

Abstract
Background
Chronic lung diseases such as chronic obstructive pulmonary disease (COPD) and epigenetic events underlie lung cancer (LC) development. The study objective was that lung tumor expression levels of specific microRNAs and their downstream biomarkers may be differentially regulated in patients with and without COPD.
Methods
In lung specimens (tumor and non-tumor), microRNAs known to be involved in lung tumorigenesis (miR-21, miR-200b, miR-126, miR-451, miR-210, miR-let7c, miR-30a-30p, miR-155 and miR-let7a, qRT-PCR), DNA methylation, and downstream biomarkers were determined (qRT-PCR and immunoblotting) in 40 patients with LC (prospective study, subdivided into LC-COPD and LC, N = 20/group).
Results
Expression of miR-21, miR-200b, miR-210, and miR-let7c and DNA methylation were greater in lung tumor specimens of LC-COPD than of LC patients. Expression of downstream markers PTEN, MARCKs, TPM-1, PDCD4, SPRY-2, ETS-1, ZEB-2, FGFRL-1, EFNA-3, and k-RAS together with P53 were selectively downregulated in tumor samples of LC-COPD patients. In these patients, tumor expression of miR-126 and miR-451 and that of the biomarkers PTEN, MARCKs, FGFRL-1, SNAIL-1, P63, and k-RAS were reduced.
Conclusions
Biomarkers of mechanisms involved in tumor growth, angiogenesis, migration, and apoptosis were differentially expressed in tumors of patients with underlying respiratory disease. These findings shed light into the underlying biology of the reported greater risk to develop LC seen in patients with chronic respiratory conditions. The presence of an underlying respiratory disease should be identified in all patients with LC as the differential biological profile may help determine tumor progression and the therapeutic response. Additionally, epigenetic events offer a niche for pharmacological therapeutic targets.
Electronic supplementary material
The online version of this article (10.1186/s13148-017-0437-0) contains supplementary material, which is available to authorized users.
Keywords: Increased susceptibility to lung cancer, Underlying chronic respiratory disease, Tumor microRNAs, DNA methylation, Lung tumorigenesis biomarkers
Background
Lung cancer (LC) continues to be the most common cause of cancer-related mortality worldwide, rising up to one third of all-cause mortality in certain regions [1]. Major respiratory conditions such as chronic obstructive pulmonary disease (COPD) have been consistently proposed as risk factors of LC development in patients [2–7]. Specifically, the presence of emphysema was demonstrated to be the most important risk factor in LC pathogenesis [2–7]. Despite the relevance of this problem, identification of the underlying biology that predisposes patients with airway obstruction and emphysema to LC remains at its infancy.
Several mechanisms such as greater levels of oxidative stress [8–11] and inflammatory markers [8, 12, 13], along with a relative predominance of Th1 cytokines and M1 macrophages, were demonstrated in the tumors and blood of patients with LC and COPD [14]. In patients with underlying chronic respiratory conditions, these biological mechanisms are likely to participate in the neoplastic transformation as they may interfere with important cell processes such as repair, angiogenesis, and apoptosis [8, 12, 13].
Epigenetic events, defined as the process whereby gene expression is regulated by heritable mechanisms that do not alter DNA sequence, significantly contribute to adaptation to environmental factors. In this regard, cigarette smoking-mediated effects were shown to induce epigenetic modifications in the airways and lungs of patients [15–17]. Alterations in several epigenetic mechanisms such as chromatin remodeling, DNA methylation, and either histone acetylation or methylation were shown to occur in the airways of COPD patients and in lung tumors of patients irrespective of COPD [18, 19]. Interestingly, in patients with COPD and LC, methylation of immune-related genes was demonstrated in their tumor samples [20]. In keeping with, several specific immune-related genes were shown to be hypermethylated in tumor samples of patients with underlying COPD, suggesting that methylation of those specific genes may contribute to the epithelial cell transformation to cancer in patients with chronic respiratory conditions [21, 22]. On the other hand, non-coding single-stranded RNA molecules (microRNAs) which regulate cellular processes, namely proliferation, cell invasion and migration, adaptation to hypoxia, apoptosis, and angiogenesis have also been proposed to underlie lung tumorigenesis in patients with chronic respiratory conditions [23–27]. Additionally, specific microRNA expression patterns may also help predict mortality and response to treatment in LC patients [25, 28]. Whether these mechanisms may take place in the actual tumor lesions of LC patients with underlying respiratory conditions remains to be fully explored.
On this basis, we hypothesized that expression levels of specific microRNAs and DNA methylation may be differentially expressed in the lung tumors of patients with underlying respiratory diseases versus those without the presence of chronic respiratory conditions. Moreover, we also hypothesized that expression of a selected panel of biomarkers known to be specifically regulated by the target microRNAs may also differ in the tumors and non-tumor lungs of patients with LC with and without COPD. These downstream biomarkers were carefully chosen on the basis of currently available literature on the biological processes that are regulated by the target LC-associated microRNAs. Hence, the specific study objectives were to assess prospectively in tumor and non-tumor lung specimens of patients with non-small cell LC (NSCLC), with and without underlying COPD: (1) expression levels of microRNAs known to underlie lung tumorigenesis and total DNA methylation and (2) levels of their downstream pathways known to promote cellular processes involved in lung tumorigenesis and progression.
Methods
Study patients
This was a prospective, controlled study in which a group of 40 Caucasian patients (33 males, Table 1) with LC, undergoing thoracotomy for their lung neoplasm, were recruited consecutively from the Lung Cancer Clinic of the Respiratory Medicine Department at Hospital del Mar (Barcelona, Spain). The patients were always recruited before having received any treatment for their lung neoplasm including chemotherapy and/or radiotherapy. Tumor and non-tumor lung specimens were obtained (n = 40), and patients were subdivided into two groups depending on the presence of underlying COPD [26, 27, 29]: (1) 20 patients with LC who also had COPD (LC-COPD group) and (2) 20 patients with LC without COPD (LC group). As part of the prospective ongoing cohort “Lung Cancer-COPD” in our center, both LC and LC-COPD patients simultaneously participated in a previous study aimed to assess Th1 and Th2 inflammatory profiles and their implications in lung tumors of patients with chronic respiratory conditions (14).
Table 1.
