

Abstract
Efficient clearance of apoptotic cells, termed efferocytosis, critically regulates normal homeostasis whereas defective uptake of apoptotic cells results in chronic and non-resolving inflammatory diseases, such as advanced atherosclerosis. Monocyte-derived macrophages recruited into developing atherosclerotic lesions initially display efficient efferocytosis and temper inflammatory responses, processes that restrict plaque progression. However, during the course of plaque development, macrophages undergo cellular reprogramming that reduces efferocytic capacity, which results in post-apoptotic necrosis of apoptotic cells and inflammation. Furthermore, defective efferocytosis in advanced atherosclerosis is a major driver of necrotic core formation, which can trigger plaque rupture and acute thrombotic cardiovascular events. In this review, we discuss the molecular and cellular mechanisms that regulate efferocytosis, how efferocytosis promotes the resolution of inflammation, and how defective efferocytosis leads to the formation of clinically dangerous atherosclerotic plaques.
Keywords: efferocytosis, atherosclerosis, inflammation resolution, macrophages, post-apoptotic necrosis
Efficient clearance of apoptotic cells, termed “efferocytosis,” is an ancient process that evolved to allow organ development, maintain homeostasis, prevent autoimmune disease, and resolve inflammatory insults (1). When efferocytosis functions efficiently, apoptotic cells are cleared before they become necrotic, anti-inflammatory cytokines and pro-resolving lipid mediators are secreted, and the release of immunogenic antigens is prevented. However, when efferocytosis becomes defective, uncleared apoptotic cells undergo post-apoptotic necrosis and release tissue-degrading enzymes, immunogenic epitopes, and pro-inflammatory mediators. Genetically modified mice show that impaired efferocytosis often develop autoimmune or chronic inflammatory diseases (2). Accordingly, there is substantial interest in understanding how efferocytosis becomes defective in chronic inflammatory diseases, such as atherosclerosis. This review will highlight the processes associated with efferocytosis and how these become dysregulated during atherosclerosis.
Finding and Binding Apoptotic Cells
Despite the fact that the macrophage population in most organs and tissues are relatively low compared with other non-immune cells, apoptotic cells are rarely detected in tissues where high levels of cellular turnover are known to occur, indicating that macrophages rapidly mobilize to areas of cell death to expeditiously remove apoptotic corpses (3). Macrophage migration toward apoptotic cells is guided by chemotactic factors secreted by dying cells either actively in an executioner caspase-dependent mechanism or passively released during self-demise. This class of mediators, known as “find-me” signals, includes the classic chemokine CX3CL1, the lipids sphingosine 1-phosphate and lysophosphatidylcholine, and the nucleotides ATP and UTP (4–7).
After having navigated tissues to arrive at apoptotic-rich areas, macrophages employ a panoply of receptors that bind either directly or indirectly, via bridging molecules, to “eat-me” signals displayed on the surface of apoptotic cells (Figure 1) (8). While several “eat-me” signals have been identified, including changes in glycosylation at the cell surface or exposure of calreticulin or ICAM-1 epitopes, externalized phosphatidylserine (PtdSer) on apoptotic cells remains the most characterized (9, 10). Macrophages bind PtdSer directly through stabilin-1, stabilin-2, the GPCR brain angiogenesis inhibitor 1 (BAI1), or through the T-cell immunoglobulin and mucin domain family of receptors Tim-1, Tim-3, and Tim-4 (11–14). Alternatively, macrophages may utilize the Tyro3–Axl–Mer (TAM) family of tyrosine kinase receptors, integrins αVβ3 and αVβ5, or CD36 to bind PtdSer indirectly through bridging molecules that interact with PtdSer (3). Gas6 and Protein S bind to TAM receptors, whereas thrombospondin-1 or MFG–E8 link PtdSer to CD36 or integrins αVβ3 and αVβ5, respectively. Some of the PtdSer-relevant receptors have well-characterized signaling capabilities, e.g., MerTK, BAI1, and integrins, while others may function primarily as tethering and adhesion molecules, e.g., the Tim family of receptors and CD36.
Figure 1.
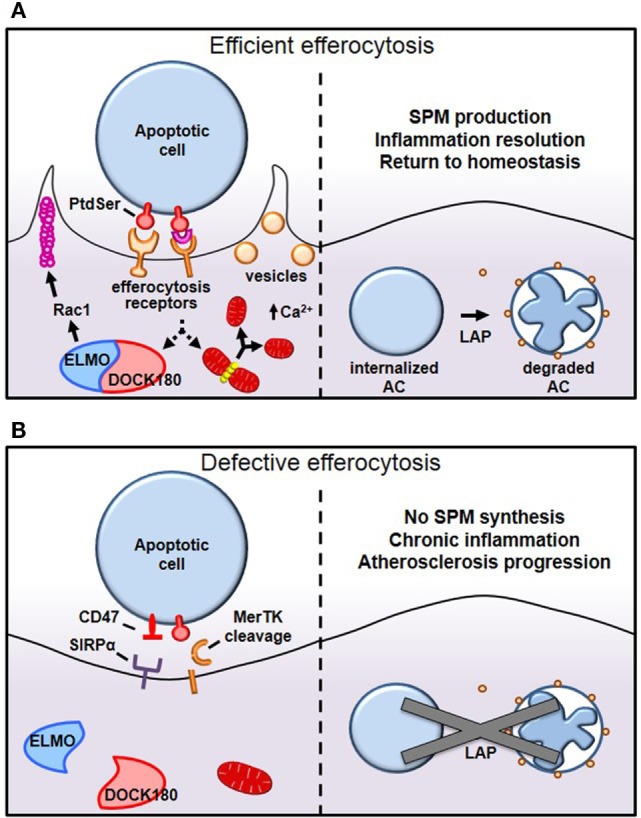
Mechanisms of efferocytosis. (A) Macrophages interact with phosphatidylserine (PtdSer) externalized on apoptotic cells either directly or indirectly, through bridging molecules. Many PtdSer receptors stimulate ELMO–DOCK180 interactions to activate Rac1 and polymerize actin around the phagosome. Simultaneously, macrophages trigger Drp1-mediated mitochondrial fission to drive calcium-dependent vesicular trafficking. Once internalized, autophagic machinery is used to conjugate lipids to LC3 bound to phagosomes, which drives phagolysosomal fusion and subsequent apoptotic cell degradation. (B) In pathological settings such as advanced atherosclerosis, one or more of these processes can become defective, leading to inefficient clearance of apoptotic cells and subsequent necrosis and inflammation. For example, in advanced atherosclerosis, apoptotic cells can inappropriately express the “don’t eat-me” signal CD47, or cell-surface receptors can get proteolytically cleaved, such as with MerTK.
