

Abstract
Proposals are described for the assignment of recently reported viruses, infecting rodents, bats and other mammalian species, to new species within the Hepacivirus and Pegivirus genera (family Flaviviridae). Assignments into 14 Hepacivirus species (Hepacivirus A–N) and 11 Pegivirus species (Pegivirus A–K) are based on phylogenetic relationships and sequence distances between conserved regions extracted from complete coding sequences for members of each proposed taxon. We propose that the species Hepatitis C virus is renamed Hepacivirus C in order to acknowledge its unique historical position and so as to minimize confusion. Despite the newly documented genetic diversity of hepaciviruses and pegiviruses, members of these genera remain phylogenetically distinct, and differ in hepatotropism and the possession of a basic core protein; pegiviruses in general lack these features. However, other characteristics that were originally used to support their division into separate genera are no longer definitive; there is overlap between the two genera in the type of internal ribosomal entry site and the presence of miR-122 sites in the 5′ UTR, the predicted number of N-linked glycosylation sites in the envelope E1 and E2 proteins, the presence of poly U tracts in the 3′ UTR and the propensity of viruses to establish a persistent infection. While all classified hepaciviruses and pegiviruses have mammalian hosts, the recent description of a hepaci-/pegi-like virus from a shark and the likely existence of further homologues in other non-mammalian species indicate that further species or genera remain to be defined in the future.
Keywords: Flaviviridae, Hepacivirus, Pegivirus, hepatitis C virus, taxonomy
Introduction
A recurrent feature of virus taxonomy is that as more information accumulates on the genetic diversity within established virus taxa such as species and genera, the discrete demarcation criteria originally applied to distinguish between them become blurred.
Hepatitis C virus (HCV) has been the only named species within the genus Hepacivirus since the genus was created in 1996. Considerable diversity exists between different isolates of HCV (Bukh et al., 1993; Simmonds et al., 1993), with 7 genotypes and 84 subtypes currently recognized (Smith et al., 2014) (https://talk.ictvonline.org/ictv_wikis/flaviviridae/w/sg_flavi/56.hcv-classification). Despite this diversity, all of these viruses are derived from human infections and are associated with acute and chronic liver disease, and there continues to be widespread agreement that they are most appropriately considered as members of a single species. A related virus, GBV-B was identified in 1995 from New World monkeys (Simons et al., 1995a) and is associated with acute liver disease, but remained unclassified. In the last few years, several more divergent viruses have been discovered with genome structures and conserved sequence motifs that are similar to those of HCV and GBV-B and isolated from a variety of host species including dog (Kapoor et al., 2011), horse (Burbelo et al., 2012), bat (Quan et al., 2013), rodent (Drexler et al., 2013; Firth et al., 2014; Kapoor et al., 2013a), Old World monkey (Lauck et al., 2013) and cow (Baechlein et al., 2015; Corman et al., 2015). These viruses differ considerably in their epidemiology and presumed route of transmission from HCV.
Similarly, the Pegivirus (pronounced peh-gee virus) genus, when first proposed (Stapleton et al., 2011), comprised two species: Pegivirus A, including the viruses GBV-A (Simons et al., 1995a) and GBV-C (Leary et al., 1996; Linnen et al., 1996; Simons et al., 1995b) isolated from primates, and Pegivirus B, including viruses derived from bats (Epstein et al., 2010). In the last few years, several papers have described viruses that share many features with Pegivirus A and Pegivirus B, but which are divergent in genome sequence and structure and infect humans (Berg et al., 2015; Kapoor et al., 2015), bats (Quan et al., 2013), horses (Chandriani et al., 2013; Kapoor et al., 2013b), rodents (Firth et al., 2014; Kapoor et al., 2013a) and pigs (Baechlein et al., 2016). In addition, viruses similar to GBV-C have been discovered in a range of primate species (Birkenmeyer et al., 1998; Sibley et al., 2014).
In the current study, we review the classification of these two genera and have revised the list of features by which the two genera can be distinguished. We additionally describe proposals for the assignment of viruses for which a complete coding sequence is available into a series of species within the two genera and provide demarcation criteria that define these assignments. We propose the creation of 13 additional Hepacivirus species and 9 additional Pegivirus species.
Results
Hepacivirus genus
Hepacivirus sequences were aligned using muscle and reduced to a set of those differing over their complete coding sequence by amino acid p-distances greater than 0.1. Since different genotypes of HCV all differ by 0.23–0.31, this cut-off would be expected to include all variants likely to represent different species. A scan of mean amino acid p-distance over the coding region revealed two regions where p-distances were consistently less than 0.6: positions 1123–1566 and 2536–2959 (numbered relative to the Hepacivirus type species, M62321 (Choo et al., 1989) (Fig. 1), and therefore most informative for phylogenetic comparisons. Phylogenies of Hepacivirus sequences in these regions were congruent apart from minor and non-bootstrap-supported rearrangements of deep branches (Fig. 2a, b). For the region 1123–1566, amino acid p-distances were greater than 0.3 apart from distances between different genotypes of HCV, which were 0.12–0.19 (Fig. 2c). A more continuous distribution of amino acid p-distances was observed for the region 2536–2959, with discontinuities centred on distances of 0.35 and 0.45.
Fig. 1.

Amino acid divergence across the Hepacivirus polyprotein. Mean amino acid p-distances were calculated for 26 aligned Hepacivirus polyprotein sequences that differed by >0.1 of amino acid positions using a sliding window of 50 amino acids incremented by 10 residues and plotted against the amino acid position of the start of the fragment. The X-axis scale is uneven because of gaps in the reference sequence (M62321). Two regions with mean p-distances consistently <0.6 are indicated by bars. A schematic representation of the Hepacivirus polyprotein is shown to scale below.
Fig. 2.

Analysis of Hepacivirus conserved regions. Maximum-likelihood trees were produced using mega6 for (a) positions 1123–1566 and (b) 2536–2959 of the virus polyprotein using the Le and Gascuel model and a gamma distribution of variation with invariant sites. Branches observed in >70 % of bootstrap replicates are indicated. Proposed Hepacivirus species assignments are indicated by single letters to the right of each branch. (c) Frequency histograms of amino acid p-distance between Hepacivirus sequences in the region 1123–1566 and (d) the region 2536–2959. The range of distances between different genotypes of Hepacivirus C (HCV) is indicated by an open arrow, between Hepacivirus G and Hepacivirus H by a shaded arrow and between Hepacivirus A and Hepacivirus C by a black arrow. The distance that demarcates different species is indicated by a dotted line.
