

Abstract
Bacteriophages likely constitute the largest biomass on Earth. However, very few phage genomes have been well-characterized, the tailed phage T4 genome being one of them. Even in T4, much of the genome remained uncharacterized. The classical genetic strategies are tedious, compounded by genome modifications such as cytosine hydroxylmethylation and glucosylation which makes T4 DNA resistant to most restriction endonucleases. Here, using the type-II CRISPR-Cas9 system, we report the editing of both modified (ghm-Cytosine) and unmodified (Cytosine) T4 genomes. The modified genome, however, is less susceptible to Cas9 nuclease attack when compared to the unmodified genome. The efficiency of restriction of modified phage infection varied greatly in a spacer-dependent manner, which explains some of the previous contradictory results. We developed a genome editing strategy by codelivering into E. coli a CRISPR-Cas9 plasmid and a donor plasmid containing the desired mutation(s). Single and multiple point mutations, insertions and deletions were introduced into both modified and unmodified genomes. As short as 50-bp homologous flanking arms were sufficient to generate recombinants that can be selected under the pressure of CRISPR-Cas9 nuclease. A 294-bp deletion in RNA ligase gene rnlB produced viable plaques, demonstrating the usefulness of this editing strategy to determine the essentiality of a given gene. These results provide the first demonstration of phage T4 genome editing that might be extended to other phage genomes in nature to create useful recombinants for phage therapy applications.
Keywords: genome engineering, T4 bacteriophage, CRISPR, Cas9 nuclease, genome editing
Graphical abstract
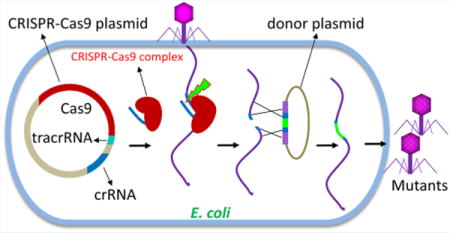
Bacteriophages inhabit all oceans, seas, rivers, and waters on Earth, and probably constitute the largest proportion of the biomass on the planet.1 A large fraction of these phages are tailed, containing an icosahedral head (capsid) that houses a linear dsDNA genome and a tail that delivers the genome into a host bacterial cell.1,2
Since their discovery in early 20th century, phages served as extraordinary models to elucidate the basic mechanisms of life and to create new avenues for genetic engineering and phage therapies.1,3–5 Felix d’Herelle, a French-Canadian scientist at Institute Pasteur and the codiscoverer of bacteriophages used cocktails of lytic phages to treat bacterial infections nearly a century ago.3,5 However, phage therapy lagged behind because of the discovery of small molecule antibiotics that provided greater breadth and potency.3,5,6 The emergence of multiantibiotic resistant bacterial pathogens and their continuing spread in the population brought new urgency to develop phage-based therapies.6,7 A striking example was the recent case in San Diego where an individual infected with the multidrug resistant Acinetobacter baumannii went into coma for nearly two months but completely recovered after intravenous administration of a mixture of phages that infect and lyse this bacterium.8
Phages have also emerged as powerful vaccine and gene therapy platforms to deliver genes and proteins into mammalian cells.9–11 We have developed one such platform using T4, a tailed phage belonging to the Myoviridae family.12–14 A unique feature of phage T4 is that its 120 × 86 nm size capsid (head) is decorated with two nonessential outer capsid proteins, Soc or small outer capsid protein (870 copies) and Hoc or highly antigenic outer capsid protein (155 copies).2,15 Antigens fused to Soc or Hoc, up to 1025 per head, can be displayed on the hoc−soc− T4 capsid with high affinity and exquisite specificity.12,13,16,17 Such nanoparticles can elicit robust immune responses and confer protection against deadly infections such as anthrax and plague.12,17 Furthermore, the interior of the capsid can be filled with foreign DNA, up to ∼170-kb, either a single long DNA molecule or multiple short plasmid DNAs.13 These nanoparticles could be targeted to specific cells to deliver combinations of genes and proteins, which could eventually lead to human therapies against genetic and infectious diseases.13
Considering the magnitude and diversity of phages, there is vast potential to harness their genomes for biomedical applications. However, other than a few phages that have been well characterized, very little has been done to unleash this potential, largely because it is tedious to manipulate phage genomes using the classical genetic strategies.18 For instance, even in the well-studied phage T4, of ∼300 potential genes in its 170-kb genome, nearly 130 of them remained uncharacterized.19,20 The ∼100 or so nonessential genes are distributed throughout the genome and the genome is circularly permuted with no unique ends, making it extremely difficult to engineer T4 as a cloning or mammalian delivery vector.19,20
Recently, the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas system was developed as an efficient tool for targeted genome editing in many organisms.21 CRISPR-Cas is an acquired immune system evolved in bacteria and archaea to counter the invasion of phages and foreign genetic elements such as plasmids. The type II CRISPR-Cas from Streptococcus pyogenes, which contains three components; crRNA derived from the unique spacer sequences present in the CRISPR region, tracrRNA that is common to all spacers, and Cas9 nuclease, is the most commonly used system for genome modification.21 When expressed in a cell, these components form a CRISPR-Cas9 complex and create a double-strand DNA break at a specific site in the genome (protospacer) that is complementary to the spacer sequence present in the crRNA. The break can then be repaired and rejoined, or recombined with a donor DNA using other DNA metabolizing enzymes present in the cell to generate mutants of interest.21 Although this system has been extensively used to modify mammalian genomes, surprisingly, there have been very few reports employing the CRISPR-Cas to engineer phage genomes.22–25 It is possible that the anti-CRISPR mechanisms evolved in phages may limit the application of CRISPR-Cas to edit phage genomes.26 The short time window of lytic phage life cycle, ∼20–30 min, might be another limitation. Furthermore, many of the phages evolved genome modifications in defense of host restriction systems. Phage T4 genome is particularly notorious because its cytosines are modified by two modifications, 5-hydroxymethylation and glucosylation.19,27,28 Consequently, the glucosyl hydroxymethyl cytosine (ghmC)-genome of phage T4 is highly resistant to most restriction endonucleases.19,27
It is unclear whether the ghmC-modifications protect the genome against attack by the CRISPR-Cas-type host defenses. Yaung et al. reported that three spacers utilized by the CRISPR-Cas9 system were functional against wild-type (WT) ghmC-modified T4 genome.28 On the other hand, Bryson et al. reported that the ghmC-modification makes the T4 genome resistant to CRISPR-Cas9 attack based on data from four spacers that prevented unmodified T4(C) mutant phage infection but not the ghmC-modified WT T4 infection.27 Here, by using a large number of spacers spanning the T4 genome, we discovered that the ghmC-modified T4 genome is vulnerable to cleavage by the Cas9 nuclease. However, it is not as susceptible to Cas9 as the unmodified genome and the efficiency of restriction of phage infection varied greatly depending on the spacer used, in part explaining the differences in previous studies.27,28 Importantly, we developed a strategy to edit either the unmodified or the ghmC-unmodified T4 genome by introducing point mutations, insertions, and deletions. We applied this editing strategy to determine whether the RNA ligase II gene rnlB is essential for phage infection. A 387-bp deletion knockout mutation in rnlB produced plaques and similar burst size as the WT T4 genome demonstrating that the rnlB function is not essential for phage infection under laboratory conditions. These results established the first phage genome editing system in T4, for both the unmodified and ghmC-modified genomes, which could potentially be extended to other phage genomes in nature to create useful recombinants for phage therapy applications.
