5) showed that the coexpressed mRNAs are enriched for translation (P = 3.3 × 10−7), immune response (P = 9.4 × 10−7), and RNA processing (P = 1.7 × 10−5).
Among the miRNA–mRNA coexpression pairs, TargetScan v7.0 (Lewis et al., 2005; Agarwal et al., 2015) predicted 3552 of the 4682 mRNAs to be potential corresponding targets for the coexpressed age‐associated miRNAs, including 406 of the 551 age‐associated mRNAs (~74%). Details of miRNAs and their coexpressed/predicted target mRNAs involved in each GO category are provided in Table S6.
Comparing miRNA age with mRNA age and DNAm age
In our previous studies, we used mRNA expression and DNA methylation data to predict age (i.e., mRNA age and DNAm age) in FHS Offspring participants (Marioni et al., 2015; Peters et al., 2015). In order to compare miRNA age with mRNA age and DNAm age, we used miRNA age calculated only in the FHS Offspring cohort. The Pearson correlation (r) of chronological age with miRNA age in the FHS Offspring cohort was 0.50 (P = 1.2 × 10−73). The r values of chronological age with mRNA age and DNAm age were 0.56 and 0.73, respectively. As shown in Fig. 4, miRNA age was positively correlated with mRNA age and DNAm age, with r values of 0.20 and 0.34, respectively. DNAm age and mRNA age were also positively correlated (r = 0.43).
Figure 4.

Comparison of miRNA Δage, mRNA age, and DNAm age. (A) miRNA age vs. DNAm age; (B) miRNA age vs. mRNA age; (C) mRNA age vs. DNAm age.
When mRNA age and DNAm age were both regressed on chronological age, the r 2 was 0.57. Regressing miRNA age, mRNA age, and DNAm age on chronological age yielded an r 2 of 0.63. Using data from individuals whose miRNA and mRNA data were both available (N = 2784), we further tested the associations of miRNA Δage with cardiometabolic traits adjusting for mRNA Δage, as shown in Table S7. The results did not change appreciably when adding mRNA Δage in the model. The miRNA Δage remained associated with CHD, hypertension, BP, and glucose. We did not include DNAm Δage in the model, because DNA methylation data were available in a smaller number of eligible individuals.
Discussion
We systematically assessed age‐associated differences in whole‐blood miRNA expression levels and developed a miRNA age predictor. We calculated the difference between miRNA predicted age and chronological age to generate Δage, which we put forth as an indicator of an individual's rate of biological aging. We found Δage to be associated with risk for all‐cause mortality and with prevalent CHD, hypertension, and glucose levels.
In comparison with previous age‐related miRNA expression studies (Noren Hooten et al., 2010, 2013; ElSharawy et al., 2012; Zhang et al., 2014), our study included > 25 times as many participants (N = 5221 vs N < 200) and a wide age range (24–92 years). As such, our study was well powered to investigate differential levels of miRNA expression in relation to age. Among the 150 whole‐blood miRNAs available for analysis in this study, we identified 127 miRNAs that were differentially expressed in relation to age at P < 0.05/150. The differential expression patterns of miRNAs were highly consistent between separate discovery and replication sample sets. The effect sizes (r 2) of individual differential miRNA expression with age were small, ranging from 0.05–0.15 (Table 2), limiting the use of single miRNAs as clinical biomarkers of age. The small effect sizes required a large sample size to identify a‐multiple miRNA age‐related biomarker.
By comparing the FHS Offspring and Third Generation cohorts, as shown in Fig. S3, the estimated beta values for 150 measured miRNAs in relation with age are consistent between cohorts. All 23 significant age‐associated miRNAs that overlapped between the two cohorts showed a concordant direction of effects. However, there were more than 50 miRNAs that were significant in the overall sample set, but not in either cohort. Because the cohort‐specific analysis reduced the available sample size by about half, the differences by cohort may have arisen from reduced power. Another possible reason for differences by cohort is that the correlation of miRNA expression with age may differ in the younger and older cohorts, which differed in age by an average of 20 years (Noren Hooten et al. 2013). However, we cannot exclude undetected technical factors contributing to differences between the cohorts.