Clinical and functional characteristics of the study patients
| Anthropometric variables | Lung cancer N = 20 | Lung cancer-COPD N = 20 |
|---|---|---|
| Age, years | 64 (12) | 65 (9) |
| Male, N/female, N | 13/7 | 20/0 |
| BMI, kg/m2 | 26 (5) | 26 (5) |
| Smoking history | ||
| Current: N, % | 12, 60 | 14, 70 |
| Ex-smoker: N, % | 5, 25 | 6, 30 |
| Never smoker: N,% | 3, 15 | 0, 0¶ |
| Pack-years | 53 (25) | 57 (20) |
| Lung function testing | ||
| vFEV1, % pred | 92 (8) | 61 (14)¶¶¶ |
| FEV1/FVC, % pred | 77 (6) | 60 (8)¶¶¶ |
| DLCO, % pred | 88 (12) | 72 (21)¶¶¶ |
| KCO, % pred | 88 (11) | 73 (17)¶¶¶ |
| TNM staging | ||
| Stage IA: N, % | 5, 25 | 5, 25 |
| Stage IB: N, % | 0, 0 | 4, 20 |
| Stage IIA: N, % | 1, 5 | 6, 30 |
| Stage IIB: N, % | 4, 20 | 2, 10 |
| Stage IIIA: N, % | 3, 15 | 3, 15 |
| Stage IIIB: N, % | 4, 20 | 0, 0 |
| Stage IV: N, % | 3, 15 | 0, 0 |
| Histological diagnosis | ||
| Squamous cell carcinoma: N, % | 6, 30 | 5, 25 |
| Adenocarcinoma: N, % | 11, 55 | 14, 70 |
| Others: N, % | 3, 15 | 1, 5 |
| Blood parameters | ||
| Total leucocytes/μL | 8.3 103 (1.9 103) | 9.2 103 (2.1 103) |
| Total neutrophils/μL | 5.7 103 (2.1 103) | 6.3 103 (1.7 103) |
| Total lymphocytes/μL | 1.7 103 (650) | 2.1·103 (617) |
| Albumin (g/dL) | 4.4 (0.4) | 4.0 (0.5)¶¶ |
| Total proteins (g/dL) | 7.4 (0.5) | 7.1 (0.6) |
| Fibrinogen (mg/dL) | 485 (99) | 409 (77)¶¶ |
| CRP (mg/dL) | 1.1 (1.1) | 1.7 (1.2) |
| GSV (mm/h) | 20 (7.4) | 30 (16.8)¶ |
| Ceruloplasmin (g/dL) | 26.2 (4.8) | 26.5 (4.8) |
| Body weight loss, kg | ||
| 0, N, % | 18, 90 | 17, 85 |
| 1–4, N, % | 2, 10 | 1, 5 |
| 5–8, N, % | 0, 0 | 2, 10 |
| 9–12, N, % | 0, 0 | 0, 0 |
Continuous variables are presented as mean (standard deviation), while categorical variables are presented as the number of patients in each group and percentage of the total population
Abbreviations: N number, kg kilograms, m meters, BMI body mass index, FEV1 forced expiratory volume in the first second, pred predicted, FVC forced vital capacity, DLco, carbon monoxide transfer, KCO Krogh transfer factor, TNM tumor, nodes, metastasis, CRP C-reactive protein, GSV globular sedimentation velocity, L liter
¶p < 0.05, ¶¶p < 0.01, ¶¶¶p < 0.001 between LC-COPD and LC patients. Comparisons of the clinical and physiological variables between LC-COPD and LC patients were assessed using the Student’s t test. Differences between the study groups in the qualitative variables were assessed using the chi-square test
Approval was obtained from the institutional Ethics Committee on Human Investigation (Hospital del Mar–IMIM, Barcelona, project number 2014/5776/1) in accordance with the World Medical Association guidelines (Helsinki Declaration of 2008) for research on human beings. Informed written consent was obtained from all patients at study entry and prior to initiation of therapies.
Anthropometrical and functional assessment
Lung function and anthropometry were evaluated in all patients as previously described [8, 14, 30–32].
Sample collection
During the surgical intervention (minimal resection or lobectomy), in patients that underwent thoracotomy (n = 40), lung samples were obtained (tumor and non-tumor specimens) by the expert lung pathologist. Non-tumor samples were collected from the distant surrounding parenchyma with respect to where the tumor was localized (7-cm minimum distance from the tumor site in all the samples), which enabled us to study non-tumor lung specimens. Briefly, non-tumor samples were carefully selected as a result of visual and manual detection (palpation) of any potential nodules that might modify disease staging. The presence of ground-glass opacities in the non-tumor specimens was always ruled out by previously analyzing the CT scan sections in all the study patients. Additionally, hematoxylin-eosin staining was performed in all non-tumor lung specimens to further confirm the absence of cancer cells. For ethical reasons, this was the only approach approved to study non-tumor lungs in COPD and non-COPD patients in our institution. In all cases, the expert lung pathologist selected a fragment of lung tumor and non-tumor specimens of approximately 10 × 10 mm2 size from the fresh samples after a careful collection of the specimens required for diagnosis purposes. On the day of thoracotomy, blood samples were obtained for conventional routine blood tests (hemogram and nutritional parameters). Importantly, a minimum amount of 50% of cancer cells was similarly identified in all tumor types from all the study patients. The remaining cell components were inflammatory and stromal cells in all the analyzed tumors. On the day of thoracotomy, blood samples were obtained for conventional routine blood tests (hemogram and nutritional parameters).
Sample preservation
Lung specimens (tumor and non-tumor) were snap-frozen in liquid nitrogen and stored at − 80 °C for further use.
Molecular biology analyses
The biology analyses were all conducted blind in our molecular biology laboratory (IMIM).
MicroRNAs and target genes
RNA and DNA isolations and quantitative real time-PCR amplification (qRT-PCR) were used to quantify in the lung specimens, the target microRNAs and downstream biomarkers (genes involved in key cellular processes related to tumorigenesis and cancer progression; Additional file 1: Tables S1 and S2, Tables 2, 3, and 4) of the microRNAs analyzed in the current study [30, 32–34].
Table 2.
