

Abstract
Tyrosine kinases are important regulators of synaptic strength. Here, we describe a key component of the synaptic vesicle release machinery, Munc18‐1, as a phosphorylation target for neuronal Src family kinases (SFKs). Phosphomimetic Y473D mutation of a SFK phosphorylation site previously identified by brain phospho‐proteomics abolished the stimulatory effect of Munc18‐1 on SNARE complex formation (“SNARE‐templating”) and membrane fusion in vitro. Furthermore, priming but not docking of synaptic vesicles was disrupted in hippocampal munc18‐1‐null neurons expressing Munc18‐1Y473D. Synaptic transmission was temporarily restored by high‐frequency stimulation, as well as by a Munc18‐1 mutation that results in helix 12 extension, a critical conformational step in vesicle priming. On the other hand, expression of non‐phosphorylatable Munc18‐1 supported normal synaptic transmission. We propose that SFK‐dependent Munc18‐1 phosphorylation may constitute a potent, previously unknown mechanism to shut down synaptic transmission, via direct occlusion of a Synaptobrevin/VAMP2 binding groove and subsequent hindrance of conformational changes in domain 3a responsible for vesicle priming. This would strongly interfere with the essential post‐docking SNARE‐templating role of Munc18‐1, resulting in a largely abolished pool of releasable synaptic vesicles.
Keywords: Munc18‐1, priming, SNARE, Src, synaptic transmission
Subject Categories: Neuroscience, Signal Transduction
Introduction
Posttranslational modifications of synaptic proteins are a powerful and well‐characterized way to modulate synaptic transmission. On the presynaptic side, phosphorylation of two proteins, Munc18‐1 and Synaptotagmin1, by protein kinase C (PKC) is required for most forms of short‐term plasticity (Wierda et al, 2007; Genc et al, 2014; de Jong et al, 2016). Many other proteins, such as Synapsins, RIM, SNAP‐25, Snapin, and Tomosyn, are phosphorylated by PKC or protein kinase A (PKA) and may modulate synaptic transmission [see for a review (de Jong & Verhage, 2009)]. Large‐scale brain‐specific phospho‐proteomics screens (Collins et al, 2005; Munton et al, 2007; Ballif et al, 2008; Tweedie‐Cullen et al, 2009; Huttlin et al, 2010) indicate that synaptic proteins are phosphorylated on many additional sites, presumably by other kinases, with unknown effects on their function.
Tyrosine kinases have long been associated with developmental processes, but are recently also recognized as modulators of synaptic plasticity (Purcell & Carew, 2003). Although most studies focus on the postsynaptic side, a role for tyrosine kinases in presynaptic neurotransmitter release is also becoming evident (Ohnishi et al, 2001; Wang, 2003; Shyu et al, 2005; Baldwin et al, 2006; Onofri et al, 2007), especially for two members of the Src tyrosine kinase (SFK) family, c‐Src, and the neuron‐specific isoform, n‐Src. The two isoforms are identical except a 6‐amino insert in the SH3 domain, which regulates activity and substrate recruitment (Martinez et al, 1987; Raulf et al, 1989). N‐Src preferably associates with presynaptic membranes (Onofri et al, 2007), while c‐Src is enriched in synaptic vesicle fractions, is upregulated after memory training, and phosphorylates the presynaptic proteins Synaptophysin, Synaptogyrin, and Synapsin (Barnekow et al, 1990; Janz & Sudhof, 1998; Janz et al, 1999; Zhao et al, 2000; Onofri et al, 2007). However, the consequences of these phosphorylation events for synaptic transmission remain largely elusive, mostly because the role of these three proteins itself appears to be already subtle (Rosahl et al, 1993; McMahon et al, 1996; Janz et al, 1999; Messa et al, 2010). A large‐scale brain phosphotyrosine proteomics screen identified several other tyrosine‐phosphorylated synaptic proteins, including Munc18‐1, Synaptotagmin1, Piccolo, Bassoon, SHANK3, and Syngap1 (Ballif et al, 2008), which may provide more likely explanations for the robust effects of tyrosine phosphorylation on synaptic transmission (Purcell & Carew, 2003).
Here, we investigated the possible role of tyrosine phosphorylation of Munc18‐1 on synaptic transmission. We identified Munc18‐1 Y473 as a substrate for several SFK members and a plausible mechanism to modulate synaptic transmission. Point mutations at this site interfere with the ability of Munc18‐1 to stimulate liposome fusion and to support synaptic vesicle priming and synaptic transmission in hippocampal neurons. We exploit recent insights into the molecular actions of Munc18‐1 domains (Hu et al, 2011; Parisotto et al, 2014; Munch et al, 2016) to elucidate how Y473 phosphorylation inhibits synaptic transmission, namely by interfering with Synaptobrevin2/VAMP2 binding and extension of helix 12 in domain 3a, hindering SNARE complex assembly. These results propose a novel and potent mechanism to inhibit synaptic transmission via tyrosine phosphorylation of Munc18‐1.
Results
Munc18‐1 is phosphorylated by Src family kinases at Y473
In large‐scale brain proteomics, two tyrosine phosphorylation sites have been identified in Munc18‐1, Y145, and Y473 (Collins et al, 2005; Ballif et al, 2008) (www.phosphosite.org). Fyn phosphorylates Munc18‐1 Y145 in HEK cells (Lim et al, 2013) and kinase prediction (scansite.mit.edu) suggests Src as kinase for the other site, Y473. Using a heterologous expression system, we found that the neuron‐specific isoform of Src, n‐Src, indeed phosphorylates Munc18‐1 and that mutating Y473 to phenylalanine reduced n‐Src‐dependent phosphorylation by ~40% (relative phosphorylation level in Y473F compared to WT: n‐Src = 0.60 ± 0.02 a.u., N = 3; Fig 1A). Other members of the Src kinase family (SFK) expressed in neurons, c‐Src and Fyn, also phosphorylated Y473 (relative phosphorylation level in Y473F compared to WT: n‐Src = 0.61 ± 0.03 a.u., c‐Src = 0.47 ± 0.04 a.u., Fyn = 0.56 ± 0.002 a.u., N = 2; Fig 1B). These results suggest that Y473 is the major, but not the only, phosphorylation site in Munc18‐1 targeted by SFKs.
Figure 1. Src family kinases target Munc18‐1 on Y473.
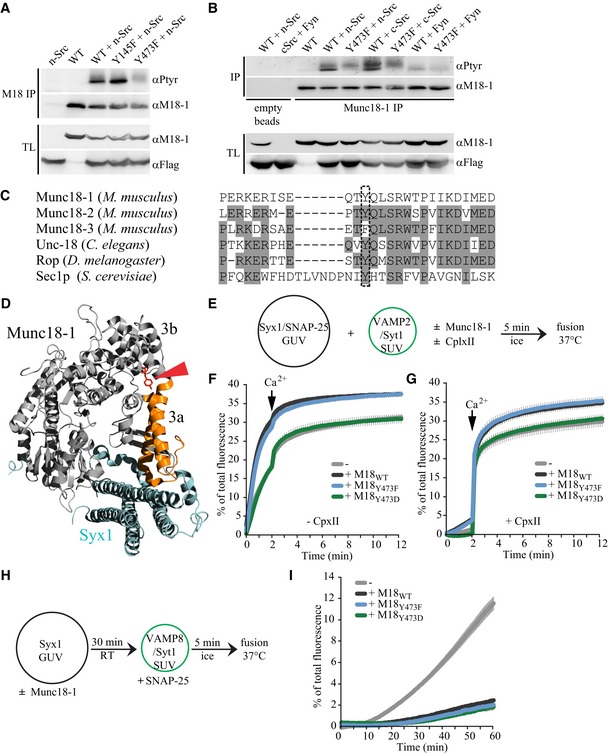
- Using denaturing immunoprecipitation (IP), Munc18‐1 was isolated from cell lysate of HEK293T cells expressing M18WT, M18Y145F, or M18Y473F together with or without n‐Src. Munc18‐1 was immunoblotted for tyrosine phosphorylation using the 4G10 antibody (αPtyr), after which the blot was stripped and reblotted for Munc18‐1. Total lysate (TL) is shown as loading control. N‐Src was tagged with a Flag‐tag for visualization.
- Same as (A) but using lysate from HEK293T cells expressing M18WT or M18Y473F together with or without n‐Src, c‐Src, or Fyn. The first two lanes are IPs performed using empty beads to control for non‐specific binding of Munc18 or kinases to the beads.
- Alignment of amino acid sequence surrounding Y473 of mouse Munc18 isoforms and of Munc18‐1 across different species. Gray‐marked residues are identical to mouse Munc18‐1.
- Crystal structure of Munc18‐1 (gray) and Syntaxin1 (cyan) adapted from Misura et al (2000) showing residue Y473 in red and helix 11–12 of domain 3a in orange.
- Incubation scheme: GUVs (14 nmol lipid) containing pre‐assembled t‐SNAREs (14 pmol Syx1/SNAP‐25) were mixed on ice with the indicated Munc18‐1 constructs (90 pmol) and subsequently SUVs (2.5 nmol lipid) containing Synaptotagmin1 (3 pmol) and Synaptobrevin/VAMP2 (8 pmol) were added. The fusion reaction was started by warming up the liposomes to 37°C. Calcium (100 μM final concentration) was added after 2 min.
- Lipid mixing was monitored by the increase of Atto 488 fluorescence for 12 min. Data were normalized to the maximum fluorescence after liposome lysis by detergent. Error bars = s.e.m., N = 3.
- Same as (E, F) but the reactions were pre‐incubated on ice for 5 min in the presence of Complexin II (CpxII 600 pmol). Error bars = s.e.m., N = 5.
- Incubation scheme: GUVs (14 nmol lipid) containing Syx1 (14 pmol) were pre‐incubated with the indicated Munc18‐1 constructs (90 pmol) at room temperature for 30 min. Subsequently, samples were incubated with soluble SNAP‐25 (128 pmol) and SUVs (2.5 nmol lipid) containing Synaptotagmin1 (3 pmol) and VAMP8 (8 pmol) for 5 min on ice. Lipid mixing was started by increasing the temperature to 37°C.
- Lipid mixing was monitored by the increase of Atto 488 fluorescence for 60 min. Data were normalized to the maximum fluorescence after liposome lysis by detergent. Error bars = s.e.m., N = 3.
Source data are available online for this figure.
The above results show that Src phosphorylates Munc18‐1 when co‐expressed in HEK cells. To gain insight in the dynamics, in vitro kinase assays were performed. Munc18‐1 phosphorylation was increased in vitro with a peak at 70 min upon incubating HEK cell lysates expressing Munc18‐1 or Src in the presence of the general protein tyrosine phosphatase (PTP) inhibitor vanadate (Fig EV1A). We then repeated the experiment using brain lysate and lysate from Src‐expressing HEK cells. In this case, neuronal Munc18‐1 was robustly phosphorylated within 10 min at room temperature (Fig EV1B). These data confirm that brain‐derived Munc18‐1 is phosphorylated by Src kinases and that the kinetics are on the minute timescale.
Figure EV1. In vitro kinase assays and Syntaxin binding of Munc18‐1 variants.
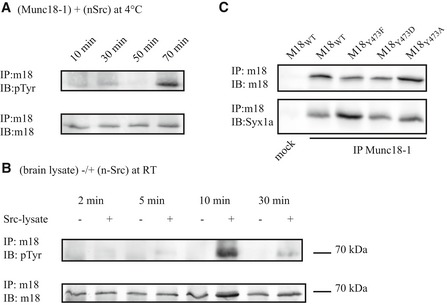
- In vitro kinase assay in which lysates from HEK cells expressing n‐Src or Munc18‐1 were combined and incubated in the presence of PTP inhibitor vanadate at 4°C. Samples were denatured after different incubation times and Munc18‐1 was immunoprecipitated and immunoblotted against phosphotyrosine.
- Same as (A) but using neuronal Munc18‐1 from brain lysate and incubation at room temperature.
- Y473 mutants all bind Syntaxin1. Munc18‐1 variants were co‐expressed with Syntaxin1 in HEK293T cells. Munc18‐1 and Munc18‐1‐bound proteins were immunoprecipitated from cell lysate and immunoblotted for detection.
Source data are available online for this figure.
Y473 is conserved across species and in the mammalian Munc18‐1 isoform Munc18‐2, but not Munc18‐3 (Fig 1C). Y473 is located on the outer surface of domain 3 in Munc18‐1 (Misura et al, 2000), away from known interaction sites for Munc18‐1's main interaction partner, Syntaxin1 (Syx1; Fig 1D). Indeed, Y473 mutations in Munc18‐1 did not affect co‐precipitation of Syntaxin1 (Fig EV1C). Hence, it appears to be unlikely that phosphorylation at Y473 affects Syntaxin1 binding affinity, and if Munc18‐1 is a relevant target for tyrosine kinases to exert their effects on synaptic strength, this probably occurs via another aspect of Munc18‐1 function.
A phosphomimetic mutant affects Munc18‐1 function in a cell‐free fusion assay
To address functional consequences of protein phosphorylation, we generated a phosphomimetic Munc18‐1 mutant by introducing a negatively charged residue (aspartate) at position 473 (M18Y473D) and a non‐phosphorylatable mutant by introducing a phenylalanine (M18Y473F). Since phenylalanine contains an aromatic ring and only diverges from tyrosine in that it lacks the hydroxyl group, structural changes are not expected.