RESULTS
Cytosine Modification of Phage T4 Genome Inhibits, But Does Not Block, Restriction by CRISPR-Cas9 Nuclease
To determine if the ghmC-modified WT T4 genome can be inactivated by CRISPR-Cas9 nuclease, we constructed 25 recombinant plasmids each containing a different 20-nt spacer sequence (Table 1) and transformed each into E. coli (Figure 1A). The spacer sequences are distributed all over the T4 genome and contained different percentages of cytosine nucleotide. (Table 1). To exclude any potential interference of DNA adenine methylation on CRISPR-Cas9 activity, none of the spacers included the adenine methylation site 5′-GATC-3′ (Table 1). The spacers were inserted into the plasmid, DS-SPCas (Addgene plasmid no. 48645),29 and kept under the control of the J23100 promoter which constitutively expresses the corresponding crRNA (see Materials and Methods for details). The crRNA would form a complex with Cas9 and tracrRNA that were also expressed constitutively from the same plasmid. If the crRNA:tracrRNA:Cas9 complex is functional, it would cleave at the protospacer sequence of the T4 genome delivered by phage infection (Figure 1B–D). Consequently, the genome will be disrupted, which likely leads to a loss of plaque forming ability, with certainty if the disruption occurred in an essential gene (Figure 1D). However, if the genome is resistant to CRISPR-Cas9 cleavage, plaques will appear at nearly the same frequency as the control plasmid lacking the spacer sequence (Figure 1E).
Table 1.
Spacer Sequencesa
| spacer no. | gene | sequence (5′ to 3′) | C (%) | G (%) | CG (%) |
|---|---|---|---|---|---|
| 1 | 23–2 | aagaacttccaaccggtaat | 25 | 15 | 40 |
| 2 | 20–1070 | gcaatatggaagatattcgt | 10 | 25 | 35 |
| 3 | Mrh.2–24 | ataatatctaaatcttcatt | 15 | 0 | 15 |
| 4 | 39.1–151 | atgttctgggctgttcttta | 15 | 25 | 40 |
| 5 | dda.1–130 | gagcagtatcgattagcttt | 15 | 25 | 40 |
| 6 | segF496 | gatttcagaaggaacttcaa | 15 | 20 | 35 |
| 7 | rnlb270 | gttgtatcttatcaagtctt | 15 | 15 | 30 |
| 8 | srh-8 | ttctgaagaagatgcttgaa | 10 | 25 | 35 |
| 9 | dexA523 | cttccaaagggaactttaga | 20 | 20 | 40 |
| 10 | cef154 | tcacgaaatacaccataaat | 25 | 5 | 30 |
| 11 | 39–37 | gaacatatcaaaaagcgtag | 15 | 20 | 35 |
| 12 | 24.2–181 | ttacagaagaaattggtgat | 5 | 25 | 30 |
| 13 | 20–995 | gcgctgcaaccaatagtctt | 30 | 20 | 50 |
| 14 | 39–96 | gcgctttatgtttggtaaat | 10 | 25 | 35 |
| 15 | 24–38 | gattgagttgctattcgttg | 10 | 30 | 40 |
| 16 | e148 | gttagtaaatcctgccacac | 30 | 15 | 45 |
| 17 | segD36 | tattcgcaattggataaatg | 10 | 20 | 30 |
| 18 | uvsy263 | agcggataaggatgttttaa | 5 | 30 | 35 |
| 19 | 23–1490 | gaatagaaggcataccgctc | 25 | 25 | 50 |
| 20 | uvsy287 | tgatacctcgttgcagtatt | 20 | 20 | 40 |
| 21 | 56–136 | aatttcgagctggtcttcgg | 20 | 30 | 50 |
| 22 | 56–63 | tttcttccgcattcattcca | 35 | 5 | 40 |
| 23 | e44 | caacgtttagaactggcact | 25 | 20 | 45 |
| 24 | IP3–28 | ggcctttactacagaagctt | 25 | 20 | 45 |
| 25 | IP3–10 | accacgggctgcattagcaa | 30 | 25 | 55 |
Sequences of the spacers used in the current study. Five spacers (spacers 1–5), which showed similar EOP for both WT and T4(C) mutant infections, are highlighted in bold font.
Figure 1.
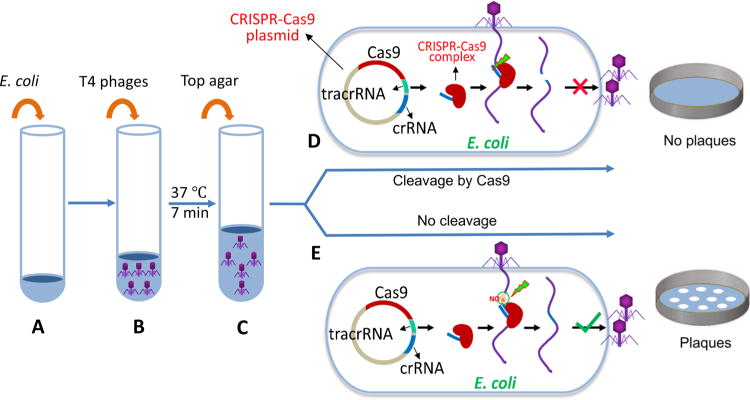
Experimental scheme for testing the effect of CRISPR-Cas on phage T4 infection. E. coli cells containing a CRISPR-Cas plasmid (∼3 × 106 cells) (A) were mixed with phage T4 (up to 106 pfu) (B). After incubation at 37 °C for 7 min, top agar was added (C), and the mixture was poured onto LB plates. The plates were incubated overnight at 37 °C. Cleavage by CRISPR-Cas9 nuclease at the protospacer sequence disrupts the phage genome resulting in loss of plaque-forming ability (D). If the genome is resistant to Cas9 cleavage, plaques will form at a frequency similar to that of the control vector plasmid (E). See Materials and Methods for more details.