Although evidence has been provided for recombination within (González-Candelas et al., 2011) and between Hepacivirus species (Thézé et al., 2015), the only known recombinant included in our dataset was the sequence KC796077 (Quan et al., 2013), which is the single known representative of its clade; exclusion of this sequence did not affect the distribution of sequence distances or phylogenetic relationships between the other species (data not shown).
The phylogenetic relationships observed for these two genome regions are consistent with the division of the Hepacivirus genus into 14 species which we propose should be named Hepacivirus A–N (Table 1). Although HCV was the first Hepacivirus to be discovered and the type species of its genus, we have chosen to assign it to Hepacivirus C rather than Hepacivirus A so as to minimize the potential for confusion. To be clear, individual isolates of this virus will still be called hepatitis C virus (HCV), but they will all belong to the species Hepacivirus C. Other species are named according to the date of publication of a complete coding sequence, with the exception of Hepacivirus B which includes GBV-B (providing a memorable link) and Hepacivirus A (canine hepacivirus/non-primate hepacivirus/equine hepacivirus). Demarcation between species is based upon amino acid p-distances of greater than 0.25 in the region 1123–1566 and greater than 0.3 in the region 2536–2959. The rationale for choosing these demarcation points is that they result in HCV and equine hepacivirus isolates being separated into two species, as seems reasonable given their different hosts, while genotypes of HCV remain as members of the same species, reflecting their shared human epidemiology and pathology. The only conflict that arises from these choices is that the rodent-derived sequences KC815310 (Kapoor et al., 2013a) and KC411784 (Drexler et al., 2013) would be considered as two species by comparison of the region 1123–1566 (amino acid p-distance 0.30), but one species by comparison of the region 2536–2959 (amino acid p-distance 0.27). Since these sequences were obtained from different rodent species in the New and Old Worlds, respectively, we prefer a demarcation point that separates these viruses into two species (Hepacivirus E and Hepacivirus F). The equivocal sequence distances of 0.30 and 0.32 in the region 2536–2959 derive from comparisons between the rodent species Hepacivirus G and Hepacivirus E and F; distances between these species in the region 1123–1566 (0.39, 0.40) are greater than those observed between Hepacivirus A and Hepacivirus C (0.35–0.38), suggesting that their demarcation into species is appropriate.
Table 1. Proposed Hepacivirus species.
| Species | Previous identifier(s) | Type isolate | Accession number | Host/region | Source | 5′ UTR | 5′ UTR miR-122 sites (CACUCC) |
K/R aa before E1 | E1/E2 N-X-S/T sites |
3′ UTR poly U (nt) |
Genome MFED (%) |
Tissue tropism |
Chronic | Disease |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hepacivirus A | Canine hepacivirus Non-primate hepacivirus Equine hepacivirus |
NZP1 | KP325401 | Horse (Dog?) | Serum, liver |
IV IRES | 1 | 34 | 4/9 | 86 | 12.8–15.2 | Liver | Yes | ? |
| Hepacivirus B | GBV-B | T-1053 | U22304 | New World primate? | Plasma | IV IRES | 2 | 20 | 2/5 | 27 | 8.7 | Liver | Rarely | Hepatitis |
| Hepacivirus C | HCV | HCV-1 | M62321 | Human | Serum, liver |
IV IRES | 2 | 31 | 4/11 | 10–108‡ | 7.3–9.9 | Liver | Yes | Hepatitis, cirrhosis, cancer |
| Hepacivirus D | Guereza hepacivirus | BWC08 | KC551800 | Old World primate | Plasma | IV IRES? | 1 | 26 | 4/4 | ? | 11.6–11.9 | ? | ? | ? |
| Hepacivirus E | Rodent hepacivirus | RHV-339 | KC815310 | New World rodent | Plasma | IV IRES | 1 | 24 | 2/4 | 3 | 4.8–5.1 | ? | ? | ? |
| Hepacivirus F | Rodent hepacivirus | NLR07-oct70 | KC411784 | Old World rodent | Serum | Pegi-like | 1 | 23† | 2/3 | ? | 6.8 | Liver | ? | ? |
| Hepacivirus G | Norway rat hepacivirus 1 | NrHV-1/NYC-C12 | KJ950938 | Global rodent | Serum, multiple organs | ? | ? | 22† | 3/3 | ? | 5.6 | Liver | ? | ? |
| Hepacivirus H | Norway rat hepacivirus 2 | NrHV-2/NYC-E43 | KJ950939 | Global rodent | Serum, multiple organs | ? | ? | 27† | 3/4 | ? | 4.5 | Liver | ? | ? |
| Hepacivirus I | Rodent hepacivirus | SAR-3 | KC411806 | Old World rodent | Serum | IV IRES | 0 | 29 | 1/2 | 5 | 1.5–1.9 | ? | ? | ? |
| Hepacivirus J | Rodent hepacivirus | RMU10-3382 | KC411777 | Old World rodent | Serum, liver, lung |
Pegi-like | 2 | 29 | 2/7 | ? | 9.8–13.6 | Liver | ? | ? |
| Hepacivirus K | Bat hepacivirus | PDB-829 | KC796074 | Old World bat | Serum | ? | ? | 34 | 1/5 | ? | 9.5 | ? | ? | ? |
| Hepacivirus L | Bat hepacivirus | PDB-112 | KC796077 | Old World bat | Serum | ? | ? | 25 | 2/4 | ? | 7.7 | ? | ? | ? |
| Hepacivirus M | Bat hepacivirus | PDB-491.1 | KC796078 | Old World bat | Serum | ? | ? | 32 | 1/4 | ? | 9.6–10.8 | ? | ? | ? |
| Hepacivirus N | Bovine hepacivirus | 463 | KP641127 | Cow | Serum | IV IRES | 1* | 25 | 2/6 | 4 | 9.5–10.8 | Liver | Yes | ? |
According to this schema, the genus Hepacivirus contains the species Hepacivirus A, including viruses first detected in dogs (canine hepacivirus) (Kapoor et al., 2011), but which subsequently have been detected more frequently in horses (non-primate hepacivirus, equine hepacivirus) (Burbelo et al., 2012). There is much greater virus diversity between equine isolates than is currently described for canine isolates (Pybus & Thézé, 2015), and several studies demonstrate transmission and pathology of infection in the horse (Pfaender et al., 2015; Ramsay et al., 2015; Scheel et al., 2015); these observations are consistent with the horse being the primary host, and for this reason we have used an equine virus (NSP1, KP325401) as the type isolate. Hepacivirus B includes GBV-B, a virus initially detected in and capable of infecting New World primates, but that has not been isolated subsequently (Simons et al., 1995a). Hepacivirus C includes all currently known genotypes and subtypes of HCV, all of which are confined to humans. Hepacivirus D includes sequences derived from colobus monkeys, but about which there is no information for tropism, chronicity or pathogenicity (Lauck et al., 2013). A similar lack of virological or biological information pertains to those species (Hepacivirus E–J) derived from rodents (Drexler et al., 2013; Firth et al., 2014; Kapoor et al., 2013a) and bats (Hepacivirus K–M) (Quan et al., 2013). We have retained KC796077 (Quan et al., 2013) as the type species of Hepacivirus L, although there is evidence that it is a recombinant (Thézé et al., 2015), since it groups separately from other species whether or not the recombinant region is included (Fig. 2), and since it is the only representative of this clade with a complete coding region sequence. Hepacivirus N is represented by viruses isolated from cows and associated with a chronic but asymptomatic liver infection (Baechlein et al., 2015; Corman et al., 2015).