Downstream targets of the microRNAs analyzed in tumor and non-tumor lungs of the study patients
| MicroRNAs | Downstream targets | LC patients | LC-COPD patients | ||
|---|---|---|---|---|---|
| Non-tumor | Tumor | Non-tumor | Tumor | ||
| miR-21 | |||||
| PTEN, gene expression | 78.5 (59.3) | 115.6 (89.7) | 82.4 (70.2) | 0.26 (0.60)*, §§§ | |
| MARCKs, gene expression | 0.4 (0.2) | 0.5 (0.2) | 0.5 (0.5) | 0.2 (0.1)*, § | |
| TPM-1, gene expression | 2.8 (2.1) | 11.0 (10.4)** | 5.8 (4.9) | 1.8 (1.5)§§ | |
| PDCD4, gene expression | 5.4 (3.4) | 5.2 (4.4) | 4.0 (2.5) | 1.4 (1.0)§ | |
| SPRY-2, gene expression | 6.0 (3.3) | 8.4 (7.1) | 3.4 (1.9) | 1.2 (1.0)§§§ | |
| miR-200b | |||||
| ETS-1, gene expression | 4.6 (3.7) | 3.3 (2.5) | 2.8 (2.5) | 0.6 (0.3)§ | |
| ZEB-2, gene expression | 2.3 (1.3) | 3.5 (2.7) | 0.7 (0.2)# | 0.4 (0.3)§§§ | |
| miR-126 | |||||
| EGFL-7, gene expression | 4.0 (2.0) | 5.0 (3.0) | 5.0 (5.0) | 1 (0.7) | |
| TOM-1, gene expression | 0.6 (0.3) | 2.2 (1.1)*** | 1.3 (1.0) | 0.8 (0.6)§§§ | |
| CRK, gene expression | 8.6 (7.1) | 21.3 (19.7)* | 6.6 (5.8) | 3.7 (3.5)§§§ | |
| Ang-2, protein content | 0.3 (0.1) | 0.3 (0.2) | 0.3 (0.07) | 0.5 (0.2)*, § | |
| Fibulin-3, protein content | 0.6 (0.2) | 0.6 (0.3) | 0.5 (0.2) | 0.4 (0.2) | |
| Fibulin-2, protein content | 0.04 (0.03) | 0.1 (0.04)* | 0.04 (0.02) | 0.04 (0.03) | |
| Fibulin-5, protein content | 0.3 (0.1) | 0.4 (0.1) | 0.5 (0.1)# | 0.5 (0.1) | |
| miR-451 | |||||
| MIF, gene expression | 0.6 (0.8) | 2.5 (2.1) ** | 1.3 (1.1) | 0.3 (0.2) §§§ | |
| RAB-14, gene expression | 6.3 (5.1) | 4.1 (1.9) | 1.5 (1.3) ## | 2.4 (2.1) | |
| miR-210 | |||||
| FGFRL-1, gene expression | 2.5 (1.8) | 3.2 (2.0) | 4.1 (2.3) | 1.3 (1.0)***, § | |
| EFNA-3, gene expression | 0.3 (0.2) | 1.0 (0.9)* | 0.2 (0.1) | 0.1 (0.08)§§ | |
| P-62, protein content | 0.1 (0.04) | 0.3 (0.1)* | 0.1 (0.02) | 0.3 (0.1)* | |
| LC3II/LC3II, protein content | 0.1 (0.05) | 0.4 (0.1)*** | 0.2 (0.1) | 0.4 (0.1)* | |
| Beclin-1, protein content | 0.3 (0.09) | 0.3 (0.05) | 0.2 (0.1) | 0.3 (0.1) | |
| BAX, protein content | 0.2 (0.06) | 0.3 (0.2) | 0.2 (0.1) | 0.3 (0.2) | |
| BCL-2, protein content | 0.04 (0.02) | 0.2 (0.1)*** | 0.1 (0.04) | 0.2 (0.05)** | |
| miR-30a-30p | |||||
| SNAIL-1, gene expression | 2.3 (1.4) | 3.4 (2.8) | 2.8 (1.7) | 0.6 (0.4)*, §§§ | |
| P53, gene expression | 0.4 (0.3) | 0.7 (0.5) | 0.5 (0.3) | 0.3 (0.2)§ | |
| CDKN2A, gene expression | 4.7 (3.9) | 1.9 (1.8)* | 9.8 (7.8)# | 19.4 (16.5)§§§ | |
| P63, gene expression | 0.1 (0.08) | 0.1 (0.05) | 0.8 (0.5)### | 0.4 (0.3)* | |
| CDKN1A, gene expression | 11.8 (10.4) | 5.7 (3.5)* | 13.5 (7.7) | 6.4 (4.8)** | |
| Ki-67 positive cells, % | 42.6 (19.0) | 96.5 (3.0)*** | 42.6 (19.0) | 96.2 (4.7)*** | |
| miR-let7c | |||||
| k-RAS, gene expression | 3.0 (2.0) | 2.2 (1.0) | 2.7 (2.1) | 1.1 (0.5)*, § | |
Variables are represented as mean (standard deviation)
Abbreviations: miR microRNA, rel.expresion relative expression, prot.content protein content, PTEN phosphatase and tensin homolog 10, MARCKS myristoylated alanine-rich protein kinase C substrate, TPM1 tropomyosin 1 (alpha), PDCD4 programmed cell death 4, SPRY2 sprouty homolog 2, ETS1 v-ets avian erythroblastosis virus E26 oncogene homolog 1, ZEB2 zinc finger E-box binding homeobox 2, EGFL7 EGF-like-domain, multiple 7, TOM1 target of myb1, CRK v-crk avian sarcoma virus CT10 oncogene homolog 17, Ang-2 angiopoietin-2, MIF macrophage migration inhibitory factor, RAB14 member RAS oncogene family 14, FGFRL-1 fibroblast growth factor receptor-like 1, EFNA3 ephrin-A3, LC3 light chain 3, BAX bcl-2 associated X protein, BCL-2 B cell lymphoma-2, SNAIL-1 snail family zinc finger 1, P53 tumor protein p53, CDKN2A cyclin-dependent kinase inhibitor 2A, P63 protein p63, CDKN1A cyclin-dependent kinase inhibitor 1A, and k-RAS Kirsten rat sarcoma viral oncogene homolog 11
§p < 0.05, §§p < 0.01, §§§p < 0.001 between tumor lungs of LC-COPD and tumor specimens of LC patients; *p < 0.05, **p < 0.01, ***p < 0.001 between tumor and non-tumor lungs in either LC or LC-COPD patients; #p < 0.05, ##p < 0.01, ###p < 0.001 between non-tumor specimens of LC-COPD and non-tumor lungs of LC patients. Absence of any statistical symbol means no differences between the study groups
Table 3.
Summary of epigenetic events in the tumor specimens of LC patients with and without the chronic respiratory condition
| Epigenetic events | LC patients | |
|---|---|---|
| Non-COPD | COPD | |
| Lung tumors | ||
| MicroRNAs | ||
| miR-21, miR-200b, miR-210, miR-let7c | ↓ | ↑ |
| miR-126, miR451, miR30a-30p, miR-155, miR-let7a | = | = |
| Histone modifications | ||
| HDAC2, Sirtuin-1 | = | = |
| DNA methylation | ↓ | ↑ |
Abbreviations: miR microRNA, HDAC2 histone deacetylase-2, = there were no significant differences for the specific tumor markers between LC-COPD and LC patients without COPD
Table 4.
Summary of biological processes represented by the downstream markers of the epigenetic events explored in the study in the tumor specimens of LC patients with and without COPD
| Downstream markers | LC patients | |
|---|---|---|
| Non-COPD | COPD | |
| Tumor suppressor genes P53, PTEN, PDCD4, TPM-1, k-RAS CDKN2A Sirtuin-1 |
↑ | ↓ |
| ↓ | ↑ | |
| = | = | |
| Cell proliferation and invasion (through ↓ SPRY-2, MIF and TOM-1) | ↓ | ↑ |
| Apoptosis (through ↓ ETS-1) | ↑ | ↓ |
| Cell differentiation (through ↓ ETS-1 and FGFRL-1) | ↑ | ↓ |
| Epithelial mesenchymal transition (through ↓ ZEB2 and SNAIL-1) | ↑ | ↓ |
| Cell- mediated immunity (through ↓ MIF) | ↓ | ↑ |
| Angiogenesis (through ↑ angiopoietin-2 and ↓ EFNA-3) | ↓ | ↑ |
| Cell adhesion and migration (through ↓ CRK, TOM-1 and MARCKs) | ↑ | ↓ |
= there were no significant differences for the specific tumor markers between LC-COPD and LC patients without COPD
Methylated DNA
Global 5-methylcytosine was quantified using the MethylFlash Methylated Kit and previous studies [30, 34].
Immunoblotting of 1D electrophoresis
Protein levels of the different molecular markers in lung specimens were explored using methodologies previously published [11, 31–33, 35].
Immunohistochemistry
Levels of ki-67 (marker of cell proliferation) were identified in the lung specimens as previously described [11, 31, 32, 35].
