

Abstract
While allosteric modulators of the cannabinoid type-1 receptor (CB1) continue to be developed and characterized, the gap between the in vitro and in vivo data is widening, raising questions regarding translatability of their effects and biological relevance. Among the CB1 allosteric modulators, PSNCBAM-1 has received little attention regarding its effects in vivo. Recently, pregnenolone was reported to act as an allosteric modulator of CB1, blocking THC’s effects in vitro and in vivo, highlighting the potential of CB1 allosteric modulators for treatment of cannabis intoxication. We investigated the pharmacological effects of PSNCBAM-1 and two structural analogs, RTICBM-15 and -28, as well as pregnenolone, in both signaling and behavioral assays including [35S]GTPγS binding, the cannabinoid tetrad and drug discrimination. While the CB1 allosteric modulator PSNCBAM-1 attenuated THC-induced anti-nociception and its structural analog RTICBM-28 reduced THC’s potency in drug discrimination, most cannabinoid effects in mice were unaffected. In contrast to the mouse studies, PSNCBAM-1 and analogs insurmountably antagonized CP55,940- and THC-stimulated [35S]GTPγS binding and exhibited negative binding cooperativity with [3H]SR141716 with similar apparent affinities. Notably, RTICBM-28, which contains a cyano substitution at the 4-chlorophenyl position of PSNCBAM-1, exhibited enhanced binding cooperativity with CP55,940. In contrast to previous findings, pregnenolone did not block THC’s effects in drug discrimination, the cannabinoid tetrad, or [35S]GTPγS. These data further highlight the difficulty in translating pharmacological effects of CB1 allosteric modulators in vivo but confirm the established pharmacology of PSNCBAM-1 and analogs in molecular assays of CB1 receptor function.
Keywords: allosteric modulation, behavioral, cannabinoids, receptor binding, signaling
1. Introduction
The endogenous cannabinoid system’s involvement in a wide range of physiological processes has led to the development of numerous potent and selective compounds with therapeutic promise in preclinical assays. Predominantly expressed in the central nervous system, the cannabinoid type-1 (CB1) receptor has shown great potential as a target for the treatment of drug addiction, pain, mood disorders, obesity/metabolic syndrome, multiple sclerosis, and other diseases; this is evidenced by a large body of preclinical data demonstrating efficacy for drugs which bind the orthosteric site of the CB1 receptor or inhibit metabolism of its endogenous ligands, anandamide (Devane et al., 1992) and 2-arachidonoylglycerol (Sugiura et al., 1995). Unfortunately, these drugs have had limited success due to their propensity to produce psychoactivity (agonists, e.g. dronabinol; Issa et al., 2014) or depression (antagonists, e.g. rimonabant; Christensen et al., 2007) or their lack of clinical efficacy (enzymatic inhibitors, e.g. PF-04457845; Huggins et al., 2012).
Fortunately, the determination that the CB1 receptor (Matsuda et al., 1990) possesses a druggable allosteric site (Laprairie et al., 2016; Price et al., 2005; Shore et al., 2014) has provided a novel means through which receptor function can be studied and exploited for the development of better pharmacotherapeutics (Abood, 2016; Ross, 2007). Allosteric modulation allows for the fine-tuning of receptor pharmacology which may facilitate signaling bias towards pathways that are more therapeutically relevant while avoiding those involved in the untoward effects. There are now a handful of molecules which exhibit allosteric properties at the CB1 receptor (for reviews see Morales et al., 2016; Nguyen et al., 2016). The majority of reported allosteric modulators differentially affect radioligand binding, exhibiting positive binding cooperativity with the CB1/CB2 agonist [3H]CP55,940 and negative binding cooperativity with the selective CB1 antagonist [3H]SR141716. The allosteric antagonists exhibit insurmountable antagonism of receptor function while the positive allosteric modulators (PAMs) enhance agonist signaling.
It has now been over 10 years since the first reported CB1 allosteric modulators (Price et al., 2005) and despite dozens of papers characterizing these and other CB1 allosteric modulators in vitro, there is a dearth of articles reporting CB1-mediated effects for these compounds in vivo. Results from the first systematic investigation into the in vivo effects of the prototypical CB1 allosteric modulator, Org27569, were largely negative with only hypophagic effects reported in mice which occurred independently of the CB1 receptor (Gamage et al., 2014). The second structural series reported two years after Org27569, PSNCBAM-1, was reported to reduce feeding in rats (Horswill et al., 2007); however, this effect has yet to be established as CB1-mediated. While Org27569 was reported to have no effect in rats on CP55,940-induced catalepsy and anti-nociception, it did attenuate its hypothermic effects (Ding et al., 2014) and later was shown to reduce drug- and cue-induced reinstatement of cocaine and methamphetamine self-administration similar to SR141716 (Jing et al., 2014). In addition to the synthetic allosteric modulators, the hormone pregnenolone was reported to act as a CB1 allosteric modulator both in vitro and in vivo, blocking the effects of THC and WIN55,212-2 (Vallee et al., 2014); however, recent attempts to observe effects in vitro have had limited success (Khajehali et al., 2015; Straiker et al., 2015), reporting only slight displacement of [3H]SR141716 binding at micromolar concentrations but no observed effect in functional assays. The first positive allosteric modulator for the CB1 receptor, ZCZ011, was recently reported to augment CB1 agonist efficacy in both cellular and molecular assays as well as in rodent models including the cannabinoid tetrad and inflammatory pain models, the latter in which it exhibited efficacy on its own through hypothesized augmentation of endogenous cannabinoid tone (Ignatowska-Jankowska et al., 2015). Of note, the anti-nociceptive effects of ZCZ-011 were shown to be blocked by administration of SR141716, demonstrating CB1 mediation.