We propose that when the next species of Hepacivirus is assigned it should be ‘P’ rather than ‘O’ in order to avoid confusion with the number 0 (zero), and that species beyond X should be named XA, XB, etc., followed by YA, YB, etc. and ZA, ZB, …, ZZ.
Pegivirus genus
A set of 26 Pegivirus sequences that differed from each other by >0.11 of amino acid positions over their complete coding sequence was used to assess amino acid sequence diversity across the genome. There were two regions where mean amino acid diversity was consistently <0.6: 888–1635 and 2398–2916 (numbered relative to U22303, Fig. 3). Phylogenetic analysis of Pegivirus sequences in these two regions produced congruent trees, providing independent evidence that these sequences are phylogenetically distinct (Fig. 4a, b). For both regions, the distribution of amino acid distances between these sequences, whether calculated using sse v1.2 as p-distances, Kimura distances or using a matrix of similarity, was distributed in a series of peaks (Fig. 4c, d) with discontinuities at 0.28–0.34 (positions 888–1635) and 0.35–0.37 (2398–2916). Using an amino acid p-distance of >0.31 for positions 888–1635 to demarcate Pegivirus species, the sequences currently described would represent 11 different species (Table 2). These individual species comprise sequences from similar hosts from either the Old or New Worlds with the exception of Pegivirus A, which includes sequences derived from New World primates and Old World bats. Two rodent sequences are both included in Pegivirus I despite having an ambiguous p-distance for the region 888–1635 (0.303), since they group together on the phylogenetic tree and both are from rodents sampled in the New World. However, if an amino acid p-distance of >0.36 for the region 2398–2916 is used to demarcate species, the amino acid p-distances between Pegivirus F, Pegivirus G and Pegivirus J would all fall below the cut-off. Higher or lower p-distance demarcation points also produce inconsistent assignments. In particular, we could not find demarcation points that divided Pegivirus A into exclusively primate or bat-derived groups of sequences.
Fig. 3.

Amino acid divergence across Pegivirus polyproteins. Mean amino acid p-distances were calculated for 26 aligned Pegivirus polyprotein sequences that differed by >0.11 of amino acid positions using a sliding window of 50 amino acids incremented by 10 residues and plotted against the amino acid position of the start of the fragment. Increments on the X-axis scale are uneven because of unnumbered gaps in the reference sequence (U22303). Two regions with distances consistently <0.6 are indicated by bars. A schematic representation of the Pegivirus polyprotein is shown to scale below.
Fig. 4.

Analysis of Pegivirus conserved regions. Maximum-likelihood trees were produced using mega6 for (a) amino acid positions 888–1635 using the Le and Gascuel model with frequencies and a gamma distribution of variation with invariant sites, and for (b) amino acid positions 2398–2916 using the Le and Gascuel model with a gamma distribution of invariant sites. Branches observed in >70 % of bootstrap replicates are indicated. Frequency histograms of amino acid p-distance between Pegivirus sequences in the region 888–1635 (c) and the region 2398–2916 (d) Amino acid p-distances between Pegivirus C sequences derived from different primate species are indicated by an open arrow, while those between primate- and bat-derived Pegivirus A sequences are indicated by a shaded arrow. The distance that demarcates different species is indicated by a broken line.
Table 2. Proposed Pegivirus species.
| Species | Previous identifier | Type isolate | Accession number | Host/region | Source | 5′ UTR | 5′ UTR miR-122 sites (CACUCC) |
K/R aa before E1 | E1/E2 N-X-S/T sites | 3′ UTR poly U (nt) |
Genome MFED (%) |
Tissue tropism |
Chronic | Disease |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pegivirus A | GBV-A | T-1053 | U22303 | New World primate, Old World bat |
Plasma | Pegi-like | 0 | 0 | 1/4 | 2 | 10.4–14.1 | ? | Yes | ? |
| Pegivirus B | GBV-D | GBV-D | GU566734 | Old World bat | Serum | ? | ? | 5* | 1/9 | ? | 9.5 | ? | ? | ? |
| Pegivirus C | GBV-C | PNF2161 | U44402 | Human, Old World primate |
Plasma | Pegi-like | 0 | 4 | 1/3 | 3 | 9.4–13.1 | Lymphocytes | Yes | No |
| Pegivirus D | Theiler’s disease-associated virus | Horse_A1 | KC145265 | Horse | Serum | ? | 0 | 1 | 2/7 | 4 | 13.1 | ? | Yes | Serum hepatitis? |
| Pegivirus E | Equine pegivirus | C0035 | KC410872 | Horse | Serum | Pegi-like | 0 | 8* | 1/4 | 4 | 11.3 | ? | Yes | ? |
| Pegivirus F | Bat pegivirus | PDB-1698 | KC796080 | New World bat | Serum | ? | 1 | 7* | 2/9 | ? | 9.3 | ? | ? | ? |
| Pegivirus G | Bat pegivirus | PDB-620 | KC796076 | Old World bat | Serum | ? | ? | 7* | 0/6 | ? | 8.6–12.0 | ? | ? | ? |
| Pegivirus H | Human hepegivirus Human pegivirus 2 | AK-790 | KT439329 | Human | Serum | IV IRES | 0 | 5 | 2/9 | 3 | 8.5 | ? | Yes | ? |
| Pegivirus I | Bat pegivirus | PDB-1715 | KC796088 | New World bat | Serum | ? | ? | 0 | 1/2 | ? | 9.4 | ? | ? | ? |
| Pegivirus J | Rodent pegivirus | CC61 | KC815311 | New World rodent | Serum, multiple organs | ? | 0 | 21* | 2/9 | 4 | 9.3 | ? | ? | ? |
| Pegivirus K | Porcine pegivirus | PPgV_903 | KU351669 | Pig | Serum | Pegi-like | 0 | 0 | 4/4 | 2 | 9.1 | ? | Yes | ? |
?, Unknown or uncertain.
*Position of AUG initiation codon uncertain.