Statistical analyses
Data are expressed as mean (standard deviation). Normality of the variables was explored using the Shapiro-Wilk test. For the quantitative variables in the lungs, differences between study groups were assessed using one-way analysis of variance (ANOVA) and Tukey’s post hoc analysis to adjust for multiple comparisons of all the study variables. Chi-square test was used to assess potential differences between the study groups for the qualitative variables. Statistical significance was established at P ≤ 0.05.
Results
Clinical characteristics
Clinical and functional characteristics of LC-COPD and LC patients recruited in the study are shown in Table 1. Age, gender, and BMI did not significantly differ between LC-COPD and LC patients (Table 1). Interestingly, smoking history was similar in both study groups (Table 1). The functional parameters forced expiratory volume in 1 s (FEV1), FEV1/forced vital capacity (FVC), and diffusion lung capacity for carbon monoxide (DLCO) were significantly lower in LC-COPD than LC patients (Table 1). Furthermore, no significant differences were found in either TNM or histological subtypes between LC-COPD and LC groups (Table 1). In LC-COPD compared to LC patients, levels of albumin and fibrinogen were significantly decreased, whereas those of globular sedimentation (GSV) were increased and C-reactive protein (CRP) levels did not differ between groups (Table 1).
Differential microRNA profile and downstream targets in lung tumors in patients with underlying respiratory disease
In LC-COPD compared to LC patients, miR-21 expression was significantly increased in the tumors, whereas that of its downstream targets PTEN, MARCKs, TPM-1, PDCD4, and SPRY-2 was significantly reduced (Fig. 1a and Table 2). However, non-tumor lung expression of miR-21, PTEN, MARCKs, TPM-1, PDCD4, and SPRY-2 did not significantly differ between LC-COPD and LC patients (Fig. 1a and Table 2). Tumor miR-200b expression was significantly greater in LC-COPD than in LC patients, while that of its downstream markers ETS-1 and ZEB-2 was significantly reduced (Fig. 1b and Table 2). Non-tumor lung expression of miR-200b also significantly increased in LC-COPD compared to LC patients, and only the expression of ZEB-2 significantly decreased (Fig. 1b and Table 2). Tumor expression levels of miR-126 did not significantly differ between LC-COPD and LC patients, whereas in the former patients, an almost significant decrease in EGFL-7 expression was observed (p = 0.073, Fig. 1c and Table 2), together with a decrease in TOM-1 and CRK expression and a rise in angiopoietin-2 content (Table 2). Tumor protein levels of fibulin-3, fibulin-2, and fibulin-5 did not differ between LC-COPD and LC patients (Table 2 and Additional file 1: Figure S1).
Fig. 1.
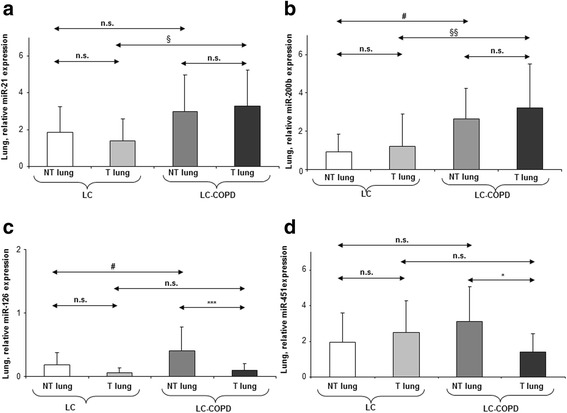
Mean values and standard deviation (relative expression) of miR-21 (a), miR-200b (b), miR-126 (c), and miR-451 (d). Definition of abbreviations: miR, microRNA. Statistical significance: *p < 0.05, ***p < 0.001 between tumor and non-tumor lungs in either LC or LC-COPD patients; #p < 0.05 between non-tumor specimens of LC-COPD and non-tumor lungs of LC patients; §p < 0.05, §§p < 0.01 between tumor lungs of LC-COPD and tumor specimens of LC patients; and n.s., non-significant differences between the study groups
Tumor and non-tumor expression of miR-451 did not differ between LC-COPD and LC patients (Fig. 1d), while MIF tumor expression was significantly lower in LC-COPD than LC patients, and RAB-14 expression was also significantly reduced in the non-tumors of the former patients (Table 2). Tumor expression of miR-210 significantly increased in LC-COPD compared to LC patients, while that of its downstream targets FGFRL-1 and EFNA-3 was reduced in the former patients (Fig. 2a and Table 2). Tumor protein levels of autophagy and apoptosis markers P62, LC3II/LC3I, beclin-1, BAX, and BCL-2 did not differ between LC-COPD and LC patients (Table 2 and Additional file 1: Figure S2). In LC-COPD, tumor expression of miR30a-30p was similar to that seen in LC patients (Fig. 2b). However, tumor expression of its downstream markers SNAIL-1 and P53 was significantly lower in LC-COPD than LC patients, while that of CDKN2A was greater and that of P63, CDKN1A, and ki-67 did not differ in tumors between patient groups (Table 2 and Additional file 1: Figure S3). Furthermore, non-tumor lung expression levels of miR-210 and miR30a-30p and their downstream markers FGFRL-1, EFNA-3, P62, LC3II/LC3I, beclin-1, BAX, BCL-2, SNAIL-1, P53, CDKN1A, and ki-67 did not significantly differ between LC-COPD and LC patients (Fig. 2a, b, Additional file 1: Figures S2, S3 and Table 2). Nonetheless, non-tumor lung expression of CDKN2A and P63 was significantly higher in LC-COPD than LC patients (Table 2). Tumor expression of miR-let7c was significantly greater in LC-COPD than LC patients (Fig. 2c), while that of its downstream marker k-RAS significantly decreased in the former patients (Table 2). Tumor expression of miR-155 and miR-let7a did not significantly differ between LC-COPD and LC patients (Figs. 2d and 3, respectively). Additionally, non-tumor lung expression levels of miR-let7c, k-RAS, miR-155, and miR-let7a markers did not significantly differ between LC-COPD and LC patients (Figs 2c, d and 3 and Table 2).
Fig. 2.
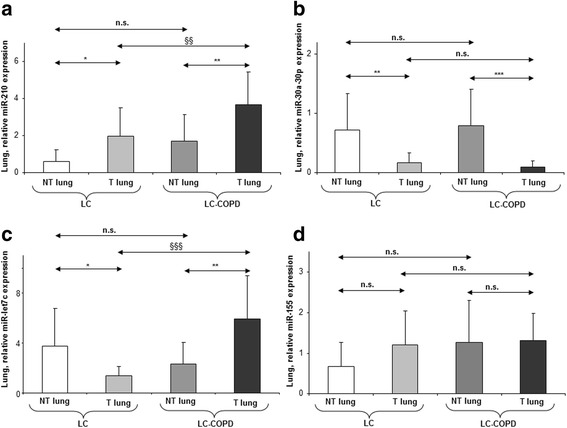
Mean values and standard deviation (relative expression) of miR-210 (a), miR-30a-30ap (b), miR-let7c (c), miR-155 (d), and miR-let7a (e) in tumor (T) and non-tumor (NT) lungs of LC and LC-COPD patients. Definition of abbreviations: miR, microRNA. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001 between tumor and non-tumor lungs in either LC or LC-COPD patients; §§p < 0.01, §§§p < 0.001 between tumor lungs of LC-COPD and tumor specimens of LC patients; and n.s., non-significant differences between the study groups
Fig. 3.