While the majority of behavioral studies involving CB1 allosteric antagonists have focused on Org27569 (Ding et al., 2014; Gamage et al., 2014; Jing et al., 2014), few have examined PSNCBAM-1 (Horswill et al., 2007) or any of its analogs. PSNCBAM-1 exhibits a very similar pharmacology to that of Org27569, exhibiting positive binding cooperativity with [3H]CP55,940 (German et al., 2014; Horswill et al., 2007) and negative binding cooperativity with [3H]SR141716 as well as insurmountable antagonism of CP55,940-stimulated [35S]GTPγS binding (Horswill et al., 2007). PSNCBAM-1 also antagonizes the CB1 receptor in other assays including depolarization-induced suppression of excitation (DSE; Straiker et al., 2015) and CP55940- and WIN55,212-induced beta-arrestin2 recruitment (Baillie et al., 2013). In the present study, PSNCBAM-1 and two of its structural analogs (Figure 1), RTICBM-15 and -28 (compounds 11 and 29 respectively in German et al., 2014), were assessed for in vitro and in vivo activity as allosteric modulators. Furthermore, we evaluated pregnenolone for its purported allosteric effects at the CB1 receptor. We hypothesized that PSNCBAM-1 and analogs would insurmountably antagonize receptor signaling in vitro (agonist-stimulated [35S]GTPγS binding) and in vivo cannabimimetic activity (cannabinoid tetrad) and drug discrimination.
Figure 1.
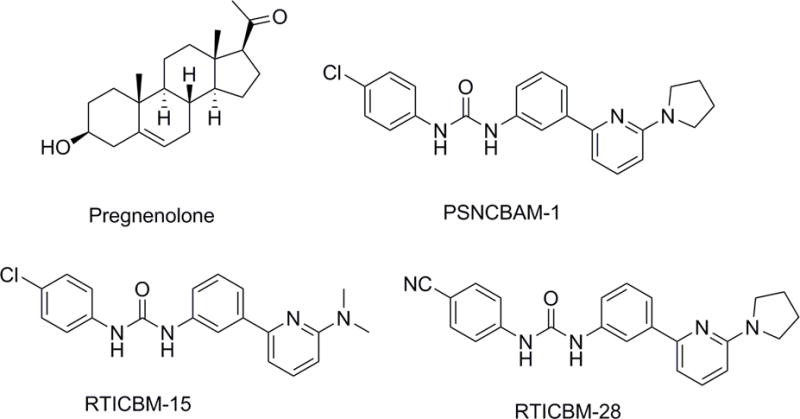
Chemical structures of PSNCBAM-1, RTICBM-15, RTICBM-28 and pregnenolone.
2. Materials and methods
2.1 Subjects
Adult male ICR mice (25–32g; Harlan, Dublin, VA) and C57/Bl6J inbred mice (20–25 g; Jackson Laboratories, Bar Harbor, ME) were housed singly in polycarbonate mouse cages. Each ICR mouse was tested with a single dose of compound in the tetrad battery. C57/Bl6J mice were used in the drug discrimination experiments. All animals were kept in a temperature-controlled (20–22°C) environment with a 12-hour light-dark cycle (lights on at 6 a.m.). ICR mice received food ad libitum when in their home cages. C57/Bl67 mice were maintained at 85–90% of free-feeding body weights by restricting daily ration of standard rodent chow. All mice received ad libitum water access when in their home cages. The in vivo studies reported in this manuscript were carried out in accordance with federal and state regulatory guidelines on the conduct of research in animals and were approved by our Institutional Care and Use Committee.
2.2 Apparatus
Measurement of spontaneous activity occurred in Plexiglas locomotor activity chambers (47 cm × 25.5 cm × 22 cm), with beam breaks (4 × 8 beam array) recorded by San Diego Instruments Photobeam Activity System software (San Diego, CA) on a computer located in the experimental room. Anti-nociception and rectal temperature were assessed with a standard tail flick device for rodents (Stoelting, Dale, IL) and a digital thermometer (Physitemp Instruments, Inc., Clifton, NJ), respectively. The ring immobility device consisted of an elevated metal ring (diameter = 5.5 cm, height = 28 cm) attached to a metal stand.