The functional effects of these point mutations were tested in vitro in a reconstituted membrane fusion assay. In such assays, Munc18‐1 is known to stimulate SNARE‐driven liposome fusion (Shen et al, 2007; Rodkey et al, 2008; Diao et al, 2010; Schollmeier et al, 2011), but also has an initial inhibitory role (Schollmeier et al, 2011), probably similar to the in vivo situation. The inhibitory and stimulatory role of Munc18‐1 relies on distinct Munc18‐1/SNARE protein interaction modes which can be selectively addressed in vitro (Schollmeier et al, 2011). Giant unilamellar vesicles (GUVs) containing pre‐assembled target(t)‐SNAREs (Syx1/SNAP‐25) were incubated with small unilamellar vesicles (SUVs) containing Synaptotagmin1 and Synaptobrevin/VAMP2 (Fig 1E) (Malsam et al, 2012). Starting with pre‐assembled t‐SNARE complexes bypasses the inhibitory function of Munc18‐1, providing a selective assay of Munc18‐1's stimulatory function. Complexin II (CpxII) was initially omitted from the assay to maximize the effect of Munc18‐1 (Parisotto et al, 2012). As expected, addition of Munc18‐1 to the GUV/SUV in vitro fusion assay increased the initial lipid‐mixing kinetics [Fig 1F, gray (−M18WT) versus black (+M18WT)]. Adding non‐phosphorylatable Munc18‐1 (M18Y473F) stimulated fusion to a similar extent as wild‐type Munc18‐1 (Fig 1F, blue). In contrast, the phosphomimetic mutant (M18Y473D) was unable to increase the fusion rate (Fig 1F, green). In the presence of Complexin II, initial Ca2+‐independent fusion is inhibited and calcium‐triggered fusion is synchronized (Malsam et al, 2012). Complexin action does not depend on the presence of Munc18‐1 (consistent with earlier findings (Parisotto et al, 2014)). Adding M18Y473F in the presence of Complexin II again stimulated the initial fusion rate similar to M18WT, while fusion kinetics in the presence of M18Y473D were similar to reactions lacking Munc18‐1 (Fig 1G, first 2 min). In both assays, Munc18‐1 also increased the maximal fusion rate. This is in line with our previous data (Parisotto et al, 2014) and attributed to an increase in the number of functional v‐ or t‐SNAREs, probably as a result of Munc18‐1 stimulating v‐/t‐SNARE complex formation or stabilizing functional t‐SNARE conformations (as also suggested by Refs Weninger et al, 2008; Baker et al, 2015). Similar results were obtained in a content‐mixing assay (Fig EV2A and B), suggesting that the lipid‐mixing assays truly report liposome fusion and not only lipid mixing.
Figure EV2. Effect of Munc18‐1 variants on content‐mixing assays.
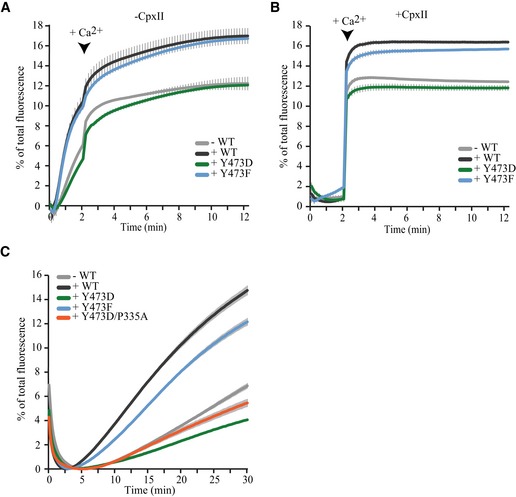
To analyze whether all mutants still retained their ability to inhibit t‐SNARE complex assembly (and exclude that M18Y473D is a completely dysfunctional protein), we next used Syx1‐GUVs with soluble SNAP‐25 instead of pre‐assembled t‐SNARE complexes. In addition, the assay was modified to eliminate the stimulatory function of the Munc18‐1‐Synaptobrevin/VAMP2 interaction by replacing Synaptobrevin/VAMP2 with VAMP8, thereby promoting the inhibitory function of Munc18‐1 (Shen et al, 2007; Schollmeier et al, 2011). All Munc18‐1 variants inhibited fusion kinetics to a similar extent (Fig 1H and I). Taken together, these data indicate that a Munc18‐1 mutation that mimics tyrosine phosphorylation at Y473 drastically inhibits SNARE‐dependent membrane fusion by interfering with Munc18's stimulatory role in fusion, not its inhibitory binding to Syntaxin1.
A phosphomimetic mutant, M18Y473D, abolishes synaptic transmission in a post‐docking step
To test the effects of Y473 phosphorylation in living neurons, munc18‐1 null neurons were rescued with lentivirus expressing M18Y473D or wild‐type Munc18‐1 as control. Neurons rescued with M18Y473D were morphologically identical to control neurons with a similar number of synapses and dendritic length as determined from confocal images using an automated image analysis routine (Fig 2A and B) (Schmitz et al, 2011). Synaptic Munc18‐1 levels showed a trend toward a reduction in M18Y473D‐expressing neurons, while somatic Munc18‐1 levels were reduced (Fig 2B). Synaptic transmission was assessed in autaptic neurons (single neurons grown on glial islands), a highly standardized, reduced model system used to study changes in release kinetics and presynaptic plasticity (Bekkers & Stevens, 1991; Cornelisse et al, 2012). Whole‐cell patch‐clamp experiments on neurons solely expressing phosphomimetic Munc18‐1 revealed a profound effect on evoked synaptic transmission, with hardly detectable excitatory postsynaptic current (EPSC) (Fig 2C).
Figure 2. Tyrosine phosphorylation of Munc18‐1 disrupts basal synaptic transmission in autaptic neurons.
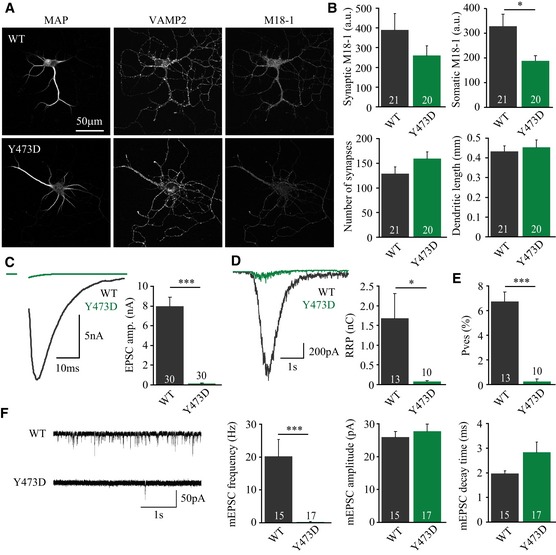
- Typical confocal images from neurons stained for MAP2, Synaptobrevin/VAMP2, and Munc18‐1.
- Average synaptic Munc18‐1 intensity (M18WT: 389 ± 85 a.u., n = 21; M18Y473D: 261 ± 49 a.u., n = 20, t‐test with Welch correction, P = 0.2000), somatic Munc18‐1 intensity (M18WT: 328 ± 49 a.u., n = 21; M18Y473D: 188 ± 21 a.u., n = 20, t‐test with Welch correction, *P = 0.0138), synapse number (M18WT: 129 ± 14, n = 21; M18Y473D: 159 ± 14, n = 20, t‐test, P = 0.1203), and dendrite length (M18WT: 0.433 ± 0.028, n = 21; M18Y473D: 0.454 ± 0.037, n = 20, t‐test, P = 0.6613).
- Left: typical examples of evoked release upon action potential stimulation. Average EPSC amplitude (M18WT: 7.96 ± 0.94 nA, n = 30; M18Y473D: 0.12 ± 0.05 nA, n = 30, Mann–Whitney U‐test, ***P < 0.0001).
- Left: typical responses to hyperosmotic sucrose application (500 mM, 3.5 s) used to assess the RRP. Average RRP charge (M18WT: 1.68 ± 0.64 nC, n = 13; M18Y473D: 0.08 ± 0.02 nC, n = 10, t‐test with Welch correction, *P = 0.0274).
- Average P ves (EPSC charge/RRP charge) (M18WT: 6.75 ± 0.77%, n = 13; M18Y473D: 0.24 ± 0.20%, n = 10; Mann–Whitney U‐test, ***P < 0.0001).
- Left: example traces of spontaneous release of single SVs (mEPSCs). Average mEPSC frequency (M18WT: 20.18 ± 5.18 Hz, n = 15; M18Y473D: 0.28 ± 0.13 Hz, n = 17, Mann–Whitney U‐test, ***P < 0.0001), mEPSC amplitude (M18WT: 26.0 ± 1.7 pA, n = 15; M18Y473D: 27.8 ± 2.2 pA, n = 17, t‐test, P = 0.5372), and mEPSC decay time (M18WT: 1.98 ± 0.10 ms, n = 15; M18Y473D: 2.84 ± 0.41 ms, n = 17, t‐test with Welch correction, P = 0.0549).
Reduced synaptic transmission can result from defects in calcium influx, readily releasable pool (RRP) size, or vesicular release probability (P ves). To further investigate this, neurons were subjected to hypertonic sucrose application to release the RRP in a calcium‐independent manner (Rosenmund & Stevens, 1996). Neurons expressing M18Y473D showed a large reduction in RRP size (Fig 2D), suggesting a strong reduction in the number of primed synaptic vesicles (SVs). P ves can be calculated by dividing the EPSC charge by the charge of the RRP. Neurons expressing M18Y473D showed a large reduction in P ves, that is, the EPSC size was reduced more than proportional to the RRP size (Fig 2E). In conclusion, the drastic inhibition of synaptic transmission in neurons expressing M18Y473D is a result of a reduction in both RRP size and P ves.
A reduced RRP is likely to also affect spontaneous release of single SVs. Indeed, these spontaneous events occurred rarely in neurons expressing M18Y473D, while common in control neurons (Fig 2F). The average mEPSC amplitude was unaffected, but mEPSCs showed a trend toward slower decay kinetics (Fig 2F). The fact that mEPSC amplitudes are normal in neurons expressing M18Y473D confirms the defects in synaptic transmission are due to presynaptic defects.
To investigate whether the reduced RRP size in neurons expressing M18Y473D is explained by a docking defect, synapse ultrastructure was assessed using electron microscopy. M18Y473D‐expressing neurons had similar synapse morphology, active zone length, and numbers of docked SVs at the active zone as M18WT‐expressing neurons (Fig 3A–E). This suggests that M18Y473D retained its known function in SV docking (Voets et al, 2001; Toonen et al, 2006). Since cryofixation can uncover docking defects that are not visible using conventional chemical fixation (Imig et al, 2014), docking was assessed in an independent experiment using cryofixed samples. In line with the chemically fixed samples, synapses expressing M18Y473D had a similar morphology and normal vesicle docking, as compared to those expressing M18WT (Fig 3F–J). Based on these results, we conclude that the phosphomimetic mutant, M18Y473D, abolishes synaptic transmission in a post‐docking step.
Figure 3. Docked synaptic vesicles are halted in a pre‐primed state.
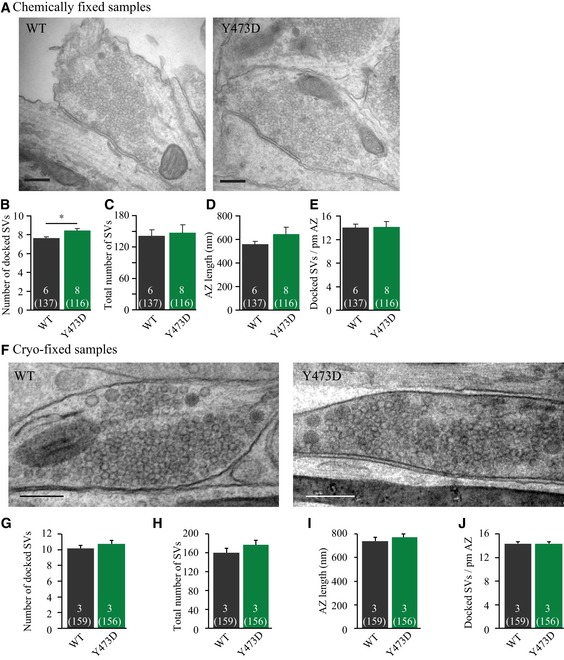
- Typical electron microscopy images from both groups. Scale bar = 100 nm.
- Average number of docked SV (M18WT: 7.63 ± 0.15 SVs; M18Y473D: 8.45 ± 0.20 docked SVs, multilevel analysis, *P = 0.047).
- Total number of SVs is not different (M18WT: 142 ± 11 SVs; M18Y473D: 148 ± 15 SVs, multilevel analysis, P = 0.456).
- The active zone (AZ) length is similar (M18WT: 561 ± 24 nm; M18Y473D: 646 ± 59 nm, multilevel analysis, P = 0.161).
- There is no change in the number of docked SVs per AZ length (M18WT: 14.0 ± 0.6 docked SVs/pm AZ; M18Y473D: 14.1 ± 0.9 docked SVs/pm AZ, multilevel analysis, P = 0.934).
- Typical electron microscopy images from both groups. Scale bar = 200 nm.
- Average number of docked SV is equal (M18WT: 10.17 ± 0.38 SVs; M18Y473D: 10.75 ± 0.40 docked SVs, multilevel analysis, P = 0.264).
- Total number of SVs is similar (M18WT: 160.2 ± 9.7 SVs; M18Y473D: 176.7 ± 9.2 SVs, multilevel analysis, P = 0.212).
- The active zone (AZ) length is similar (M18WT: 740.2 ± 29.7 nm; M18Y473D: 771.4 ± 28.1 nm, multilevel analysis, P = 0.871).
- There is no change in the number of docked SVs per AZ length (M18WT: 14.36 ± 0.32 docked SVs/pm AZ; M18Y473D: 14.34 ± 0.32 docked SVs/pm AZ, multilevel analysis, P = 0.969).
High‐frequency stimulation temporarily enhances synaptic transmission in M18Y473D‐expressing neurons
During trains of action potentials, we observed that M18Y473D‐expressing neurons developed robust facilitation, which led to a transient increase in synchronous release during sustained stimulation (Fig 4A and B). M18WT‐expressing neurons showed synaptic depression under the same circumstances. At the highest frequency (40 Hz), this difference in facilitation/depression ratio between M18Y473D‐ and M18WT‐expressing neurons led to a full rescue of synaptic transmission after approximately 1.5 s (Fig 4B). At these high frequencies, synaptic transmission is largely asynchronous after the first responses (Fig 4B), and therefore, EPSC charge instead of amplitude was used to quantify the amount of current induced by transmitter release during such intense stimulation. These results indicate that the loss‐of‐function phenotype upon mutating a key residue for tyrosine phosphorylation in Munc18‐1 can be temporarily bypassed by high‐frequency stimulation (HFS). This sustained stimulation‐induced rescue of synaptic transmission is very similar to rescue previously observed for neurons lacking Munc13‐1 (Rosenmund et al, 2002), CAPS1/2 (Jockusch et al, 2007), or RIM‐BP (Liu et al, 2011).