The plating efficiencies of the WT T4 phage and the T4(C) mutant (similar to T4gt mutant30) were determined for each of the 25 spacer plasmids by plaque assay (Figure 2). The T4(C) mutant has amber mutations in the cytosine hydroxymethylase (g42) and dCTPase (g56) genes. When grown on sup0 E. coli, the T4(C) mutant produces phage particles containing unmodified C-genomes. As shown in Figure 2A, the WT phage containing the modified genome gave higher plating efficiencies when compared to the mutant phage containing the unmodified genome. For most of the spacers, the difference was greater than three logs, many showing four to five log difference. These data thus demonstrate that the T4(C) mutant phage was highly restricted by various spacers, whereas the restriction of WT phage infection varied greatly depending on the spacer used. For instance, spacers 23–2 and 20–1070, both in the essential genes encoding the major capsid protein gp23 and the portal protein gp20, respectively, highly restricted both ghmC-modified and unmodified phage infections (plating efficiency, ∼10−6). On the other hand, two other spacers, 23–1490 and 20–995, in the same genes, showed high restriction for the unmodified phage (plating efficiency, ∼10−5) but poor restriction for the ghmC-modified phage (plating efficiency, ∼10−1). These differences could not be due to differences in the C or GC content of the spacer sequences because no such correlation has been observed (Table 1). Furthermore, when the experiments were repeated in the genetic background of E. coli DH5α (sup2) which suppresses the amber mutations, the restriction of T4(C) mutant was reduced by as much as four logs in most cases (Figure 2B), in many cases approaching that seen with the WT phage (e.g., spacers segD36, uvsY263).
Figure 2.
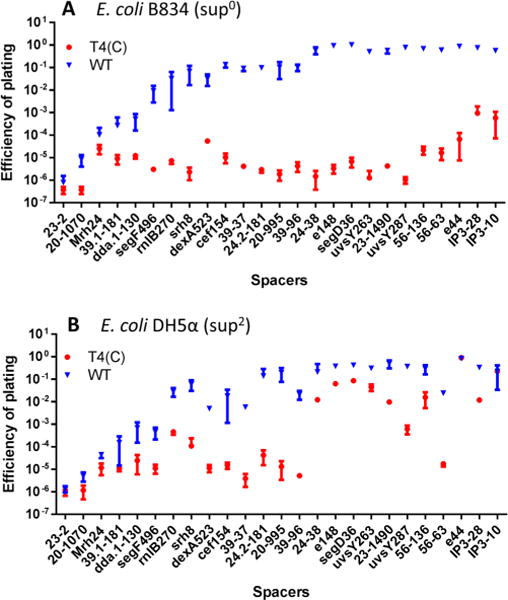
Restriction of phage T4 infection by CRISPR-Cas. The spacer-containing E. coli cells were infected with WT T4 or T4(C) mutant as per the basic scheme shown in Figure 1. See Materials and Methods for more details. The restriction of phage T4 infection was determined by the efficiency of plating (EOP), as determined by plaque assay. EOP was calculated by dividing the number of pfu produced on the spacer containing E. coli by the number of input pfu. EOP of modified (WT, blue symbols) or unmodified mutant (T4(C), red symbols) phages was determined on E. coli strains B834 (sup0) (A) or DH5α (sup2) (B). The labels on the X-axis denote the spacer. The sequences of the spacers are listed in Table 1. The experiments were done in triplicate.
Preliminary sequencing of the plaques produced from phage infections as above contained mutations in the CRISPR editing region, which presumably allowed the virus to escape the Cas9 nuclease attack. Indeed, the plaques produced from the T4(C) mutant infections all had mutations in the PAM (Protospacer Adjacent Motif) sequence (Figure 3). In the case of Streptococcus pyogenes Cas9, PAM is a three nucleotide 5′-NGG-3′ sequence immediately downstream of the protospacer sequence that is strictly required for binding of the CRISPR-Cas9 complex. On the other hand, the “CRISPR-escaped” plaques from the WT T4 phage infection showed a different pattern. The plaques generated when the plating efficiency was low (spacer 23–2) contained mutations in PAM or protospacer sequences, similar to that observed with the T4(C) mutant phage infections (Figure 3A). However, the plaques generated when the plating efficiency was high (spacer 20–995) contained largely WT sequences (8 out of 10) and a few with mutations in PAM sequences (2 out of 10) (Figure 3B). These results suggest that either the loss of the Cas9 recognition of the PAM sequence or the mismatches in the spacer sequence resulted in the escape of Cas9 cleavage and appearance of plaques. In the case of the WT phage containing ghmC-modified genome, escape could also occur even when there is no mutation in the protospacer or PAM sequences, provided that the protospacer was resistant to Cas9 cleavage. The latter likely involves complex mechanisms that require a detailed investigation, which is in progress.
Figure 3.

Alignment of CRISPR-Cas escape mutant sequences. The WT and T4(C) mutant phage infections were carried out as per the basic scheme shown in Figure 1. Single plaques that appeared on E. coli containing spacer 23–2 (g23) (A) or 20–995 (g20) (B) were isolated and the DNA flanking the protospacer and PAM sequences was amplified by PCR and sequenced. The blue arrows show the 5′ to 3′ direction of the spacer sequences. The spacer sequences are marked with red boxes and the PAM sequences are marked with blue boxes. The asterisks (∗) indicate the WT sequences.
Creation of Specific Mutants by Editing of T4 Genome Using CRISPR-Cas9
If the modified genome is susceptible to CRISR-Cas9 cleavage, as the above data suggest, it should be possible to edit the genome at the cleaved site. Specifically, could we incorporate site-directed mutations, for example, an amber mutation, at a given position? To answer this question, we codelivered, along with the CRISPR-Cas spacer plasmid, a second donor plasmid with an amber mutation in the protospacer sequence into the same E. coli (Figure 4). We predicted that the amber mutation in the protospacer sequence, at codon 468 of the major capsid protein gp23, will make the donor resistant to Cas9 attack (Figure 4A,B). To make it even more stringent, we introduced two additional silent mutations near the amber mutation to completely block Cas9 cleavage of the donor plasmid (Figure 4F). Consistent with our prediction, we encountered no difficulty in transforming or maintaining the donor plasmid under the CRISPR-Cas9 background. Upon infection of these E. coli cells by WT T4 phage (or T4(C) mutant), the delivered genome will be cleaved resulting in inactivation of the genome and loss of plaque forming ability (Figure 4A). However, since a donor DNA is present, recombination between the cleaved ends and the donor can result in the transfer of the amber mutation from donor to T4 genome, which, in addition, would restore the integrity of the genome (Figure 4B). To allow the amber mutant genome to survive and produce a plaque, the spacer and donor plasmids were cotransformed into E. coli B40 (sup1) that constitutively expresses a serine tRNA suppressor which allows translation of full-length and functional gp23. E. coli cells containing either the donor g23 mutant plasmid or the CRISPR-Cas9 plasmid alone were used as controls and the infection was carried out as depicted in Figure 1 using the WT T4 phage.
Figure 4.