Mice in the drug discrimination experiment were trained and tested in mouse operant chambers (Coulbourn Instruments, Whitehall, PA), housed within light- and sound-attenuating cubicles. Each chamber contained two nose-poke apertures, with stimulus lights over each aperture, and a separate house light. A food dispenser delivered 20-mg food pellets (Bioserv Inc., Frenchtown, NJ) into a food cup (with a light) centered between the two aperture. Illumination of lights, delivery of food pellets, and recording of aperture responses were controlled by a computer-based system (Coulbourn Instruments, Graphic State Software, v 3.03, Whitehall, PA).
2.3 Chemicals
Δ9-THC (NIDA Drug Supply Program, Bethesda, MD), SR141716/rimonabant (NIDA), PSNCBAM-1 and its analogs (synthesized in our laboratories), pregnenolone (Steraloids, Newport, RI), otenabant (Toronto Research Chemicals, Toronto, Canada), and the open ring degradant of the synthetic cannabinoid XLR-11 (1-[1-(5-fluoropentyl)-1H-indol-3-yl]-3,3,4-trimethyl-4-penten-1-one; Cayman Chemical, Ann Arbor, MI) were dissolved in a vehicle of 7.8% Polysorbate 80 N.F. (VWR, Marietta, GA) and 92.2% sterile saline USP (Butler Schein, Dublin, OH). For in vitro studies, Δ9-THC, CP55,940 (NIDA), [3H]SR141716 (24 Ci/mmol; NIDA), [3H]CP55,940 (81.1 Ci/mmol; NIDA) and unlabeled SR141716 were dissolved in absolute ethanol whereas PSNCBAM-1, RTICBM-15, RTICBM-28, and pregnenolone were dissolved in 100% DMSO. All drugs were stored at −80°C as 10 mM stocks and diluted to final concentration of 0.1–0.2% solvent. GDP (Sigma Aldrich, St. Louis, MO), unlabeled GTPγS (Sigma Aldrich, St. Louis, MO), and [35S]GTPγS (1250 Ci/mmol; Perkin Elmer Life Sciences, Boston, MA) were dissolved in distilled water, aliquotted and stored at −80°C. Adenosine deaminase (Sigma Aldrich, St. Louis, MO) was diluted in distilled water and stored at 4°C.
2.4 Receptor Binding and Function
Cerebella from adult male ICR mice were dissected on ice, snap frozen in liquid nitrogen, and stored at −80°C until the day of the experiment. Cerebella were homogenized by polytron in membrane buffer (50 mM Tris, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4) on ice, centrifuged for 10 min at 40,000×g at 4°C. The supernatant was discarded and the pellet was suspended in membrane buffer, homogenized, and centrifuged again for 10 min at 40,000×g. The pellet was resuspended in membrane buffer and protein quantified by Bradford method. For receptor binding, reactions were carried out in assay buffer (membrane buffer containing 1 mg/ml bovine serum albumin; BSA) and membranes were incubated for 90 min at 30°C in 1 nM [3H]SR141716A (KD=0.52±0.11 nM) and varying concentrations of allosteric modulators. Non-specific binding was determined by excess cold ligand (1 μM) in the absence and presence of test compounds as these were found to affect non-specific binding. Total bound of [3H]SR141716A was less than 10% of total added (minimal ligand depletion).
For receptor signaling, membranes were preincubated in assay buffer for 10 min with 3 units/ml adenosine deaminase then incubated for 60 min at 30°C with 30 μM GDP, 0.1 nM [35S]GTPγS, and non-specific binding was determined by adding 30 μM unlabeled GTPγS. Concentration response curves for the synthetic CB1/CB2 agonist CP55,940 were conducted in the absence and presence of multiple concentrations of test compounds.
2.5 Tetrad tests
Each mouse was tested in the “cannabinoid tetrad,” four assays in which cannabinoid agonists produce a profile of in vivo effects: suppression of locomotor activity, decreased rectal temperature, anti-nociception, and catalepsy. Prior to injection, baseline values were obtained for rectal temperature and in the tail flick test in each mouse. In the latter procedure, the mouse’s tail was placed under an intense light (radiant heat) and the latency (s) to remove it was recorded. In order to minimize tail damage, a maximal latency (10 s) was employed. After baseline measurements were taken, mice were injected intraperitoneally (i.p.) with vehicle or10 mg/kg of an allosteric modulator or 3 mg/kg rimonabant 10 min before being injected i.p. with vehicle or a cannabinoid agonist (30 mg/kg THC or 5.6 mg/kg the open ring degradant of XLR-11, a tetramethylcyclopropyl synthetic cannabinoid with high affinity and efficacy at CB1 receptors (Thomas et al., 2017). Thirty min after injection with the agonist (40 min after allosteric modulator administration), mice were placed into individual activity chambers for a 10 min session. Immediately upon removal from the chambers, tail-flick latency and rectal temperature were measured again followed by placement on the elevated ring apparatus at 50 min post-injection. The amount of time the animals remained motionless on the ring during a 5 min period was recorded. If a mouse fell off the ring during the catalepsy test, it was immediately placed back on and timing was continued for up to 9 falls. After the 10th fall, the test was terminated for the mouse.