Tethering and internalization are two separate but intimately linked events and operate first through interaction with weak and low avidity “eat-me” signals to ensure adhesion, followed by stereospecific interaction of externalized PtdSer to PtdSer receptors to drive cytoskeletal reorganization around the apoptotic cell. While externalized PtdSer on apoptotic cells binds to receptors on macrophages to mediate tethering, this process alone is insufficient to trigger internalization (15). However, coupling of PtdSer/receptor interaction with other receptors trigger the switch from adhesion to internalization, a process referred to as “tether and tickle” (16). Live cells may also express PtdSer and yet are spared from efferocytosis, primarily because live cells present the “don’t eat-me” signals CD31 and CD47 on their cell surface, which actively suppresses efferocytosis. CD31 is expressed on viable cells and cues repulsion or detachment when making homophilic interactions in trans with efferocytes (17). Interestingly, CD31 on macrophages may interact with apoptotic cells using the extracellular matrix protein fibronectin as a bridging molecule (18). When this occurs, integrin α5β1 becomes activated and subsequently promotes phagocytosis of apoptotic cells (18). Therefore, selective CD31 interactions not only prevents accidental internalization of viable cells but may also actively promote efferocytosis (Figure 1B). The other major “don’t eat-me” signal, CD47, is expressed on live cells and interacts with cell-surface signal-regulatory protein α on macrophages to inactivate myosin assembly and thereby prevent cytoskeletal rearrangement around the phagosome (19).
Internalization of apoptotic cells requires macrophages to dynamically reorganize their actin cytoskeleton to drive F-actin formation around apoptotic cells, forming a so-called phagocytic cup, which then promotes mechanical retraction of the phagosome into the cell (Figure 1A) (20). Since the Rho-family of small GTPases, Rac1, Cdc42, and RhoA rearrange the cytoskeleton to mechanically drive migration of cells, it is not surprising that they are also involved in mechanically internalizing phagosomes containing apoptotic cells. Using Forster resonance energy transfer biosensors, it was discovered that these small GTPase family members work in a temporally regulated fashion in which Rac1 and Cdc42 are activated early to facilitate phagocytic cup formation through actin polymerization followed by Rho activation, which drives mechanical retraction and phagosome internalization (21). Constitutive activation of Rac1 may decrease phagocytosis, because Rac1 must be rapidly inactivated to permit engulfment (21). However, when RhoA effectors are inhibited, to tilt the Rac1/RhoA axis toward Rac1 activation, uncontrolled phagocytosis occurs (22). A shared Rac1 activation pathway that is conserved among several of the apoptotic cell receptors involves association with the adaptor protein ELMO to the RacGEF DOCK180 (23). This ELMO/DOCK180 complex then activates Rac1 to initiate phagocytic cup formation, which then leads to apoptotic cell internalization (24). Accordingly, inhibition of ELMO/DOCK180 signaling prevents efferocytosis (13, 24).
While cytoskeletal remodeling is required for apoptotic cell internalization, the role of membrane trafficking in efferocytosis becomes astoundingly evident given that cells internalize ~50% of their entire surface, yet, plasma membrane surface area does not change as a result of phagocytosis (25). This finding suggests that internal membranes are rapidly recruited to the cell surface to complete closure of the phagosome and to replenish cell membranes utilized during efferocytosis. This concept can be demonstrated in tetanus or botulinum B toxin-microinjected cells, which show defective exocytosis and reduced membrane delivery to incoming phagosomes (26). New work has shown that these critical vesicular trafficking events require efferocytes to undergo Drp1-dependent mitochondrial fission (27). Mitochondrial fission causes endoplasmic reticulum calcium to be released into the cytosol rather than into the mitochondria, and this increase in cytosolic calcium then drives vesicular trafficking (27). Importantly, Drp1-deficient macrophages, which cannot undergo mitochondrial fission upon encountering apoptotic cells, are unable to move vesicles to the site of apoptotic cell attachment, which significantly delays both initial apoptotic cell phagosome sealing and, more notably, compromises the ability of the efferocytes to take up a second apoptotic cell (27).
AC Corpse Degradation
Once apoptotic cells have been internalized, certain autophagy-related proteins are recruited to conjugate LC3-family proteins to lipids at the phagosome membrane, a process called LC3-associated phagocytosis (LAP) (Figure 1A) (28). LAP promotes phagosome fusion to lysosomes to drive hydrolytic degradation of apoptotic cell constituents (28). Importantly, defects in LC3 conjugation to phagosomal membranes delay or even prevent phagosome fusion with lysosomes, resulting in failure to acidify the phagosome and an inability to degrade apoptotic cells (29). After apoptotic cells are degraded in phagolysosomes, macrophages become overloaded with macromolecular constituents and therefore have evolved elegant mechanisms to either use or efflux this cargo. For instance, the burden of cholesterol from degraded apoptotic cells activates members of the peroxisome proliferator-activated receptor (PPAR) and liver X receptor (LXR) families of nuclear receptors and drive ABCA1 and ABCG1 expression, which mediate cholesterol efflux from the cells (30). Furthermore, PPARγ and LXR agonists further enhance efferocytosis (31, 32). To handle the large amount of chromosomal DNA derived from degraded apoptotic cells, macrophage lysosomes contain DNase II that cleaves this phagocytosed DNA. Macrophages lacking DNase II accumulate undigested DNA fragments, and mice lacking DNase II exhibit polyarthritis, an autoimmune disease similar to rheumatoid arthritis in humans (33).
Atherosclerosis
Although the last several decades have seen significant medical advances in the diagnosis and treatment of cardiovascular disease, atherosclerosis remains the major cause of morbidity and mortality worldwide (34). Atherosclerosis begins when circulating apolipoprotein B-containing lipoproteins accumulate in focal areas in the subendothelium matrix of medium-sized and large arteries (35). These subendothelial lipoproteins, particularly after oxidation, generate an inflammatory stimulus that drives leukocyte influx into the vessel wall (36–39). Primary among these infiltrating cells are monocyte-derived macrophages, which internalize cholesterol-rich lipoproteins and give rise to foam cells. Foam cells secrete extracellular matrix that further promotes lipoprotein retention as well as pro-inflammatory cytokines that augment the recruitment of additional monocytes, T cells, and neutrophils. In the face of persistent inflammatory stimuli and other cytotoxic factors, many lesional cells become apoptotic. Early on, apoptotic cells are efficiently cleared by neighboring macrophages in an attempt to limit overall lesion cellularity (40). However, efferocytosis can fail as plaques progress, leading to the accumulation of secondarily necrotic cells and the formation of a highly inflammatory “necrotic core” (41–43). Large necrotic cores are a hallmark of advanced atherosclerotic disease and have been associated with the types of atherosclerotic plaque that give rise to heart attack and stroke (44, 45). Therefore, the efficient clearance of dead and dying cells plays a key role in preventing the development of clinically significant atherosclerotic plaques.