Many of our identified miRNAs were previously shown to be differentially expressed in relation to age in whole blood (ElSharawy et al., 2012), PBMCs (Noren Hooten et al., 2010), and serum (Noren Hooten et al., 2013; Zhang et al., 2014; Smith‐Vikos et al., 2016). For example, miR‐130b‐5p (the second most significant miRNA in our study) was previously reported to be negatively associated with age in serum (Zhang et al., 2014). In addition, miR‐340‐3p was reported to be significantly downregulated in long‐lived individuals vs. short‐lived individuals (Smith‐Vikos et al., 2016). In mouse studies, miR‐130b‐5p was shown to regulate cholesterol and triglyceride homeostasis (Wagschal et al., 2015). One of our top 25 miRNAs, miR‐24, was reported to be negatively correlated with age in whole blood (ElSharawy et al., 2012) and PBMCs (Noren Hooten et al., 2010). One study found that increased expression of miR‐24 in T‐cell lines downregulated the expression of histone H2A family member X, which plays a key role in DNA damage response (Brunner et al., 2012). We also found that most of the age‐associated miRNAs are downregulated in older individuals, consistent with previous findings.
miRNAs play a pivotal role in post‐transcriptional regulation of protein‐coding genes. Therefore, we hypothesized that age‐associated mRNAs might be regulated by age‐associated miRNAs. Our miRNA–mRNA coexpression and target prediction analysis revealed that the coexpressed/targeted mRNAs for the identified age‐associated miRNAs were enriched for previously reported age‐associated mRNAs (enrichment P < 1.0 × 10−16) (Peters et al., 2015). This result suggests that age‐associated changes in miRNA expression levels may alter the expression of their targeted protein‐coding genes, which play vital roles in aging processes. GO analysis results were consistent with effects on known aging mechanisms including regulation of transcription, translation, and immune response.
The regulation of transcription and translation of protein‐coding genes is essential to aging processes. In C. elegans studies, reducing the levels of key proteins involved in translation, such as ribosomal proteins, ribosomal‐protein S6 kinase, and translation initiation factors, increased the lifespan of C. elegans (Hansen et al., 2007; Pan et al., 2007). Ribosomal‐protein S6 kinase also regulates lifespan in mammalian models (Selman et al., 2009). miRNAs in Drosophila have been shown to block the eIF4F translation initiation complex assembly, thereby inhibiting overall translation of protein‐coding genes (Fukaya et al., 2014). Similarly, a recent study reported that miR‐139 represses the translation initiation factor EIF4G2 and thereby reduces overall protein synthesis (Emmrich et al., 2016). Our results found that many miRNAs (e.g., miR‐139 and miR‐140) were coexpressed with and targeted many ribosomal genes (e.g., RPL11 and RPL30) as well as translation initiation and elongation genes (e.g., EEF2 and EIF4B) (Table S6). Our results call for further functional studies to explore the specific mechanisms by which age‐associated miRNAs exert their effects on aging and age‐related diseases through regulation of key transcription and translation genes.
Many miRNAs have been found to be involved in immune pathways. For example, previous studies showed that miR‐181a regulates local immune balance (Liu et al., 2012) and T‐cell sensitivity (Li et al., 2007), although its mechanistic action remains unclear. We found that miR‐181a‐3p was coexpressed with and predicted to target three immune response genes, namely CXCL16, RAB27A, and SPON2. RAB27A is also involved in the T‐cell activation pathway. Other identified miRNAs such as miR‐193b‐3p target immune function‐related genes, including CTSW, ETS1, FAIM3, ICOS, TGFBR3, DPP4, and KIF13B. Similarly, miR‐31‐5p targets CD27, FASLG, INPP5D, TCF7, TGFBR3, and DPP4 and may merit further investigation.
Heritability analysis revealed that miRNA Δage is a heritable trait (h 2=0.38). We did not, however, find genome‐wide significant associations with miRNA Δage, likely due to an insufficient sample size for GWAS of this trait.
We also showed that individuals with a predicted miRNA age greater than their chronological age (i.e., higher miRNA Δage) exhibit higher blood pressure and glucose levels, a higher prevalence of CHD and hypertension, as well as increased risk of death from all causes. In comparison, DNAm Δage was previously shown to be associated with all‐cause mortality, but not with prevalent CHD or hypertension (Marioni et al., 2015; Horvath et al., 2016). mRNA Δage was reported to be associated with blood pressure, glucose, and HDL and total cholesterol, but not with all‐cause mortality (Peters et al., 2015). In comparing miRNA age with DNAm age and mRNA age in FHS Offspring participants, miRNA age showed modest correlations with DNAm age (r = 0.3) and mRNA age (r = 0.2), suggesting that miRNA age may complement mRNA and epigenetic age prediction models and can capture unique aspects of the molecular mechanisms of aging and age‐related diseases.