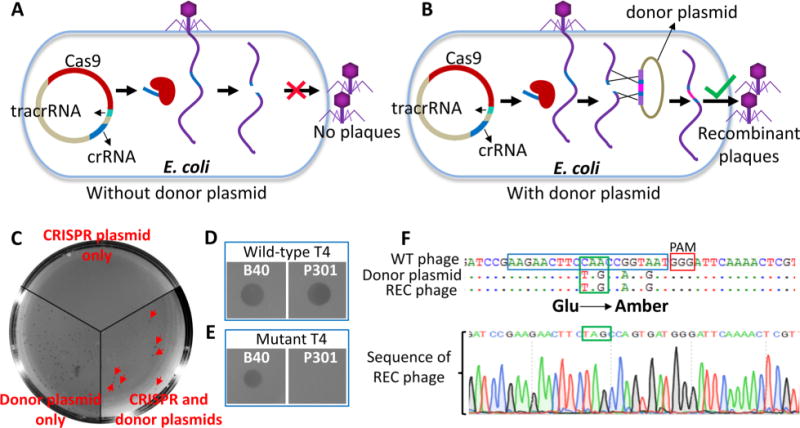
Genome editing of phage T4 using CRISPR-Cas9. Experimental scheme for T4 genome editing. T4 phage infections were performed according to the basic scheme shown in Figure 1. (A) E. coli containing only the spacer plasmid leads to restriction of phage infection. (B) E. coli containing both the spacer plasmid and a Cas9-resistant donor plasmid allows recombination between the genomic ends generated by Cas9 cleavage and the homologous arms of the donor plasmid DNA. The resultant recombinant phages are released following lysis. (C) Plating of the phage lysates from various infections. (D) Spot-test of a random plaque generated from E. coli containing only the donor plasmid. (E) Spot-test of a random plaque generated from E. coli containing both the CRISPR plasmid and the donor plasmid. Spot tests were done on lawns of E. coli B40 (sup1) and P301 (sup0) strains. (F) Sequencing of a random plaque generated from E. coli containing both the CRISPR plasmid and the donor plasmid. It shows the presence of amber mutation and two silent mutations in the recombinant plaque. Protospacer and PAM sequences are marked with blue and red boxes, respectively.
No plaques were produced when E. coli containing only the CRISPR-Cas9 plasmid was infected with 500 plaque forming units (pfu) of WT T4 phage (Figure 4A,C). This means that, consistent with the data in Figure 2, the T4 genome was inactivated by CRISPR-Cas9 cleavage. At the other extreme, a large number of plaques (∼500 pfu, nearly the same number as the input) were produced when E. coli containing only the donor plasmid was infected (Figure 4C). About 50 random plaques were picked from this infection and tested on E. coli B40 (sup1) and E. coli P301 (sup0). Each of them was WT since they grew on both the strains (Figure 4D). This is expected because there would be no restriction of T4 genome without the CRISPR-Cas9 plasmid. Although the donor plasmid can recombine with the delivered genome, this would be rare relative to the parental phage background. Furthermore, the donor plasmid will be degraded within minutes after T4 infection by the phage-derived denA and denB nucleases.19,31
A significant numbers of plaques were produced when E. coli cells containing both the CRISPR and donor plasmids were infected (∼50-fold lower than that produced on donor plasmid only) (Figure 4C). Importantly, all the 20 random plaques picked from this infection could survive only on E. coli B40 (sup1) but not on E. coli P301 (sup0) (Figure 4E), the phenotype of amber mutants. PCR-amplification and sequencing of g23 flanking the spacer sequence from individual plaques (after two rounds of plaque purification) showed that each of these plaques contained the amber mutation as well as the two additional silent mutations introduced into the protospacer sequence of the donor plasmid (Figure 4F). These data show that the recombinant genome was selected due to restriction of the infecting genome by CRISPR-Cas9 cleavage. Hence, no significant background of WT plaques was observed. In a separate experiment, we have successfully repeated the same result using the portal protein gene (g20) by codelivering the CRISPR-Cas plasmid containing the spacer 20–1070 (Table 1) and the corresponding donor plasmid containing the amber and silent mutations (Figure S1).
The above sets of data demonstrated that the T4 genome can be efficiently edited and the desired mutants can be selected with virtually no parental phage background under the strong selection pressure of CRISP-Cas9.
The Length of the Homologous Arms Correlates with Editing Efficiency
In the above experiments, the amber mutation was introduced near the center of the gene flanked by ∼1.2 kb DNA on either side. To determine the relationship between the length of the flanking arms and editing efficiency, five donor plasmids with different lengths of homologous arms ranging from 50 bp to 1200 bp containing the same mutation at the center were constructed and cotransformed into E. coli B40 along with the CRISPR-Cas9 plasmid (Figure 5A). The T4 infection assay was carried out as described above. We found that all the donor plasmids produced plaques while there were no plaques from the control that contained only the CRISPR-Cas9 plasmid. The number of plaques increased with increasing length of the arms (Figure 5B,C). At the shortest length, 50-bp, plaques were generated at a frequency of 0.03% of the input phage, which increased up to 2–3% when the length of the arms increased to 1200 bp (Figure 5C). These data show that while longer homologous arms would be preferred, relatively short arms are sufficient to generate edited mutants, especially since there was no background under the CRISPR-Cas9 pressure.
Figure 5.

Effect of the length of the homologous arms on editing efficiency. (A) Schematic of the donor templates with different lengths of homologous arms flanking the amber mutation site. (B) Plating of the phage lysates from the donor DNA templates shown in panel A. (C) The number of recombinant plaques produced using donor templates of different lengths. “∗” and “∗∗” indicate p < 0.05 and <0.01, respectively (ANOVA).
Genome Editing Using Two Adjacent Spacers
Often, it is necessary to edit the genome by introducing multiple mutations at a site or replace it with a new sequence of interest. CRISPR-Cas allows for a unique strategy to accomplish this. By expressing two crRNAs corresponding to adjacent proto-spacers, the CRISPR-Cas9 complex can be directed to make two cuts on the genome thereby excising the intervening sequence (Figure 6A). Recombination between the end sequences of the genome and the homologous arms of the donor plasmid will then replace the excised sequence with the mutant sequence and also restore genome integrity. A recombinant plasmid containing two spacer sequences flanking a 53 nucleotide sequence was constructed such that both the crRNAs will be constitutively expressed (Figure 6C). The donor plasmid was designed by including a series of twenty-eight mutations in a sequence spanning 112 nucleotides flanked by 500 bp homologous arms (Figure 6B,C). The donor and the CRISPR-Cas9 plasmids were cotransformed into E. coli and the T4 infection was performed as described above. Plaques at an editing efficiency of ∼1% of the input phage were generated. Sequencing of one random plaque showed that the genome sequence was precisely replaced by the donor sequence (Figure 6C).
Figure 6.