Pegivirus A includes GBV-A and other isolates from New World monkeys (U22303, U94421, AF023425 and AF023424) (Leary et al., 1997; Simons et al., 1995a) as well as viruses obtained from African bats (KC796085, KC796082, KC796086, KC796081, KC796075 and KC796089) (Quan et al., 2013). Pegivirus B includes viruses (GBV-D) derived from bats in Asia (GU566735 and GU566734) (Epstein et al., 2010) and Africa (KC796073 and KC796083) (Quan et al., 2013). Pegivirus C is proposed as a new species to include GBV-C/hepatitis G virus (Leary et al., 1996; Linnen et al., 1996) and related viruses isolated from Old World primates (Bailey et al., 2015; Birkenmeyer et al., 1998; Kapusinszky et al., 2015; Sibley et al., 2014). Within this species, the virus phylogeny corresponds closely to that of the host (Bailey et al., 2015; Sharp & Simmonds, 2011; Sibley et al., 2014) with separate lineages for human (78 complete genome sequences), chimpanzee (AF070476), yellow baboon (KR996153, KR996142, KR996146, KR996144, KR996152, KR996151, KR996150, KR996149, KR996148, KR996147, KR996145, KR996143, KP890673 and KP890672), olive baboon (KF234530), red-tailed guenon (KF234529, KF234528, KF234526, KF234525 and KF234527), red colobus (KF234523, KF234524, KF234507,KF234522, KF234521, KF234520, KF234519, KF234518, KF234517, KF234516, KF234515, KF234514, KF234513, KF234512, KF234511, KF234510, KF234509, KF234508, KF234506, KF234505, KF234504, KF234503, KF234502, KF234501, KF234500 and KF234499) and African green monkey (KP296858). The proposed species Pegivirus D (KC145265) (Chandriani et al., 2013) and Pegivirus E (KC410872) (Kapoor et al., 2013b) both include single complete coding region sequences derived from horses. Pegivirus F, G and I all include viruses derived from Old and New World bats (Quan et al., 2013), Pegivirus H includes viruses described as human pegivirus 2 and human hepegivirus (Berg et al., 2015; Kapoor et al., 2015), while Pegivirus J includes viruses derived from rodents (Firth et al., 2014; Kapoor et al., 2013a). Pegivirus K is a recently described virus isolated from pigs (Baechlein et al., 2016). Some of the proposed species identifiers used will assist association with previous isolate names or designations (Pegivirus A: GBV-A, Pegivirus C: GBV-C, Pegivirus E: equine pegivirus and Pegivirus H: human pegivirus 2).
A division of the Pegivirus genus into two clades based on phylogenetic relationships (Kapoor et al., 2015) could also be observed in our analyses [Pegivirus A, B, C, D, E, I and K (clade 1) and Pegivirus F, G, H and J (clade 2)]. Further investigation may support the suggestion that these clades differ from each other in internal ribosomal entry site (IRES) type; the reported correlation between these groupings with frequencies of N-linked glycosylation in the E1 and E2 proteins was not sustained (Table 2).
Demarcation between genera
Several characteristics have been used to differentiate members of the Hepacivirus and Pegivirus genera (Stapleton et al., 2011). This expanded survey of diversity within these genera weakens some of these associations. For example, the number of N-linked glycosylation sites in the E1 and E2 glycoproteins was thought to be higher in members of the Hepacivirus genus, but with the expanded number of species considered here, this trend is no longer apparent either for E1 and E2 combined or when considered separately (Fig. 5). Similarly, a poly U tract of at least 10 residues is present in the 3′ UTR of some members of the Hepacivirus genus (Bukh et al., 1999; Kolykhalov et al., 1996; Scheel et al., 2015; Tanaka et al., 1995), but not others (Table 1). Some members of the Hepacivirus genus are hepatotropic and induce hepatitis, but for many species this information is unknown, while Pegivirus D (Theiler’s disease-associated virus) has been reported to be associated with serum hepatitis in horses (Chandriani et al., 2013). Persistent infection can occur with members of either genus, but in many cases this information is lacking. The same difficulty applies to the characterization of virus IRES types; such regions are often lacking or incomplete despite the coding region being complete. In addition, in most cases no detailed molecular biology has been undertaken to confirm proposed secondary structures. However, even with these caveats, it is already clear that viruses with a similar type IV IRES (e.g. HCV) occur in members of both the Hepacivirus and Pegivirus genera (Tables 1, 2). The presence of ordered secondary structures across the genome as measured by mean free energy difference (MFED) values does not differ between the two genera.
Fig. 5.
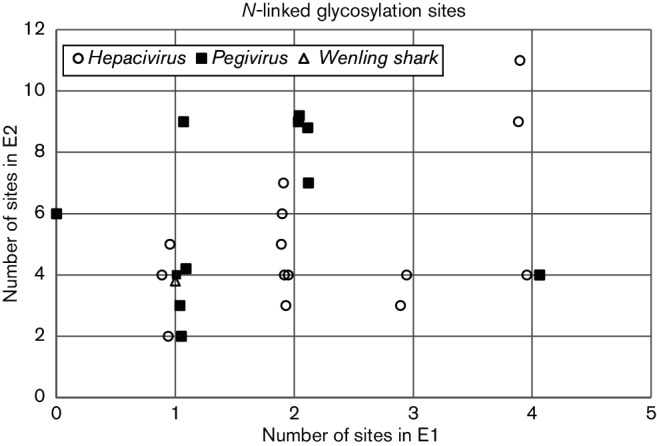
Predicted number of N-glycosylation sites. Scatter plots show the number of Asn-X-Ser/Thr glycosylation sites predicted for E1 (X-axis) or E2 (Y-axis) using NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/) for individual species of Pegivirus (■), Hepacivirus (○) and for Wenling shark virus (∆). Points have been jittered on the X-axis to improve legibility.
Nevertheless, there remains a clear demarcation between the two genera in their phylogenetic relationships (Fig. 6). In addition, all members of the Hepacivirus genus have a long basic core region with between 20 and 34 lysine or arginine residues in the 156–216 residues between the presumed initiation codon and the presumed E1 cleavage site. Most members of the Pegivirus genus have a shorter and less basic sequence in this region or no identifiable coding sequence upstream of E1. However, a long and relatively basic regions is predicted in Pegivirus J.
Fig. 6.

Comparative analysis of Wenling shark virus. (a) Mean amino acid p-distance across the virus polyprotein between single representatives of each Hepacivirus and Pegivirus species with Wenling shark virus was plotted against polyprotein position (numbered relative to M62321). The positions of two regions where mean p-distances were <0.6 are indicated by solid bars. A schema of the boundaries between virus proteins is given below. (b) Phylogenetic analysis of representatives of each Hepacivirus (filled circles) and Pegivirus (open circles) species was analysed together with Wenling shark virus (filled triangle) using amino acid sequences between positions (b) 1216–1534 and (c) 2560–2745. Trees were constructed from maximum-likelihood distances using a Le and Gascuel model with a gamma distribution of variation with a class of invariant sites. Branches supported by >70 % of bootstrap replicates are indicated.