One limitation of this study is that we assessed miRNA expression in whole blood, which consists of multiple cell types and plasma. To address this, we tested whether the proportions of individual blood cell types influenced the association of age with miRNA expression levels. In comparing results with and without cell type adjustment, we observed only minimal changes due to cell type composition. In our previous miRNA expression QTL study, we also did not find substantial cell type effects on the correlation between genetic variants and miRNA expression. Another limitation is that we were unable to perform external replication of our results. To our knowledge, no other study has published extensive analyses of miRNA expression in relation to age in a large sample size with a wide age range. In addition, by splitting our sample set into independent discovery and replications sets, we demonstrated a high degree of replicability of our age‐related miRNAs.
Experimental procedures
Study population
The FHS Offspring cohort was initially recruited in 1971 and included 5124 offspring (and their spouses) from the FHS Original cohort. The FHS Third Generation cohort was recruited from 2002 to 2005 and included 4095 adult children of the Offspring cohort participants (Feinleib et al., 1975; Splansky et al., 2007). The samples used for miRNA analysis in the present study (N = 5221) included 2295 participants from the FHS Offspring cohort who attended the eighth examination cycle (Exam 8, 2005–2008) and 2926 participants from the FHS Third Generation cohort who attended the second examination cycle (Exam 2, 2008–2011). Because the samples from the Offspring and Third Generation cohorts share familial relatedness, we merged the samples from the two cohorts together. Next, the entire FHS sample set was split 1:1 by pedigrees into separate discovery and replication sets. To ensure that the samples in the discovery set did not share relatedness with samples in the replication set, a given pedigree was assigned either to the discovery or to the replication set, but not in both.
Gene (mRNA) expression data were obtained from 2446 FHS Offspring (Exam 8) and 3180 Third Generation (Exam 2) participants (Huan et al., 2015a). DNA methylation data were obtained from 2377 FHS Offspring cohort participants (Exam 8). miRNA and mRNA expression data were available for 5012 participants, and there were 2079 participants with miRNA, mRNA, and DNA methylation data. All participants provided written consent for genetic research.
miRNA expression profiling and normalization
Peripheral whole‐blood samples were collected in PAXgene tubes (Asuragen, Inc., Austin, TX, USA). Purified RNA was extracted using the PAXgene Blood RNA System Kit (Qiagen, Venlo, Netherlands). The WT‐Ovation Pico RNA Amplification System (NuGEN, San Carlos, CA, USA) was used to amplify 50 ng RNA samples according to the manufacturer's recommended protocols. RNA quality was measured using an Agilent 2100 Bioanalyzer and RNA concentration was quantified using a NanoDrop ND‐1000 spectrophotometer. miRNA profiling was carried out at the high‐throughput Gene Expression and Biomarker Core Laboratory at the University of Massachusetts Medical School using TaqMan chemistry‐based miRNA assays by using Dynamic Arrays on BioMark System (Fluidigm, South San Francisco, CA, USA).
The initial miRNA list encompassed all TaqMan miRNA assays available at the start of the study, including 754 miRNAs that were profiled in ~600 FHS individuals. 346 miRNAs expressed in > 20% samples were further profiled in 2445 FHS Offspring and 3245 Third Generation cohort participants. Quantification of miRNA expression was based on cycle threshold (Ct), where lower Ct values signify higher miRNA expression levels. miRNAs with Ct values ≥ 27 indicated that they were not expressed in the sample. Outlier miRNAs with Ct values ≥ 5 standard deviations from the mean Ct value were categorized as missing. We excluded miRNAs expressed in < 5000 samples and samples with > 10% of miRNAs having missing values from analysis. A total of 150 miRNAs and 5221 samples remained for analysis, and 1.2% of remaining missing values were replaced with Ct = 27.
As previously described, raw miRNA Ct values were adjusted for four technical variables: isolation batch (50 batches), RNA concentration, RNA quality (defined as RNA integrity number [RIN]), and RNA 260/280 ratio (ratio of absorbance at 260 and 280 nm; measured using a spectrophotometer). This normalization model explained 20% to 60% of variability of raw miRNA measurements for 80% of miRNAs.
mRNA expression profiling
Messenger RNA (mRNA) expression profiling of whole blood‐derived RNA was performed using the Affymetrix Human Exon 1.0 ST GeneChip platform including 17 318 transcripts. Robust multichip average (RMA) methods were used to normalize mRNA expression values (log‐2‐transformed expression intensity) with quality control measures as previously described (Joehanes et al., 2013).