Figure 4. Synaptic transmission is partly restored during sustained stimulation.
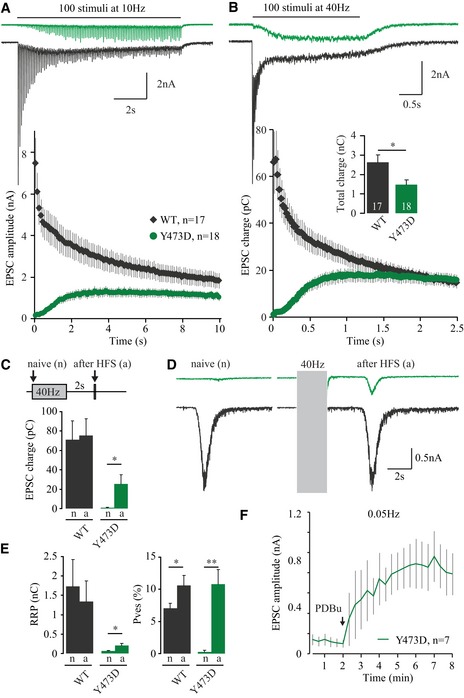
- Rundown of EPSC amplitude during 100 pulses at 10 Hz. Top: example traces of the current evoked by 40‐Hz stimulation.
- EPSC charge during 100 pulses at 40 Hz. Top: example traces of the current evoked by 40‐Hz stimulation. Insert shows total charge transferred during entire train (M18WT: 2.63 ± 0.38 nC, n = 18; M18Y473D: 1.48 ± 0.24 nC, n = 18; t‐test with Welch correction, *P = 0.0177).
- Synaptic transmission was assessed before [naive (n)] and 2 s after (a) 40‐Hz stimulation. Average EPSC charge M18WT (naive: 70.9 ± 19.5 pC; after HFS: 75.4 ± 17.4 pC, n = 12, paired t‐test, P = 0.4049). Average EPSC charge M18Y473D (naive: 0.888 ± 0.342 pC, after HFS: 25.6 ± 9.3 pC, n = 9, paired t‐test, *P = 0.0271).
- A hypertonic sucrose solution (500 mM, 3.5 s) was applied to release the RRP before [naive (n)] or 2 s after (a) HFS (100 pulses at 40 Hz, gray box).
- Quantification of RRP and P ves (EPSC charge/RRP charge * 100) from (C and D). Average RRP size M18WT (naive: 1.73 ± 0.69 nC, after HFS: 1.34 ± 0.54 nC, n = 12, paired t‐test, P = 0.0557). Average RRP size M18Y473D (naive: 0.061 ± 0.019 nC, after HFS: 0.207 ± 0.050 nC, n = 9, paired t‐test, *P = 0.0246). Average P ves M18WT (naive: 7.07 ± 0.76%, after HFS: 10.6 ± 1.6%, n = 12, paired t‐test, *P = 0.0201). Average P ves M18Y473D (naive: 0.27 ± 0.22%, after HFS: 10.8 ± 2.2%, n = 9, paired t‐test, **P = 0.0012).
- PDBu application (1 μM) enhanced EPSC size in neurons expressing M18Y473D.
To gain further insight in this mechanism, synaptic transmission after high‐frequency stimulation (HFS) was studied in more detail. First, EPSC size was tested at the start and 2 s after HFS. While neurons expressing M18WT returned to their naive EPSC size 2 s after HFS, the EPSC size was augmented in neurons expressing M18Y473D (Fig 4C). Second, neurons were subjected to hypertonic sucrose before (naive) and after HFS to assess the RRP size and vesicular release probability (P ves, Fig 4D and E). Neurons expressing M18Y473D showed a pronounced increase in P ves and a modest increase in RRP size after HFS (Fig 4E). This suggests that increased release probability is the main basis for augmentation in M18Y473D‐expressing neurons.
Activation of the diacylglycerol (DAG) pathway is a well‐known modulatory pathway to enhance release probability (Lou et al, 2008; de Jong & Verhage, 2009). We tested whether this pathway could still be activated in neurons expressing M18Y473D using the phorbol ester PDBu. Bath application of PDBu enhanced the EPSC size (Fig 4F). Hence, both HFS and phorbol ester activation can enhance synaptic transmission in the loss‐of‐function M18Y473D mutant.
Promoting extension of helix 12 largely restores synaptic transmission in M18Y473D‐expressing neurons
The debate about the positive role of Munc18‐1 in exocytosis has recently converged onto conformational changes in helix 12 of domain 3a of Munc18‐1 (Fig 5A), which is thought to prime vesicles by clashing with the closed configuration of Syntaxin1 and forming a template for Synaptobrevin/VAMP2 (Hu et al, 2011; Parisotto et al, 2014). This property might be similar to the recently elucidated role of a related SM protein, Vps33, in SNARE complex assembly [“SNARE‐templating” (Baker et al, 2015)]. This hypothesis is now supported in vivo in mouse adrenal chromaffin cells, where extension of helix 12 promotes vesicle priming (Munch et al, 2016). In view of these developments, we tested whether the priming‐defective phosphomimetic M18Y473D affected the low‐affinity interaction with Synaptobrevin/VAMP2. Indeed, pull‐down assays using immobilized purified Munc18 constructs demonstrated that M18Y473D reduces Synaptobrevin/VAMP2 binding by ~80% (Fig 5B). This suggests that Y473 phosphorylation fails to produce a Munc18‐1/Synaptobrevin/VAMP2 template to promote SNARE complex formation.
Figure 5. Promoting helix 12 extension partly rescues the inhibitory effect of Y473D.
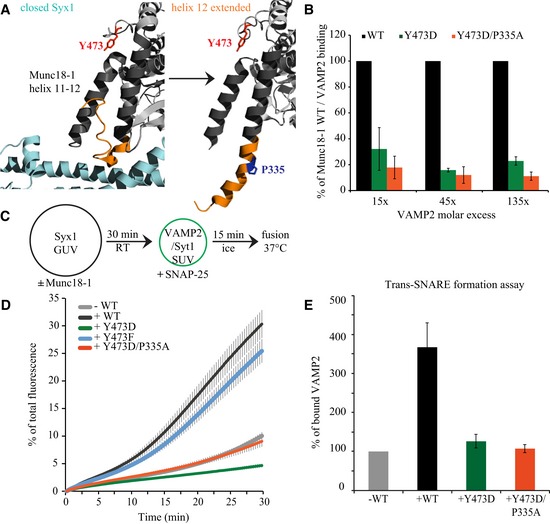
- GST‐Munc18‐1 constructs were immobilized on glutathione beads and incubated with the indicated molar excess of Synaptobrevin/VAMP2 (over Munc18‐1) for 1.5 h at 4°C. Bound Synaptobrevin/VAMP2 was analyzed by Western blot and immune decorating with an anti‐Synaptobrevin/VAMP2 antibody and quantified by the LI‐COR system and ImageJ software. Synaptobrevin/VAMP2 bound to the GST‐Munc18‐1 mutants is represented as percentage of Synaptobrevin/VAMP2 bound to GST‐Munc18‐1 WT. Error bars indicate s.e.m., n = 3.
- GUVs (14 nmol lipid) containing Syx1 (14 pmol) were pre‐incubated with the indicated Munc18‐1 constructs (90 pmol) at room temperature for 30 min. Subsequently, samples were incubated with soluble SNAP‐25 (128 pmol) and SUVs (2.5 nmol lipid) containing Synaptotagmin1 (3 pmol) and Synaptobrevin/VAMP2 (8 pmol) for 15 min on ice. Lipid mixing was started by increasing the temperature to 37°C.
- Lipid mixing was monitored by the increase of Atto 655 fluorescence for 30 min. Data were normalized to the maximum fluorescence after liposome lysis by detergent. The minimum fluorescence was set to 0%. Error bars = s.e.m., N = 3.
- Trans‐SNARE formation assay: t‐ and v‐SNARE SUVs were incubated in the presence or absence of different Munc18‐1 constructs for 30 min at 4°C. After solubilization and precipitation of the t‐SNAREs using nickel beads, presence of full‐length VAMP2 in the precipitates was probed by immunoblotting, which was used as an indicator for trans‐SNARE assembly between SUVs. Data are represented as mean ± s.e.m. N = 3 replicates.
Source data are available online for this figure.
Extension of helix 12 can be artificially promoted by mutating a proline hinge into alanine, P335A (Fig 5A), leading to a gain‐of‐function phenotype (Parisotto et al, 2014; Munch et al, 2016). We therefore tested whether promoting extension of helix 12 using the P335A mutation would restore SNARE complex formation and synaptic transmission in M18Y473D. To detect the effect on SNARE complex assembly, we used Syntaxin1 GUVs, soluble SNAP‐25, and VAMP2/Synaptotagmin1 SUVs in our reconstituted liposome assay (Fig 5C). The double mutant M18Y473D/P335A did not increase Synaptobrevin/VAMP2 binding (Fig 5B), but improved liposome fusion compared to M18Y473D (Fig 5C and D). As expected, M18WT stimulated lipid mixing and M18Y473D retained its prominent inhibitory effect (Fig 5D), consistent with Fig 1F–I. Similar results were obtained in a membrane mixing assay (Fig EV2C).
The use of monomer Syntaxin, instead of assembled t‐SNAREs, allows the formation of inhibitory Syntaxin‐Munc18‐1 dimers. As reported previously (Schollmeier et al, 2011), subsequent incubation with SNAP‐25 and VAMP2‐SNARE liposomes reverses the inhibitory effect of Munc18‐1 and allows Munc18‐1 to stimulate fusion. The double mutant M18Y473D/P335A could have improved liposome function by reversing the inhibitory effect of M18Y473D or by (partly) restoring the stimulatory effect of Munc18‐1. To distinguish between these options, a trans‐SNARE formation assay was performed in which t‐SNARE SUVs were mixed with v‐SNARE SUVs and incubated with or without Munc18‐1 (Fig 5E). Surprisingly, while M18WT promoted VAMP2 binding to t‐SNAREs by 350%, M18Y473D and M18Y473D/P335A did not. Hence, a mutation that promotes helix 12 extension and SNARE complex formation partially rescues the inhibitory effect of the phosphomimetic M18Y473D mutation in vitro, probably by rendering M18Y473D unable to inhibit fusion or by reversing the inhibitory effect of Munc18‐1.
To test whether the M18Y473D/P335A double mutant also rescues synaptic transmission, we expressed M18Y473D, M18Y473D/P335A, or M18WT in munc18‐1 null hippocampal neurons. Indeed, the defect in synaptic transmission in neurons expressing M18Y473D was largely restored in neurons expressing the double mutant M18Y473D/P335A (Fig 6A–C). In addition, while neurons expressing M18Y473D showed facilitation during HFS, normal synaptic depression was largely restored in neurons expressing M18Y473D/P335A, exhibiting similar kinetics as neurons expressing M18WT (Fig 6D). Taken together, these data suggest that while Y473 phosphorylation interferes with VAMP2 binding and SNARE complex assembly, synaptic transmission could be (partly) restored by helix 12 extension.
Figure 6. Promoting helix 12 extension partly rescues synaptic transmission.
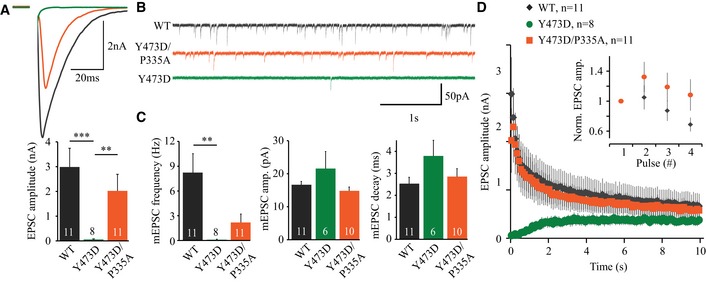
- Typical examples of single EPSCs. EPSC amplitude (M18WT: 2.98 ± 0.76 nA, n = 11; M18Y473D: 0.057 ± 0.022 nA, n = 8, M18Y473D/P335A: 2.02 ± 0.67 nA, n = 11, Kruskal–Wallis test, P < 0.0002, Dunn's multiple comparison test, M18WT versus M18Y473D/P335A = P > 0.05, M18WT versus M18Y473D = ***P < 0.001, M18Y473D versus M18Y473D/P335A = **P < 0.01).
- Typical traces of spontaneous release.
- mEPSC frequency (M18WT: 8.23 ± 2.29 Hz, n = 11; M18Y473D: 0.096 ± 0.056 Hz, n = 8, M18Y473D/P335A: 2.18 ± 1.02 Hz, n = 11, Kruskal–Wallis test, P = 0.2390), mEPSC amplitude (M18WT: 16.7 ± 1.0 pA, n = 11; M18Y473D: 21.6 ± 5.2 pA, n = 6, M18Y473D/P335A: 14.8 ± 1.2 pA, n = 10, ANOVA, P = 0.1465), and decay time (M18WT: 2.53 ± 0.29 ms, n = 11; M18Y473D: 3.79 ± 0.71 ms, n = 6, M18Y473D/P335A: 2.85 ± 0.36 pA, n = 10, Kruskal–Wallis test, P < 0.0003, Dunn's multiple comparison test, M18WT versus M18Y473D/P335A = P > 0.05, M18WT versus M18Y473D = ***P < 0.001, M18Y473D versus M18Y473D/P335A = P > 0.05).
- Depression of EPSC amplitude during a 10‐Hz train. Inset shows first four pulses normalized to the first EPSC.
A non‐phosphorylatable mutant, M18Y473F, does not affect synaptic transmission
To address whether the opposite situation, preventing tyrosine phosphorylation, also affects synaptic transmission, we tested the ability of a non‐phosphorylatable Munc18‐1 mutant, M18Y473F, to support synaptic transmission in munc18‐1 null neurons. The EPSC amplitude was unaffected in the non‐phosphorylatable mutant (Fig 7A), as was the amplitude and frequency of spontaneous release events (Fig 7B). Short‐term plasticity during 10‐ or 40‐Hz train stimulation was comparable in neurons expressing M18Y473F or M18WT (Fig 7C and D). Also, the EPSC recovery after RRP depletion by a 40‐Hz train was similar (Fig 7E). To conclude, synaptic transmission is unaltered in the non‐phosphorylatable M18Y473F mutant, suggesting that tyrosine phosphorylation of Munc18‐1 at Y473 does not play a role in basal synaptic transmission.