Mechanisms of Impaired Efferocytosis in Atherosclerosis
Why does efferocytosis fail in advanced atherosclerosis? Because efferocytosis is a high-capacity process, it is unlikely that overwhelming lesional apoptosis is the primary cause. Rather, efferocytosis itself becomes defective and/or lesional apoptotic cells become poor substrates for efferocytosis. As an example of the latter, CD47 expression is significantly increased in human atherosclerotic plaque cells, presumably via a TNFα-dependent mechanism, and for the reasons explained earlier (19), these cells are poorly internalized by lesional efferocytes (46) (Figure 1B). In keeping with this concept, administration of CD47-blocking antibodies to atheroprone mice led to improved lesional efferocytosis and smaller necrotic cores. Other findings suggest that dead cells in lesions express lower amounts of the “eat-me” signal calreticulin (47). For example, Apoe−/− mice lacking Cdkn2b show decreased levels of calreticulin, and apoptotic bodies from these animals show resistance to being engulfed (47). When fed a Western diet, these mice have an increased overall lesion size as well as increased necrotic core size (47) (Table 1). Interestingly, human carriers of the cardiovascular risk allele at the chromosome 9p21 GWAS locus were found to have lower intraplaque expression of calreticulin, suggesting that defective efferocytosis may contribute to cardiovascular disease in these patients (48).
Table 1.
Efferocytosis pathway molecules shown to have a causative role in atherosclerosis.
| Molecule | Function | Animal model | Effect on aortic lesion area | Effect on necrotic core size | Other findings | Reference |
|---|---|---|---|---|---|---|
| MerTK | Receptor | ↑ ACs | ||||
| ↓ In situ efferocytosis | ||||||
| MerTKKD Apoe−/− mice | No change | ↑ | ↑ Lesional macrophages | (49) | ||
| MerTK−/− marrow → Ldlr−/− mice | ↑ | ↑ | ↓ In situ efferocytosis | (50) | ||
| MerTKCR Ldlr−/− mice | No change | ↓ | ↑ Collagen cap thickness | (51) | ||
| ↑ T regulatory cells | ||||||
| ↑ Specialized pro-resolving mediators | ||||||
| Lipoprotein receptor-related protein 1 (LRP1) | Receptor | Macrophage LRP1−/− marrow → Ldlr−/− mice Macrophage LRP1−/− Apoe−/− mice |
↑ | ↑ | ↑ ACs ↓ In situ efferocytosis ↑ Lesional macrophages ↑ MMP9 levels |
(52, 53) |
| ↑ | ↑ | ↑ ACs ↓ In situ efferocytosis ↑ Lesional macrophages |
(54) | |||
| SR-B1 | Receptor | SR-B1−/− ApoE−/− marrow → Apoe−/− mice SR-B1−/− marrow → Ldlr−/− mice |
↑ | ↑ | ↑ ACs ↓ In situ efferocytosis ↓ Lesional macrophages ↓ Collagen lesion area and cap thickness ↑ ACs ↓ In situ efferocytosis ↓ Lesional macrophages ↓ Collagen lesion area and cap thickness |
(55) |
| Tim-1/Tim-4 | Receptor | Ldlr−/− treated with Tim-1 or Tim-4 blocking antibodies | ↑ | Not tested | ↑ ACs ↓ In situ efferocytosis ↑ Lesional T cells |
(56) |
| Mineralo-corticoid receptor | Non-efferocytosis nuclear receptor | Myeloid MRKO−/− marrow → Ldlr−/− mice | ↓ | ↓ | ↓ ACs ↑ In situ efferocytosis ↓ Lesional macrophages ↓ Foam cell formation ↑ Collagen lesion area |
(57) |
| MFG-E8 | Bridging molecule | MFG-E8−/− marrow → Ldlr−/− mice | ↑ | ↑ | ↑ ACs ↑ Collagen cap thickness |
(58) |
| C1q | Bridging molecule | C1q−/− Ldlr−/− | ↑ | Not tested | ↑ ACs ↑ Lesional macrophages In vitro: incubating macrophage cell line with C1q enhances C19-mediated efferocytosis |
(59) |
| (60) | ||||||
| Transglutaminase 2 (TG2) | Bridging molecule | TG2−/− marrow → Ldlr−/− mice | ↑ | ↑ | ↑ Lesional macrophages In vitro: TG2−/− macrophages have decreased efferocytosis |
(61) |
| Gas6 | Bridging molecule | Gas6−/− Apoe−/− mice | No change | ↓ | ↑ Collagen content | (62) |
| CX3CL1 | Find-me signal | CX3CL1−/− Apoe−/− mice CX3CL1−/− Ldlr−/− mice |
Inconsistent change at aortic root, ↓ at brachiocephalic artery ↓At aortic root and brachiocephalic artery |
Not tested Not tested |
↓ Lesional macrophages ↓ Lesional macrophages |
(63) |
| Fas/Fas ligand | Mediates find-me signaling | Fas−/− Apoe−/− mice Gld Apoe−/− mice |
↑ ↑ |
Not tested Not tested |
↑ ACs ↑ ACs ↑ Lesional macrophages ↑ T cells ↓ In situ efferocytosis in lymph tissue |
(64) |
| (65) | ||||||
| Calreticulin | Eat-me signal | Cdkn2b−/− Apoe−/− mice have low levels of calreticulin | ↑ | ↑ | ↓ Collagen content and cap thickness ↓ In situ efferocytosis by semi-quantitative measure In vitro: loss of CKDN2B leads to ↓ efferocytosis |
(47) |
| CD47 | Don’t eat-me signal | Apoe−/− mice treated with CD47-blocking antibody | ↓ | ↓ | ↓ ACs ↑ In situ efferocytosis |
(46) |
| miR-21 | MicroRNA | miR21−/− marrow → Ldlr−/− mice | ↑ | ↑ | ↑ ACs ↓ Lesional macrophages ↓ Collagen cap area with no change in lesional collagen content |
(66) |
| In vitro: miR21−/− macrophages have ↓MerTK and ↓ efferocytosis | (66, 67) | |||||
Efferocytosis may also be compromised by competition for apoptotic cell binding. As atherosclerosis progresses, lesions continue to accumulate lipids and ROS, leading to increased levels of oxidized phospholipids. These lipids can bind to efferocytosis receptors and may compete for apoptotic cell recognition (68). Similarly, autoantibodies against oxLDL and other oxidized phospholipids are able to bind to and potentially mask “eat-me” ligands on the surface of dying cells in the lesions (69, 70). Further, oxLDL increases the expression of and signaling through toll-like receptor 4 (TLR4), leading to increased secretion of the pro-atherogenic cytokines TNFα and IL-1β while reducing the anti-inflammatory cytokines TGFβ and IL-10 (71). This pro-inflammatory environment impairs efferocytosis by reducing the expression of various key efferocytosis molecules, as discussed below, and promotes increased lipid uptake at the expense of phagocytosis (72).