Wenling shark virus
Using the 14 Hepacivirus species and 11 Pegivirus species as references, we produced an amino acid alignment with the addition of Wenling shark virus (Shi et al., 2015). The genome sequence possesses a predicted single ORF encoding 3087 amino acids, similar in length to polyproteins of hepaciviruses and pegiviruses. The 5′ UTR was short (131 bases) and potentially incomplete, but showed no identifiable regions of sequence homology with equivalent regions of hepaciviruses or pegiviruses. This suggests possible possession of an IRES type distinct from mammalian viruses. The 3′ end was 262 nucleotides in length, without the poly(U/C) tract observed in some hepaciviruses. The region could be predicted to possess RNA secondary structure with a series of stem-loops comparable to those of other members of the Flaviviridae family. The coding region of the genome possesses a predicted structured RNA with a mean MFED value of 18 %, higher than that of either hepaciviruses or pegiviruses.
The structural genes were predicted using the SignalP server to identify signalase cleavage sites characteristic of hepaciviruses and pegiviruses in this region, along with tentative alignment of cleavage sites between core/E1 and E1/E2 of hepaciviruses. This analysis predicts a relatively long core gene of 300 amino acids (nucleotide positions 132–1031), but with several regions of identifiable amino acid sequence homology towards the carboxy terminus of the protein. It also possesses 52 basic residues at the amino terminus consistent with an RNA packaging function. The predicted E1 protein (nucleotide positions 1032–1586, 185 amino acids) was similarly identifiably homologous to those of hepaciviruses with one predicted N-linked glycosylation site. The predicted E2 protein spans 272 amino acids (nucleotide positions 1587-2402) with four predicted N-linked sites, lower than for the E2 protein of most hepaciviruses.
Mean amino acid divergence between Wenling shark virus and either Hepacivirus or Pegivirus species was consistently less than 0.6 in two regions of the genome: between positions 1216 and 1534 and between 2560 and 2745 (numbered relative to M62321, Fig. 6). Phylogenetic analysis suggests that this virus is distinct from either genus, since each forms a separate clade in the region 1216–1534 (the Hepacivirus clade is supported by 68 % of bootstrap replicates), while for the region 2560–2745, the branch structure of the Hepacivirus clade, that includes Wenling shark virus, is poorly supported by bootstrap resampling. Overall, this analysis identifies a potentially greater overall similarity of Wenling shark virus to hepaciviruses, based both on the existence of predicted structural proteins homologous to those of hepaciviruses and the evidence for the grouping of Wenling shark virus with hepaciviruses on phylogenetic analysis of non-structural protein sequences between amino acid positions 2560–2745 (Fig. 6). However, without further information concerning the biology and molecular biology of this virus, we suggest it remains as an unclassified species within the Flaviviridae.
Discussion
In recent years, there has been a remarkable increase in our knowledge of diversity within the Hepacivirus and Pegivirus genera. This change is largely due to the application of primer-independent deep-sequencing techniques to a wide range of mammalian host species. In 2011 there was evidence for two species in each genus (Stapleton et al., 2011), whereas we now propose that the Hepacivirus genus should encompass 14 species (Table 1), while the Pegivirus genus should include 11 species (Table 2). The proposed criteria for demarcating between different species are divergence in two different regions of the virus genome. Very similar phylogenetic relationships have been reported for Bayesian or maximum-likelihood analysis based upon complete coding regions or subgenomic regions of subsets of these sequences (Baechlein et al., 2015, 2016; Corman et al., 2015; Kato et al., 1990; Pfaender et al., 2014; Quan et al., 2013; Sibley et al., 2014; Thézé et al., 2015). We have chosen to base species demarcation criteria on amino acid p-distances for two defined regions of the virus genome (portions of the NS2/NS3 [protease] and NS5B [RNA-dependent RNA polymerase] proteins), since these regions can be easily aligned around conserved motifs with distances quickly computed using standard software.
In almost all cases, these distinctions are correlated with the known host range. One exception is Hepacivirus A, which was first described from dogs and described as canine hepacivirus (Kapoor et al., 2011), but has since been reported at a high frequency in horses (Burbelo et al., 2012; Kapoor et al., 2013b; Lyons et al., 2012; Postel et al., 2015) and in only one subsequent report in dogs (El-Attar et al., 2015). Phylogenetic analysis suggests that sequences derived from dogs are nested within those derived from horses, consistent with the detection of this virus in dogs being a secondary event, conceivably through the administration of vaccines manufactured using equine serum (Pybus & Thézé, 2015). The other species with a complex host range is Pegivirus A (GBV-A), which has been detected in New World primates and Old World bats. Virus variants within this species segregate with host species in the case of both primates (Bukh & Apgar, 1997) and bats (Quan et al., 2013), consistent with a long period of co-evolution.
Our analysis does not include viruses represented only by partial genome sequences, although it is likely that these viruses include potential species within the Hepacivirus and Pegivirus genera additional to our proposed classification scheme (Drexler et al., 2013; Quan et al., 2013). In particular, we note that a large, diverse clade of bat-derived pegiviruses described from partial genome sequences (Quan et al., 2013) is therefore excluded from our proposals. The reason for this decision is that, even though our phylogenetic analysis is based on subgenomic sequences, we do not feel that it is appropriate to propose species names for viruses for which the complete coding sequence remains uncharacterized and for which genome organization is only partly determined. The technical challenge of obtaining complete genome sequences of viruses, even without prior isolation, is considerably reduced following the advent of next-generation sequencing and, we believe, now justifies this requirement. Indeed, without this information, some of the important characters such as the presence of a basic core protein and the number of N-linked glycosylation sites may be unavailable and this might reduce the confidence of taxonomic assignments. Another reason for excluding subgenomic coding sequences from taxonomic proposals is that there is evidence for recombination between different species of Hepacivirus (Thézé et al., 2015), this could not be properly assessed on subgenomic sequences. We have been less stringent with regard to the presence of non-coding regions of the genome since, although these contain features relevant to virus taxonomy, difficulties are sometimes experienced in sequencing these terminal regions of the genome. An example of the difficulties that could be produced by relying on subgenomic coding region sequences is provided by Hepacivirus E and Hepacivirus F, which might have been classified as a single species if comparisons had been limited to the region 2536–2959 (Fig. 2).