Figure 7. Preventing tyrosine phosphorylation on Y473 has no effect on synaptic transmission.
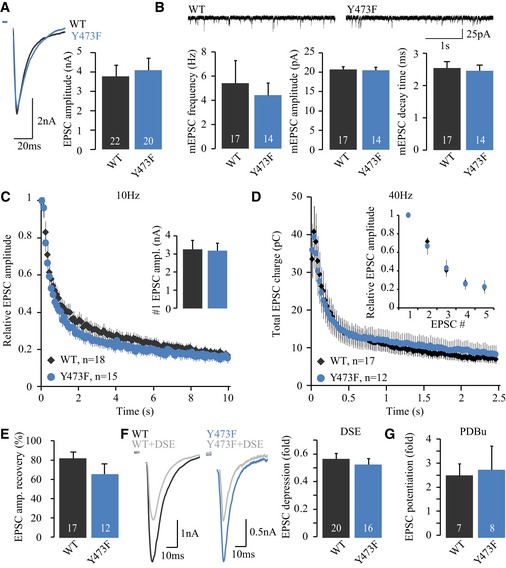
- Evoked release upon action potential stimulation. Average EPSC amplitude (M18WT: 3.78 ± 0.58 nA, n = 22; M18Y473F: 4.10 ± 0.61 nA, n = 20, Mann–Whitney U‐test, P = 0.7915). Typical responses are depicted on the top.
- Average mEPSC frequency (M18WT: 5.42 ± 1.86 Hz, n = 17; M18Y473F: 4.42 ± 1.01 Hz, n = 14, t‐test with Welch correction, P = 0.6424), mEPSC amplitude (M18WT: 20.72 ± 0.7 pA, n = 17; M18Y473F: 20.5 ± 0.9 pA, n = 14, t‐test, P = 0.8373), and mEPSC decay time (M18WT: 2.54 ± 0.20 ms, n = 17; M18Y473F: 2.46 ± 0.18 ms, n = 14, t‐test, P = 0.7550). Example traces of spontaneous release are depicted on top.
- EPSC depression during 10‐Hz stimulation is normal.
- Depression of EPSC charge during 40‐Hz stimulation is normal. Inset shows the first five pulses.
- EPSC amplitude recovery 2 s after an RRP depleting 40‐Hz train. Average EPSC recovery (M18WT: 82.2 ± 6.7%, n = 17; M18Y473F: 65.5 ± 11.0%, n = 12, t‐test, P = 0.1805).
- The amount of DSE‐triggered EPSC depression was quantified by the ratio of the average EPSC amplitude (taken over four pulses given at 0.2 Hz) before or immediately following 10‐s depolarization to 0 mV. Typical EPSCs before and following DSE induction are depicted on the left. Average DSE (M18WT: 0.57 ± 0.04 fold, n = 20; M18Y473F: 0.53 ± 0.04 fold, n = 16, t‐test, P = 0.5227).
- PDBu was bath‐applied during 0.05‐Hz stimulation. Maximum potentiation of EPSC amplitude (M18WT: 2.56 ± 0.48 fold, n = 7; M18Y473F: 2.82 ± 0.98 fold, n = 8, t‐test with Welch correction, P = 0.8168).
To address this issue further, neurons were incubated for 1 h with Src family kinase inhibitor PP2 (20 μM) or its inactive form PP3 as a negative control. In both neurons expressing M18WT and M18Y473F, PP2 did not affect EPSC size (Fig EV3A). In addition, PP2 treatment did not modify the paired‐pulse ratio between two subsequent pulses, suggestive of unchanged release probability (Fig EV3B). Thus, inhibiting Src activity did not affect basal synaptic transmission. Conversely, we aimed to test the consequences of increased Src kinase activity on synaptic transmission by overexpressing Flag‐tagged Src in cultured neurons. Indeed, n‐Src overexpression led to higher cellular levels (Fig EV3C). Surprisingly, total tyrosine phosphorylation levels in the cells did not increase (except for a single band at the height of Src kinase itself) and no detectable increase in tyrosine phosphorylation of Munc18‐1 was observed (Fig EV3C). These results suggest that Src activity is under tight homeostatic regulation, consistent with previous conclusions that Src is largely inactive under basal conditions (Roskoski, 2005) and that physiologically relevant phosphorylation of SFK substrates may be highly local and/or transient.
Figure EV3. Src activity in cultured neurons.
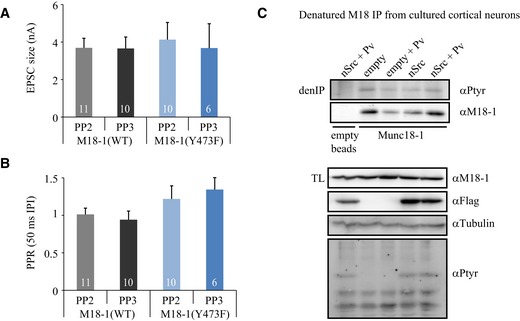
- Autaptic excitatory munc18‐1 neurons expressing M18WT or M18Y473F were subjected to 1‐h incubation with Src family kinase inhibitor PP2 or its inactive form PP3. EPSC size was unaffected by drug treatment (M18WT + PP2: 3.69 ± 0.51 nA, n = 11; M18WT + PP3: 3.65 ± 0.62 nA, n = 10; M18Y473F + PP2: 4.13 ± 0.92 nA, n = 10; M18Y473F + PP3: 3.68 ± 1.30 nA, n = 6).
- Two pulses were given with an interpulse interval of 50 ms. Paired‐pulse ratio (PPR: EPSC#2/EPSC#1) was not affected by drug treatment (M18WT + PP2, n = 11: 1.01 ± 0.08; M18WT + PP3: 0.94 ± 0.12, n = 10; M18Y473F + PP2: 1.22 ± 0.17, n = 10; M18Y473F + PP3: 1.34 ± 0.16, n = 6).
- Neuronal cultures were subjected to protein tyrosine phosphatase inhibitor vanadate and/or overexpression of Flag‐tagged n‐Src. Munc18‐1 tyrosine phosphorylation was determined by denatured Munc18‐1 IP and immunoblotting with a phosphotyrosine antibody.
Munc18‐1 phosphorylation at Y473 provides a plausible explanation for forms of inhibitory synaptic plasticity, such as depolarization‐induced suppression of excitation (DSE). This form of synaptic plasticity has been observed in autaptic hippocampal neurons in response to prolonged depolarization and depends on retrograde endocannabinoid signaling (Straiker & Mackie, 2005). However, applying a ten‐second depolarization step to munc18‐1 KO neurons expressing M18Y473F or M18WT led to a pronounced but similar reduction in EPSC size in both groups (Fig 7F), suggesting that tyrosine phosphorylation of Munc18‐1 does not play a role in DSE.
Finally, we investigated whether PKC and Src phosphorylation on Munc18‐1 were interdependent. For this purpose, phorbol ester‐induced potentiation was monitored in the M18Y473F mutant. Both M18WT‐ and M18Y473F‐expressing neurons showed similar enhancement of synaptic transmission upon application of 1 μM PDBu (Fig 7G).
Other structural modifications at Y473 affect synaptic transmission similar to Y473D
As indicated by the data in Figs 1, 2, 3, 4, 5, structural changes at Y473 (phosphorylation) appear to affect the function of helix 12. To strengthen this case further, we removed the aromatic ring at Y473 using the neutral amino acid alanine, M18Y473A. As expected, this substitution also reduced phosphorylation of Munc18‐1 by SFKs when overexpressed in HEK293T cells (phosphoM18/totalM18 normalized to M18WT: M18Y473A: 0.26 ± 0.03 a.u., N = 3, Fig 8A). When expressed in munc18‐1 KO neurons, M18Y473A rescued neuronal viability and morphology as efficiently as M18WT (Fig 8B and C). Evoked release was strongly reduced, resulting from a reduction in RRP size and P ves (Fig 8D and E). Although these effects are pronounced, the reduction in RRP size and P ves was slightly less severe than observed for M18Y473D (Fig 2C–E). Spontaneous release frequency was also strongly affected, but not the amplitude or decay time (Fig 8F). Furthermore, ultrastructural morphometry showed that the amount of vesicles docked at the active zone was reduced in M18Y473A‐expressing neurons compared to controls, also taken into account a small reduction in the active zone length in synapses expressing M18Y473A (Fig 8G). This docking defect appears to be an unlikely explanation for the larger reduction in RRP size and almost complete inhibition of spontaneous release. Hence, the M18Y473A mutation appears to impair both docking and post‐docking functions of Munc18‐1.
Figure 8. Structural modifications to Tyr473 strongly affect synaptic transmission.
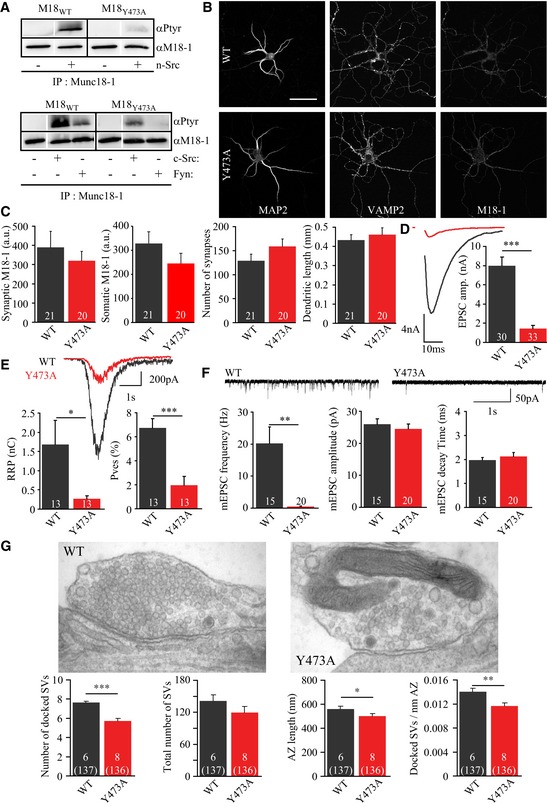
- Using denatured IP, Munc18‐1 was pulled down from cell lysate of HEK293T cells expressing M18WT or M18Y473A together with SFK kinases or an empty vector as control. Munc18‐1 was then immunoblotted for tyrosine phosphorylation using the 4G10 antibody. The total amount of Munc18‐1 was detected after stripping and reblotting for Munc18‐1.
- Typical confocal images of neurons stained for MAP2, Synaptobrevin/VAMP2, and Munc18‐1. Scale bar = 50 μm.
- Quantification of confocal images. Average synaptic Munc18‐1 intensity (M18WT: 389 ± 85 a.u., n = 21; M18Y473A: 321 ± 48 a.u., n = 20, t‐test with Welch correction, P = 0.4868), somatic Munc18‐1 intensity (M18WT: 328 ± 49 a.u., n = 21; M18Y473A: 245 ± 42 a.u., n = 20, t‐test, P = 0.2810), synapse number (M18WT: 129 ± 14, n = 21; M18Y473A: 159 ± 16, n = 20, t‐test, P = 0.1548), and dendrite length (M18WT: 0.433 ± 0.028, n = 21; M18Y473A: 0.461 ± 0.037, n = 20, t‐test, P = 0.5473).
- Evoked release upon action potential stimulation. Average EPSC amplitude (M18WT: 7.96 ± 0.94 nA, n = 30; M18Y473A: 1.44 ± 0.34 nA, n = 33, t‐test with Welch correction, ***P < 0.0001). Typical responses are depicted on the right.
- Release by hyperosmotic sucrose application (500 mM, 3.5 s) was used to assess the RRP. Left: RRP charge (M18WT: 1.68 ± 0.64 nC, n = 13; M18Y473A: 0.27 ± 0.08 nC, n = 13, t‐test with Welch correction, *P = 0.0487). Typical responses are depicted in the middle. Right: vesicular release probability per neuron (EPSC charge/RRP charge). Mean P ves (M18WT: 6.75 ± 0.77%, n = 13; M18Y473A: 1.98 ± 0.72%, n = 13, t‐test, ***P = 0.0001).
- Top: example traces of spontaneous release of single vesicles (mEPSCs). Average mEPSC frequency (M18WT: 20.18 ± 5.18 Hz, n = 15; M18Y473A: 0.48 ± 0.15 Hz, n = 20, t‐test with Welch correction, **P = 0.0019), amplitude (M18WT: 26.0 ± 1.7 pA, n = 15; M18Y473A: 24.5 ± 1.6 pA, n = 20, t‐test, P = 0.5334), and decay time (M18WT: 1.98 ± 0.10 ms, n = 15; M18Y473A: 2.13 ± 0.17 ms, n = 20, t‐test with Welch correction, P = 0.4499).
- Typical electron microscopy images from rescued autaptic hippocampal Munc18‐1 null neurons (scale bar = 100 nm). Neurons expressing M18Y473A have less synaptic vesicles (SV) docked at the active zone. Average number of docked SV (M18WT: 7.63 ± 0.15 SVs; M18Y473A: 5.71 ± 0.28 SVs, multilevel analysis, ***P < 0.001), total number of SVs (M18WT: 142 ± 11 SVs; M18Y473A: 120 ± 12 SVs, multilevel analysis, P = 0.077), active zone (AZ) length (M18WT: 561 ± 24 nm; M18Y473A: 501 ± 21 nm, multilevel analysis, *P = 0.015), and docked SVs per AZ length (M18WT: 0.0140 ± 0.0006 docked SVs/nm AZ; M18Y473A: 0.0117 ± 0.0005 docked SVs/nm AZ, multilevel analysis, **P = 0.003). M18WT: N = 6 autaptic neurons, n = 137 synapses; M18Y473A: N = 8 autaptic neurons, n = 136 synapses.