Finally, accumulating experimental evidence has demonstrated that the expression and function of efferocytosis receptors and their bridging molecules are deficient in advanced atherosclerosis. One such example is MerTK and its associated bridging molecule, Gas6. MerTK is expressed by macrophages in both murine and human plaques (50, 51). As lesions progress, MerTK levels on the macrophage surface decline, and this decrease is associated with cleavage of the receptor by the metalloproteinase ADAM17 (Figure 1B). Multiple athero-relevant inflammatory stimuli have been shown to promote ADAM17 activity and MerTK cleavage (73–75). Indeed, levels of the soluble fragment of the receptor (solMer) accumulate within the aortas of mice and in human carotid plaques (51). In vitro, solMer has been shown to inhibit efferocytosis by competing with Gas6, suggesting that this may amplify the deleterious effects on efferocytosis (73). Using a genetically engineered mouse in which the cleavage domain of MerTK has been rendered resistant, Cai and colleagues demonstrated that cleavage of the MerTK receptor is a causal factor in the development of necrotic cores in atherosclerotic lesions (51). Consistent with this protective role for MerTK activity in atherosclerosis, loss of MerTK, either by genetic deletion or through models in which MerTK has been replaced by a version with an inactive kinase domain, results in increased lesion size and larger necrotic cores (49, 50) (Table 1). Interestingly, deletion of the related TAM family member, Axl, in bone marrow cells of Ldlr−/− mice did not affect lesional efferocytosis or plaque necrosis in advanced atherosclerosis (76).
Low-density lipoprotein receptor-related protein 1 (LRP1) is a receptor that is activated by calreticulin on the surface of apoptotic cells (77). The macrophage receptor LRP1 can also be downregulated in response to TLR4 signaling and inactivated by ADAM17-mediated proteolytic cleavage (78, 79). Several studies have demonstrated that the loss of Lrp1 in macrophages or in bone marrow cells leads to increased lesion area and necrotic core size in an Apoe−/− mice (52–54) (Table 1). A particular bridging molecule that is reduced in atherosclerotic lesions is milk fat globule-epidermal growth factor 8 (MFG-E8), which functions to tether apoptotic cells to efferocytes by interacting with αVβ3 integrins and the transglutaminase 2 (TG2) co-receptor on phagocytes (80, 81). MFG-E8 is expressed in atherosclerotic plaques, but its expression declines in advanced plaque, potentially owing to downregulation by inflammatory stimuli (82). In an Ldlr−/− mouse model lacking Mfge8 in bone marrow cells, larger plaque area and necrotic cores were observed (58). In addition, Ldlr−/− mice lacking TG2 in bone marrow also show increased plaque area and necrotic core size (81). Another bridging molecule, complement component 1q (C1q), has also been shown to be important in atherosclerosis. In vitro, macrophages can produce large amounts of C1q, which promotes both cell survival and efferocytosis (60). Loss of C1q from Ldlr−/− mice led to larger lesion area and an increase in apoptotic cells, consistent with defective apoptotic cell clearance (59). As another possible mechanism for defective efferocytosis, the pro-inflammatory molecule high-mobility group box 1 (HMGB1) is increased in human and murine atherosclerosis (83, 84), and the secreted form has been shown to interact with integrin αVβ3 and PtdSer to block efferocytosis (85, 86). Apoe−/− mice administered an anti-HMGB1 antibody developed smaller atherosclerotic plaques, although necrotic core size was not reported (84). Silencing of HMGB1 in peritoneal macrophages in vitro leads to increased efferocytosis, and partially rescues the efferocytosis defect observed in SR-B1−/− macrophages (55).
Recently, microRNAs have been found to have a novel role in the regulation of efferocytosis. Das and colleagues found that macrophages undergoing efferocytosis increase their expression of miR-21 in a TLR4-dependent manner in vitro (67). Further, when treated with an miR-21 mimetic in vitro, the rate of efferocytosis by bone marrow-derived macrophages increased (67). Transplantation of miR21−/− marrow into Ldlr−/− mice increased plaque area and necrotic core size. One study reported that loss of miR-21 in macrophages decreases MerTK expression, providing a mechanism for the increased necrotic core size in these mice (66). Additional work is necessary to determine the mechanism by which miR-21 regulates MerTK expression. Another miR, miR-33, is also known to regulate the outcome of atherosclerosis. Murine primary macrophages treated with anti-miR-33 enhanced efferocytosis in vitro and treatment of Ldlr−/− mice with anti-miR-33 decreased necrotic cores compared with the anti-miR control (87). Together, these results suggest that specific miRs play important roles in regulating efferocytosis in atherosclerosis.
Efferocytosis and Inflammation Resolution
To successfully terminate an inflammatory process, the active process of inflammation resolution is required (88). This process is mediated by various endogenous molecules, including bioactive lipids such as lipoxins, resolvins, protectins, and maresins, which are often referred to as specialized pro-resolving mediators (SPMs); proteins such as annexin A1 and interleukin-10; and gasses such as hydrogen sulfide (88). When the resolution program fails inflammation persists, and this mechanism is now understood to be an underlying factor in the pathogenesis of many chronic inflammatory diseases, including atherosclerosis (89). Emerging evidence has defined an important role for resolution and SPMs in both murine and human atherosclerotic disease (89–91). Gene variants encoding proteins and enzymes necessary for SPM biosynthesis, including 5-lipoxygenase (5-LOX), have been associated with atherosclerosis, stroke, and myocardial infarction in selected populations (92–96). Patients with coronary artery disease have lower plasma SPMs than do healthy patients (97), and one SPM, aspirin-triggered lipoxin A4, was found to be significantly associated with peripheral and coronary atherosclerosis in humans even after correction for age, sex, and C-reactive protein levels (98). A recent paper showed that stable regions of human atherosclerotic plaque have a higher SPM:leukotriene ratio when compared with more advanced, vulnerable regions having larger necrotic cores and thinner collagen caps (99). Similarly, early murine lesions from Western diet-fed Ldlr−/− mice had a higher SPM:leukotriene ratio when compared with advanced lesions (90, 99). In several mouse models of atherosclerosis, treatment of mice with various pro-resolving ligands including annexin 1, Ac2-26, IL-10, resolvin D1 (RvD1), resolvin D2, or maresin 1 decreased lesional necrosis, suggesting improvements in efferocytosis by lesional phagocytes (90, 99, 100–102).