DNA methylation
Buffy coat preparations were obtained from peripheral whole‐blood samples. Genomic DNA was extracted from buffy coat using the Gentra Puregene DNA extraction kit (Qiagen, Venlo, Netherlands) and bisulfite‐converted using EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA). Samples underwent whole‐genome amplification, fragmentation, array hybridization, and single base pair extension. DNA methylation quantification was conducted in two laboratory batches at the Johns Hopkins Center for Inherited Disease Research (lab batch #1, N = 576) and University of Minnesota Biomedical Genomics Center (lab batch #2, N = 2270).
Methylation beta values were generated using the DASEN methodology implemented in the wateRmelon package in R version 3.0, which includes background adjustment and quantile normalization. Sample exclusion criteria included poor SNP matching of control positions, missing rate > 1%, poor single nucleotide polymorphism (SNP) matching to the 65 SNP control probe locations, outliers from multidimensional scaling (MDS), and sex mismatch. Probes were excluded if missing rate > 20%, previously identified to map to multiple locations, or having an underlying SNP (minor allele frequency > 5% in European ancestry (EUR) 1000 Genomes Project data) at the CpG site or within 10 bp of the single base extension. A total of 2377 samples and 443 252 CpG probes remained for analysis.
Imputing cell counts
The cell count proportions of whole blood were measured in 2138 Third Generation FHS participants (Exam 2), but not for all samples used in this study. Cell counts were imputed using a partial least‐squares regression method (Abdi, 2010) applied to mRNA expression. The estimated cell count proportion values imputed were generally consistent with the measured values in the 2138 samples with cross‐validated estimates of prediction accuracy r 2 > 0.8 for white blood cell, red blood cell, platelet, lymphocyte percent, monocyte percent, and eosinophil percent, and r 2 = 0.25 for basophil percent. miRNA expression analysis accounting for cell counts effects was performed in the 5012 samples.
Identifying differentially expressed miRNAs in relation to age
A linear mixed‐effects model was used to model miRNA expression Ct values (150 miRNAs in total) as the dependent variable and chronological age as an explanatory variable, adjusting for sex, technical variables (batch, RNA concentration, RNA quality score, and 260/280 ratio), and family structure. In a sensitivity analysis, additional adjustments were made for imputed cell counts. The statistical analysis was implemented in the lmekin() R function (http://cran.r-project.org/web/packages/kinship/) (Almasy & Blangero, 1998). Correcting for 150 tests (the number of miRNAs), Bonferroni‐corrected P < 3.3 × 10−4 was used as the significant threshold.
miRNA expression age prediction
The standardized residuals of miRNA expression Ct values (150 miRNAs) were obtained by adjusting raw Ct values for three technical covariates (RNA concentration, RNA quality score, and 260/280 ratio) and sex. Because miRNA expression measurements for the FHS Offspring and Third Generation cohorts were performed in independent batches, we did not adjust Ct values for batch effect. In a sensitivity analysis, additional adjustments were made for imputed cell counts.
We used an elastic net regression model (implemented in the R package glmnet function (Friedman et al., 2010)) to regress chronological age on miRNAs. Elastic net regression is a combination of traditional Lasso and ridge regression methods, emphasizing model sparsity while appropriately balancing the contributions of coexpressed miRNAs. To ensure an unbiased validation, the prediction model was trained in the discovery set. Optimal regularization parameters were estimated via 10‐fold cross‐validation. The alpha parameter of glmnet was set to 0.5, and the lambda value from the best prediction model was set to 0.2. glmnet automatically selected miRNAs for building an age predictor and reported effect size for each miRNA. miRNA predicted age (i.e., miRNA age) was calculated in the replication set using the predictor trained in the discovery set. miRNA Δage was defined as miRNA age minus chronological age.
Estimation of the additive heritability of miRNA Δage
The narrow‐sense heritability estimate of miRNA Δage (denoted as was the proportion of the additive polygenic genetic variance of the total phenotypic variance of miRNA Δage: , where denotes the additive polygenic genetic variance and denotes the total phenotypic variance of a gene expression trait. The estimate was obtained using variance‐component methodology implemented in the lmekin() function of Kinship Package in R (Almasy & Blangero, 1998). Heritability estimation for miRNA Δage was performed using all FHS samples.