A similar argument could be made in terms of our incomplete knowledge of the biological and molecular properties (persistence, tissue tropism and pathogenicity) of viruses for which all that is known is their presumed primary host and complete genome sequence (Tables 1, 2). However, such additional information can be labour intensive and difficult to obtain, especially in viruses initially identified in wild fauna; in many cases this information may never be obtained. The requirement for a complete genome sequence at least makes it possible for future biological studies to be performed through assembly of synthetic infectious clones, as has been reported for Hepacivirus A, Hepacivirus B, Hepacivirus C (Bukh et al., 1999; Kolykhalov et al., 1997; Scheel et al., 2015; Yanagi et al., 1997) and Pegivirus C (Xiang et al., 2000).
We have applied a more stringent test for Wenling shark virus, since although a complete coding sequence has been obtained and the host is known, phylogenetic analysis does not place this virus clearly within either the Hepacivirus or Pegivirus genera, although the arrangement of structural genes is most consistent with its eventual assignment as a Hepacivirus. Until biological and molecular information is obtained about this and other hepaci- or pegi-like viruses from non-mammalian hosts, we believe it is most prudent that this virus remains unassigned to either an existing or novel genus.
This survey of diversity within the Hepacivirus and Pegivirus genera somewhat obscures the demarcation criteria proposed to differentiate these genera (Stapleton et al., 2011). Considerable overlap was observed between genera in 5′ UTR IRES type, the frequency of N-linked glycosylation (Fig. 5), the presence of poly U in the 3′ UTR, the persistence of infection and liver tropism (Tables 1, 2). At present the demarcation between these genera relies on the results of phylogenetic analysis (Fig. 6), the presence of one or more miR-122 sites (CACUCC) in the 5′ UTR followed by a basic core region, hepatotropism and liver pathology. We note that similar obscuration of demarcation criteria is likely to arise within these genera as additional complete genome sequences are obtained from a widening sample of host species by next-generation sequencing technology. It would be a mistake to regard such an outcome as a deficiency in current methods of taxonomy; genera and species are man-made categories imposed by us on a diverse virus fauna as a tool to organizing information and should be judged as such.
The evolutionary history of the Hepacivirus and Pegivirus genera is obviously complex and has been associated with multiple shifts of host species and genome mosaicism in the case of IRES sequences. Notable species-specific associations amongst primates are observed for Pegivirus A (Bukh & Apgar, 1997) and Pegivirus C (Sharp & Simmonds, 2011; Sibley et al., 2014), consistent with co-speciation, whereas the presence of multiple Pegivirus species in bats and of multiple Hepacivirus species in rodents, as well as the relatedness of bat and primate-derived Pegivirus A isolates, is not. The primarily sequence-based species assignments proposed in the current study nevertheless divide the two genera into groups of viruses with a number of shared biological properties, either demonstrated or inferred, that will be of value in the future epidemiological, clinical and virological characterization of these viruses.
Methods
Nucleotide sequences (other than HCV) assigned to the Hepacivirus genus and >6000 nucleotides in length were retrieved from GenBank and, together with single representatives of HCV genotypes 1–7, were aligned in their coding regions using muscle (Edgar, 2004) as implemented in sse v1.2 (Simmonds, 2012). Sequences differing by amino acid p-distances of <0.1 were then removed; this is a conservative cut-off since genotypes 1–7 of HCV differ by amino acid p-distances of 0.23–0.31. The final alignment consisted of sequences with the GenBank accession numbers M62321, D00944, D17763, Y11604, Y13184, Y12083, EF108306, KC411784, KC815310, KJ950939, KJ950938, KC411806, KC411777, KP325401, KP641127, KC551800, KC796078, KC796074, KC796077 and U22304. Scans of mean amino acid p-distances between groups of sequences were performed in sse (window size 50 amino acid residues, shifted by 10). Regions where mean amino acid p-distances were consistently <0.6 were adjusted visually to remove terminal poorly unaligned regions, and then used to produce histograms of amino acid distances. The optimal amino acid substitution model for each conserved region was assessed using mega6 (Tamura et al., 2013) and used to produce maximum-likelihood trees. Frequency histograms were based on the p-distance between pairs of sequences.
The same process was carried out to produce an alignment of 160 genome sequences representing different Pegivirus variants. The final alignment, after removal of sequences differing by amino acid p-distances of less than 0.11, consisted of GenBank accession numbers U22303, U44402, AF070476, KR996153, KP296858, KF234529, KF234523, U94421, AF023425, AF023424, KT439329, KC410872, KC145265, KJ950934, KC815311, KC796085, KC796082, KC796086, KC796081, KC796088, GU566734, KC796073, KC796083, KC796087, KC796093, KC796076 and KC796084; we also included KU351669, a novel pegivirus recently isolated from a pig (Baechlein et al., 2016). Separate analyses included the Hepacivirus and Pegivirus sets together with Wenling shark virus (KR902729).
N-linked glycosylation sites were predicted by analysis of envelope E1 and E2 fragments from examples of each species using NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/). Cleavage sites in structural protein regions were independently predicted using the SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP/). MFED values were calculated by comparing folding energies of consecutive fragments of nucleotide sequence to random sequence order controls using the program Folding Energy Scan in the sse package (Simmonds, 2012). Values represent the percentage difference between the MFE of the native sequence from that of the mean value of 50 sequence order randomized controls.
Acknowledgements
This work was supported by grants from the Wellcome Trust to the Centre for Immunity, Infection and Evolution at the University of Edinburgh (095831/Z/11/Z) and to the Nuffield Department of Medicine, University of Oxford (WT108418AIA). The authors are grateful to Troels Scheel (Copenhagen Hepatitis C Program, University of Copenhagen and Department of Infectious Diseases, Hividovre Hospital, Copenhagen, Denmark) for his helpful comments on the manuscript, and to Oliver Pybus and Julien Thézé (Department of Zoology, University of Oxford, UK) for sharing their unpublished analysis of Wenling shark virus.