Efferocytosis plays a major mechanistic role in the resolution of inflammation. First, expeditious clearance of dead cells prevents their secondary necrosis. Second, the act of efferocytosis itself triggers several different anti-inflammatory and pro-resolving signaling pathways. Engagement and activation of the TAM family of efferocytosis receptors, including MerTK and Axl, induces the expression of suppressor of cytokine signaling-1 and 3 (SOCS-1 and 3), leading to the inhibition of signaling pathways triggered by cytokines and toll-like receptor ligands (103, 104). Efferocytosis has also been shown to actively increase the secretion of anti-inflammatory cytokines, including TGF-β and IL-10, and decreased secretion of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-8 (105, 106). Further, uptake of apoptotic cells enhances the synthesis of SPMs, while concomitantly reducing the production of pro-inflammatory leukotrienes (107, 108). Recently, a specific mechanistic link between the efferocytosis receptor MerTK and SPM production was revealed (108). In response to engagement of the MerTK receptor, the key biosynthetic enzyme 5-LOX translocates from the nucleus to the cytoplasm, where it drives the production of the pro-resolving mediator lipoxin A4. When MerTK is inactivated either genetically or proteolytically, 5-LOX is restricted to the nuclear membrane, where it instead favors the production of the pro-inflammatory leukotriene B4 (108). Mice whose myeloid cells express a cleavage-resistant variant of MerTK (MerTKCR mice) have higher rates of efferocytosis than their wild type counterparts, and macrophages from these mice demonstrate enhanced production of LXA4 and RvD1. In addition, when Ldlr−/− mice are transplanted with bone marrow from MerTKCR mice and fed an atherogenic diet for 16 weeks, the aortas contained an increased SPM:leukotriene ratio (51). The process of resolution in atherosclerosis can also enhance efferocytosis. A recent study showed that administration of RvD1 to Western diet-fed Ldlr−/− mice significantly increased the SPM:leukotriene ratio, while also decreasing plaque necrosis and enhancing lesional efferocytosis (99). These studies suggest a positive feedback cycle between resolution and efferocytosis, which, if interrupted, can lead to an amplification loop of inflammation and necrosis that promotes advanced atherosclerotic plaque progression.
Summary and Conclusion
Defective clearance of apoptotic cells in atherosclerotic lesions drives post-apoptotic necrosis of lesional cells and inflammation triggered by the release of cellular debris from these necrotic cells (2). Moreover, active cell signaling programs of inflammation suppression and inflammation resolution in efferocytes are often lost when apoptotic cells are not properly cleared (89). As a result, defective efferocytosis can transform stable, asymptomatic atherosclerotic lesions into necrotic, inflammatory, and non-resolving plaques that are prone to rupture (41). Although we do not know for certain why efferocytosis fails in advanced atherosclerosis, studies thus far suggest complementary mechanisms that involve both poor recognition of lesional apoptotic cells, e.g., due to inappropriate expression of CD47, coupled with defects in the efferocytes themselves; e.g., due to proteolytically cleavage of MerTK (46, 51, 108).
How might this knowledge suggest new types of therapies to prevent atherothrombotic vascular disease? Therapies that lower LDL in the blood are the mainstay of therapy to prevent atherosclerotic disease, and there is reason to posit that this type of therapy can indirectly prevent processes in plaques, such as inflammation and oxidative stress, that may ultimately contribute to defective efferocytosis. However, to the extent that many subjects at risk are not able to lower their LDL to a level low enough to completely suppress atherosclerotic disease, there is a place for complementary therapies (109). For example, recent success of the CANTOS trial demonstrated that lowering inflammation, through administering an anti-IL-1β antibody, successfully reduced recurrent cardiovascular events independently of lipid lowering (110). One type of new approach that may successfully target defective efferocytosis is antibodies that block CD47. However, anti-CD47 antibodies also causes anemia owing to inappropriate clearance of red blood cells (46, 111, 112). Another type of approach is to enhance the function of efferocytes themselves by preventing proteolysis of efferocytosis receptors, e.g., by blocking ADAM17-mediated cleavage of MerTK, or by enhancing the ability of efferocytes to clear multiple apoptotic cells, e.g., by boosting the mitochondrial fission-calcium mechanism that enables macrophages to efficiently ingest secondarily encountered apoptotic cells (27, 51, 108). Yet another approach would be tilting the SPM:leukotriene ratio to favor SPM production, such as through the administration of RVD1, which has been shown to enhance macrophage–apoptotic cell interactions and to increase lesional efferocytosis (51, 99). Finally, glucocorticoids generate anti-inflammatory molecules and are therefore routinely used for managing inflammatory diseases. One such glucocorticoid product, annexin A1, enhances efferocytosis, resolves inflammation, and delays atherosclerosis in mice (113–115). Indeed, the combination enhancing efferocytosis while at the same time restoring resolution mediators in lesions may offer the most promising therapeutic strategy to combat atherosclerotic cardiovascular disease.
Author Contributions
All authors contributed to drafting and editing the review. AY designed the graphic in Figure 1 and AD designed Table 1.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This review was supported by the American Federation for Aging Research, Research Grant for Junior Faculty (GF, A16034); American Heart Association Fellow-to-Faculty grant (AD, 17FTF33660643) and Post-Doctoral Fellowship grant (BC, 15POST25620024); and NIH grants T32 HL007343-28 (AY), R00 HL119587 (GF), and R01s HL075662, HL127464, and HL132412 (IT).