All‐cause mortality ascertainment
Outcome analyses included all deaths that occurred prior to January 1, 2014 (about 6 years of follow‐up). Survival status was ascertained using multiple strategies, including routine contact with participants for health history updates, surveillance at the local hospital, review of obituaries in the local newspaper, and queries to the National Death Index. We requested death certificates, hospital and nursing home records prior to death, and autopsy reports. When cause of death was undeterminable, the next of kin were interviewed. The date and cause of death were reviewed by an endpoint panel of three investigators.
Associations between miRNA Δage and mortality were tested using Cox proportional hazards regression models utilizing the coxph() function in the ‘survival’ R library (https://stat.ethz.ch/R-manual/R-devel/library/survival/html/coxph.html), adjusting for age at sample collection and sex. Potential cofounders that were included in the fully adjusted model included systolic blood pressure, diastolic blood pressure, BMI, HDL cholesterol, total cholesterol, triglycerides, fasting blood glucose, smoking status, lipid treatment, diabetes treatment, hypertension treatment, prevalent cardiovascular disease, prevalent cancer, and prevalent diabetes. Hazard ratios for miRNA Δage were expressed as annual risk of death over 6 years of follow‐up. Survival curves were drawn by tertiles of miRNA Δage.
Association analysis of miRNA Δage with prevalent CHD, prevalent diabetes, prevalent hypertension, and cardiometabolic traits
To avoid the cofounding effects of drug treatment, all association tests of miRNA Δage and cardiometabolic traits were performed in individuals who were not receiving antihypertensive, lipids, or diabetes treatment (n = 2993). Linear mixed‐effects models were used to test associations between miRNA Δage (dependent variable) and cardiometabolic traits (independent variable), including systolic blood pressure, diastolic blood pressure, total cholesterol, HDL cholesterol, triglycerides, fasting blood glucose, and BMI using the lmekin() R function. All association tests were adjusted for chronological age, sex, and familial relatedness. To account for the effects of obesity on cardiovascular disease and other traits, we additionally adjusted for BMI (except in analyses of BMI).
Linear mixed‐effects models (R package lmekin() function) were used to test associations between miRNA Δage (dependent variable) and prevalent CHD, diabetes, and hypertension (independent variable, coding ‘1’ for cases and ‘0’ for controls), adjusting for age, sex, BMI, and familial relatedness. Association analysis of miRNA Δage with CHD was performed in all available samples (n = 5221, including 250 prevalent CHD cases). Diabetes and hypertension analyses were performed in individuals who were not receiving antidiabetic treatment (n = 4909, including 164 type‐II diabetes cases) and antihypertensive treatment (n = 3481, including 379 hypertension cases), respectively.
miRNA–mRNA coexpression analysis
The coexpression analysis was performed on FHS participants in whom miRNA and mRNA data were both available. Linear mixed‐effects models (R package lmekin() function) were used to conduct pairwise coexpression analyses for all profiled mRNAs (N = 17 318) and 150 miRNAs, with fixed effects including age, sex, technical covariates, imputed cell types, surrogate variables (SV), and a random effect to account for family structure. As described above, the mRNA expression was quantified by log‐2‐transformed expression intensities. For miRNA expression, higher Ct values reflect lower miRNA expression levels. Adjustment was made for technical covariates (11 for mRNA expression and four for miRNA expression). Surrogate variables (SVs) were computed from the mRNA expression data using the R package ‘SVA’ (Leek & Storey, 2007), and 51 SVs associated with at least one miRNA at Bonferroni‐corrected P < 3.3 × 10−4 (0.05/150) were included in the statistical model. The Benjamini–Hochberg method was used to compute false discovery rate (FDR). Significant miRNA–mRNA coexpression pairs were selected using FDR < 0.05.
Predicting miRNA targets
For the coexpressed miRNA–mRNA pairs, we used TargetScan v7.0 (Lewis et al., 2005; Agarwal et al., 2015) to predict whether the mRNAs were the corresponding targets for the miRNAs. TargetScan predicts mRNA targets of miRNAs by searching for the presence of 8‐mer, 7‐mer, and 6‐mer sites that match the seed region of each miRNA. The sequences from 3′UTR, 5′UTR, and coding regions of each mRNA were downloaded from the University of California Santa Cruz (UCSC) Table Browser (https://genome.ucsc.edu/). The miRNA seed regions were downloaded from miRbase v21 (http://www.mirbase.org/).