As was observed for M18Y473D (Fig 4A and B), EPSCs also facilitated in M18Y473A‐expressing neurons during sustained stimulation (Fig 9A–C) and were augmented immediately after a train in a frequency‐dependent manner (Fig 9D). As was described above for M18Y473D‐expressing neurons, this augmentation resulted from a pronounced increase in P ves and modest increase in RRP size (Fig 9E). In conclusion, removing the aromatic ring at the tyrosine phosphorylation site of Munc18‐1 produces a strong loss‐of‐function phenotype, which is, however, not as severe as the addition of a negative charge that mimics the phosphorylated state (M18Y473D). Similar to M18Y473D‐expressing neurons, M18Y473A‐expressing neurons show strong enhancement of synaptic transmission upon high‐frequency stimulation by increasing both P ves and RRP size.
Figure 9. Altered short‐term plasticity in neurons expressing M18Y473A .
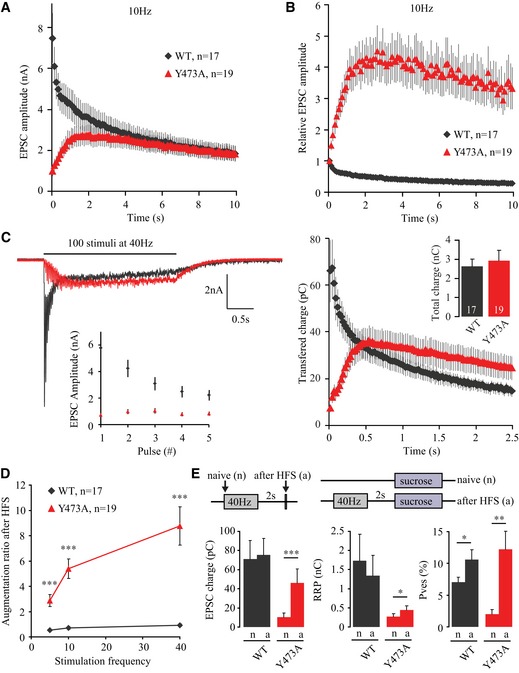
- Absolute EPSC amplitudes during a 10‐Hz train.
- EPSC amplitudes during a 10‐Hz train normalized to the first EPSC.
- Left, upper panel: example traces of the currents evoked by 40‐Hz stimulation. Left, lower panel: first five pulses of the 40‐Hz train. Right: transferred charge during 40‐Hz train. Insert shows total charge transferred during entire 40‐Hz train (M18WT: 2.63 ± 0.38 nC, n = 17; M18Y473A: 2.93 ± 0.54 nC, n = 19, t‐test, P = 0.6630).
- EPSC augmentation is calculated by dividing the EPSC amplitude of a single pulse after a stimulation train by the amplitude of the first EPSC within this train. Mean augmentation after 5‐Hz train (M18WT: 0.54 ± 0.05, n = 8; M18Y473A: 2.88 ± 0.47, n = 10; Unpaired t‐test with Welch correction, ***P = 0.0008), 10‐Hz train (M18WT: 0.70 ± 0.10, n = 8; M18Y473A: 5.39 ± 0.77, n = 10; unpaired t‐test with Welch correction, ***P = 0.0002), and 40‐Hz train (M18WT: 0.93 ± 0.11, n = 8; M18Y473A: 8.77 ± 1.52, n = 10; unpaired t‐test with Welch correction, ***P = 0.0006).
- A single action potential or sucrose application (500 mM, 3.5 s) was given before (naive) or 2 s after HFS (100 pulses at 40 Hz). Average EPSC charge M18WT (naive: 70.9 ± 19.5 pC, after HFS: 75.4 ± 17.4 pC, n = 12, paired t‐test, P = 0.4049). Average EPSC charge M18Y473A (naive: 10.3 ± 4.5 pC, after HFS: 46.2 ± 14.5 pC, n = 13, Wilcoxon matched‐pairs signed‐ranks test, ***P = 0.0005). Average RRP size M18WT (naive: 1.73 ± 0.69 nC, after HFS: 1.34 ± 0.54 nC, n = 12, paired t‐test, P = 0.0557). Average RRP size M18Y473A (naive: 0.268 ± 0.075 nC, after HFS: 0.439 ± 0.107 nC, n = 13, paired t‐test, *P = 0.0100). Average P ves M18WT (EPSC charge/RRP charge * 100) (naive: 7.07 ± 0.76%, after HFS: 10.6 ± 1.6%, n = 12, paired t‐test, *P = 0.0201). Average P ves M18Y473A (naive: 1.98 ± 0.72%, 12.2 ± 2.8%, n = 13, paired t‐test, **P = 0.0021).
Discussion
This study confirms that Munc18‐1 is tyrosine‐phosphorylated at Y473, as previously observed in open screens (Ballif et al, 2008), and identified neuronal SFK members n‐Src, c‐Src, and Fyn as effective kinases. We found that replacing Munc18‐1 with a non‐phosphorylatable version supports normal synaptic transmission, but expression of a phosphomimetic mutant produces a > 98% reduction in synaptic transmission due to a drastic reduction in the number of primed vesicles and impaired release probability of the rare primed vesicles. Hence, tyrosine phosphorylation of Munc18‐1 is a potent way to shut down synaptic transmission upon SFK activation. The block on synaptic transmission could be partly restored by high‐frequency stimulation and by promoting the extension of helix 12 of Munc18‐1.
Src family kinases are mainly known for their postsynaptic role in LTP (Salter & Kalia, 2004), but are also expressed in presynaptic terminals (Onofri et al, 2007) and were found to inhibit glutamate release (Ohnishi et al, 2001; Baldwin et al, 2006; Onofri et al, 2007), although a stimulatory role has also been observed (Shyu et al, 2005). These studies were performed using Src inhibitors in synaptosomes and the resolution/timescale of such experiments does not exclude indirect effects. Insights into the physiological triggers of (presynaptic) Src activity remain elusive. SFKs are typically in a dormant state and tonically inhibited via multiple endogenous inhibitors and reversing the dormant state contributes to cancer growth. Physiological SFK activation (in mitotic cells) requires a multistep process, termed unlatch‐unclamp‐switch (Roskoski, 2015). This tight regulation of SFK kinases is in line with our finding that increased SFK levels do not produce enhanced kinase activity (Fig EV3C), as shown before (Onofri et al, 1997; Kotani et al, 2007), or only within a short time frame (Zhao et al, 2000). This is further complicated by the fact that Src activation is likely to be compartmentalized, as exemplified by Synapsin‐mediated Src activation on purified synaptic vesicles (Onofri et al, 1997, 2007). Hence, it remains uncertain under which exact conditions SFKs are activated in the brain and the nerve terminal. However, it is beyond doubt that SFK regulation is relevant for the brain (Purcell & Carew, 2003; Kalia et al, 2004) and we have confirmed that native brain Munc18‐1 is phosphorylated by SFKs. When that happens, synaptic transmission is potently inhibited.
The > 98% reduction in synaptic transmission in neurons expressing the phosphomimetic Y473D mutation might in principle be explained by a loss of functional Munc18‐1 protein, since the munc18‐1 null mutant shows a complete loss of synaptic transmission (Verhage et al, 2000). However, M18Y473D protein still retained several aspects of its normal function. First, M18Y473D expression rescued the lethal munc18‐1 null phenotype (Heeroma et al, 2004). Second, a stable protein was purified from heterologous cells and bound Syntaxin1, indicating that the mutant protein has normal overall tertiary structure. Third, Munc18‐1 protein is correctly targeted to synapses (Fig 2A and B). Fourth, the reconstituted membrane fusion assay indicates that the inhibitory function of Munc18‐1, when the t‐SNARE complex is not pre‐assembled, is unaffected by the Y473D mutation (Fig 1H and I). Together, these arguments indicate that the Y473D mutation does not produce a completely dysfunctional protein, but a mutant protein that is correctly folded, stable, and targeted to the correct locations binds its natural binding partner and retains some aspects of its natural function, but has a selective and highly efficient inhibitory effect on synaptic transmission.
Replacing Munc18‐1 Y473 with alanine led to a profound defect in synaptic transmission, although less severe than M18Y473D, while replacing with phenylalanine, a non‐phosphorylatable substitution similar to tyrosine (M18Y473F) did not. In previous studies on Tyr phosphorylation of other proteins, acidic amino acid (glutamate, aspartate) and alanine substitutions for tyrosine also produced effects in the same direction (Potter et al, 2005; Hussain et al, 2007; Anthis et al, 2009). Alanine and aspartate both lack the bulky, hydrophobic phenol group typical for phenylalanine and tyrosine. Removing the phenol group is probably sufficient to introduce local changes in Munc18‐1 and inhibit synaptic transmission, while addition of the highly charged PO3 − group by tyrosine phosphorylation enhances this effect. Although amino acid substitutions are a widely used option to investigate the molecular and cellular impact of tyrosine phosphorylation and have produced many important insights in the physiology of kinases, such substitutions do not fully mimic phosphotyrosine since the position of the negative charge is different. However, structural models of Munc18‐1 and other SM proteins (Misura et al, 2000; Baker et al, 2015) suggest that phosphorylation on position Y473 interferes with the VAMP2 binding groove. This is exactly what we observed experimentally for the phospho‐mimic mutant: M18Y473D does not bind VAMP/Synaptobrevin (Fig 5B). Hence, this functional effect of the phospho‐mimic suggests that this amino acid substitution is a valid approach to probe the cellular effects of Munc18‐1 phosphorylation.
The loss of synaptic transmission in neurons expressing M18Y473D is mainly caused by a drastic reduction in the number of primed vesicles (Fig 2D), while electron microscopy using different fixation methods (chemical fixation and cryofixation) indicates that vesicle docking is unaffected (Fig 3). This suggests that Tyr phosphorylation of Munc18‐1 inhibits a step downstream of docking/tethering in the supply or retention of primed (readily releasable) vesicles. However, neurons expressing M18Y473D (and M18Y473A) also have a decreased vesicular release probability and a disproportionally large reduction in spontaneous release. Thus, in addition to the main priming defect, our data suggest that Tyr phosphorylation of Munc18‐1 also inhibits the release probability of the remaining primed vesicles.
Evidence is accumulating that extension of helix 12 in domain 3a of Munc18‐1 acts as a molecular switch arranging the transition from the inhibitory Munc18‐1/Syntaxin1 dimer to a conformation that allows Synaptobrevin/VAMP2 interaction, SNARE complex formation, and vesicle priming (Xu et al, 2010; Hu et al, 2011; Parisotto et al, 2014; Munch et al, 2016). Y473 is not located inside the flexible region of domain 3a (aa 324–339, Fig 1C), but at the interface between domain 3b and 3a, close enough to potentially hinder conformational changes in domain 3a. This suggestion is supported by three observations: (i) M18Y473D inhibits the interaction with Synaptobrevin/VAMP2 (Fig 5B). (ii) Y473 substitutions produce a phenocopy of L348R, a single point mutation in helix 12, which also blocks Synaptobrevin/VAMP2 interaction and inhibits priming (Parisotto et al, 2014; Munch et al, 2016). (iii) P335A, which promotes helix 12 extension, compensates for the inhibitory effects of the M18Y473D phosphomimetic mutant (Fig 6). Hence, interfering with Synaptobrevin/VAMP2 binding and/or the extension of helix 12 in domain 3a is a plausible explanation for the inhibition of vesicle priming upon Munc18 Y473 phosphorylation. On the other hand, when the stimulatory function of Munc18‐1 was probed with a trans‐SNARE formation assay (using pre‐assembled t‐SNAREs), the double mutant P335A/Y473D still showed an impaired stimulatory function. This is consistent with the observation that the double mutant did not restore VAMP2 binding, which is critical for the stimulatory function of Munc18‐1 in vitro (Schollmeier et al, 2011; Parisotto et al, 2014). We conclude that P335A either renders Y473D unable to inhibit fusion, which is likely because the P335A single mutant also stimulates fusion under conditions that normally inhibit fusion (Parisotto et al, 2014), or was not able to reverse the inhibitory effect of Munc18‐1.
While the initial primed pool was drastically reduced in neurons expressing M18Y473D or M18Y473A, sustained stimulation led to strong facilitation during HFS and frequency‐dependent augmentation after HFS. This suggests that activity‐dependent priming known to be activated by sustained stimulation (Neher & Sakaba, 2008) is still intact in neurons expressing these mutants and produces a primed vesicle pool. Interestingly, facilitation and frequency‐dependent augmentation were also observed in neurons lacking two other essential priming proteins, Munc13‐1 and CAPS1/2 (Rosenmund et al, 2002; Jockusch et al, 2007), and in neurons lacking RIM‐BP (Liu et al, 2011). This suggests that the Munc18‐1 mutants described in this study might exert (part of) their phenotype by interfering with Munc13, CAPS, and/or RIM‐BP functions. Functional interdependencies between Munc18‐1, CAPS, and Munc13 have been suggested before (Daily et al, 2010; Ma et al, 2011). However, the inability of M18Y473D to stimulate fusion in our reconstituted membrane fusion assay, which lacks CAPS, Munc13, and RIM‐BP, suggests that an inherent Munc18‐1 function is (also) inhibited (Fig 1E and F). Diacylglycerol (DAG) activates Munc13 (Rhee et al, 2002; Basu et al, 2007) and PKC, which phosphorylates Munc18‐1 (Wierda et al, 2007; Genc et al, 2014) and Synaptotagmin1 (de Jong et al, 2016) and increases release rates probably by lowering the energy barrier for fusion (Basu et al, 2007; Schotten et al, 2015). Our results suggest that tyrosine phosphorylation of Munc18‐1 does the opposite: In the phosphorylated state, the energy barrier for fusion appears to be increased, leading to lower vesicular release probability and low spontaneous fusion rates.