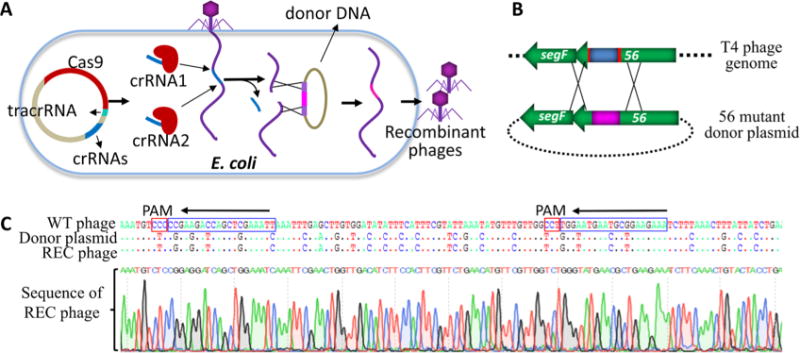
Genome editing using two adjacent spacers. (A) Schematic of gene replacement by CRISPR-Cas editing using two spacers. (B) Protospacers in phage T4 genome are shown in red and the region targeted for replacement is shown in blue. The two CRISPR-Cas9 complexes formed with the two different crRNAs will create breaks in the T4 genome at the corresponding protospacer sequences in g56. This results in excision of the intervening sequence. Recombination of the genome ends with the donor DNA flanking the inset will lead to replacement of the original sequence with that present in the donor DNA (shown in pink). (C) Sequencing of a random plaque showing the transfer of the new sequence into phage genome as a result of CRISPR-Cas mediated genome editing. Protospacer and PAM sequences are marked with blue and red boxes, respectively.
Determining the Essentiality of rnlB Gene by Genome Editing
T4 genome encodes about 300 potential genes.20 However, despite decades of classical genetic experiments, nearly 130 of these genes still remain uncharacterized. Of these, the essentiality of several genes including the gene rnlB, an RNA ligase, remain unknown.19 The T4 RNA ligase has been extensively used in recombinant DNA technology to ligate single stranded RNA (or DNA) molecules.32
T4 produces two RNA ligases encoded by genes 63 and rnlB.32,33 While 63 RNA ligase I is essential in E. coli strains containing the prr locus,33 the importance of rnlB RNA ligase II is unknown. In E. coli strains containing the prr locus, the tRNALys is cleaved 5′ to the wobble position by an anticodon nuclease (suicide function) induced upon T4 phage infection. The g63 RNA ligase together with the T4 polynucleotide kinase (pnk) repairs the tRNA allowing productive phage infection.34 If Pnk and RNA ligase I are not present, the synthesis of viral proteins and phage production is impaired by depletion of tRNALys.34 Whether the rnlB RNA ligase II also plays a role in phage survival is unknown.34
To determine if the above CRISPR-Cas9 T4 genome editing strategy could be used to determine the essentiality of the rnlB gene, we first constructed an rnlB spacer plasmid (rnlB270, Table 1) and a donor plasmid with an amber codon at amino acid 98. The plasmids were cotransformed into E. coli and plaques were selected using the same experimental design described in Figure 4. The sequencing data showed that the T4 genome was successfully edited by transferring the amber mutation from the donor plasmid into the CRISPR-Cas9 cleaved T4 genome. Spot testing of individual edited plaques showed that the rnlB amber phage can survive both on E. coli CR63 (sup1) and B834 (Sup0), indicating that the rnlB gene is not essential for phage T4 viability under the laboratory conditions used to grow the phage.
Since an amber mutation can spontaneously revert at some frequency, it would be necessary to delete a portion of the gene to completely knock out function. Further, the ability to create deletion mutants would be critical to engineer phage genomes for phage therapy applications where background would not be tolerated. To knockout the rnlB gene, we constructed a deletion donor plasmid in which a 437-bp fragment containing the last 143-bp corresponding to the 3′-end of gene 24.2 and the 294-bp of rnlB gene and its upstream region were deleted. Consequently, the DNA sequence corresponding to the promoter, the translation initiation site, and the first 98 amino acids of rnlB gene were removed. Two stop codons were also introduced right before the amino acid 99 to further prevent any run-on translation from an upstream sequence (Figure 7A). The donor and CRIPSR-Cas9 plasmids (24.2–181, Table 1) were cotransformed into E. coli strains B834 (sup0), which were then infected by phage T4(C). Five progeny plaques were randomly picked and tested for their ability to grow on E. coli strains CR63 (sup1) and B834 (sup0). All the plaques survived on both the E. coli strains, and the representative results of spot tests are shown in Figure 7B. The recombinant phages were plaque-purified and the mutated region was amplified using flanking primers rnlBFW and 24.3XbaBW with appropriate controls. The amber mutant from above and the WT T4 had the same size PCR product, ∼1.5kb, whereas the deletion mutant phage had ∼400 bp shorter band demonstrating a deletion in the rnlB gene of T4 genome (Figure 7C). A one-step growth experiment was performed with the rnlB.del phage and compared with the WT phage. The data showed that the rnlB deletion mutant generated a similar burst size of progeny phage as the parental WT T4(C) but the eclipse time is about 5 min shorter (Figure 7F). These results demonstrated that the T4 rnlB gene is not essential for phage T4 infection under the laboratory conditions.
Figure 7.
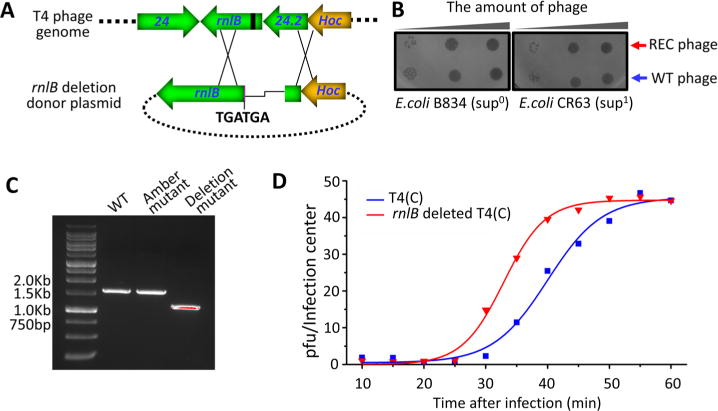
The phage T4 rnlB is a nonessential gene. (A) Schematic showing the introduction of a 437-bp deletion into rnlB region using CRISPR-Cas9 genome editing. Protospacer is shown in black and Hoc gene is shown in orange. Two stop codons were also introduced following the deletion. The line shows the deleted region. (B) Spot-testing of an rnlB deletion mutant plaque shows that it grows on both E. coli B834 (sup0) and CR63 (sup1). WT phage was used as a control (blue arrow). (C) PCR demonstrates that the rnlB mutant plaque has a deletion of appropriate size. (D) One-step growth curves of rnlB deleted T4(C) and T4(C) phages on E. coli DH5α.
DISCUSSION
Phage T4 is one of the most well characterized viruses. The atomic structures of essentially all the key components of the virus including the head, tail, fibers, and the DNA packaging machine have been determined.2,35–40 Genetic and biochemical pathways were elucidated in 60s and 70s that revealed common principles of virus assembly.19 Combined with the unique features of the T4 outer capsid proteins Hoc and Soc and the promiscuous nature of the DNA packaging machine, a platform to deliver genes and proteins into mammalian cells has been developed.12,13,41 However, it has been difficult to engineer the T4 genome owing to its modified genome that is refractory to most restriction enzymes.19,27 Lack of a clustered nonessential region in the genome that can be replaced with foreign DNA posed another barrier to use T4 as a cloning or protein/gene delivery vector.20 Overcoming such barriers would be essential to unleash the potential of T4 and other phages for biomedical applications. Our studies reported here demonstrate that some of these barriers could be overcome by CRISPR-Cas genome editing, which could potentially be extended to phages in general.