2.6 Drug Discrimination
Training in the mouse discrimination procedure was similar to that described previously (Vann et al., 2009). Briefly, two groups of mice were trained in a drug discrimination procedure. Each mouse was placed in a standard operant conditioning chamber with two nose-poke apertures. Mice were trained to respond on one of the two apertures following i.p. administration of 5.6 mg/kg THC and to respond on the other aperture following i.p. vehicle injection according to a fixed ratio 10 (FR10) schedule of food reinforcement, under which 10 consecutive responses on the correct (injection-appropriate) aperture resulted in delivery of a food pellet. Responses on the incorrect aperture reset the ratio requirement on the correct aperture. Daily injections were administered on a double alternation sequence of THC and vehicle (e.g., drug, drug, vehicle, vehicle). Daily 15 min training sessions were held Monday–Friday until the mice consistently met three criteria: (1) the first completed FR10 was on the correct aperture, (2) ≥ 80% of the total responding occurred on the correct aperture, and (3) response rate must have been ≥ 0.17 responses/s. When the criteria were met, acquisition of the discrimination was established and substitution testing began.
Stimulus substitution tests were typically conducted on Tuesdays and Fridays during 15 min test sessions, with maintenance of training continuing on intervening days. During test sessions, 10 consecutive responses on either aperture delivered reinforcement. If a mouse responded the other aperture prior to completing 10 responses on a single aperture, the ratio requirement on the original aperture was reset. To be tested in the experiment, mice must have completed the first FR10 on the correct aperture, ≥ 80% of the total responding must have occurred on the correct aperture, and response rate must have been ≥ 0.17 responses/s during the prior day’s training session. In addition, the mouse must have met these same criteria during previous training sessions with the alternate training compound (THC or vehicle). In the first discrimination group, substitution tests were conducted with THC followed by tests with the following compounds (alone and in combination with THC): compound RTICBM-15, pregnenolone, rimonabant (CB1 receptor antagonist/inverse agonist), and SR144528 (CB2 receptor antagonist/inverse agonist). In the second discrimination group, a dose-effect curve was determined with THC. Subsequently, the following compounds (alone and in combination with THC) were assessed: PSNCBAM-1, RTICBM-28, otenabant, and rimonabant. THC was administered i.p. 30 min before the start of the discrimination test. With the exception of pregnenolone, all allosteric modulators and antagonists were administered i.p. 40 min pre-session (i.e., 10 min before THC or vehicle injection). Pregnenolone was injected s.c. immediately before THC or vehicle injection at 30 min pre-session. All compounds were administered at a volume of 10 ml/kg.
2.7 Data Analysis
[35S]GTPγS data were fit to either 3 parameter non-linear regression with data normalized to percent maximal stimulation by CP55,940 or THC to calculate changes in Emax or functional data were fit the allosteric operational model (see below). For pIC50 comparisons, inhibition curves were calculated and normalized for each individual concentration of CP55,940 (i.e. VEH = 100%) and were fit to 3 parameter non-linear regression, with bottom and top constrained to 0 and 100 respectively. pIC50 values were considered significantly different when 95% confidence intervals did not overlap. Emax data were analyzed by One-Way ANOVA with Bonferroni post-hoc tests. For [3H]SR141716 receptor binding data, test compounds were fit to the allosteric ternary complex model (ATCM) (see equation 1) using Graphpad Prism 6 for the derivation of cooperativity factors (logα) and test compound affinities (pKB) with hot ligand ([3H]SR141716) KA constrained to 0.52 nM and the concentration constrained to 1 nM. For [3H]CP55,940 binding, data were fit to 3 parameter nonlinear regression to compare maximal increases in binding between modulators due to the inability of the ATCM to account for changes in Bmax (May et al., 2007). For curve fitting of functional data and determination of binding cooperativity (α) and the efficacy modifier (β) for each allosteric modulator, data were fit to the allosteric operational model data using equation 2. Emax was constrained to 100 and slope (n) to 1. LogKA was constrained to -6.6 for and TA was constrained to 14 which was the mean average of calculated values for each set of curves. LogKB was constrained to corresponded affinity values for allosteric modulators determined from radioligand binding data. TB was constrained to 0 as none of the tested compound exhibited efficacy.
| Equation 1 |
| Equation 2 |
Spontaneous activity was measured as total number of photocell beam interruptions during the 10 min session. Anti-nociception was expressed as the percent maximum possible effect (MPE) using a 10 s maximum test latency as follows: [(test−control)/(10−control)]×100. Rectal temperature values were expressed as the difference between control temperature (before injection) and temperature following drug administration (Δ°C). For catalepsy, the total amount of time (s) that the mouse remained motionless on the ring apparatus (except for breathing and whisker movement) was used as an indication of catalepsy-like behavior. This value was divided by 300 s and multiplied by 100 to obtain a percent immobility. For each of the four measures, a separate two-way (allosteric modulator/antagonist X agonist) ANOVA was conducted. Significant ANOVAs were further analyzed with Tukey post hoc tests (α = 0.05) to determine differences between means.