Genome‐wide association study of miRNA Δage
DNA was isolated from buffy coat or from immortalized lymphoblast cell lines in FHS participants. Genotyping was conducted with the Affymetrix 500K mapping array and the Affymetrix 50K gene‐focused MIP array, using previously described quality control procedures (Levy et al., 2009). Genotypes were imputed to the 1000 Genomes Project panel of approximately 36.3 million variants using MACH (Li et al., 2010). We filtered out SNPs with MAF < 0.05 and imputation quality ratio < 0.3 (the imputation quality ratio is denoted by the ratio of the variances of the observed and the estimated allele counts). About 9 million variants remained after the filter. The association of miRNA Δage with each SNP was tested by a linear mixed‐effects model that was implemented in the lmekin() R function. miRNA Δage was used as dependent variable, and each SNP as an explanatory variable adjusting for chronological age and sex.
Gene ontology and pathway enrichment analysis
Coexpressed or predicted targeted mRNAs for age‐associated miRNAs were combined as gene sets and classified using Gene Ontology (GO) databases to identify potentially relevant biological processes. Fisher's exact test was used to calculate enrichment P values. DAVID Bioinformatics resources 6.7 (https://david.ncifcrf.gov/) (Huang et al., 2009), an online tool for GO analysis, was used for this analysis. The significant threshold was set at FDR < 0.05.
Data access
The microRNA expression, mRNA expression, and DNA methylation data used in this article are available online in dbGaP (http://www.ncbi.nlm.nih.gov/gap; accession number phs000007).
Authors’ contributions
D.L. and J.M.M. designed, directed, and supervised the project. T. H., G.C., and D.L. drafted the manuscript. M.G.L. directed and supervised the statistical analyses. T.H., C.L., J.R., and A.B. conducted the analyses. J.E.F. and K.T. conducted the miRNA expression experiments. All authors participated in revising and editing the manuscripts. All authors have read and approved the final version of the manuscript.
Funding
The Framingham Heart Study is funded by National Institutes of Health contract N01‐HC‐25195 and HHSN268201500001I. The laboratory work for this investigation was funded by The Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health. The analytical component of this project was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, and The Center for Information Technology, National Institutes of Health, Bethesda, MD and R56AG029451 (M.G.L.). The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Conflict of interests
The authors declare that they have no conflict of interests.
Supporting information
Fig. S1 Analysis flowchart.
Fig. S2 Effect size and P values of differentially expressed miRNAs in relation to chronological age in the discovery and replication sets.
Fig. S3 Effect size and P values of differentially expressed miRNAs in relation to chronological age in the FHS Offspring and Third Generation sets.
Fig. S4 miRNA Δage vs. miRNA age.
Fig. S5 miRNA age vs. chronological age.
Fig. S6 Manhattan plot of genome‐wide associations with miRNA Δage
Fig. S7 Comparison of effect size and P value of differentially expressed miRNAs in relation to chronological age before and after cell type adjustment.
Table S1 Differentially expressed miRNAs in relation to chronological age at Bonferroni.
Table S2 Differentially expressed miRNAs in relation to chronological age in the FH.
Table S3 Differentially expressed miRNAs in relation to chronological age in the FHS Th.
Table S4 miRNA age prediction formula.
Table S5 Gene ontology results.
Table S6 Coexpressed miRNAs and mRNAs in each GO category.
Table S7 Associations of miRNA Δage with prevalent CHD, diabetes, hypertension, and cardiometabolic traits, adjusting for mRNA Δage
Contributor Information
Tianxiao Huan, Email: tianxiao.huan@nih.gov.
Joanne M. Murabito, Email: murabito@bu.edu
Daniel Levy, Email: levyd@nih.gov.