According to the Vps33/Nyv1 crystal structure (Baker et al, 2015), the R‐SNARE binding groove on SM proteins consists of two parts, a lower groove for the N‐terminal and an upper groove for the C‐terminal part of the R‐SNARE. If these interactions are indeed conserved among SM proteins, as suggested by Baker et al (2015) and supported by Sitarska et al (2017), phosphorylation of Munc18‐1 at Y473 is predicted to occlude the upper binding groove, thereby preventing Munc18‐1 from forming a template for SNARE complex assembly. This is supported by the reduced Synaptobrevin/VAMP2 binding efficiency of M18Y473D. Access of the R‐SNARE N‐terminus to the lower binding groove might be controlled by helix 12 extension as proposed by Parisotto et al (2014). However, while the double mutant M18Y473D/P335A showed largely normal synaptic transmission, Synaptobrevin/VAMP2 binding affinity and stimulation of membrane fusion were still impaired in the reconstituted membrane fusion assay, which does not contain the full set of regulatory components. Hence, Synaptobrevin/VAMP2 binding and the stimulatory role of Munc18‐1 in membrane fusion appear to be largely dispensable for synaptic transmission. Alternatively, binding of Synaptobrevin/VAMP2 to the upper C‐terminus binding groove promotes/stabilizes helix 12 extension, which is now bypassed by P335A. Taken together, our findings support a model in which tyrosine phosphorylation of Munc18‐1 directly occludes a Synaptobrevin/VAMP2 binding groove and as a consequence hinders conformational changes in domain 3a, thereby inhibiting SNARE complex assembly (“SNARE‐templating”).
Materials and Methods
Animals
Munc18‐1‐deficient mice were generated as described previously (de Vries et al, 2000). Munc18‐1 null mutant mice are stillborn and can be easily distinguished from wild‐type or heterozygous littermates. E18 embryos were obtained by cesarean section of pregnant females from timed mating of heterozygous mice. Newborn P0‐P1 pups from pregnant female Wistar rats were used for glia preparations. Animals were housed and bred according to institutional, Dutch, and U.S. governmental guidelines.
Constructs
Mouse n‐Src, c‐Src, and Fyn clones were ordered from Imagines, ligated into pCRblunt, and subcloned into pCMV‐3tag expression vectors. Single amino acid substitutions on Y473 in Munc18‐1 were generated using Quickchange (Stratagene) and verified by sequencing. For protein expression in bacteria, constructs were then subcloned into pGEX 4T3 (GE Healthcare Life Sciences). For expression in mammalian cells, constructs were subcloned into pLenti vectors, and viral particles were produced as described (Naldini et al, 1996). Transduction efficiencies of lentivirus containing Munc18‐1 were assessed on HEK293T cells using a concentration range, and were taken into account when viruses were applied to neuronal cultures. For this purpose, HEK293T cells were infected with lentivirus 1 day after plating in DMEM containing 10% fetal calf serum (FCS; Gibco) and 1% penicillin/streptomycin (Gibco). Upon reaching confluence, infected cells were counted based on EGFP expression.
Protein purification
Recombinant mammalian His6‐ or GST (glutathione‐S‐transferase)‐tagged proteins were expressed in Escherichia coli BL21 (DE3) bacteria (Stratagene). Protein purification of Synaptobrevin/VAMP2, t‐SNARE (Syntaxin1‐SNAP25), Synaptotagmin1 (Syt1), Complexin II (CpxII), and Munc18‐1 was performed as described previously via Ni2+‐NTA (nitrilotriacetic acid) (Qiagen) or glutathione Sepharose 4 fast flow (GE Healthcare Biosciences) affinity chromatography and subsequent ion exchange chromatography (Weber et al, 1998; Malsam et al, 2012; Parisotto et al, 2014).
Protein reconstitution into liposomes
Proteins were reconstituted into SUVs/GUVs as described previously (Malsam et al, 2012; Parisotto et al, 2014). All lipids were from Avanti Polar lipids with the exception of 3H‐DPPC (Amersham Biosciences) and Atto488/Atto550‐DPPE (ATTO‐TEC).
For the v‐SNARE/Syt1 SUVs, the following lipid composition was used: 29 mol% POPC (1‐palmitoyl‐2‐oleoyl‐SN‐glycero‐3‐phosphocholine), 15 mol% DOPS (1,2‐dioleoyl‐SN‐glycero‐3‐phosphoserine), 25 mol% POPE (1‐ hexadecanoyl‐2‐octadecenoyl‐SN‐glycero‐3‐phosphoethanolamine), 5 mol% liver PI (L‐α‐phosphatidylinositol), 25 mol% cholesterol (from ovine wool), 0.5 mol% Atto488‐DPPE (1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphoethanolamine), 0.5 mol% Atto550‐DPPE and trace amounts of 3H‐DPPC (1,2‐dipalmitoyl‐phosphatidylcholine), 3 μmol total lipid. For some experiments, the Atto488/Atto550‐DPPE was exchanged with 2% Atto655‐DPPE (self‐quenching concentration). SUVs were formed using the previously described technique of dilution and dialysis followed by a Nycodenz gradient centrifugation (Weber et al, 1998). Synaptobrevin/VAMP2 and VAMP8 were incorporated in a protein‐to‐lipid ratio of 1:300 and Synaptotagmin1 1:800.
For content mixing, 25 mM sulforhodamine B was incorporated into the SUVs by a single freeze–thaw cycle. Free dye was removed by a Nycodenz gradient. For the Syntaxin1/t‐SNARE SUVs/GUVs, the lipid composition consisted of 36 mol% POPC, 15 mol% DOPS, 20 mol% POPE, 3 mol% liver PI, 1 mol% brain PI(4,5)P2 (L‐α‐phosphatidylinositol‐4,5‐bisphosphate), 25 mol% cholesterol and trace amounts of 3H‐DPPC, 5 μmol total lipid. Syntaxin1 or t‐SNARE (Syx1/SNAP‐25) was reconstituted at a protein‐to‐lipid ratio of 1:1,000. t‐SNARE SUVs were used to produce t‐SNARE GUVs by electro‐swelling as described previously (Malsam et al, 2012) with the following modifications: (i) the 2nd desalting step was done using again a PD10 column (GE Healthcare); (ii) platinum‐coated glass slides (GeSiM) were used instead of ITO‐coated ones; and (iii) GUV buffer was 1 mM EPPS‐KOH (pH 8.0), 0.24 M sucrose (Ca2+ free from FLUKA), and 1 mM DTT (DL‐dithiothreitol).
Protein‐to‐lipid ratios in the reconstituted liposomes were determined by measuring the lipid amounts via 3H‐DPPE and by quantifying the reconstituted proteins after resolving them by SDS–PAGE and staining by Coomassie Blue. A BSA (bovine serum albumin) protein standard and ImageJ software were used for protein quantification.
SUV/GUV fusion assay
The lipid‐mixing assay was performed as described previously (Malsam et al, 2012; Parisotto et al, 2014) with the following modifications. Briefly, t‐SNARE GUVs were mixed with Synaptobrevin/VAMP2/Syt1‐SUVs, in the presence or absence of Munc18‐1 (900 nM) in a total volume of 104 μl fusion buffer (25 mM HEPES‐KOH, pH 7.4, 135 mM KCl, 0.1 mM EGTA, 0.5 mM MgCl2, 1 mM DTT) on ice. The fusion reaction was immediately started by transferring 100 μl of the mixture into a pre‐warmed 96‐well reaction plate (37°C). Atto488 fluorescence (excitation 485/20 nm, emission 525/20 nm) was measured with a Synergy 4 plate reader (BioTek Instruments GmbH) at 37°C in 10‐s intervals. After 2 min, Ca2+ was added to a final concentration of 100 μM. The fluorescence signal was normalized to the maximal signal obtained after lysis of the liposomes by addition of 0.7% (total concentration each) SDS and dodecylmaltoside. The fluorescence signals of control reactions of GUVs pre‐incubated with the soluble part of Synaptobrevin/VAMP21–94 were subtracted from individual measurement sets. For reactions containing CpxII, t‐SNARE GUVs were pre‐incubated together with CpxII (6 μM), Synaptobrevin/VAMP2/Syt1‐SUVs, ±Munc18‐1 for 5 min on ice before starting of the fusion reaction. For content mixing, the same setup was used, but sulforhodamine B fluorescence (excitation 530/25 nm, emission 590/35 nm) was measured.
To test the stimulatory function of Munc18‐1 on t‐SNARE assembly (Fig 5C and D), a fusion assay similar to the one described above was used, with the following modifications. Syntaxin1 GUVs were pre‐incubated with or without Munc18‐1 for 30 min at RT. After cooling down the sample on ice, soluble SNAP25 (ninefold excess to Syntaxin1) and Synaptobrevin/VAMP2/Syt1‐SUVs were added, and the mixture was incubated for additional 15 min on ice. The reaction was started as described above and Atto655 fluorescence (excitation 645/15 nm, emission 680/30 nm) for lipid mixing or sulforhodamine B fluorescence (excitation 530/25 nm, emission 590/35 nm) for content mixing was measured for a time period of 30 min. For normalization of this set of experiments, the lowest fluorescence signals were set to 0%, and the maximal signals reached after detergent addition were set to 100% fluorescence as described previously (Weber et al, 1998).
For analyzing the inhibitory function of Munc18‐1, VAMP8/Syt1‐SUVs were used instead of Synaptobrevin/VAMP2/Syt1‐SUVs and the incubation on ice was only 5 min.
Munc18‐1—Synaptobrevin/VAMP2 binding assay
The binding assay was performed as described previously (Parisotto et al, 2014) with a few modifications; ~5 μg GST‐Munc18‐1 (54 pmol) or the corresponding molar amount of GST (generated by thrombin cleavage of GST‐Munc18‐1 to remove Munc18‐1) was bound to 20 μl of GSH‐Sepharose 4 fast flow beads and subsequently washed 3× with fusion buffer (25 mM HEPES‐KOH, pH 7.4, 135 mM KCl, 1 mM DTT) and 3× with binding buffer (same as fusion buffer but including 5% (w/v) glycerol and 0.5% (v/v) Triton X‐100). Immobilized GST‐Munc18 was incubated with a molar excess of full‐length Synaptobrevin/VAMP2 (15× – 135×) for 1–1.5 h at 4°C on a rotating wheel in a final volume of 300 μl. To block unspecific binding, the incubation with Synaptobrevin/VAMP2 was performed in binding buffer containing E. coli lysate (total of 1.5 mg protein). Subsequently, beads were washed with 4 × 1.5 ml binding buffer. 30 μl of Laemmli buffer was added, and the beads were incubated for 5 min at 98°C. For analyzing Synaptobrevin/VAMP2 binding, proteins (8 μl of each reaction) were separated by SDS–PAGE and transferred onto a PVDF membrane via semidry blotting. Synaptobrevin/VAMP2 was detected by immunostaining with a primary rabbit antibody directed against the Synaptobrevin/VAMP2 N‐terminus and visualized by a fluorescently labeled secondary goat anti‐rabbit antibody using the Odyssey imaging system (LI‐COR Biosciences). Fluorescence intensities were quantified by ImageJ software.
Trans‐SNARE formation assay
To examine the positive effect of Munc18‐1 on the formation of trans‐SNARE complexes, a trans‐SNARE formation assay similar to the one described by Shen et al (2015) was used.
Briefly, reconstituted t‐SNARE SUVs (33 pmol t‐SNARE) were mixed with v‐SNARE SUVs (48 pmol VAMP2) and incubated in the presence or absence of 1.2 μM Munc18‐1 for 30 min at 4°C. After addition of ninefold excess amount of inhibitory VAMP2 CD, the SUVs were solubilized with 1% CHAPS. The t‐SNAREs were than precipitated using nickel beads (through binding to His6‐SNAP‐25), and the binding of full‐length VAMP2 to the t‐SNARE was probed by immunoblotting using monoclonal anti‐VAMP2 antibodies (1:2,000 dilution; Synaptic Systems, Cat#104 211).
Immunoblotting
HEK293T cells were plated in DMEM containing 10% FCS (Gibco) and 1% penicillin/streptomycin (Gibco) at equal density 1 day before transfection. Cells were transfected with calcium phosphate transfection at 80% confluence and were allowed to grow for approximately 36 h before harvesting. For calcium transfection, 150 μl 250 mM CaCl2 was added to 150 μl filter sterilized 2× HEBS solution [274 mM NaCl, 10 mM KCl, 1.4 mM Na2HPO4·2H2O, 1 mM dextrose, and 42 mM HEPES in H2O (pH 7.05)] mixed with 3 μg DNA under constant shaking. Precipitate was allowed to settle for 20 min after which it was added to a single well on a 6‐well plate. Medium was replaced the next morning.
For denatured IPs, cells were lysed with Laemmli sample buffer (LSB) containing 2% SDS, 10% glycerol, 2% β‐mercaptoethanol, 60 mM Tris (pH 6.8). Samples were boiled for 5 min and DNA was sheared with an insulin syringe; 8% was taken for total protein levels and stored at −20°C while the rest of the sample was diluted 15× in IP buffer (50 mM Tris pH7.5, 1% Triton X‐100, 1.5 mM MgCl2, 5.0 mM EDTA, and 100 mM NaCl) supplemented with 1 mM Na3VO4. IPs were performed overnight at 4°C using polyclonal rabbit Munc18‐1 antibody (3 μl, described in de Vries et al, 2000) conjugated to protein A agarose/sepharose beads (Sigma). Beads were washed 5× with alternating regular and high salt PKB buffer (high salt buffer contains double the amount of NaCl as regular PKB buffer). Proteins were eluted by boiling in LSB and analyzed by Western blotting.
For Western blotting, proteins were separated on 10% SDS polyacrylamide gels and transferred to PVDF membranes using the standard SDS–PAGE/Western blot. Membranes were blocked in TBST [TBS (pH 7.4) containing 0.1% Tween‐20] supplemented with 3% BSA in case of phospho‐detection or 2% milk powder and 0.5% BSA to reduce unspecific antibody binding. The following primary antibodies were applied overnight at 4°C: anti‐Munc18‐1 (monoclonal mouse (Ms), 1:5,000, BD Biosciences), anti‐phosphotyrosine (monoclonal Ms, 1:2,000, clone 4G10), and anti‐Flag (monoclonal Ms, 1:5,000, Sigma). After washing, alkaline phosphatase‐conjugated secondary antibodies (1:10,000, Dako) were applied for 1 h at 4°C. Blots were washed again, incubated with ECF (GE Healthcare) or AttoPhos substrate for 5 min, and scanned on a Fujifilm FLA‐5000 Reader. If needed, blots were stripped with stripping buffer (2 × 30 min in 0.1 M glycine pH 2.5–3), blocked, and reused for immunostaining. Results were analyzed using the Gel Analyzer tool in ImageJ (NIH, Bethesda, MD, USA).