For each drug discrimination session, percentage of responses on the drug-assigned aperture and response rate (responses/s) were calculated. Since mice that responded less than 10 times during a test session did not respond on either aperture a sufficient number of times to earn a reinforcer, their data were excluded from analysis of drug aperture selection, but response rate data were included. Response-rate data were analyzed using separate repeated-measures ANOVAs for each dose-effect curve. Significant ANOVAs were further analyzed with Tukey post hoc tests (α = 0.05) to specify differences between means.
3. Results
3.1 In Vitro Receptor Binding and [35S]GTPγS Assay
PSNCBAM-1, RTICBM-15 and -28 exhibited complete insurmountable antagonism of CP55,940 stimulated [35S]GTPγS binding in contrast to the orthosteric CB1 antagonist/inverse agonist, SR141716, which produced a dextral shift of CP55,940’s concentration response curve (Figure 2A–D). There was a significant reduction in Emax for CP55,940 (Table 1) in the presence of increasing concentrations of PSNCBAM-1 [F(5,12)=34.9, p<0.001], RTICBM-28 [F(5,12)=40.7, p<0.001], and RTICBM-15 [F(5,12)=61.2, p<0.001], consistent with insurmountable antagonism. Fitting of the allosteric operational model revealed differences in binding cooperativity (α) for all three modulators with RTICBM-28 exhibiting the greatest binding cooperativity with CP55,940 (Table 2). CP55,940 inhibition curves were constructed for each compound and for SR141716 to determine potency shifts. All three compounds exhibited significantly greater potency at higher concentrations of CP55,940 (e.g. 10 nM vs. 1 μM; Figure 2F–H) as exhibited by non-overlapping 95% confidence intervals, consistent with an allosteric mechanism wherein the modulator increases probe binding affinity (Table 3). Consistent with greater binding cooperativity, RTICBM-28 produced a larger maximal increase in specific binding of [3H]CP55,940 compared to PSNCBAM-1 (Table 4). In contrast to the allosteric modulators, SR141716A exhibited rightward shifts in potency with increasing concentrations of CP55,940 consistent with competitive antagonism (Figure 2E). Schild analysis of these data resulted in a pA2 value of −9.3 (CL: −8.9 – −9.9) for SR141716, similar to the log KD of −9.28 (0.52 nM) obtained from saturation binding for [3H]SR141716. In addition to antagonism of CP55,940, the allosteric modulators PSNCBAM-1 [F(5,12)=23.19, p<0.0001], RTICBM-15 [F(5,12)=12.67, p<0.001] and RTICBM-28 [F(5,12)=21.14, p<0.0001] all produced insurmountable antagonism of THC-stimulated [35S]GTPγS binding (Figure 3A–C; Table 1). In contrast, pregnenolone (1 – 10 μM) did not affect THC-stimulated [35S]GTPγS binding at any concentration, though a small but non-significant trend to reduce the pEC50 of THC was observed at 10 μM [VEH: pEC50=7.31±0.19 (CL: 6.52 – 7.31); 10 μM: pEC50=6.41±0.16 (CL: 6.07 – 6.74); Figure 3D]. For radioligand binding, PSNCBAM-1, RTICBM-15 and RTICBM-28 exhibited negative binding cooperativity with [3H]SR141716A with similar apparent affinities and cooperativity (Table 5). Interestingly, inclusion of 30 μM GDP in the binding assay appeared to reduce the apparent affinity of all three allosteric modulators when displacing [3H]SR141716, suggesting that differences in potency for inhibiting CP55,940’s effects are likely not due to increased affinity for the allosteric modulators for the receptor by GDP.
Figure 2.