References
- Abdi H (2010) Partial least squares regression and projection on latent structure regression (PLS Regression). Wiley Interdiscip. Rev. Comput. Stat. 2, 97–106. [Google Scholar]
- Agarwal V, Bell GW, Nam J‐W, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasy L, Blangero J (1998) Multipoint quantitative‐trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62, 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm M, Slack F (2005) A developmental timing microRNA and its target regulate life span in C. elegans. Science 310, 1954–1957. [DOI] [PubMed] [Google Scholar]
- Brunner S, Herndler‐Brandstetter D, Arnold CR, Wiegers GJ, Villunger A, Hackl M, Grillari J, Moreno‐Villanueva M, Bürkle A, Grubeck‐Loebenstein B (2012) Upregulation of miR‐24 is associated with a decreased DNA damage response upon etoposide treatment in highly differentiated CD8 + T cells sensitizing them to apoptotic cell death. Aging Cell 11, 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Durik M, Baker DJ, van Deursen JM (2015) Cellular senescence in aging and age‐related disease: from mechanisms to therapy. Nat. Med. 21, 1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, Christensen K (2016) DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell 15, 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes KR, Srivastava D (2009) MicroRNA regulation of cardiovascular development. Circ. Res. 104, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElSharawy A, Keller A, Flachsbart F, Wendschlag A, Jacobs G, Kefer N, Brefort T, Leidinger P, Backes C, Meese E (2012) Genome‐wide miRNA signatures of human longevity. Aging Cell 11, 607–616. [DOI] [PubMed] [Google Scholar]
- Emmrich S, Engeland F, El‐Khatib M, Henke K, Obulkasim A, Schöning J, Katsman‐Kuipers J, Zwaan CM, Pich A, Stary J (2016) miR‐139‐5p controls translation in myeloid leukemia through EIF4G2. Oncogene 35, 1822–1831. [DOI] [PubMed] [Google Scholar]
- Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP (1975) The Framingham offspring study. Design and preliminary data. Prev. Med. 4, 518–525. [DOI] [PubMed] [Google Scholar]
- Fontana L, Partridge L, Longo VD (2010) Extending healthy life span—from yeast to humans. science 328, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R (2010) Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1. [PMC free article] [PubMed] [Google Scholar]
- Fukaya T, Iwakawa H‐O, Tomari Y (2014) MicroRNAs block assembly of eIF4F translation initiation complex in Drosophila. Mol. Cell 56, 67–78. [DOI] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science 333, 1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan J‐B, Gao Y (2013) Genome‐wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C (2007) Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6, 95–110. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–60. [DOI] [PubMed] [Google Scholar]
- Hayes J, Peruzzi PP, Lawler S (2014) MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol. Med. 20, 460–469. [DOI] [PubMed] [Google Scholar]
- Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol. 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Gurven M, Levine ME, Trumble BC, Kaplan H, Allayee H, Ritz BR, Chen B, Lu AT, Rickabaugh TM (2016) An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 17, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T, Liu C, Joehanes R, Zhang X, Chen BH, Johnson AD, Yao C, Courchesne P, O'Donnell CJ, Munson PJ (2015a) A systematic heritability analysis of the human whole blood transcriptome. Hum. Genet. 134, 343–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T, Rong J, Liu C, Zhang X, Tanriverdi K, Joehanes R, Chen BH, Murabito JM, Yao C, Courchesne P (2015b) Genome‐wide identification of microRNA expression quantitative trait loci. Nat. Commun. 6, 6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T, Rong J, Tanriverdi K, Meng Q, Bhattacharya A, McManus DD, Joehanes R, Assimes TL, McPherson R, Samani NJ (2015c) Dissecting the roles of microRNAs in coronary heart disease via integrative genomic analyses. Arterioscler. Thromb. Vasc. Biol. 35, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Ibáñez‐Ventoso C, Yang M, Guo S, Robins H, Padgett RW, Driscoll M (2006) Modulated microRNA expression during adult lifespan in Caenorhabditis elegans. Aging Cell 5, 235–246. [DOI] [PubMed] [Google Scholar]
- Joehanes R, Ying S, Huan T, Johnson AD, Raghavachari N, Wang R, Liu P, Woodhouse KA, Sen SK, Tanriverdi K, Courchesne P, Freedman JE, O'Donnell CJ, Levy D, Munson PJ (2013) Gene expression signatures of coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 33, 1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Ambros V (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science 294, 862–864. [DOI] [PubMed] [Google Scholar]
- Lee R, Feinbaum R, Ambros V (2004) A short history of a short RNA. Cell 116, S89–S92. [DOI] [PubMed] [Google Scholar]