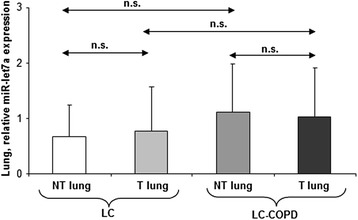
Mean values and standard deviation (relative expression) of miR-let7a in tumor (T) and non-tumor (NT) lungs of LC and LC-COPD patients. Definition of abbreviations: miR, microRNA. Statistical significance: n.s. non-significant differences between the study groups
Differential profile of histone deacetylases and DNA methylation in lung tumors of patients with underlying chronic respiratory disease
Tumor expression levels of the markers SIRT-1 and histone deacetylase-2 (HDAC2) did not differ between the two study groups (Fig. 3a, b and Additional file 1: Figure S4). Nevertheless, in LC-COPD compared to LC patients, non-tumor SIRT-1 expression was greater, whereas HDAC2 non-tumor protein levels did not differ between the study groups (Fig. 4a, b and Additional file 1: Figure S4). Total DNA methylation levels were significantly greater in both tumor and non-tumor lungs of LC-COPD than in LC patients (Fig. 4c).
Fig. 4.
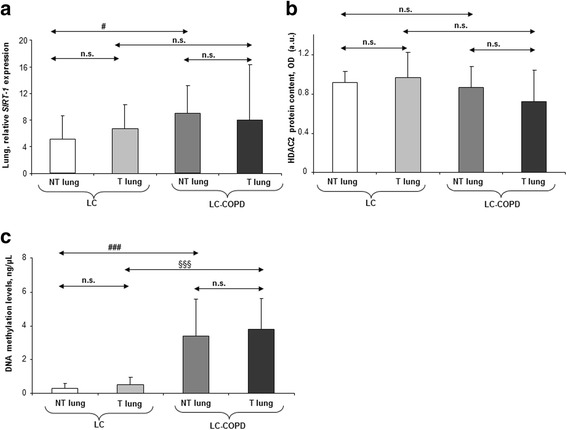
Mean values and standard deviation (relative expression) of SIRT-1 (a), HDAC2 (b), and DNA methylation levels (c) in tumor (T) and non-tumor (NT) lungs of LC and LC-COPD patients. Definition of abbreviations: miR, microRNA; SIRT-1, sirtuin-1; and HDAC2, histone deacetylase 2. Statistical significance: #p < 0.05, ###p < 0.001 between non-tumor specimens of LC-COPD and non-tumor lungs of LC patients; §§§p < 0.001 between tumor lungs of LC-COPD and tumor specimens of LC patients; and n.s., non-significant differences between the study groups
Discussion
In the current study, the study hypothesis has been confirmed. Importantly, the most relevant findings encountered in the investigation were that expression levels of miR-21, miR-200b, miR-210, miR-let7c, and DNA methylation were significantly greater in lung tumors of patients with underlying COPD than in those without this chronic condition (Table 3). Furthermore, expression levels of the downstream markers of the corresponding microRNAs, namely PTEN, MARCKs, TPM-1, PDCD4, SPRY-2, ETS-1, ZEB-2, FGFRL-1, EFNA-3, and k-RAS were downregulated in the lung tumor lesions of patients with the underlying condition compared to non-COPD patients (Table 4). Importantly, levels of the tumor suppressor gene P53 were also downregulated in the tumors of patients with COPD. These are biological markers of the underlying processes that are involved in tumorigenesis and tumor progression. Remarkably, these are novel findings that may partly explain the underlying biology of the reported greater risk of patients with COPD to develop lung tumors.
Downregulation of the study genes may entail alterations in the target pathways such as increased cell proliferation, migration, adhesion, invasion and angiogenesis, potential development of metastatic tumors, and adaptation of the cancer cells to hypoxia [25]. Clearly, all these biological processes promote tumor growth and favor disease progression in the patients. Previous investigations have demonstrated that levels of miR-21, miR-200b, miR-210, and miR-let7c were also upregulated in the tumor lesions of lung cancer cells in several experimental models and in patients [25, 36–42]. Interestingly, in other investigations, cancer cells were also shown to be more resistant to radiation or chemotherapy when those markers were elevated [43, 44]. In the present investigation, these results suggest that upregulation of miR-21, miR-200b, miR-210, and miR-let7c expression and that of DNA methylation may partly underlie the increased susceptibility of COPD patients to develop lung tumors. In keeping with, aberrant methylation status of tumor suppressor (CDKN2A) and DNA repair genes has already been identified in tumors of patients with LC and COPD [45, 46]. Furthermore, it has also been proposed that cancer cells express specific patterns of microRNA profiles that may contribute to the process of tumorigenesis [46]. Whether these epigenetic events may also confer a greater resistance to cancer treatment in patients with COPD will remain a matter of future research.
As the approach used in the study enabled us to analyze the target markers also in the surrounding non-tumor parenchyma in both groups of patients with LC, it is worth mentioning that levels of expression of miR-200b, miR-126, and DNA methylation were significantly greater in non-tumor specimens of patients with COPD than in those without this chronic condition. Additionally, expression levels of the downstream markers ZEB-2 and RAB-14 were downregulated, while those of the tumor suppressors CDKN2A, P63, and SIRT-1 were upregulated in the non-tumor specimens of LC patients with underlying respiratory disease compared to non-COPD patients. These are relevant findings that reveal the underlying biology of the lung parenchyma in patients with COPD. These results also imply that in COPD, a differential epigenetic regulation of cell cycle and proliferation also takes place in the lung parenchyma regardless of the presence of cancer cells. In fact, it has been clearly demonstrated that transition of cancer cells at the invasive site to a mesenchyme-like phenotype, commonly known as epithelial-to-mesenchymal transition, is crucial for invasion and distant metastasis in cancer progression [47]. In the current investigation, expression levels of the epithelial-to-mesenchymal transition inducers SNAIL-1 and ZEB-2 were significantly downregulated in tumors of patients with LC-COPD compared to those without underlying COPD. Thus, these results may imply that epithelial-to-mesenchymal transition may not play a major role in lung tumorigenesis in those patients, probably as result of areas of lung destruction. Clearly, future avenues of research should specifically address this question.
A third group of novel findings observed in this investigation were that within each study group of patients, in the lung tumors compared to non-tumor specimens, expression levels of miR-126 and miR-451 decreased in LC-COPD patients, while those of miR-30a-30p were reduced in both groups of patients. Compared to non-tumor specimens, levels of miR-let7c were also lower in lung tumors of patients without COPD, while a rise was seen in those with underlying COPD. The last findings are somehow counter to those previously reported in patients with COPD, who exhibited a decrease in miR-let7c expression in the sputum macrophages and bronchial airway epithelial cells [24]. As miR-let7c acts a tumor suppressor in tumor cells, it is likely that COPD induces a positive feedback on its expression in order to offset the deleterious effects of cancer biology. Levels of miR-let7c, which negatively correlated with metastasis, vascular invasion, and poor survival, were also downregulated in patients with NSCLC in another investigation [42]. However, whether patients also had underlying chronic respiratory disease was not explored in that study [42].