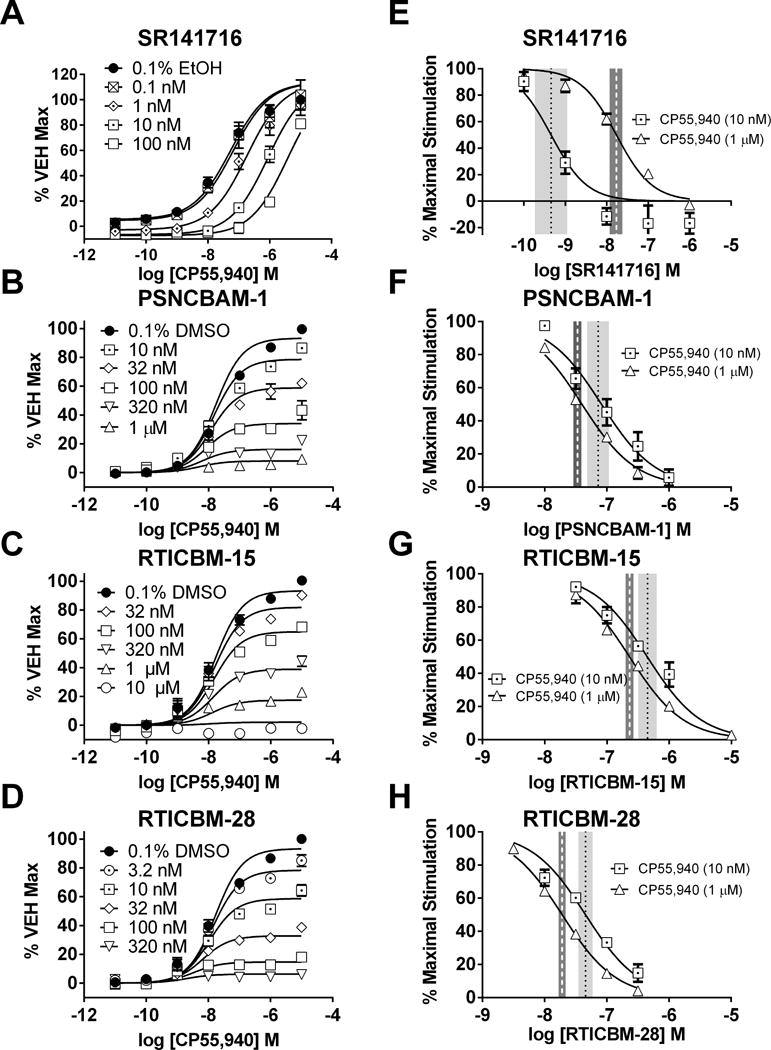
Insurmountable antagonism of CP55,940-stimulated [35S]GTPγS binding in mouse cerebellar membranes by PSNCBAM-1 and analogs. (A) The CB1 antagonist SR141716 produced dextral shifts in CP55,940’s dose response consistent with competitive antagonism whereas (B) PSNCBAM-1, (C) RTICBM-15, and (D) RTICBM-28 produced insurmountable antagonism. The logIC50 value for (E) SR141716 shifted rightward with a greater concentration of CP55,940, consistent with competitive binding, whereas the logIC50 values for (F) PSNCBAM-1, (G) RTICBM-15, and (H) RTICBM-28 demonstrated greater inhibitory potency for these compounds when the concentration of CP55,940 was higher, consistent with positive binding cooperativity. Data are mean±SEM of N=6 and normalized to maximal CP55,940 stimulation in presence of VEH. Dotted lines for E-H indicate logIC50 values with dark shaded areas covering 95% confidence intervals for CP55,940 (1 μM) and light shaded areas covering 95% confidence intervals for CP55,940 (10 nM). VEH = vehicle (0.1% DMSO)
Table 1.
Allosteric modulators exhibit insurmountable antagonism of CP55,940- and THC-stimulated [35S]GTPγS binding
| Compound | Allosteric Modulator Concentration
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Vehicle | 3.2 nM | 10 nM | 32 nM | 100 nM | 320 nM | 1 μM | 10 μM | |
| Emax ± SEM | Emax ± SEM | Emax ± SEM | Emax ± SEM | Emax ± SEM | Emax ± SEM | Emax ± SEM | Emax ± SEM | |
|
CP55,940
| ||||||||
| PSNCBAM-1 | 97.29 ± 3.7 | ND | 71.58 ± 12.6 | 51.19 ± 4.2** | 32.92 ± 2.7*** | 10.25 ± 5.1**** | 3.396 ± 1.8**** | ND |
| RTICBM-15 | 102.8 ± 3.2 | ND | ND | 93.24 ± 3.5 | 71.55 ± 3.3* | 50.93 ± 4.4*** | 31.31 ± 9.6**** | 0.9833 ± 1.0**** |
| RTICBM-28 | 102.5 ± 1.1 | 88.47 ± 5.9 | 74.54 ± 7.5 | 42.42 ± 4.6*** | 19.05 ± 9.5**** | 11.99 ± 9.5**** | ND | ND |
| THC | ||||||||
|
|
||||||||
| PSNCBAM-1 | 104.1 ± 9.0 | ND | 74.14 ± 6.8* | 68.15 ± 9.5* | ND | 45.77 ± 6.1*** | 29.45 ± 5.8**** | 6.165 ± 5.2**** |
| RTICBM-15 | 99.49 ± 7.7 | ND | ND | 97.09 ± 8.9 | 87.08 ± 12.4 | 65.87 ± 5.9 | 52.5 ± 7.1 | 23.52 ± 6.1 |
| RTICBM-28 | 96.64 ± 7.6 | ND | 75.79 ± 10.7 | 60.2 ± 9.6* | ND | 21.78 ± 6.4*** | 7.228 ± 4.7**** | ND |
p < 0.05,
p < 0.01,
p < 0.001,
p < 0.0001 compared to Vehicle
ND = Not determined
Table 2.