Our studies led to several new findings. First, the infection data from 25 different spacers spanning the T4 genome showed that the WT phage T4 genome containing the ghmC-modified DNA, is vulnerable to CRISPR-Cas9 attack. However, it is not as highly susceptible to Cas9 cleavage as the T4(C) mutant genome containing the unmodified C-DNA. While the T4(C) mutant phage genome showed very low plating efficiency, on the order of ∼10−5 of the input phage, the plating efficiency of the WT T4 phage varied greatly, between 10−1 to 10−6 of the input phage. Preliminary sequencing of the plaques that arose in WT or T4(C) mutant infections showed CRISPR-Cas escape mutations in the PAM trinucleotide sequence or the protospacer sequence. However, more work is underway to delineate the mechanisms involved in the selection of escape mutants.
Second, our data demonstrate that CRISPR-Cas9 cleavage allowed the selection of edited T4 genomes. This required cointroduction into the same E. coli cell of both the CRISPR-Cas spacer plasmid and a donor plasmid containing the desired mutation(s). Conveniently, these mutation(s) also resulted in mismatch between the spacer-derived crRNA and the protospacer sequence of the donor, thereby sparing the donor plasmid from cleavage by Cas9 nuclease. This allowed recombination between the cleaved ends of the delivered genome and the donor sequence. That such recombinants arose at high frequency (up to 2–3% of the input) means that the ends of the Cas9 cleaved genome remained competent for recombination and not degraded by nucleases. Whether this rescue was carried out by the E. coli recombinase or the highly efficient phage T4 recombination system42 requires further investigation. A notable aspect of the CRISPR-Cas mediated recombinational editing was that there was essentially no parental phage background among the progeny phage, presumably due to the strong pressure of CRISPR-Cas9 nuclease which selects out the parental background.
Third, the T4 genome editing strategy could be extended to constructing complex mutants involving multiple point mutations, insertions, and deletions, including the simultaneous use of two spacers. The frequency of survived mutant progeny decreased with increasing length of the mutant region and increased with increasing length of the flanking homologous arms. A limitation of the strategy, however, is that the editing site must be associated with the PAM sequence for the CRISPR-Cas9 complex to recognize the target and cleave the adjacent spacer sequence. However, this could be overcome by using mutant Cas9 proteins that exhibit increased breadth of the PAM recognition sequences.43
Finally, we demonstrate that the CRISPR editing allows functional characterization of the phage genome, especially of the nonessential genes, that has been otherwise difficult by the classical genetic strategies.44 We could readily introduce a 437-bp deletion into the T4 RNA ligase II gene rnlB using CRISPR-Cas editing. The rnlB knockout mutant not only formed plaques but also its burst size is similar to that of the parental phage. However, the knockout phage exhibited a shorter eclipse time compared to the parental phage, the reason for which is not clear. These data suggest that the RNA ligase function of rnlB is not essential for phage infection, at least under the laboratory growth conditions at 37 °C. However, it is possible that rnlB might provide survival advantage(s) under certain E. coli genetic backgrounds, or in the absence of g63 RNA ligase I, which also has a second essential function, attachment of tail fibers to the baseplate.45 The edited mutants such as the rnlB phage will now allow detailed characterizations of the complex functional and evolutionary relationships among the phage genes as well as the host–virus interactions.
In conclusion, our studies for the first time established a CRISPR-Cas editing strategy to engineer both modified and unmodified genomes of phage T4. Selection of edited mutants under the strong selective pressure of CRISPR-Cas9 nuclease makes it a powerful strategy to modify the phage T4 genome for functional characterizations as well as to accelerate the development of T4 as a gene/protein delivery vehicle. This editing strategy could potentially be applied to other phage genomes to harness the vast potential of the naturally occurring phages for various biotechnology applications and phage therapies.
METHODS
Bacterial and Bacteriophage Strains
E. coli strains DH5α (hsdR17(rK−mK+) sup2), CR63 (sup1λr), B834 (hsdRB hsdMB met thi sup0), P301 (sup0), and B40 (sup1) were used in the experiments described below. WT T4 was propagated on E. coli P301 as described previously.46 T4(C) is a mutant containing an amber mutation at amino acid 58 of gene 42 that codes for dCMP hydroxymethylase and an amber mutation at amino acid 124 of gene 56.30 To prevent accumulation of spontaneous revertants, the T4(C) mutant was propagated on E. coli B834 for only one generation. The phage stocks containing revertant phage at a frequency of <10−6 were used for all the experiments. For some experiments, the T4(C) phage was propagated on suppressor-plus E. coli strain CR63 to produce phage with modified cytosines in the genome.
Plasmids
CRISPR-Cas9 spacer plasmids were constructed by cloning spacer sequences into the streptomycin-resistant plasmid DS-SPCas (Addgene no. 48645).29 Sequences of the spacers are shown in Table 1. The homologous donor plasmids were constructed by cloning the donor DNA into the pET28b vector. The donor plasmid containing an amber mutation in g23 at amino acid 468 was constructed by two rounds of cloning. First, the full length g23 was amplified with primers 23FW and 23BW using T4 genomic DNA and cloned into the pET28b DNA linearized with BglII and BamHI restriction enzymes to generate pET-23. A DNA fragment containing partial genes of segD and gp23 with the amber mutation was amplified from T4 genome DNA using primers, 23am FW and segD BW, and cloned into pET-23 DNA linearized with NdeI and XhoI to generate the g23 donor plasmid. The donor plasmid was then used as PCR template to amplify homologous arms of different lengths (50–500 bp). These were inserted into the pET28b DNA linearized with BglII and XhoI. The primer sequences are shown in Supporting Information, Table S1.
A DNA fragment containing a multiple mutations in the g56 gene between amino acids 121 and 158 was synthesized by Genscript (Piscataway, NJ). The DNA was inserted into the pET-28b DNA linearized with BglII and BamHI to generate the g56 donor plasmid.
The rnlB donor plasmids were constructed by two rounds of cloning. First, the full length rnlB was amplified with primers rnlB FW and rnlB BW using the T4 genome DNA as template. The DNA was then cloned into pET28b linearized with XhoI and EcoRI to generate pET-rnlB. To introduce an amber mutation, a DNA fragment containing the first 300 bp of rnlB and gene 24.2 was amplified from T4 genome using primers 24.3Xba BW and rnlBamDSFW into which an amber mutation was introduced at amino acid 95. The PCR product was then cloned into the pET-rnlB DNA linearized with EcoRI and XbaI to generate pET-rnlB amber. To construct the rnlB deletion donor plasmid, a DNA fragment between nt639 of Hoc and nt145 of gene 24.2 was amplified using the T4 genome as a template and primers 24.2EcoR FW and HocXba BW. The DNA was then cloned into pET-rnlB DNA linearized with EcoRI and XbaI to generate the pET-rnlB deletion. The primers are shown in Table S1. The accuracy of all the constructs was confirmed by DNA sequencing.