References
- Baechlein C., Fischer N., Grundhoff A., Alawi M., Indenbirken D., Postel A., Baron A. L., Offinger J., Becker K., et al. (2015). Identification of a novel Hepacivirus in domestic cattle from Germany. J Virol 897007–7015. 10.1128/JVI.00534-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baechlein C., Grundhoff A., Fischer N., Alawi M., Hoeltig D., Waldmann K. H., Becher P.(2016). Pegivirus infection in domestic pigs, Germany. Emerg Infect Dis 221312–1314. 10.3201/eid2207.160024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A. L., Lauck M., Mohns M., Peterson E. J., Beheler K., Brunner K. G., Crosno K., Mejia A., Mutschler J., et al. (2015). Durable sequence stability and bone marrow tropism in a macaque model of human pegivirus infection. Sci Transl Med 7305ra144. 10.1126/scitranslmed.aab3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg M. G., Lee D., Coller K., Frankel M., Aronsohn A., Cheng K., Forberg K., Marcinkus M., Naccache S. N., et al. (2015). Discovery of a novel human pegivirus in blood associated with Hepatitis C virus co-infection. PLoS Pathog 11e1005325. 10.1371/journal.ppat.1005325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenmeyer L. G., Desai S. M., Muerhoff A. S., Leary T. P., Simons J. N., Montes C. C., Mushahwar I. K.(1998). Isolation of a GB virus-related genome from a chimpanzee. J Med Virol 5644–51. 10.1002/(SICI)1096-9071(199809)56:1<44::AID-JMV8>3.0.CO;2-N [DOI] [PubMed] [Google Scholar]
- Bukh J., Purcell R. H., Miller R. H.(1993). At least 12 genotypes of hepatitis C virus predicted by sequence analysis of the putative E1 gene of isolates collected worldwide. Proc Natl Acad Sci U S A 908234–8238. 10.1073/pnas.90.17.8234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukh J., Apgar C. L.(1997). Five new or recently discovered (GBV-A) virus species are indigenous to new world monkeys and may constitute a separate genus of the Flaviviridae. Virology 229429–436. 10.1006/viro.1997.8461 [DOI] [PubMed] [Google Scholar]
- Bukh J., Apgar C. L., Yanagi M.(1999). Toward a surrogate model for Hepatitis C virus: an infectious molecular clone of the GB virus-B hepatitis agent. Virology 262470–478. 10.1006/viro.1999.9941 [DOI] [PubMed] [Google Scholar]
- Burbelo P. D., Dubovi E. J., Simmonds P., Medina J. L., Henriquez J. A., Mishra N., Wagner J., Tokarz R., Cullen J. M., et al. (2012). Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J Virol 866171–6178. 10.1128/JVI.00250-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandriani S., Skewes-Cox P., Zhong W., Ganem D. E., Divers T. J., Van Blaricum A. J., Tennant B. C., Kistler A. L.(2013). Identification of a previously undescribed divergent virus from the Flaviviridae family in an outbreak of equine serum hepatitis. Proc Natl Acad Sci U S A 110E1407–E1415. 10.1073/pnas.1219217110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo Q. L., Kuo G., Weiner A. J., Overby L. R., Bradley D. W., Houghton M.(1989). Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244359–362. 10.1126/science.2523562 [DOI] [PubMed] [Google Scholar]
- Corman V. M., Grundhoff A., Baechlein C., Fischer N., Gmyl A., Wollny R., Dei D., Ritz D., Binger T., et al. (2015). Highly divergent hepaciviruses from African cattle. J Virol 895876–5882. 10.1128/JVI.00393-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler J. F., Corman V. M., Müller M. A., Lukashev A. N., Gmyl A., Coutard B., Adam A., Ritz D., Leijten L. M., et al. (2013). Evidence for novel hepaciviruses in rodents. PLoS Pathog 9e1003438. 10.1371/journal.ppat.1003438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C.(2004). muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 321792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Attar L. M., Mitchell J. A., Brooks Brownlie H., Priestnall S. L., Brownlie J.(2015). Detection of non-primate hepaciviruses in UK dogs. Virology 48493–102. 10.1016/j.virol.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J. H., Quan P. L., Briese T., Street C., Jabado O., Conlan S., Ali Khan S., Verdugo D., Hossain M. J., et al. (2010). Identification of GBV-D, a novel GB-like flavivirus from old world frugivorous bats (Pteropus giganteus) in Bangladesh. PLoS Pathog 6e1000972. 10.1371/journal.ppat.1000972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth C., Bhat M., Firth M. A., Williams S. H., Frye M. J., Simmonds P., Conte J. M., Ng J., Garcia J., et al. (2014). Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. MBio 5e01933–14. 10.1128/mBio.01933-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Candelas F., López-Labrador F. X., Bracho M. A.(2011). Recombination in hepatitis C virus. Viruses 32006–2024. 10.3390/v3102006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Simmonds P., Gerold G., Qaisar N., Jain K., Henriquez J. A., Firth C., Hirschberg D. L., Rice C. M., et al. (2011). Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci U S A 10811608–11613. 10.1073/pnas.1101794108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Simmonds P., Scheel T. K. H., Hjelle B., Cullen J. M., Burbelo P. D., Chauhan L. V., Duraisamy R., Sanchez Leon M., et al. (2013a). Identification of rodent homologs of hepatitis C virus and pegiviruses. MBio 4e00216–13. 10.1128/mBio.00216-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Simmonds P., Cullen J. M., Scheel T. K. H., Medina J. L., Giannitti F., Nishiuchi E., Brock K. V., Burbelo P. D., et al. (2013b). Identification of a pegivirus (GB virus-like virus) that infects horses. J Virol 877185–7190. 10.1128/JVI.00324-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Kumar A., Simmonds P., Bhuva N., Singh Chauhan L., Lee B., Sall A. A., Jin Z., Morse S. S., et al. (2015). Virome analysis of transfusion recipients reveals a novel human virus that shares genomic features with Hepaciviruses and Pegiviruses. MBio 6e01466–15. 10.1128/mBio.01466-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusinszky B., Mulvaney U., Jasinska A. J., Deng X., Freimer N., Delwart E.(2015). Local virus extinctions following a host population bottleneck. J Virol 898152–8161. 10.1128/JVI.00671-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N., Hijikata M., Ootsuyama Y., Nakagawa M., Ohkoshi S., Sugimura T., Shimotohno K.(1990). Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc Natl Acad Sci U S A 879524–9528. 10.1073/pnas.87.24.9524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolykhalov A. A., Feinstone S. M., Rice C. M.(1996). Identification of a highly conserved sequence element at the 3' terminus of hepatitis C virus genome RNA. J Virol 703363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolykhalov A. A., Agapov E. V., Blight K. J., Mihalik K., Feinstone S. M., Rice C. M.(1997). Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277570–574. 10.1126/science.277.5325.570 [DOI] [PubMed] [Google Scholar]