In vitro kinase assays
HEK 293T cells were seeded to 60% confluency on the day of transfection and transfected with munc18‐1 wt and/or Flag‐n‐Src WT. After 24 h, cells were lysed in IP buffer (supplemented with 1 mM Na3VO4) and denatured by adding LSB after 0–90 min of tumbling at 4°C or at room temperature. After denaturing (boiling for 5 min), the samples were diluted 15× in IP buffer and a denatured IP was performed as described above, only now protein A agarose beads (vector shield) were added after 16 h.
For Fig EV1B, brain lysate was used in addition to HEK 293T cell lysate expressing n‐Src. Adult WT mouse brain was homogenized in IP buffer (supplemented with 1 mM Na3VO4 and 1 mM NaF) using a tissue homogenizer (IKA Ultra Turrax T18 basic) at 18,000 1/min for 1 min. Half of the homogenate was isolated by centrifugation at 20,000 g for 10 min. In the other half of the sample, cell debri was removed by gravitation. Both supernatants were combined to form the brain lysate. Src‐transfected HEK cells were lysed in IP buffer and the supernatant was cleared by centrifugation. Brain lysates and lysis buffer with or without n‐Src containing HEK cell lysate were divided over tubes and denatured according to the different time points.
Dissociated neuronal cultures
Hippocampi from munc18‐1 null mice were collected in ice‐cold Hanks’ buffered salt solution (HBSS; Sigma) buffered with 7 mM HEPES (Invitrogen). After removal of the meninges, neurons were incubated in Hanks–HEPES containing 0.25% trypsin (from 10× stock, Invitrogen) for 20 min at 37°C. After washing, neurons were triturated using a fire‐polished Pasteur pipette and counted in a Fuchs‐Rosenthal chamber. Neurons were plated in pre‐warmed Neurobasal medium supplemented with 2% B‐27, 1.8% HEPES, 0.25% glutamax, and 0.1% Pen/Strep (all Invitrogen) and infected with lentiviral particles encoding Munc18‐1 variants several hours after plating.
To achieve autaptic cultures, hippocampal munc18‐1 null neurons were plated on micro‐islands of rat glia at a density of 6K per well in a 12‐well plate. To generate these micro‐islands, glass coverslips (Menzel) were etched in 1 M HCl for at least 2 h and neutralized with 1 M NaOH for maximum 1 h, washed thoroughly with MilliQ water, and washed once with 70% ethanol. Coverslips were stored in 96% ethanol and coated with agarose type II‐A (0.0015% in H2O, Sigma) prior to microdot application. Coating was done by spreading a thin layer of agarose solution (heated in microwave and kept at 55°C during use) with a cotton swab over the entire coverslip. Microdots were created using a custom‐made rubber stamp (dot diameter 250 μm) to apply solution consisting of 0.1 mg/ml poly‐D‐lysine (Sigma), 0.7 mg/ml rat tail collagen (BD Biosciences), and 10 mM acetic acid [Sigma) by stamping from a wet filter paper (3‐mm cellulose chromatography paper (Whatman)]. Coverslips were UV‐sterilized for 20 min before further use. Astrocytes were plated at 6–8 K/well in pre‐warmed DMEM (Invitrogen) supplemented with 10% FCS, 1% nonessential amino acids (NAA), and 1% penicillin/streptomycin (all Gibco).
For confocal microscopy experiments, hippocampal neurons were plated at 25 K/well in 12‐well plates containing glass coverslips disinfected with 96% ethanol and coated with 0.5 milli‐percent poly‐l‐ornithine (Sigma) and 2 μg/ml laminin (Sigma) in PBS overnight and thoroughly washed.
Tyrosine phosphorylation of Munc18‐1 in dissociated neuronal cultures
Wild‐type cortical neurons were prepared as described above and grown at high density (150 K) on a glia layer in PDL/collagen sprayed 6‐well plates. Half of the medium was replaced every week. Neurons were infected with SFV particles encoding IRES‐EGFP or n‐Src‐IRES‐EGFP at DIV17, treated with or without 1 mM vanadate for 30 min, and lysed in LSB buffer 8 h after SFV particles were added. Denatured IPs were performed as described above for HEK cells with the following changes: (i) Neurons were washed 1× with PBS before lysis; (ii) samples were precleared by adding empty beads for 1 h at 4°C before IP; and (iii) beads were blocked with chicken egg albumin to reduce background.
Electrophysiological recordings
Autaptic cultures of munc18‐1 null neurons were grown for 13–18 days before measuring. Whole‐cell voltage‐clamp recordings (V m = −70 mV) were performed at room temperature with borosilicate glass pipettes (2.5–4.5 mOhm) filled with 125 mM K+‐gluconic acid, 10 mM NaCl, 4.6 mM MgCl2, 4 mM K2‐ATP, 15 mM creatine phosphate, 10 U/ml phosphocreatine kinase, and 1 mM EGTA (pH 7.30). External solution contained the following (in mM): 10 HEPES, 10 glucose, 140 NaCl, 2.4 KCl, 4 MgCl2, and 4 CaCl2 (pH = 7.30, 300 mOsmol). Inhibitory neurons were identified and excluded based on the decay of postsynaptic currents. Recording was acquired with an Axopatch 200A amplifier (Molecular Devices), Digidata 1322A, and Clampex 9.0 software (Molecular Devices). After whole‐cell mode was established, only cells with an access resistance of < 12 MΩ and leak current of < 300 pA were accepted for analysis. EPSCs were elicited by a 1‐ms depolarization to 30 mV. RRP size was assessed by hypertonic sucrose application (500 mM, 3.5 s) (Rosenmund & Stevens, 1996).
For PDBu experiments, PDBu was bath‐applied to a final concentration of 1 μM PDBu (Calbiochem). DSE was induced by depolarizing neurons for 10 s to 0 mV. DSE was calculated by dividing the average of four EPSCs immediately after DSE with the average of four EPSCs preceding DSE.
Offline analysis was performed using Clampfit v9.0 (Axon Instruments), Mini Analysis Program v6.0 (Synaptosoft), and custom‐written software routines in Matlab R2011a (Mathworks).
Immunocytochemistry
Neurons were allowed to develop for 10 days before fixation. Cultures were fixed with 3.7% formaldehyde (Electron Microscopy Sciences). After washing with PBS, cells were permeated with 0.1% Triton X‐100 for 5 min and incubated in 2% normal goat serum for 20 min to block non‐specific binding. Cells were incubated for 1 h at room temperature in a primary antibody mixture of monoclonal mouse anti‐VAMP (1:1,000, SySy), polyclonal chicken anti‐MAP2 (1:10,000, Abcam), and polyclonal rabbit anti‐Munc18‐1 (1:500, described in de Vries et al, 2000) antibodies. After washing, cells were incubated for 1 h at room temperature with secondary antibodies conjugated to Alexa dyes (1:1,000, Molecular Probes) and washed again. Coverslips were mounted with DABCO‐Mowiol (Invitrogen), and images were acquired with a confocal microscope (LSM 510, Carl Zeiss) using a 40× oil immersion objective (NA = 1.3) with 0.7× zoom at 1,024 × 1,024 pixels and averaged over two scans. Confocal settings were kept the same for all scans within an experiment. Neuronal morphology and protein levels were analyzed using automated image analysis routine (Schmitz et al, 2011).
Chemical fixation electron microscopy
Autaptic hippocampal cultures of munc18‐1 null mutant mice (E18) obtained from three different litters were fixed at DIV14‐16 for 45 min at room temperature with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) (de Wit et al, 2006; Wierda et al, 2007). As for electrophysiology, only glia islands containing a single neuron were used for analysis. After fixation, cells were washed three times for 5 min with 0.1 M cacodylate buffer (pH 7.4), postfixed for 2 h at room temperature with 1% osmium tetroxide/1% potassium ferrocyanide in bidest, washed, and stained with 1% uranyl acetate for 40 min in the dark. Following dehydration through a series of increasing ethanol concentrations, cells were embedded in Epon and polymerized for 24 h at 60°C. After polymerization of the Epon, the coverslip was removed by alternately dipping it in liquid nitrogen and hot water. Cells of interest were selected by observing the flat Epon‐embedded cell monolayer under the light microscope, and mounted on pre‐polymerized Epon blocks for thin sectioning. Ultrathin sections (~90 nm) were cut parallel to the cell monolayer and collected on single‐slot, formvar‐coated copper grids, and stained in uranyl acetate and lead citrate. Hippocampal synapses were randomly selected at low magnification using an electron microscope (JEOL1010), and the operator was blinded for the genotype. For each condition, the number of docked SVs, total SV number, and active zone length were measured on digital images taken at 80,000‐fold magnification using custom‐written semiautomatic image analysis software running in Matlab (Mathworks). For all morphological analyses, we selected clearly recognizable synapses with intact synaptic plasma membranes with a recognizable pre‐ and postsynaptic area and defined SV membranes. SVs were defined as docked if there was no distance visible between the SV membrane and the active zone membrane.
Cryofixation electron microscopy
Dissociated hippocampal neurons (20,000/well) from munc18‐1 null mice were seeded on pre‐grown cultures of rat glia on sapphire disks (Leica Microsystems) to form micro‐networks of 2–10 neurons per sapphire disk. This method is an adaptation of micro‐islands cultures described above. Prior to seeding the glia, sapphire disks were coated by carbon and subsequently a mixture of 0.1 mg/ml poly‐d‐lysine (Sigma), 0.7 mg/ml rat tail collagen (BD Biosciences), and 10 mM acetic acid (Sigma) and placed in an agarose‐coated 12‐well plate to form glia monolayer islands selectively on sapphire disks. On DIV0 of the neuron culture, the neurons were infected with lentivirus encoding Munc18‐1 WT‐IRES‐EGFP or Munc18‐1 Y473D‐IRES‐EGFP. All cells observed at DIV17 expressed GFP and the sapphire disks were cryofixed in an EM‐PACT2 (Leica Microsystems) high‐pressure freezer in 10% trehalose/20% BSA cryoprotectant. Frozen samples were postfixed in 0.1% OsO4 in acetone at −90°C and brought to 0°C at 2°/h, holding for 8 h at −60 and −30°C. After several washes with ice‐cold acetone, the sapphire disks were washed with propylene oxide and with an increasing Epon concentration series. The samples were embedded in fresh Epon overnight and left to polymerize at 65°C for 48 h. Sapphire disks were removed from the Epon by dipping the samples in boiling water and liquid nitrogen and regions with micro‐networks were selected by light microscopy. These regions were cut out and mounted on pre‐polymerized Epon blocks for ultrathin sectioning. Ultrathin sections (80 nm) were cut parallel to the sapphire disk, collected on single‐slot, formvar‐coated copper grids, and stained in uranyl acetate and lead citrate. Synapse selection and analysis were performed as described above for chemical fixation electron microscopy.
Data analysis
Data are presented as mean values ± s.e.m., with n referring to the number of cells from each group unless stated otherwise. Statistical analysis was performed with Instat v3.05 software (GraphPad Software). Data samples were first tested for normality with the Kolmogorov and Smirnov test and for heterogeneity of variance with the method of Bartlett. If data allowed, an unpaired t‐test (with Welch correction if standard deviations are not equal) was used to determine statistical significance. If data failed to pass the normality test, the nonparametric Mann–Whitney U‐test was used. For the analysis of electron microscopy data, a multilevel comparison was used to accommodate nested data (synapses originating from the same neuron) (Aarts et al, 2014). P‐values below 0.05 are considered significant and are indicated as following: *P < 0.05, **P < 0.01, ***P < 0.001.
Author contributions
MM, MV, RFT, and THS designed the project and interpreted the results. MM performed physiology experiments, immunocytochemistry, and HEK cell biochemistry and analyzed the data. BD performed SUV/GUV fusion assay experiments and in vitro binding experiments and analyzed and interpreted the data. CB and HCAL aided with physiology data acquisition and analysis. JRTvW analyzed and interpreted EM data. MM and MV wrote the manuscript with inputs from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 5
Source Data for Figure 8
Acknowledgements
We thank Robbert Zalm for virus production, Desiree Schut for primary culture preparation, Ingrid Saarloos for cloning and performing in vitro kinase assays, and Jurjen Broeke and Emmeke Aarts for expert help with analysis. We would like to thank Jörg Malsam and Kerstin Rink for contributing purified proteins to this study. We thank Christiaan van der Meer for animal breeding and maintenance. Rien Dekker is acknowledged for performing electron microscopy. This work was supported by the EU (EUSynapse project 019055 to MV, EUROSPIN project HEALTH‐F2‐2009‐241498 to MV, HEALTH‐F2‐2009‐242167 SynSys project to MV, and ERC Advanced Grant 322966 to MV), the Netherlands Organization for Scientific Research, NWO (Pionier/VICI 900‐01‐001 and ZonMW 903‐42‐095 to MV), the NeuroBsik Mouse Phenomics Consortium (BSIK03053), and the German Research Foundation (DFG) (SFB/TRR 83 to BD and THS).