- Leek JT, Storey JD (2007) Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3, e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T (2009) Genome‐wide association study of blood pressure and hypertension. Nat. Genet. 41, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- Li Q‐J, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P (2007) miR‐181a is an intrinsic modulator of T cell sensitivity and selection. Cell 129, 147–161. [DOI] [PubMed] [Google Scholar]
- Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR (2010) MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 34, 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wang Y, Fan H, Zhao X, Liu D, Hu Y, Kidd AR, Bao J, Hou Y (2012) MicroRNA‐181a regulates local immune balance by inhibiting proliferation and immunosuppressive properties of mesenchymal stem cells. Stem Cells 30, 1756–1770. [DOI] [PubMed] [Google Scholar]
- López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez‐Saavedra E, Lamb J, Peck D, Sweet‐Cordero A, Ebert BL, Mak RH, Ferrando AA (2005) MicroRNA expression profiles classify human cancers. nature 435, 834–838. [DOI] [PubMed] [Google Scholar]
- Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR (2015) DNA methylation age of blood predicts all‐cause mortality in later life. Genome Biol. 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H, Fraifeld VE (2013) The role of DNA damage and repair in aging through the prism of Koch‐like criteria. Ageing Res. Rev. 12, 661–684. [DOI] [PubMed] [Google Scholar]
- Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK (2010) microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 5, e10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren Hooten N, Fitzpatrick M, Wood WH 3rd, De S, Ejiogu N, Zhang Y, Mattison JA, Becker KG, Zonderman AB, Evans MK (2013) Age‐related changes in microRNA levels in serum. Aging 5, 725–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P (2007) Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell 6, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MJ, Joehanes R, Pilling LC, Schurmann C, Conneely KN, Powell J, Reinmaa E, Sutphin GL, Zhernakova A, Schramm K (2015) The transcriptional landscape of age in human peripheral blood. Nat. Commun. 6, 8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus Z, Smith‐Vikos T, Slack FJ (2011) MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet. 7, e1002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al‐Qassab H, Carmignac D, Ramadani F (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EM, Olson EN (2011) Pervasive roles of microRNAs in cardiovascular biology. Nature 469, 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith‐Vikos T, Liu Z, Parsons C, Gorospe M, Ferrucci L, Gill TM, Slack FJ (2016) A serum miRNA profile of human longevity: findings from the Baltimore Longitudinal Study of Aging (BLSA). Aging (Albany NY) 8, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, D'Agostino RB, Fox CS, Larson MG, Murabito JM (2007) The third generation cohort of the National Heart, Lung, and Blood Institute's Framingham Heart Study: design, recruitment, and initial examination. Am. J. Epidemiol. 165, 1328–1335. [DOI] [PubMed] [Google Scholar]
- Tomaru U, Takahashi S, Ishizu A, Miyatake Y, Gohda A, Suzuki S, Ono A, Ohara J, Baba T, Murata S (2012) Decreased proteasomal activity causes age‐related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 180, 963–972. [DOI] [PubMed] [Google Scholar]
- Wagschal A, Najafi‐Shoushtari SH, Wang L, Goedeke L, Sinha S, Black JC, Ramírez CM, Li Y, Tewhey R, Hatoum I (2015) Genome‐wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 21, 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yang H, Zhang C, Jing Y, Wang C, Liu C, Zhang R, Wang J, Zhang J, Zen K (2014) Investigation of microRNA expression in human serum during the aging process. J. Gerontol. A Biol. Sci. Med. Sci., 70, 102–109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Analysis flowchart.
Fig. S2 Effect size and P values of differentially expressed miRNAs in relation to chronological age in the discovery and replication sets.
Fig. S3 Effect size and P values of differentially expressed miRNAs in relation to chronological age in the FHS Offspring and Third Generation sets.
Fig. S4 miRNA Δage vs. miRNA age.
Fig. S5 miRNA age vs. chronological age.
Fig. S6 Manhattan plot of genome‐wide associations with miRNA Δage
Fig. S7 Comparison of effect size and P value of differentially expressed miRNAs in relation to chronological age before and after cell type adjustment.
Table S1 Differentially expressed miRNAs in relation to chronological age at Bonferroni.
Table S2 Differentially expressed miRNAs in relation to chronological age in the FH.
Table S3 Differentially expressed miRNAs in relation to chronological age in the FHS Th.
Table S4 miRNA age prediction formula.
Table S5 Gene ontology results.
Table S6 Coexpressed miRNAs and mRNAs in each GO category.
Table S7 Associations of miRNA Δage with prevalent CHD, diabetes, hypertension, and cardiometabolic traits, adjusting for mRNA Δage