Posttranslational modifications of histones regulate gene transcription in normal and disease-related cellular processes. Levels of markers regulated by histone acetylation such as PTEN, MARCKs, EGFL-7 (almost significant decrease), FGFRL-1, SNAIL-1, P63, and k-RAS were significantly reduced in the lung tumors compared to non-tumors only in patients with LC-COPD. Indeed, proliferation markers and k-RAS mutations were reduced in lung tumors among COPD patients [48]. However, expression levels of downstream markers TPM-1, TOM-1, CRK, fibulin-2, MIF, and EFNA-3 were greater in the lung tumors than in non-tumor specimens only in patients without COPD. Interestingly, in the tumors of both groups of patients, the autophagy markers P62 and LC3, the anti-apoptotic BCL-2, and the proliferation marker ki-67 were increased compared to non-tumor specimens, while those of the inhibitor of cell proliferation CDKN1A were reduced. These findings also confirm that epigenetics play a significant role in the regulation of cellular mechanisms such as apoptosis, angiogenesis, and cell migration and adhesion that underlie tumorigenesis itself in both groups of patients, with a specific pattern of expression in patients with COPD.
Study critique
Since epigenetic events may be influenced by cigarette smoking, results might differ between smokers and non-smokers. Nonetheless, as no differences in smoking history or pack-years were observed between the two study groups, the results encountered in the investigation are likely to be attributed to the presence of the lung tumor and COPD in the patients with the underlying condition. Another factor that might have affected the study results is aging. Nevertheless, as age did not significantly differ between the two patient groups, the encountered findings can also be mostly recognized to the presence of the neoplasm and to COPD in the patients with this disease. Another limitation in the study refers to the relatively small number of lung specimens analyzed from both patient groups. Nevertheless, for ethical reasons, tumor and non-tumor lung specimens could only be obtained from patients undergoing thoracotomy for the treatment of their lung neoplasm (approximately 25% of all patients with LC). It should also be acknowledged that in three patients of the LC group, the lung neoplasms were in stage IV. These patients underwent surgery, chemotherapy, and/or radiotherapy for the treatment of the single metastasis always following thoracotomy for their primary lung neoplasm. It should also be mentioned that the approach used in the investigation enabled us to assess the effects of COPD itself without the cancer lesions on the epigenetic targets as distant non-tumor samples were also obtained from both groups of study patients. This is a unique approach commonly used in our group and the only acceptable from an ethical standpoint as also previously reported [11, 14]. Finally, future studies may be conducted in which the markers analyzed in this study will be validated in large-scale cohorts of patients. Nonetheless, it should also be emphasized that the results reported in the present investigation have been obtained within the frame of a hypothesis-driven prospective study.
Clinical impact of the study findings and future perspectives
Epigenetic regulation of cellular processes may explain disease pathogenesis. In the study, LC patients with an underlying respiratory condition exhibited a differential expression profile of several epigenetic markers, namely DNA methylation and microRNA expression along with that of downstream markers that are involved in tumorigenesis and tumor suppression. The clinical relevance of these findings is based on the following principles: (1) the target biological processes analyzed in the study may be eventually used as potential biomarker panels for the early diagnosis of LC in patients with COPD (the vast majority of patients with LC); (2) the epigenetic mechanisms could be used as potential biomarkers of disease progression (clinical staging) and prognosis, especially in patients with chronic respiratory diseases; (3) microRNAs such as miR-21 and miR-let7c may be targeted pharmacologically [49, 50], and thus, they may act as suitable biomarkers for the treatment of LC, which in turn may also help to gauge the response to therapy in these patients; and (4) the presence of an underlying respiratory disease should be identified in all patients with LC as the differential biological profile may help determine tumor progression and the therapeutic response. Future investigations should focus on the elucidation of whether microRNAs may serve as biomarkers of tumor progression, clinical staging, and recurrence in patients with underlying respiratory diseases.
Conclusions
A differential epigenetic regulation characterized by increased expression of several microRNAs and DNA methylation that control cellular processes involved in lung tumorigenesis exists in patients with LC and underlying respiratory disease. Fingerprint biomarkers of mechanisms involved in tumor growth, angiogenesis, migration, and apoptosis were also differentially expressed in tumors of patients with COPD. These findings shed light into the underlying biology of the reported greater risk to develop LC in patients with chronic respiratory conditions. Furthermore, they also have potential therapeutic implications as epigenetic events may be specifically targeted pharmacologically.
Acknowledgements
The authors are thankful to Ms. Arianne Bercowsky for her help with the ki-67 counting, to Ms. Mireia Admetlló for her help with the patient clinical assessment, and Mr. Sergi Mojal for his help with the comprehensive statistical analyses.
Funding
This study has been supported by Instituto de Salud Carlos III (Ministry of Economy and Competitiveness (Spain): FIS 11/02029 (FEDER), FIS 14/00713 (FEDER), PT13/0010/0005 (FEDER), and CIBERES (Lung Cancer Program, which is a member of the International Association for the Study of Lung Cancer, Instituto de Salud Carlos III), SEPAR 2016, FUCAP 2016, unrestricted grant from Menarini SA 2014 (Laboratorios Menarini S.A.), and the Xarxa de Bancs de tumors sponsored by Pla Director d'Oncologia de Catalunya (XBTC), Catalan Government.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ANG-2
Angiopoietin-2
- ANOVA
One-way analysis of variance
- BAX
Bcl-2-associated X protein
- BCL-2
B cell lymphoma-2
- BMI
Body mass index
- CDKN1A
Cyclin-dependent kinase inhibitor 1A
- CDKN2A
Cyclin-dependent kinase inhibitor 2A
- COPD
Chronic obstructive pulmonary disease
- CRK
v-crk avian sarcoma virus CT10 oncogene homolog 17
- CRP
C-reactive protein
- DLco
Carbon monoxide transfer
- EFNA3
Ephrin-A3
- EGFL7
EGF-like-domain, multiple 7
- ETS1
v-ets avian erythroblastosis virus E26 oncogene homolog 1
- FEV1
Forced expiratory volume in the first second
- FGFRL-1
Fibroblast growth factor receptor-like 1
- FVC
Forced vital capacity
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GSV
Globular sedimentation velocity
- HDAC2
Histone deacetylase-2
- KCO