The EMBO Journal (2018) 37: 300–320
References
- Aarts E, Verhage M, Veenvliet JV, Dolan CV, van der Sluis S (2014) A solution to dependency: using multilevel analysis to accommodate nested data. Nat Neurosci 17: 491–496 [DOI] [PubMed] [Google Scholar]
- Anthis NJ, Haling JR, Oxley CL, Memo M, Wegener KL, Lim CJ, Ginsberg MH, Campbell ID (2009) Beta integrin tyrosine phosphorylation is a conserved mechanism for regulating talin‐induced integrin activation. J Biol Chem 284: 36700–36710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM (2015) A direct role for the Sec1/Munc18‐family protein Vps33 as a template for SNARE assembly. Science 349: 1111–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin ML, Cammarota M, Sim AT, Rostas JA (2006) Src family tyrosine kinases differentially modulate exocytosis from rat brain nerve terminals. Neurochem Int 49: 80–86 [DOI] [PubMed] [Google Scholar]
- Ballif BA, Carey GR, Sunyaev SR, Gygi SP (2008) Large‐scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J Proteome Res 7: 311–318 [DOI] [PubMed] [Google Scholar]
- Barnekow A, Jahn R, Schartl M (1990) Synaptophysin: a substrate for the protein tyrosine kinase pp60c‐src in intact synaptic vesicles. Oncogene 5: 1019–1024 [PubMed] [Google Scholar]
- Basu J, Betz A, Brose N, Rosenmund C (2007) Munc13‐1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J Neurosci 27: 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF (1991) Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA 88: 7834–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MO, Yu L, Coba MP, Husi H, Campuzano I, Blackstock WP, Choudhary JS, Grant SG (2005) Proteomic analysis of in vivo phosphorylated synaptic proteins. J Biol Chem 280: 5972–5982 [DOI] [PubMed] [Google Scholar]
- Cornelisse LN, Tsivtsivadze E, Meijer M, Dijkstra TM, Heskes T, Verhage M (2012) Molecular machines in the synapse: overlapping protein sets control distinct steps in neurosecretion. PLoS Comput Biol 8: e1002450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily NJ, Boswell KL, James DJ, Martin TF (2010) Novel interactions of CAPS (Ca2+‐dependent activator protein for secretion) with the three neuronal SNARE proteins required for vesicle fusion. J Biol Chem 285: 35320–35329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Su Z, Lu X, Yoon TY, Shin YK, Ha T (2010) Single‐vesicle fusion assay reveals Munc18‐1 binding to the SNARE core is sufficient for stimulating membrane fusion. ACS Chem Neurosci 1: 168–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genc O, Kochubey O, Toonen RF, Verhage M, Schneggenburger R (2014) Munc18‐1 is a dynamically regulated PKC target during short‐term enhancement of transmitter release. Elife 3: e01715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeroma JH, Roelandse M, Wierda K, van Aerde KI, Toonen RF, Hensbroek RA, Brussaard A, Matus A, Verhage M (2004) Trophic support delays but does not prevent cell‐intrinsic degeneration of neurons deficient for munc18‐1. Eur J Neuorsci 20: 623–634 [DOI] [PubMed] [Google Scholar]
- Hu SH, Christie MP, Saez NJ, Latham CF, Jarrott R, Lua LH, Collins BM, Martin JL (2011) Possible roles for Munc18‐1 domain 3a and Syntaxin1 N‐peptide and C‐terminal anchor in SNARE complex formation. Proc Natl Acad Sci USA 108: 1040–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A, Cao D, Peng J (2007) Identification of conserved tyrosine residues important for gibberellin sensitivity of Arabidopsis RGL2 protein. Planta 226: 475–483 [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP (2010) A tissue‐specific atlas of mouse protein phosphorylation and expression. Cell 143: 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig C, Min SW, Krinner S, Arancillo M, Rosenmund C, Sudhof TC, Rhee J, Brose N, Cooper BH (2014) The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 84: 416–431 [DOI] [PubMed] [Google Scholar]
- Janz R, Sudhof TC (1998) Cellugyrin, a novel ubiquitous form of synaptogyrin that is phosphorylated by pp60c‐src. J Biol Chem 273: 2851–2857 [DOI] [PubMed] [Google Scholar]
- Janz R, Sudhof TC, Hammer RE, Unni V, Siegelbaum SA, Bolshakov VY (1999) Essential roles in synaptic plasticity for synaptogyrin I and synaptophysin I. Neuron 24: 687–700 [DOI] [PubMed] [Google Scholar]
- Jockusch WJ, Speidel D, Sigler A, Sorensen JB, Varoqueaux F, Rhee JS, Brose N (2007) CAPS‐1 and CAPS‐2 are essential synaptic vesicle priming proteins. Cell 131: 796–808 [DOI] [PubMed] [Google Scholar]
- de Jong AP, Meijer M, Saarloos I, Cornelisse LN, Toonen RF, Sorensen JB, Verhage M (2016) Phosphorylation of synaptotagmin‐1 controls a post‐priming step in PKC‐dependent presynaptic plasticity. Proc Natl Acad Sci USA 113: 5095–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong AP, Verhage M (2009) Presynaptic signal transduction pathways that modulate synaptic transmission. Curr Opin Neurobiol 19: 245–253 [DOI] [PubMed] [Google Scholar]
- Kalia LV, Gingrich JR, Salter MW (2004) Src in synaptic transmission and plasticity. Oncogene 23: 8007–8016 [DOI] [PubMed] [Google Scholar]
- Kotani T, Morone N, Yuasa S, Nada S, Okada M (2007) Constitutive activation of neuronal Src causes aberrant dendritic morphogenesis in mouse cerebellar Purkinje cells. Neurosci Res 57: 210–219 [DOI] [PubMed] [Google Scholar]
- Lim SH, Moon J, Lee M, Lee JR (2013) PTPRT regulates the interaction of Syntaxin‐binding protein 1 with Syntaxin 1 through dephosphorylation of specific tyrosine residue. Biochem Biophys Res Comm 439: 40–46 [DOI] [PubMed] [Google Scholar]
- Liu KS, Siebert M, Mertel S, Knoche E, Wegener S, Wichmann C, Matkovic T, Muhammad K, Depner H, Mettke C, Buckers J, Hell SW, Muller M, Davis GW, Schmitz D, Sigrist SJ (2011) RIM‐binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 334: 1565–1569 [DOI] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N, Schneggenburger R (2008) Phorbol esters modulate spontaneous and Ca2+‐evoked transmitter release via acting on both Munc13 and protein kinase C. J Neurosci 28: 8257–8267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, Rizo J (2011) Munc13 mediates the transition from the closed syntaxin‐Munc18 complex to the SNARE complex. Nat Struct Mol Biol 18: 542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Söllner TH (2012) Complexin arrests a pool of docked vesicles for fast Ca2+‐dependent release. EMBO J 31: 3270–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R, Mathey‐Prevot B, Bernards A, Baltimore D (1987) Neuronal pp60c‐src contains a six‐amino acid insertion relative to its non‐neuronal counterpart. Science 237: 411–415 [DOI] [PubMed] [Google Scholar]
- McMahon HT, Bolshakov VY, Janz R, Hammer RE, Siegelbaum SA, Sudhof TC (1996) Synaptophysin, a major synaptic vesicle protein, is not essential for neurotransmitter release. Proc Natl Acad Sci USA 93: 4760–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messa M, Congia S, Defranchi E, Valtorta F, Fassio A, Onofri F, Benfenati F (2010) Tyrosine phosphorylation of synapsin I by Src regulates synaptic‐vesicle trafficking. J Cell Sci 123: 2256–2265 [DOI] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI (2000) Three‐dimensional structure of the neuronal‐Sec1‐syntaxin 1a complex. Nature 404: 355–362 [DOI] [PubMed] [Google Scholar]
- Munch AS, Kedar GH, van Weering JR, Vazquez‐Sanchez S, He E, Andre T, Braun T, Söllner TH, Verhage M, Sorensen JB (2016) Extension of helix 12 in Munc18‐1 induces vesicle priming. J Neurosci 36: 6881–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munton RP, Tweedie‐Cullen R, Livingstone‐Zatchej M, Weinandy F, Waidelich M, Longo D, Gehrig P, Potthast F, Rutishauser D, Gerrits B, Panse C, Schlapbach R, Mansuy IM (2007) Qualitative and quantitative analyses of protein phosphorylation in naive and stimulated mouse synaptosomal preparations. Mol Cell Proteomics 6: 283–293 [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T (2008) Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59: 861–872 [DOI] [PubMed] [Google Scholar]
- Ohnishi H, Yamamori S, Ono K, Aoyagi K, Kondo S, Takahashi M (2001) A src family tyrosine kinase inhibits neurotransmitter release from neuronal cells. Proc Natl Acad Sci USA 98: 10930–10935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onofri F, Giovedi S, Vaccaro P, Czernik AJ, Valtorta F, De Camilli P, Greengard P, Benfenati F (1997) Synapsin I interacts with c‐Src and stimulates its tyrosine kinase activity. Proc Natl Acad Sci USA 94: 12168–12173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onofri F, Messa M, Matafora V, Bonanno G, Corradi A, Bachi A, Valtorta F, Benfenati F (2007) Synapsin phosphorylation by SRC tyrosine kinase enhances SRC activity in synaptic vesicles. J Biol Chem 282: 15754–15767 [DOI] [PubMed] [Google Scholar]
- Parisotto D, Malsam J, Scheutzow A, Krause JM, Söllner TH (2012) SNAREpin assembly by Munc18‐1 requires previous vesicle docking by synaptotagmin 1. J Biol Chem 287: 31041–31049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisotto D, Pfau M, Scheutzow A, Wild K, Mayer MP, Malsam J, Sinning I, Söllner TH (2014) An extended helical conformation in domain 3a of Munc18‐1 provides a template for SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) complex assembly. J Biol Chem 289: 9639–9650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter MD, Barbero S, Cheresh DA (2005) Tyrosine phosphorylation of VE‐cadherin prevents binding of p120‐ and beta‐catenin and maintains the cellular mesenchymal state. J Biol Chem 280: 31906–31912 [DOI] [PubMed] [Google Scholar]
- Purcell AL, Carew TJ (2003) Tyrosine kinases, synaptic plasticity and memory: insights from vertebrates and invertebrates. Trends Neurosci 26: 625–630 [DOI] [PubMed] [Google Scholar]
- Raulf F, Robertson SM, Schartl M (1989) Evolution of the neuron‐specific alternative splicing product of the c‐src proto‐oncogene. J Neurosci Res 24: 81–88 [DOI] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Sudhof TC, Takahashi M, Rosenmund C, Brose N (2002) Beta phorbol ester‐ and diacylglycerol‐induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108: 121–133 [DOI] [PubMed] [Google Scholar]
- Rodkey TL, Liu S, Barry M, McNew JA (2008) Munc18a scaffolds SNARE assembly to promote membrane fusion. Mol Biol Cell 19: 5422–5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Sudhof TC (1993) Short‐term synaptic plasticity is altered in mice lacking synapsin I. Cell 75: 661–670 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16: 1197–1207 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS (2002) Differential control of vesicle priming and short‐term plasticity by Munc13 isoforms. Neuron 33: 411–424 [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr (2005) Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Comm 331: 1–14 [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr (2015) Src protein‐tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res 94: 9–25 [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5: 317–328 [DOI] [PubMed] [Google Scholar]
- Schmitz SK, Hjorth JJ, Joemai RM, Wijntjes R, Eijgenraam S, de Bruijn P, Georgiou C, de Jong AP, van Ooyen A, Verhage M, Cornelisse LN, Toonen RF, Veldkamp WJ (2011) Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J Neurosci Methods 195: 185–193 [DOI] [PubMed] [Google Scholar]
- Schollmeier Y, Krause JM, Kreye S, Malsam J, Söllner TH (2011) Resolving the function of distinct Munc18‐1/SNARE protein interaction modes in a reconstituted membrane fusion assay. J Biol Chem 286: 30582–30590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotten S, Meijer M, Walter AM, Huson V, Mamer L, Kalogreades L, ter Veer M, Ruiter M, Brose N, Rosenmund C, Sorensen JB, Verhage M, Cornelisse LN (2015) Additive effects on the energy barrier for synaptic vesicle fusion cause supralinear effects on the vesicle fusion rate. Elife 4: e05531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ (2007) Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128: 183–195 [DOI] [PubMed] [Google Scholar]
- Shen C, Rathore SS, Yu H, Gulbranson DR, Hua R, Zhang C, Schoppa NE, Shen J (2015) The trans‐SNARE‐regulating function of Munc18‐1 is essential to synaptic exocytosis. Nat Commun 6: 8852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu KG, Jow GM, Lee YJ, Wang SJ (2005) PP2 inhibits glutamate release from nerve endings by affecting vesicle mobilization. NeuroReport 16: 1969–1972 [DOI] [PubMed] [Google Scholar]
- Sitarska E, Xu J, Park S, Liu X, Quade B, Stepien K, Sugita K, Brautigam CA, Sugita S, Rizo J (2017) Autoinhibition of Munc18‐1 modulates synaptobrevin binding and helps to enable Munc13‐dependent regulation of membrane fusion. Elife 6: e24278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mackie K (2005) Depolarization‐induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol 569: 501–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Wierda K, Sons MS, de Wit H, Cornelisse LN, Brussaard A, Plomp JJ, Verhage M (2006) Munc18‐1 expression levels control synapse recovery by regulating readily releasable pool size. Proc Natl Acad Sci USA 103: 18332–18337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie‐Cullen RY, Reck JM, Mansuy IM (2009) Comprehensive mapping of post‐translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J Proteome Res 8: 4966–4982 [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287: 864–869 [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M (2001) Munc18‐1 promotes large dense‐core vesicle docking. Neuron 31: 581–591 [DOI] [PubMed] [Google Scholar]
- de Vries KJ, Geijtenbeek A, Brian EC, de Graan PN, Ghijsen WE, Verhage M (2000) Dynamics of munc18‐1 phosphorylation/dephosphorylation in rat brain nerve terminals. Eur J Neuorsci 12: 385–390 [DOI] [PubMed] [Google Scholar]
- Wang SJ (2003) A role for Src kinase in the regulation of glutamate release from rat cerebrocortical nerve terminals. NeuroReport 14: 1519–1522 [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92: 759–772 [DOI] [PubMed] [Google Scholar]
- Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT (2008) Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP‐25 complex. Structure 16: 308–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M (2007) Interdependence of PKC‐dependent and PKC‐independent pathways for presynaptic plasticity. Neuron 54: 275–290 [DOI] [PubMed] [Google Scholar]
- de Wit H, Cornelisse LN, Toonen RF, Verhage M (2006) Docking of secretory vesicles is syntaxin dependent. PLoS One 1: e126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Su L, Rizo J (2010) Binding of Munc18‐1 to synaptobrevin and to the SNARE four‐helix bundle. Biochemistry 49: 1568–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Cavallaro S, Gusev P, Alkon DL (2000) Nonreceptor tyrosine protein kinase pp60c‐src in spatial learning: synapse‐specific changes in its gene expression, tyrosine phosphorylation, and protein‐protein interactions. Proc Natl Acad Sci USA 97: 8098–8103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 5
Source Data for Figure 8