Plaque assays
The efficiency of individual spacer plasmids to restrict T4 phage infection was determined by plaque assay (Figure 1). The CRISPR-Cas plasmids with different spacers were transformed into E. coli strains DH5α or B834. Up to ∼106 PFU of either WT T4 or T4 (C) in 100 μL was added to 300 μL of E. coli (∼108 cells/ml) containing the CRISPR-Cas plasmid. After incubation for 7 min at 37 °C, 3 mL of 0.7% top agar with streptomycin (50 μg/mL) was added into each tube, mixed, and poured onto LB-streptomycin plates. The plates were incubated at 37 °C overnight. The efficiency of plating (EOP) was calculated by dividing the pfu produced from infection of E. coli containing a spacer by the input pfu.
DNA Sequencing of Single Plaques
Single plaques were picked using a sterile Pasteur glass pipet and transferred into a 1.5 mL Eppedoff tube containing 200 μL of Pi-Mg buffer (26 mM Na2HPO4, 68 mM NaCl, 22 mM KH2PO4, 1 mM MgSO4, pH 7.5) plus 2 μL of chloroform. After 1 h incubation at room temperature with mixing every few min, 4 μL of the sample was used as a template for PCR using Phusion High-Fidelity PCR Master Mix (Thermo Fisher). Prior to starting PCR, the phage was denatured at 95 °C for 10 min. The amplified DNA was purified by agarose gel electrophoresis using QIAquick Gel Extraction Kit (Qiagen) and was sequenced (Retrogene).
Genome Editing
The donor plasmid and the corresponding CRISPR-Cas plasmid were cotransformed into a suppressor containing E. coli strain such as B40 (sup1). B40 cells transformed with either the donor plasmid or the CRISPR plasmid were used as controls. The cells were infected with WT or T4(C) mutant as described above. The progeny plaques produced were analyzed for genetic markers or the genome was amplified and sequenced as described above.
One-Step Growth Experiment
Log phase E. coli cells (∼2 × 108 cells per mL) grown on 1:1 LB/M9CA medium were infected with phage at a multiplicity of infection (m.o.i.) of 1 at 37 °C. Five minutes after infection, 100 μL of the 105-times diluted mixture was plated to determine the number of infected cells. Samples were withdrawn from the same mixture every 5 min until 60 min. The phage titer was determined by plaque assay following serial dilutions. Burst size is defined as the number of progeny phage produced per infection center.
Statistics
Each experiment was repeated at least three times. The data were initially analyzed by a two-way analysis of variance (ANOVA), followed by a Bonferonni post hoc test to compare individual groups using Prism Graphpad software.
Supplementary Material
Acknowledgments
This work was supported by the NIAID/NIH Grants AI111538 and AI081726 to V.B.R.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00179.
Generation of a g20 amber mutant by CRISPR-Cas editing; primers used for donor construction (PDF)
ORCID
Pan Tao: 0000-0002-0082-2229
Author Contributions
P.T. and V.R. designed the experiments; P.T., X.W., W.T., and J.Z. performed the experiments; P.T. and V.R. wrote the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Salmond GP, Fineran PC. A century of the phage: past, present and future. Nat Rev Microbiol. 2015;13:777–786. doi: 10.1038/nrmicro3564. [DOI] [PubMed] [Google Scholar]
- 2.Rao VB, Black LW. Structure and assembly of bacteriophage T4 head. Virol J. 2010;7:356. doi: 10.1186/1743-422X-7-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chanishvili N. Phage therapy–history from Twort and d’Herelle through Soviet experience to current approaches. Adv Virus Res. 2012;83:3–40. doi: 10.1016/B978-0-12-394438-2.00001-3. [DOI] [PubMed] [Google Scholar]
- 4.Karam JD. Bacteriophages: the viruses for all seasons of molecular biology. Virol J. 2005;2:19. doi: 10.1186/1743-422X-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wittebole X, De Roock S, Opal SM. A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence. 2014;5:226–235. doi: 10.4161/viru.25991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu TK, Koeris MS. The next generation of bacteriophage therapy. Curr Opin Microbiol. 2011;14:524–531. doi: 10.1016/j.mib.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 7.Viertel TM, Ritter K, Horz HP. Viruses versus bacteria-novel approaches to phage therapy as a tool against multidrug-resistant pathogens. J Antimicrob Chemother. 2014;69:2326–2336. doi: 10.1093/jac/dku173. [DOI] [PubMed] [Google Scholar]
- 8.LaFee S, Buschman H. Novel Phage Therapy Saves Patient with Multidrug-Resistant Bacterial Infection. UC San Diego News Center; San Diego: 2017. [Google Scholar]
- 9.Bakhshinejad B, Sadeghizadeh M. Bacteriophages as vehicles for gene delivery into mammalian cells: prospects and problems. Expert Opin Drug Delivery. 2014;11:1561–1574. doi: 10.1517/17425247.2014.927437. [DOI] [PubMed] [Google Scholar]
- 10.Pranjol MZ, Hajitou A. Bacteriophage-derived vectors for targeted cancer gene therapy. Viruses. 2015;7:268–284. doi: 10.3390/v7010268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sunderland KS, Yang M, Mao C. Phage-Enabled Nanomedicine: From Probes to Therapeutics in Precision Medicine. Angew Chem, Int Ed. 2017;56:1964–1992. doi: 10.1002/anie.201606181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tao P, Mahalingam M, Kirtley ML, van Lier CJ, Sha J, Yeager LA, Chopra AK, Rao VB. Mutated and bacteriophage T4 nanoparticle arrayed F1-V immunogens from Yersinia pestis as next generation plague vaccines. PLoS Pathog. 2013;9:e1003495. doi: 10.1371/journal.ppat.1003495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tao P, Mahalingam M, Marasa BS, Zhang Z, Chopra AK, Rao VB. In vitro and in vivo delivery of genes and proteins using the bacteriophage T4 DNA packaging machine. Proc Natl Acad Sci U S A. 2013;110:5846–5851. doi: 10.1073/pnas.1300867110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tao P, Mahalingam M, Rao VB. Highly Effective Soluble and Bacteriophage T4 Nanoparticle Plague Vaccines Against Yersinia pestis. Methods Mol Biol. 2016;1403:499–518. doi: 10.1007/978-1-4939-3387-7_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishii T, Yanagida M. The two dispensable structural proteins (soc and hoc) of the T4 phage capsid; their purification and properties, isolation and characterization of the defective mutants, and their binding with the defective heads in vitro. J Mol Biol. 1977;109:487–514. doi: 10.1016/s0022-2836(77)80088-0. [DOI] [PubMed] [Google Scholar]