Allosteric operational model parameters for PSNCBAM-1, RTICBM-15, and RTICBM-28 in CP55,940-stimulated [35S]GTPγS binding.
| Compound | Calculated affinity and cooperativity factors
|
|||
|---|---|---|---|---|
| alpha ± SEM | 95% C.I. | beta ± SEM | 95% C.I. | |
|
|
|
|||
| PSNCBAM-1 | 55.01 ± 3.76 | (47.65 – 62.37) | 0.003 ± 0.001 | (0.0003 – 0.005) |
| RTICBM-15 | 20.71 ± 1.43 | (17.91 – 23.51) | ~0 | ND |
| RTICBM-28 | 201.1 ± 11.95 | (177.7 – 224.5) | 0.001 ± 0.001 | (0 – 0.003) |
Data were fit to allosteric operational model (see equation 2) with parameters constrained as follows: logKA=−6.6; Emax=100; TA=14; TB=0; n=1; α>0; β>0. LogKB was constrained individually to values obtained from [3H]SR141716 binding data (Table 4). Values considered significant by non-overlapping 95% confidence intervals. ND = not determined.
Table 3.
Allosteric modulators exhibit greater inhibitory potency when tested at higher concentrations of CP55,940
| Compound | CP55,940 Concentration
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 10 nM | 100 nM | 1 μM | 10 μM | |||||
| pIC50 ± SEM | 95% C.I. | pIC50 ± SEM | 95% C.I. | pIC50 ± SEM | 95% C.I. | pIC50 ± SEM | 95% C.I. | |
| PSNCBAM-1 | 7.08 ± 0.07 | (6.93 – 7.24) | 7.31 ± 0.03 | (7.23 – 7.38) | 7.40 ± 0.03 | (7.33 – 7.47) | 7.30 ± 0.03 | (7.25 – 7.36) |
| RTICBM-15 | 6.35 ± 0.07 | (6.22 – 6.49) | 6.70 ± 0.06 | (6.57 – 6.70) | 6.64 ± 0.03 | (6.58 – 6.70) | 6.59 ± 0.06 | (6.47 – 6.72) |
| RTICBM-28 | 7.34 ± 0.05 | (7.23 – 7.46) | 7.51 ± 0.04 | (7.42 – 7.60) | 7.72 ± 0.03 | (7.66 – 7.77) | 7.59 ± 0.03 | (7.53 – 7.65) |
Values considered significant by non-overlapping 95% confidence intervals. ND = not determined.
Table 4.
RTICBM-28 produces greater increase in specific binding of [3H]CP55,940 compared to PSNCBAM-1, consistent with enhanced binding cooperativity
| Compound | Calculated affinity and binding maxima
|
|||
|---|---|---|---|---|
| pEC50 ± SEM | 95% C.I. | Top ± SEM | 95% C.I. | |
|
|
|
|||
| PSNCBAM-1 | 6.53 ± 0.05 | (6.43 – 6.63) | 419.1 ± 8.75 | (400.9 – 437.3) |
| RTICBM-15 | 6.43 ± 0.07 | (6.21 – 6.66) | 317.2 ± 15.03** | (285.6 – 348.7) |
| RTICBM-28 | 6.61 ± 0.07 | (6.46 – 6.76) | 477.5 ± 15.14* | (446.1 – 509.0) |
Data fit to 3 parameter non-linear regression.
p<0.05,
p<0.01 compared to PSNCBAM-1
Figure 3.
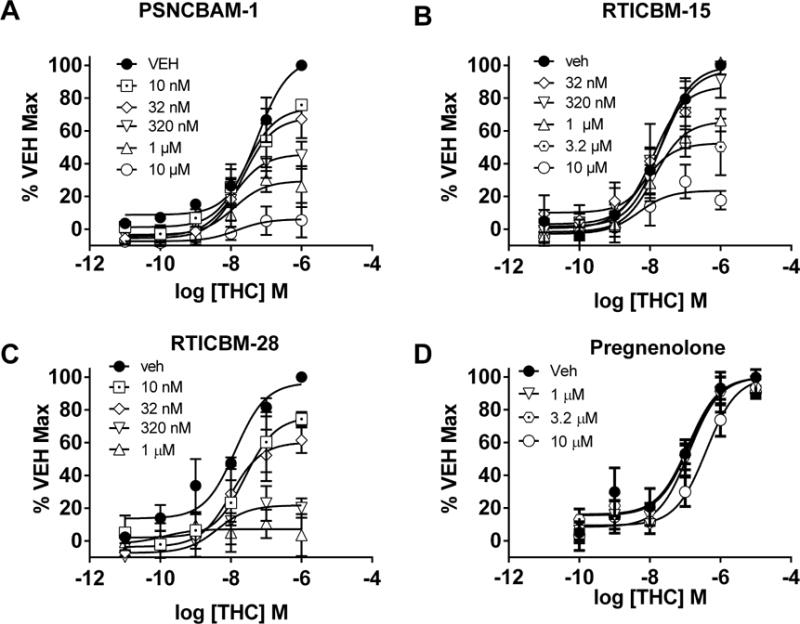
Insurmountable antagonism of THC-stimulated [35S]GTPγS binding in mouse cerebellar membranes by PSNCBAM-1, RTICBM-15 and RTICBM-28. The prototypical CB1 allosteric modulator (A) PSNCBAM-1, and its analogs (B) RTICBM-15 and (C) RTICBM-28, insurmountably antagonized THC-stimulated [35S]GTPγS binding. (D) Pregnenolone did not affect THC-stimulated [35S]GTPγS binding. Data are mean±SEM of N=3 for A, B and C. N=4 for D. VEH = vehicle (0.1% DMSO)
Table 5.