References
- 1.Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol (2015) 16(9):907–17. 10.1038/ni.3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol (2014) 14(3):166–80. 10.1038/nri3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol (2013) 5(1):a008748. 10.1101/cshperspect.a008748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood (2008) 112(13):5026–36. 10.1182/blood-2008-06-162404 [DOI] [PubMed] [Google Scholar]
- 5.Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J (2008) 22(8):2629–38. 10.1096/fj.08-107169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mueller RB, Sheriff A, Gaipl US, Wesselborg S, Lauber K. Attraction of phagocytes by apoptotic cells is mediated by lysophosphatidylcholine. Autoimmunity (2007) 40(4):342–4. 10.1080/08916930701356911 [DOI] [PubMed] [Google Scholar]
- 7.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature (2009) 461(7261):282–6. 10.1038/nature08296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W. Eat-me signals: keys to molecular phagocyte biology and “appetite” control. J Cell Physiol (2012) 227(4):1291–7. 10.1002/jcp.22815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol (1992) 148(7):2207–16. [PubMed] [Google Scholar]
- 10.Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem (2001) 276(2):1071–7. 10.1074/jbc.M003649200 [DOI] [PubMed] [Google Scholar]
- 11.Park SY, Jung MY, Lee SJ, Kang KB, Gratchev A, Riabov V, et al. Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J Cell Sci (2009) 122(Pt 18):3365–73. 10.1242/jcs.049569 [DOI] [PubMed] [Google Scholar]
- 12.Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ (2008) 15(1):192–201. 10.1038/sj.cdd.4402242 [DOI] [PubMed] [Google Scholar]
- 13.Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature (2007) 450(7168):430–4. 10.1038/nature06329 [DOI] [PubMed] [Google Scholar]
- 14.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev (2010) 235(1):172–89. 10.1111/j.0105-2896.2010.00903.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci U S A (2011) 108(48):19246–51. 10.1073/pnas.1114799108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, et al. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol (2001) 155(4):649–59. 10.1083/jcb.200108080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature (2002) 418(6894):200–3. 10.1038/nature00811 [DOI] [PubMed] [Google Scholar]
- 18.Vernon-Wilson EF, Auradé F, Brown SB. CD31 promotes beta1 integrin-dependent engulfment of apoptotic Jurkat T lymphocytes opsonized for phagocytosis by fibronectin. J Leukoc Biol (2006) 79(6):1260–7. 10.1189/jlb.1005571 [DOI] [PubMed] [Google Scholar]
- 19.Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol (2008) 180(5):989–1003. 10.1083/jcb.200708043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castellano F, Montcourrier P, Chavrier P. Membrane recruitment of Rac1 triggers phagocytosis. J Cell Sci (2000) 113(Pt 17):2955–61. [DOI] [PubMed] [Google Scholar]
- 21.Nakaya M, Kitano M, Matsuda M, Nagata S. Spatiotemporal activation of Rac1 for engulfment of apoptotic cells. Proc Natl Acad Sci U S A (2008) 105(27):9198–203. 10.1073/pnas.0803677105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SY, Kim S, Bae DJ, Park SY, Lee GY, Park GM, et al. Coordinated balance of Rac1 and RhoA plays key roles in determining phagocytic appetite. PLoS One (2017) 12(4):e0174603. 10.1371/journal.pone.0174603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu M, Ravichandran KS. Dock180-ELMO cooperation in Rac activation. Methods Enzymol (2006) 406:388–402. 10.1016/S0076-6879(06)06028-9 [DOI] [PubMed] [Google Scholar]
- 24.Albert ML, Kim JI, Birge RB. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol (2000) 2(12):899–905. 10.1038/35046549 [DOI] [PubMed] [Google Scholar]
- 25.Mellman IS, Plutner H, Steinman RM, Unkeless JC, Cohn ZA. Internalization and degradation of macrophage Fc receptors during receptor-mediated phagocytosis. J Cell Biol (1983) 96(3):887–95. 10.1083/jcb.96.3.887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hackam DJ, Rotstein OD, Sjolin C, Schreiber AD, Trimble WS, Grinstein S. v-SNARE-dependent secretion is required for phagocytosis. Proc Natl Acad Sci U S A (1998) 95(20):11691–6. 10.1073/pnas.95.20.11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Subramanian M, Yurdagul A, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, et al. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell (2017) 171:331–45.e22. 10.1016/j.cell.2017.08.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol (2015) 17(7):893–906. 10.1038/ncb3192 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A (2011) 108(42):17396–401. 10.1073/pnas.1113421108 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Kidani Y, Bensinger SJ. Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol Rev (2012) 249(1):72–83. 10.1111/j.1600-065X.2012.01153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity (2009) 31(2):245–58. 10.1016/j.immuni.2009.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lea S, Plumb J, Metcalfe H, Spicer D, Woodman P, Fox JC, et al. The effect of peroxisome proliferator-activated receptor-γ ligands on in vitro and in vivo models of COPD. Eur Respir J (2014) 43(2):409–20. 10.1183/09031936.00187812 [DOI] [PubMed] [Google Scholar]
- 33.Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature (2006) 443(7114):998–1002. 10.1038/nature05245 [DOI] [PubMed] [Google Scholar]
- 34.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics – 2015 update: a report from the American Heart Association. Circulation (2015) 131(4):e29–322. 10.1161/CIR.0000000000000152 [DOI] [PubMed] [Google Scholar]
- 35.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol (1995) 15(5):551–61. 10.1161/01.ATV.15.5.551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yurdagul A, Orr AW. Blood brothers: hemodynamics and cell-matrix interactions in endothelial function. Antioxid Redox Signal (2016) 25:415–34. 10.1089/ars.2015.6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yurdagul A, Finney AC, Woolard MD, Orr AW. The arterial microenvironment: the where and why of atherosclerosis. Biochem J (2016) 473(10):1281–95. 10.1042/BJ20150844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yurdagul A, Sulzmaier FJ, Chen XL, Pattillo CB, Schlaepfer DD, Orr AW. Oxidized LDL induces FAK-dependent RSK signaling to drive NF-κB activation and VCAM-1 expression. J Cell Sci (2016) 129(8):1580–91. 10.1242/jcs.182097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yurdagul A, Green J, Albert P, McInnis MC, Mazar AP, Orr AW. α5β1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol (2014) 34(7):1362–73. 10.1161/ATVBAHA.114.303863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol (2005) 25(11):2255–64. 10.1161/01.ATV.0000184783.04864.9f [DOI] [PubMed] [Google Scholar]
- 41.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol (2010) 10(1):36–46. 10.1038/nri2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol (2005) 25(6):1256–61. 10.1161/01.ATV.0000166517.18801.a7 [DOI] [PubMed] [Google Scholar]
- 43.Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J (2016) 80(11):2259–68. 10.1253/circj.CJ-16-0924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med (2015) 278(5):483–93. 10.1111/joim.12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yahagi K, Kolodgie FD, Otsuka F, Finn AV, Davis HR, Joner M, et al. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat Rev Cardiol (2016) 13(2):79–98. 10.1038/nrcardio.2015.164 [DOI] [PubMed] [Google Scholar]