- Lauck M., Sibley S. D., Lara J., Purdy M. A., Khudyakov Y., Hyeroba D., Tumukunde A., Weny G., Switzer W. M., et al. (2013). A novel hepacivirus with an unusually long and intrinsically disordered NS5A protein in a wild old world primate. J Virol 878971–8981. 10.1128/JVI.00888-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary T. P., Muerhoff A. S., Simons J. N., Pilot-Matias T. J., Erker J. C., Chalmers M. L., Schlauder G. G., Dawson G. J., Desai S. M., Mushahwar I. K.(1996). Sequence and genomic organization of GBV-C: a novel member of the flaviviridae associated with human non-A-E hepatitis. J Med Virol 4860–67. 10.1002/(SICI)1096-9071(199601)48:1<60::AID-JMV10>3.0.CO;2-A [DOI] [PubMed] [Google Scholar]
- Leary T. P., Desai S. M., Erker J. C., Mushahwar I. K.(1997). The sequence and genomic organization of a GB virus A variant isolated from captive tamarins. J Gen Virol 782307–2313. 10.1099/0022-1317-78-9-2307 [DOI] [PubMed] [Google Scholar]
- Linnen J., Wages J., Zhang-Keck Z.-Y., Fry K. E., Krawczynski K. Z., Alter H., Koonin E., Gallagher M., Alter M., et al. (1996). Molecular cloning and disease association of hepatitis G virus: a transfusion-transmissible agent. Science 271505–508. 10.1126/science.271.5248.505 [DOI] [PubMed] [Google Scholar]
- Lyons S., Kapoor A., Sharp C., Schneider B. S., Wolfe N. D., Culshaw G., Corcoran B., McGorum B. C., Simmonds P.(2012). Nonprimate hepaciviruses in domestic horses, United kingdom. Emerg Infect Dis 181976–1982. 10.3201/eid1812.120498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaender S., Brown R. J., Pietschmann T., Steinmann E.(2014). Natural reservoirs for homologs of hepatitis C virus. Emerg Microbes Infect 3e21. 10.1038/emi.2014.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaender S., Cavalleri J. M., Walter S., Doerrbecker J., Campana B., Brown R. J., Burbelo P. D., Postel A., Hahn K., et al. (2015). Clinical course of infection and viral tissue tropism of hepatitis C virus-like nonprimate hepaciviruses in horses. Hepatology 61447–459. 10.1002/hep.27440 [DOI] [PubMed] [Google Scholar]
- Postel A., Cavalleri J. M., Pfaender S., Walter S., Steinmann E., Fischer N., Feige K., Haas L., Becher P.(2015). Frequent presence of hepaci and pegiviruses in commercial equine serum pools. Vet Microbiol 1828–14. 10.1016/j.vetmic.2015.10.032 [DOI] [PubMed] [Google Scholar]
- Pybus O. G., Thézé J.(2015). Hepacivirus cross-species transmission and the origins of the hepatitis C virus. Curr Opin Virol 161–7. 10.1016/j.coviro.2015.10.002 [DOI] [PubMed] [Google Scholar]
- Quan P. L., Firth C., Conte J. M., Williams S. H., Zambrana-Torrelio C. M., Anthony S. J., Ellison J. A., Gilbert A. T., Kuzmin I. V., et al. (2013). Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc Natl Acad Sci U S A 1108194–8199. 10.1073/pnas.1303037110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay J. D., Evanoff R., Wilkinson T. E., Divers T. J., Knowles D. P., Mealey R. H.(2015). Experimental transmission of equine hepacivirus in horses as a model for hepatitis C virus. Hepatology 611533–1546. 10.1002/hep.27689 [DOI] [PubMed] [Google Scholar]
- Scheel T. K., Kapoor A., Nishiuchi E., Brock K. V., Yu Y., Andrus L., Gu M., Renshaw R. W., Dubovi E. J., et al. (2015). Characterization of nonprimate hepacivirus and construction of a functional molecular clone. Proc Natl Acad Sci U S A 1122192–2197. 10.1073/pnas.1500265112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P. M., Simmonds P.(2011). Evaluating the evidence for virus/host co-evolution. Curr Opin Virol 1436–441. 10.1016/j.coviro.2011.10.018 [DOI] [PubMed] [Google Scholar]
- Shi M., Lin X.-D., Vasilakis N., Tian J.-H., Li C.-X., Chen L.-J., Eastwood G., Diao X.-N., Chen M.-H., et al. (2015). Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the flaviviridae and related viruses. J Virol 90659–669. 10.1128/JVI.02036-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley S. D., Lauck M., Bailey A. L., Hyeroba D., Tumukunde A., Weny G., Chapman C. A., O'Connor D. H., Goldberg T. L., Friedrich T. C.(2014). Discovery and characterization of distinct simian pegiviruses in three wild African old world monkey species. PLoS One 9e98569. 10.1371/journal.pone.0098569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P., Holmes E. C., Cha T. A., Chan S. W., McOmish F., Irvine B., Beall E., Yap P. L., Kolberg J., Urdea M. S.(1993). Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J Gen Virol 742391–2399. 10.1099/0022-1317-74-11-2391 [DOI] [PubMed] [Google Scholar]
- Simmonds P.(2012). SSE: a nucleotide and amino acid sequence analysis platform. BMC Res Notes 550. 10.1186/1756-0500-5-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons J. N., Pilot-Matias T. J., Leary T. P., Dawson G. J., Desai S. M., Schlauder G. G., Muerhoff A. S., Erker J. C., Buijk S. L., Chalmers M. L.(1995a). Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc Natl Acad Sci U S A 923401–3405. 10.1073/pnas.92.8.3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons J. N., Leary T. P., Dawson G. J., Pilot-Matias T. J., Muerhoff A. S., Schlauder G. G., Desai S. M., Mushahwar I. K.(1995b). Isolation of novel virus-like sequences associated with human hepatitis. Nat Med 1564–569. 10.1038/nm0695-564 [DOI] [PubMed] [Google Scholar]
- Smith D. B., Bukh J., Kuiken C., Muerhoff A. S., Rice C. M., Stapleton J. T., Simmonds P.(2014). Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59318–327. 10.1002/hep.26744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton J. T., Foung S., Muerhoff A. S., Bukh J., Simmonds P.(2011). The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol 92233–246. 10.1099/vir.0.027490-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S.(2013). mega6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 302725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Kato N., Cho M. J., Shimotohno K.(1995). A novel sequence found at the 3' terminus of hepatitis C virus genome. Biochem Biophys Res Commun 215744–749. 10.1006/bbrc.1995.2526 [DOI] [PubMed] [Google Scholar]
- Thézé J., Lowes S., Parker J., Pybus O. G.(2015). Evolutionary and phylogenetic analysis of the hepaciviruses and pegiviruses. Genome Biol Evol 72996–3008. 10.1093/gbe/evv202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J., Wünschmann S., Schmidt W., Shao J., Stapleton J. T.(2000). Full-length GB virus C (Hepatitis G virus) RNA transcripts are infectious in primary CD4-positive T cells. J Virol 749125–9133. 10.1128/JVI.74.19.9125-9133.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi M., Purcell R. H., Emerson S. U., Bukh J.(1997). Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci U S A 948738–8743. 10.1073/pnas.94.16.8738 [DOI] [PMC free article] [PubMed] [Google Scholar]