- 16.Li Q, Shivachandra SB, Leppla SH, Rao VB. Bacteriophage T4 capsid: a unique platform for efficient surface assembly of macromolecular complexes. J Mol Biol. 2006;363:577–588. doi: 10.1016/j.jmb.2006.08.049. [DOI] [PubMed] [Google Scholar]
- 17.Shivachandra SB, Rao M, Janosi L, Sathaliyawala T, Matyas GR, Alving CR, Leppla SH, Rao VB. In vitro binding of anthrax protective antigen on bacteriophage T4 capsid surface through Hoc-capsid interactions: a strategy for efficient display of large full-length proteins. Virology. 2006;345:190–198. doi: 10.1016/j.virol.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 18.Pires DP, Cleto S, Sillankorva S, Azeredo J, Lu TK. Genetically Engineered Phages: a Review of Advances over the Last Decade. Microbiol Mol Biol Rev. 2016;80:523–543. doi: 10.1128/MMBR.00069-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karam JD, Drake JW, Kreuzer KN, Mosig G, Hall DH, Eiserling FA, Black LW, Spicer EK, Kutter E, Carlson K, Miller ES. Molecular biology of bacteriophage T4. American Society for Microbiology; Washington, DC: 1994. [Google Scholar]
- 20.Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. Bacteriophage T4 genome. Microbiol Mol Biol Rev. 2003;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353 doi: 10.1126/science.aad5147. aad5147. [DOI] [PubMed] [Google Scholar]
- 22.Box AM, McGuffie MJ, O’Hara BJ, Seed KD. Functional Analysis of Bacteriophage Immunity through a Type I-E CRISPR-Cas System in Vibrio cholerae and Its Application in Bacteriophage Genome Engineering. J Bacteriol. 2016;198:578–590. doi: 10.1128/JB.00747-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiro R, Shitrit D, Qimron U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014;11:42–44. doi: 10.4161/rna.27766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemay ML, Tremblay DM, Moineau S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth Biol. 2017 doi: 10.1021/acssynbio.6b00388. [DOI] [PubMed] [Google Scholar]
- 25.Martel B, Moineau S. CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014;42:9504–9513. doi: 10.1093/nar/gku628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, Davidson AR. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell. 2016;167:1829. doi: 10.1016/j.cell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryson AL, Hwang Y, Sherrill-Mix S, Wu GD, Lewis JD, Black L, Clark TA, Bushman FD. Covalent Modification of Bacteriophage T4 DNA Inhibits CRISPR-Cas9. mBio. 2015;6:e00648. doi: 10.1128/mBio.00648-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yaung SJ, Esvelt KM, Church GM. CRISPR/Cas9-mediated phage resistance is not impeded by the DNA modifications of phage T4. PLoS One. 2014;9:e98811. doi: 10.1371/journal.pone.0098811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson GG, Young KY, Edlin GJ, Konigsberg W. High-frequency generalised transduction by bacteriophage T4. Nature. 1979;280:80–82. doi: 10.1038/280080a0. [DOI] [PubMed] [Google Scholar]
- 31.Carlson K, Krabbe M, Nystrom AC, Kosturko LD. DNA determinants of restriction. Bacteriophage T4 endonuclease II-dependent cleavage of plasmid DNA in vivo. J Biol Chem. 1993;268:8908–8918. [PubMed] [Google Scholar]
- 32.Ho CK, Shuman S. Bacteriophage T4 RNA ligase 2 (gp24.1) exemplifies a family of RNA ligases found in all phylogenetic domains. Proc Natl Acad Sci U S A. 2002;99:12709–12714. doi: 10.1073/pnas.192184699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jabbar MA, Snyder L. Genetic and physiological studies of an Escherichia coli locus that restricts polynucleotide kinase-and RNA ligase-deficient mutants of bacteriophage T4. J Virol. 1984;51:522–529. doi: 10.1128/jvi.51.2.522-529.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amitsur M, Morad I, Kaufmann G. In vitro reconstitution of anticodon nuclease from components encoded by phage T4 and Escherichia coli CTr5X. EMBO J. 1989;8:2411–2415. doi: 10.1002/j.1460-2075.1989.tb08371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arisaka F, Yap ML, Kanamaru S, Rossmann MG. Molecular assembly and structure of the bacteriophage T4 tail. Biophys Rev. 2016;8:385–396. doi: 10.1007/s12551-016-0230-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Black LW, Rao VB. Structure, assembly, and DNA packaging of the bacteriophage T4 head. Adv Virus Res. 2012;82:119–153. doi: 10.1016/B978-0-12-394621-8.00018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao VB, Feiss M. The bacteriophage DNA packaging motor. Annu Rev Genet. 2008;42:647–681. doi: 10.1146/annurev.genet.42.110807.091545. [DOI] [PubMed] [Google Scholar]
- 38.Sun S, Kondabagil K, Draper B, Alam TI, Bowman VD, Zhang Z, Hegde S, Fokine A, Rossmann MG, Rao VB. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell. 2008;135:1251–1262. doi: 10.1016/j.cell.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun S, Rao VB, Rossmann MG. Genome packaging in viruses. Curr Opin Struct Biol. 2010;20:114–120. doi: 10.1016/j.sbi.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yap ML, Rossmann MG. Structure and function of bacteriophage T4. Future Microbiol. 2014;9:1319–1327. doi: 10.2217/fmb.14.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z, Kottadiel VI, Vafabakhsh R, Dai L, Chemla YR, Ha T, Rao VB. A promiscuous DNA packaging machine from bacteriophage T4. PLoS Biol. 2011;9:e1000592. doi: 10.1371/journal.pbio.1000592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mosig G, Gewin J, Luder A, Colowick N, Vo D. Two recombination-dependent DNA replication pathways of bacteriophage T4, and their roles in mutagenesis and horizontal gene transfer. Proc Natl Acad Sci U S A. 2001;98:8306–8311. doi: 10.1073/pnas.131007398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh JR, Aryee MJ, Joung JK. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selick HE, Kreuzer KN, Alberts BM. The bacteriophage T4 insertion/substitution vector system. A method for introducing site-specific mutations into the virus chromosome. J Biol Chem. 1988;263:11336–11347. [PubMed] [Google Scholar]
- 45.Runnels JM, Soltis D, Hey T, Snyder L. Genetic and physiological studies of the role of the RNA ligase of bacteriophage T4. J Mol Biol. 1982;154:273–286. doi: 10.1016/0022-2836(82)90064-x. [DOI] [PubMed] [Google Scholar]
- 46.Tao P, Li Q, Shivachandra SB, Rao VB. Bacteriophage T4 as a Nanoparticle Platform to Display and Deliver Pathogen Antigens: Construction of an Effective Anthrax Vaccine. Methods Mol Biol. 2017;1581:255–267. doi: 10.1007/978-1-4939-6869-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.