- 46.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature (2016) 536(7614):86–90. 10.1038/nature18935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kojima Y, Downing K, Kundu R, Miller C, Dewey F, Lancero H, et al. Cyclin-dependent kinase inhibitor 2B regulates efferocytosis and atherosclerosis. J Clin Invest (2014) 124(3):1083–97. 10.1172/JCI70391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nanda V, Downing KP, Ye J, Xiao S, Kojima Y, Spin JM, et al. CDKN2B regulates TGFbeta signaling and smooth muscle cell investment of hypoxic neovessels. Circ Res (2016) 118(2):230–40. 10.1161/CIRCRESAHA.115.307906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol (2008) 28(8):1421–8. 10.1161/ATVBAHA.108.167197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol (2008) 28(8):1429–31. 10.1161/ATVBAHA.108.169078 [DOI] [PubMed] [Google Scholar]
- 51.Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B, et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest (2017) 127:564–8. 10.1172/JCI90520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, et al. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and Akt activation. Arterioscler Thromb Vasc Biol (2010) 30(4):787–95. 10.1161/ATVBAHA.109.202051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ Res (2007) 100(5):670–7. 10.1161/01.RES.0000260204.40510.aa [DOI] [PubMed] [Google Scholar]
- 54.Yancey PG, Ding Y, Fan D, Blakemore JL, Zhang Y, Ding L, et al. Low-density lipoprotein receptor-related protein 1 prevents early atherosclerosis by limiting lesional apoptosis and inflammatory Ly-6Chigh monocytosis: evidence that the effects are not apolipoprotein E dependent. Circulation (2011) 124(4):454–64. 10.1161/CIRCULATIONAHA.111.032268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tao H, Yancey PG, Babaev VR, Blakemore JL, Zhang Y, Ding L, et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res (2015) 56(8):1449–60. 10.1194/jlr.M056689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foks AC, Engelbertsen D, Kuperwaser F, Alberts-Grill N, Gonen A, Witztum JL, et al. Blockade of Tim-1 and Tim-4 enhances atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol (2016) 36(3):456–65. 10.1161/ATVBAHA.115.306860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen ZX, Chen XQ, Sun XN, Sun JY, Zhang WC, Zheng XJ, et al. Mineralocorticoid receptor deficiency in macrophages inhibits atherosclerosis by affecting foam cell formation and efferocytosis. J Biol Chem (2017) 292(3):925–35. 10.1074/jbc.M116.739243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ait-Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, et al. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation (2007) 115(16):2168–77. 10.1161/CIRCULATIONAHA.106.662080 [DOI] [PubMed] [Google Scholar]
- 59.Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol (2007) 170(1):416–26. 10.2353/ajpath.2007.060406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pulanco MC, Cosman J, Ho MM, Huynh J, Fing K, Turcu J, et al. Complement protein C1q enhances macrophage foam cell survival and efferocytosis. J Immunol (2017) 198(1):472–80. 10.4049/jimmunol.1601445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boisvert WA, Rose DM, Boullier A, Quehenberger O, Sydlaske A, Johnson KA, et al. Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler Thromb Vasc Biol (2006) 26(3):563–9. 10.1161/01.ATV.0000203503.82693.c1 [DOI] [PubMed] [Google Scholar]
- 62.Lutgens E, Tjwa M, Garcia de Frutos P, Wijnands E, Beckers L, Dahlback B, et al. Genetic loss of Gas6 induces plaque stability in experimental atherosclerosis. J Pathol (2008) 216(1):55–63. 10.1002/path.2381 [DOI] [PubMed] [Google Scholar]
- 63.Teupser D, Pavlides S, Tan M, Gutierrez-Ramos JC, Kolbeck R, Breslow JL. Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc Natl Acad Sci U S A (2004) 101(51):17795–800. 10.1073/pnas.0408096101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng X, Li H, Rumbin AA, Wang X, La Cava A, Brechtelsbauer K, et al. ApoE-/-Fas-/- C57BL/6 mice: a novel murine model simultaneously exhibits lupus nephritis, atherosclerosis, and osteopenia. J Lipid Res (2007) 48(4):794–805. 10.1194/jlr.M600512-JLR200 [DOI] [PubMed] [Google Scholar]
- 65.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, et al. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med (2004) 199(8):1121–31. 10.1084/jem.20031557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Canfran-Duque A, Rotllan N, Zhang X, Fernandez-Fuertes M, Ramirez-Hidalgo C, Araldi E, et al. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol Med (2017) 9(9):1244–62. 10.15252/emmm.201607492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Das A, Ganesh K, Khanna S, Sen CK, Roy S. Engulfment of apoptotic cells by macrophages: a role of microRNA-21 in the resolution of wound inflammation. J Immunol (2014) 192(3):1120–9. 10.4049/jimmunol.1300613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gillotte-Taylor K, Boullier A, Witztum JL, Steinberg D, Quehenberger O. Scavenger receptor class B type I as a receptor for oxidized low density lipoprotein. J Lipid Res (2001) 42(9):1474–82. [PubMed] [Google Scholar]
- 69.Chang MK, Bergmark C, Laurila A, Horkko S, Han KH, Friedman P, et al. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci U S A (1999) 96(11):6353–8. 10.1073/pnas.96.11.6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shaw PX, Horkko S, Tsimikas S, Chang MK, Palinski W, Silverman GJ, et al. Human-derived anti-oxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler Thromb Vasc Biol (2001) 21(8):1333–9. 10.1161/hq0801.093587 [DOI] [PubMed] [Google Scholar]
- 71.Bae YS, Lee JH, Choi SH, Kim S, Almazan F, Witztum JL, et al. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ Res (2009) 104(2):210–8, 21p following 218. 10.1161/CIRCRESAHA.108.181040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem (2003) 278(3):1561–8. 10.1074/jbc.M209634200 [DOI] [PubMed] [Google Scholar]
- 73.Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, et al. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood (2007) 109(3):1026–33. 10.1182/blood-2006-05-021634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J Biol Chem (2011) 286(38):33335–44. 10.1074/jbc.M111.263020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garbin U, Baggio E, Stranieri C, Pasini A, Manfro S, Mozzini C, et al. Expansion of necrotic core and shedding of Mertk receptor in human carotid plaques: a role for oxidized polyunsaturated fatty acids? Cardiovasc Res (2013) 97(1):125–33. 10.1093/cvr/cvs301 [DOI] [PubMed] [Google Scholar]
- 76.Subramanian M, Proto JD, Matsushima GK, Tabas I. Deficiency of AXL in bone marrow-derived cells does not affect advanced atherosclerotic lesion progression. Sci Rep (2016) 6:39111. 10.1038/srep39111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123(2):321–34. 10.1016/j.cell.2005.08.032 [DOI] [PubMed] [Google Scholar]
- 78.Costales P, Castellano J, Revuelta-Lopez E, Cal R, Aledo R, Llampayas O, et al. Lipopolysaccharide downregulates CD91/low-density lipoprotein receptor-related protein 1 expression through SREBP-1 overexpression in human macrophages. Atherosclerosis (2013) 227(1):79–88. 10.1016/j.atherosclerosis.2012.12.021 [DOI] [PubMed] [Google Scholar]