Krogh transfer factor
- k-RAS
Kirsten rat sarcoma viral oncogene homolog 11
- L
Liter
- LC
Lung cancer
- LC3
Light chain 3
- MARCKS
Myristoylated alanine-rich protein kinase C substrate
- MIF
Macrophage migration inhibitory factor
- miR
MicroRNA
- N
Number
- NSCLC
Non-small cell lung cancer
- P53
Tumor protein p53
- P63
Protein p63
- PDCD4
Programmed cell death 4
- PTEN
Phosphatase and tensin homolog 10
- qRT-PCR
Quantitative real time polymerase chain reaction
- RAB14
Member RAS oncogene family 14
- SD
Standard deviation
- SIRT-1
Sirtuin-1
- SNAIL-1
Snail family zinc finger 1
- SPRY2
Sprouty homolog 2
- TNM
Tumor, nodes, metastasis
- TOM1
Target of myb1
- TPM1
Tropomyosin 1 (alpha)
- ZEB2
Zinc finger E-box binding homeobox 2
Additional file
Detailed information on the methodologies used in the study as well as the immunoblot images of the analysed markers. (DOCX 1563 kb)
Authors’ contributions
EB, VC, and LP contributed to the study conception and design. VC, MMJ, ARF, RA, ASF, and LP contributed to the patient assessment and recruitment and sample collection. LP contributed to the pathological diagnosis. MMJ and EB contributed to the molecular biology analyses. EB, MMJ, and VC contributed to the statistical analyses and data interpretation. EB, MMJ, VC, ARF, RA, and JG contributed to the manuscript drafting and intellectual input. EB contributed to the manuscript writing of the final version. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Approval was obtained from the institutional Ethics Committee on Human Investigation (Hospital del Mar–IMIM, Barcelona, project number 2014/5776/1) in accordance with the World Medical Association guidelines (Helsinki Declaration of 2008) for research on human beings. Informed written consent was obtained from all patients at study entry and prior to initiation of therapies.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13148-017-0437-0) contains supplementary material, which is available to authorized users.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Portillo K, Perez-Rodas N, Garcia-Olive I, Guasch-Arriaga I, Centeno C, Serra P, et al. Lung cancer in patients with combined pulmonary fibrosis and emphysema and idiopathic pulmonary fibrosis. A descriptive study in a Spanish series. Arch Bronconeumol. 2016; 10.1016/j.arbres.2016.10.004. [DOI] [PubMed]
- 3.Alvarez FV, Trueba IM, Sanchis JB, Lopez-Rodo LM, Rodriguez Suarez PM, de Cos Escuin JS, et al. Recommendations of the Spanish Society of Pneumology and Thoracic Surgery on the diagnosis and treatment of non-small-cell lung cancer. Arch Bronconeumol. 2016;52(Suppl 1):2–62. doi: 10.1016/S0300-2896(16)30198-3. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez J, de Torres JP. Lung cancer and emphysema. Arch Bronconeumol. 2016; 10.1016/j.arbres.2016.07.010. [DOI] [PubMed]
- 5.Sanchez-Salcedo P, Berto J, de Torres JP, Campo A, Alcaide AB, Bastarrika G, et al. Lung cancer screening: fourteen year experience of the Pamplona early detection program (P-IELCAP) Arch Bronconeumol. 2015;51(4):169–176. doi: 10.1016/j.arbres.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez-Salcedo P, Wilson DO, de Torres JP, Weissfeld JL, Berto J, Campo A, et al. Improving selection criteria for lung cancer screening. The potential role of emphysema. Am J Respir Crit Care Med. 2015;191(8):924–931. doi: 10.1164/rccm.201410-1848OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de CJ S, Hernandez JH, Lopez MF, Sanchez SP, Gratacos AR, Porta RR. SEPAR guidelines for lung cancer staging. Arch Bronconeumol. 2011;47(9):454–465. doi: 10.1016/j.arbres.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 8.Barreiro E, Fermoselle C, Mateu-Jimenez M, Sanchez-Font A, Pijuan L, Gea J, et al. Oxidative stress and inflammation in the normal airways and blood of patients with lung cancer and COPD. Free Radic Biol Med. 2013;65:859–871. doi: 10.1016/j.freeradbiomed.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Gupta A, Srivastava S, Prasad R, Natu SM, Mittal B, Negi MP, et al. Oxidative stress in non-small cell lung cancer patients after chemotherapy: association with treatment response. Respirology. 2010;15(2):349–356. doi: 10.1111/j.1440-1843.2009.01703.x. [DOI] [PubMed] [Google Scholar]
- 10.Masri FA, Comhair SA, Koeck T, Xu W, Janocha A, Ghosh S, et al. Abnormalities in nitric oxide and its derivatives in lung cancer. Am J Respir Crit Care Med. 2005;172(5):597–605. doi: 10.1164/rccm.200411-1523OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mateu-Jimenez M, Sanchez-Font A, Rodriguez-Fuster A, Aguilo R, Pijuan L, Fermoselle C, et al. Redox imbalance in lung cancer of patients with underlying chronic respiratory conditions. Mol Med. 2016; 10.2119/molmed.2015.00199. Pubmed PMID: PMC5004710 [DOI] [PMC free article] [PubMed]
- 12.Cho WC, Kwan CK, Yau S, So PP, Poon PC, Au JS. The role of inflammation in the pathogenesis of lung cancer. Expert Opin Ther Targets. 2011;15(9):1127–1137. doi: 10.1517/14728222.2011.599801. [DOI] [PubMed] [Google Scholar]
- 13.O'Byrne KJ, Dalgleish AG. Chronic immune activation and inflammation as the cause of malignancy. Br J Cancer. 2001;85(4):473–483. doi: 10.1054/bjoc.2001.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mateu-Jimenez M, Curull V, Pijuan L, Sanchez-Font A, Rivera-Ramos H, Rodriguez-Fuster A, et al. Systemic and tumor Th1 and Th2 inflammatory profile and macrophages in lung cancer: influence of underlying chronic respiratory disease. J Thorac Oncol. 2017;12(2):235–248. doi: 10.1016/j.jtho.2016.09.137. [DOI] [PubMed] [Google Scholar]
- 15.Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88(4):450–457. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics. 2015;7:113. doi: 10.1186/s13148-015-0148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Elgizouli M, Schottker B, Holleczek B, Nieters A, Brenner H. Smoking-associated DNA methylation markers predict lung cancer incidence. Clin Epigenetics. 2016;8:127. doi: 10.1186/s13148-016-0292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sundar IK, Mullapudi N, Yao H, Spivack SD, Rahman I. Lung cancer and its association with chronic obstructive pulmonary disease: update on nexus of epigenetics. Curr Opin Pulm Med. 2011;17(4):279–285. doi: 10.1097/MCP.0b013e3283477533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Wang Z. Epigenetic aberrant methylation of tumor suppressor genes in small cell lung cancer. J Thorac Dis. 2013;5(4):532–537. doi: 10.3978/j.issn.2072-1439.2013.08.21. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Chen C, Hua H, Han C, Cheng Y, Cheng Y, Wang Z, et al. Prognosis value of MGMT promoter methylation for patients with lung cancer: a meta-analysis. Int J Clin Exp Pathol. 2015;8(9):11560–11564. [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki M, Wada H, Yoshino M, Tian L, Shigematsu H, Suzuki H, et al. Molecular characterization of chronic obstructive pulmonary disease-related non-small cell lung cancer through aberrant methylation and alterations of EGFR signaling. Ann Surg Oncol. 2010;17(3):878–888. doi: 10.1245/s10434-009-0739-3. [DOI] [PubMed] [Google Scholar]
- 22.Wauters E, Janssens W, Vansteenkiste J, Decaluwe H, Heulens N, Thienpont B, et al. DNA methylation profiling of non-small cell lung cancer reveals a COPD-driven immune-related signature. Thorax. 2015;70(12):1113–1122. doi: 10.1136/thoraxjnl-2015-207288. [DOI] [PubMed] [Google Scholar]
- 23.Houghton AM. Mechanistic links between COPD and lung cancer. Nat Rev Cancer. 2013;13(4):233–245. doi: 10.1038/nrc3477. [DOI] [PubMed] [Google Scholar]
- 24.Van Pottelberge GR, Mestdagh P, Bracke KR, Thas O, van Durme YM, Joos GF, et al. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(7):898–906. doi: 10.1164/rccm.201002-0304OC. [DOI] [PubMed] [Google Scholar]
- 25.Liloglou T, Bediaga NG, Brown BR, Field JK, Davies MP. Epigenetic biomarkers in lung cancer. Cancer Lett. 2014;342(2):200–212. doi: 10.1016/j.canlet.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 26.Miravitlles M, Soler-Cataluna JJ, Calle M, Molina J, Almagro P, Quintano JA, et al. Spanish guidelines for management of chronic obstructive pulmonary disease (GesEPOC) 2017. Pharmacological treatment of stable phase. Arch Bronconeumol. 2017;53(6):324–335. doi: 10.1016/j.arbres.2017.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, Bourbeau J, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Arch Bronconeumol. 2017;53(3):128–149. doi: 10.1016/j.arbres.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Blandino G, Fazi F, Donzelli S, Kedmi M, Sas-Chen A, Muti P, et al. Tumor suppressor microRNAs: a novel non-coding alliance against cancer. FEBS Lett. 2014;588(16):2639–2652. doi: 10.1016/j.febslet.2014.03.033. [DOI] [PubMed] [Google Scholar]
- 29.Miravitlles M. What was the impact of the Spanish COPD guidelines (GesEPOC) and how can they be improved? Arch Bronconeumol. 2016;52(1):1–2. doi: 10.1016/j.arbres.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Puig-Vilanova E, Ausin P, Martinez-Llorens J, Gea J, Barreiro E. Do epigenetic events take place in the vastus lateralis of patients with mild chronic obstructive pulmonary disease? PLoS One. 2014;9(7):e102296. doi: 10.1371/journal.pone.0102296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puig-Vilanova E, Rodriguez DA, Lloreta J, Ausin P, Pascual-Guardia S, Broquetas J, et al. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic Biol Med. 2015;79:91–108. doi: 10.1016/j.freeradbiomed.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 32.Puig-Vilanova E, Martinez-Llorens J, Ausin P, Roca J, Gea J, Barreiro E. Quadriceps muscle weakness and atrophy are associated with a differential epigenetic profile in advanced COPD. Clin Sci (Lond) 2015;128(12):905–921. doi: 10.1042/CS20140428. [DOI] [PubMed] [Google Scholar]
- 33.Chacon-Cabrera A, Gea J, Barreiro E. Short- and long-term hindlimb immobilization and reloading: profile of epigenetic events in gastrocnemius. J Cell Physiol. 2016; 10.1002/jcp.25635. [DOI] [PubMed]
- 34.Puig-Vilanova E, Aguilo R, Rodriguez-Fuster A, Martinez-Llorens J, Gea J, Barreiro E. Epigenetic mechanisms in respiratory muscle dysfunction of patients with chronic obstructive pulmonary disease. PLoS One. 2014;9(11):e111514. doi: 10.1371/journal.pone.0111514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mateu-Jimenez M, Fermoselle C, Rojo F, Mateu J, Pena R, Urtreger AJ, et al. Pharmacological approaches in an experimental model of non-small cell lung cancer: effects on tumor biology. Curr Pharm Des. 2016;22(34):5300–5310. doi: 10.2174/1381612822666160623065523. [DOI] [PubMed] [Google Scholar]
- 36.Fasanaro P, D'Alessandra Y, Di SV, Melchionna R, Romani S, Pompilio G, et al. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem. 2008;283(23):15878–15883. doi: 10.1074/jbc.M800731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schliekelman MJ, Gibbons DL, Faca VM, Creighton CJ, Rizvi ZH, Zhang Q, et al. Targets of the tumor suppressor miR-200 in regulation of the epithelial-mesenchymal transition in cancer. Cancer Res. 2011;71(24):7670–7682. doi: 10.1158/0008-5472.CAN-11-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC) Clin Chim Acta. 2010;411(11–12):846–852. doi: 10.1016/j.cca.2010.02.074. [DOI] [PubMed] [Google Scholar]
- 39.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J Biol Chem. 2007;282(19):14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 40.Trang P, Medina PP, Wiggins JF, Ruffino L, Kelnar K, Omotola M, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010;29(11):1580–1587. doi: 10.1038/onc.2009.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang PY, Sun YX, Zhang S, Pang M, Zhang HH, Gao SY, et al. Let-7c inhibits A549 cell proliferation through oncogenic TRIB2 related factors. FEBS Lett. 2013;587(16):2675–2681. doi: 10.1016/j.febslet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Zhao B, Han H, Chen J, Zhang Z, Li S, Fang F, et al. MicroRNA let-7c inhibits migration and invasion of human non-small cell lung cancer by targeting ITGB3 and MAP4K3. Cancer Lett. 2014;342(1):43–51. doi: 10.1016/j.canlet.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 43.Oh JS, Kim JJ, Byun JY, Kim IA. Lin28-let7 modulates radiosensitivity of human cancer cells with activation of K-Ras. Int J Radiat Oncol Biol Phys. 2010;76(1):5–8. doi: 10.1016/j.ijrobp.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 44.Zhong M, Ma X, Sun C, Chen L. MicroRNAs reduce tumor growth and contribute to enhance cytotoxicity induced by gefitinib in non-small cell lung cancer. Chem Biol Interact. 2010;184(3):431–438. doi: 10.1016/j.cbi.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 45.Guzman L, Depix MS, Salinas AM, Roldan R, Aguayo F, Silva A, et al. Analysis of aberrant methylation on promoter sequences of tumor suppressor genes and total DNA in sputum samples: a promising tool for early detection of COPD and lung cancer in smokers. Diagn Pathol. 2012;7(87) 10.1186/1746-1596-7-87. Pubmed PMID: PMC3424112 [DOI] [PMC free article] [PubMed]
- 46.Wang DC, Shi L, Zhu Z, Gao D, Zhang Y. Genomic mechanisms of transformation from chronic obstructive pulmonary disease to lung cancer. Semin Cancer Biol. 2017;42:52–59. doi: 10.1016/j.semcancer.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Courtney JM, Spafford PL. The role of epithelial-mesenchymal transition in chronic obstructive pulmonary disease. Cells Tissues Organs. 2017;203(2):99–104. doi: 10.1159/000450919. [DOI] [PubMed] [Google Scholar]
- 48.Saber A, van der Wekken AJ, Kerner GS, van den Berge M, Timens W, Schuuring E, et al. Chronic obstructive pulmonary disease is not associated with KRAS mutations in non-small cell lung cancer. PLoS One. 2016;11(3):e0152317. doi: 10.1371/journal.pone.0152317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tufman A, Tian F, Huber RM. Can microRNAs improve the management of lung cancer patients? A clinician’s perspective. Theranostics. 2013;3(12):953–963. doi: 10.7150/thno.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Markou A, Zavridou M, Lianidou ES. miRNA-21 as a novel therapeutic target in lung cancer. Lung Cancer (Auckl ) 2016;7:19–27. doi: 10.2147/LCTT.S60341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.