- 79.Gorovoy M, Gaultier A, Campana WM, Firestein GS, Gonias SL. Inflammatory mediators promote production of shed LRP1/CD91, which regulates cell signaling and cytokine expression by macrophages. J Leukoc Biol (2010) 88(4):769–78. 10.1189/jlb.0410220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature (2002) 417(6885):182–7. 10.1038/417182a [DOI] [PubMed] [Google Scholar]
- 81.Tóth B, Garabuczi E, Sarang Z, Vereb G, Vámosi G, Aeschlimann D, et al. Transglutaminase 2 is needed for the formation of an efficient phagocyte portal in macrophages engulfing apoptotic cells. J Immunol (2009) 182(4):2084–92. 10.4049/jimmunol.0803444 [DOI] [PubMed] [Google Scholar]
- 82.Komura H, Miksa M, Wu R, Goyert SM, Wang P. Milk fat globule epidermal growth factor-factor VIII is down-regulated in sepsis via the lipopolysaccharide-CD14 pathway. J Immunol (2009) 182(1):581–7. 10.4049/jimmunol.182.1.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kalinina N, Agrotis A, Antropova Y, DiVitto G, Kanellakis P, Kostolias G, et al. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arterioscler Thromb Vasc Biol (2004) 24(12):2320–5. 10.1161/01.ATV.0000145573.36113.8a [DOI] [PubMed] [Google Scholar]
- 84.Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S, et al. High-mobility group box protein 1 neutralization reduces development of diet-induced atherosclerosis in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol (2011) 31(2):313–9. 10.1161/ATVBAHA.110.218669 [DOI] [PubMed] [Google Scholar]
- 85.Friggeri A, Yang Y, Banerjee S, Park YJ, Liu G, Abraham E. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol (2010) 299(6):C1267–76. 10.1152/ajpcell.00152.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu G, Wang J, Park YJ, Tsuruta Y, Lorne EF, Zhao X, et al. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol (2008) 181(6):4240–6. 10.4049/jimmunol.181.6.4240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ouimet M, Ediriweera H, Afonso MS, Ramkhelawon B, Singaravelu R, Liao X, et al. MicroRNA-33 regulates macrophage autophagy in atherosclerosis. Arterioscler Thromb Vasc Biol (2017) 37(6):1058–67. 10.1161/ATVBAHA.116.308916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510(7503):92–101. 10.1038/nature13479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fredman G, Tabas I. Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am J Pathol (2017) 187(6):1211–21. 10.1016/j.ajpath.2017.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res (2016) 119:1030–8. 10.1161/CIRCRESAHA.116.309492 [DOI] [PubMed] [Google Scholar]
- 91.Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J (2008) 22(10):3595–606. 10.1096/fj.08-112201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Helgadottir A, Manolescu A, Helgason A, Thorleifsson G, Thorsteinsdottir U, Gudbjartsson DF, et al. A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction. Nat Genet (2006) 38(1):68. 10.1038/ng1692 [DOI] [PubMed] [Google Scholar]
- 93.Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H, Thorsteinsdottir U, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nature (2004) 36:233–9. 10.1038/ng1311 [DOI] [PubMed] [Google Scholar]
- 94.Kajimoto K, Shioji K, Ishida C, Iwanaga Y. Validation of the association between the gene encoding 5-lipoxygenase-activating protein and myocardial infarction in a Japanese population. Circ J (2005) 69:1029–34. 10.1253/circj.69.1029 [DOI] [PubMed] [Google Scholar]
- 95.Wang G, Wang Y, Sun H, Cao W, Zhang J. Variants of the arachidonate 5-lipoxygenase-activating protein (ALOX5AP) gene and risk of ischemic stroke in Han Chinese of eastern China. J Biomed Res (2011) 25:319–27. 10.1016/S1674-8301(11)60043-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zintzaras E, Rodopoulou P. Variants of the arachidonate 5-lipoxygenase-activating protein (ALOX5AP) gene and risk of stroke: a HuGE gene-disease association review and meta-analysis. Am J Epidemiol (2009) 169:523–32. 10.1093/aje/kwn368 [DOI] [PubMed] [Google Scholar]
- 97.Elajami TK, Colas RA, Dalli J, Chiang N, Serhan CN, Welty FK. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J (2016) 30(8):2792–801. 10.1096/fj.201500155R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AH, Pande R, et al. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol (2010) 177(4):2116–23. 10.2353/ajpath.2010.091082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun (2016) 7:12859. 10.1038/ncomms12859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fredman G, Kamaly N, Spolitu S. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med (2015) 7:275ra20. 10.1126/scitranslmed.aaa1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kamaly N, Fredman G, Fojas JJR, Subramanian M, Choi WI, Zepeda K, et al. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano (2016) 10:5280–92. 10.1021/acsnano.6b01114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kusters DH, Chatrou ML, Willems BA, De Saint-Hubert M, Bauwens M, van der Vorst E, et al. Pharmacological treatment with annexin A1 reduces atherosclerotic plaque burden in LDLR-/- mice on western type diet. PLoS One (2015) 10:e0130484. 10.1371/journal.pone.0130484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell (2007) 131(6):1124–36. 10.1016/j.cell.2007.10.034 [DOI] [PubMed] [Google Scholar]
- 104.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390(6658):350–1. 10.1038/37022 [DOI] [PubMed] [Google Scholar]
- 105.Alciato F, Sainaghi PP, Sola D, Castello L, Avanzi GC. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J Leukoc Biol (2010) 87(5):869–75. 10.1189/jlb.0909610jlb.0909610 [DOI] [PubMed] [Google Scholar]
- 106.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101(4):890–8. 10.1172/JCI1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood (2012) 120(15):e60–72. 10.1182/blood-2012-04-423525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A (2016) 113(23):6526–31. 10.1073/pnas.1524292113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sampson UK, Fazio S, Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep (2012) 14(1):1–10. 10.1007/s11883-011-0219-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377(12):1119–31. 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 111.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science (2000) 288(5473):2051–4. 10.1126/science.288.5473.2051 [DOI] [PubMed] [Google Scholar]
- 112.Burger P, Hilarius-Stokman P, de Korte D, van den Berg TK, van Bruggen R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood (2012) 119(23):5512–21. 10.1182/blood-2011-10-386805 [DOI] [PubMed] [Google Scholar]
- 113.Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol (2009) 9(1):62–70. 10.1038/nri2470 [DOI] [PubMed] [Google Scholar]
- 114.Sugimoto MA, Ribeiro ALC, Costa BRC, Vago JP, Lima KM, Carneiro FS, et al. Plasmin and plasminogen induce macrophage reprogramming and regulate key steps of inflammation resolution via annexin A1. Blood (2017) 129(21):2896–907. 10.1182/blood-2016-09-742825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tzelepis F, Verway M, Daoud J, Gillard J, Hassani-Ardakani K, Dunn J, et al. Annexin1 regulates DC efferocytosis and cross-presentation during Mycobacterium tuberculosis infection. J Clin Invest (2015) 125(2):752–68. 10.1172/JCI77014 [DOI] [PMC free article] [PubMed] [Google Scholar]