Allosteric modulators exhibit comparable affinity and negative cooperativity with [3H]SR141716 when binding data are fit to allosteric ternary complex model
| Compound | Calculated affinity and cooperativity factors
|
|||
|---|---|---|---|---|
| pKB ± SEM | 95% C.I. | logα ± SEM | 95% C.I. | |
|
|
|
|||
| PSNCBAM-1 | 6.71 ± 0.18 | (6.3 – 7.1) | −0.73 ± 0.12 | (−0.99 – −0.47) |
| RTICBM-15 | 6.50 ± 0.12 | (6.20 – 6.50) | −0.85 ± 0.11 | (−1.07 – −0.63) |
| RTICBM-28 | 6.65 ± 0.11 | (6.42 – 6.87) | −1.13 ± 0.18 | (−1.50 – −0.76) |
| Calculated affinity and cooperativity in presence of 30 μM GDP | ||||
|---|---|---|---|---|
|
|
|
|||
| Compound | pKB ± SEM | 95% C.I. | logα ± SEM | 95% C.I. |
|
|
|
|||
| PSNCBAM-1 | 5.74 ± 0.10 | (5.52 – 5.95) | ND | ND |
| RTICBM-15 | 5.89 ± 0.11 | (5.68 – 6.11) | ND | ND |
| RTICBM-28 | 5.86 ± 0.10 | (5.66 – 6.05) | ND | ND |
ND = not determined, calculated values very low with very wide confidence intervals
3.2 Tetrad Battery Results
Figure 4 shows the effects of the allosteric modulators in combination with vehicle and 30 mg/kg THC in the cannabinoid tetrad assay. When followed by vehicle injection, 10 mg/kg of each modulator or 3 mg/kg rimonabant did not have any effect on locomotor activity (Figure 4A), anti-nociception (Figure 4B), rectal temperature (Figure 4C), or catalepsy (Figure 4D), each as compared to the vehicle/vehicle condition. In contrast, the 30 mg/kg dose of THC produced rimonabant-reversible antinociception (Figure 4B), hypothermia (Figure 4C), and catalepsy (Figure 4D). The 30 mg/kg dose of THC did not affect locomotor activity when administered with vehicle, but the combination of 3 mg/kg rimonabant and 30 mg/kg THC increased activity (Figure 4A). The 10 mg/kg doses of PSNCBAM-1, RTICBM-15 and RTICBM-28 did not alter THC’s effects on activity (Figure 4A), hypothermia (Figure 4C), or catalepsy (Figure 4D); however, PSNCBAM-1, but not RTICBM-15 or RTICBM-28, decreased THC’s anti-nociceptive effect (Figure 4B). Although statistically significant [F(4,50)=7.31, p<0.05], the magnitude of this decrease was modest, as the degree of anti-nociception still exceeded that observed after vehicle injection. Results of combination tests with 5.6 mg/kg of the open ring degradant of XLR-11 revealed that none of the three allosteric modulators (10 mg/kg doses of PSNCBAM-1, RTICBM-15, and RTICBM-28) had any effect on the XLR-11 degradant-induced locomotor suppression, anti-nociception, hypothermia or catalepsy (data not shown).
Figure 4.
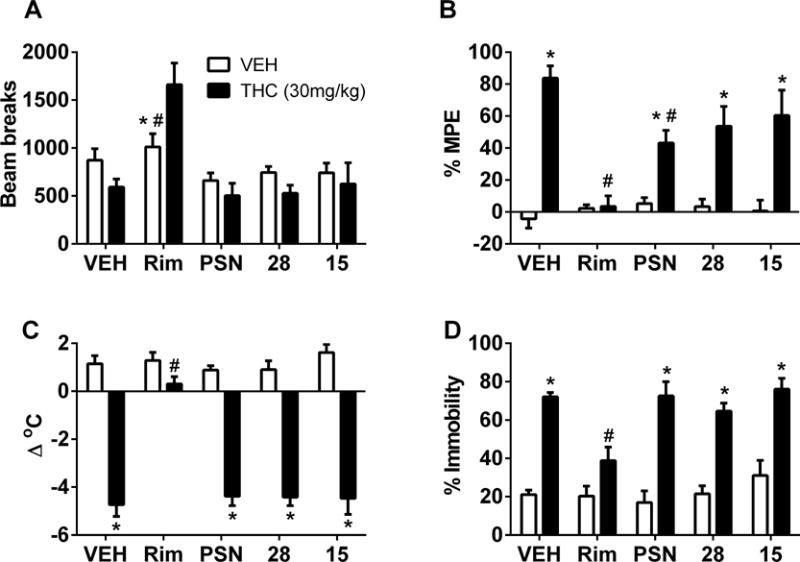
Effects of vehicle, rimonabant (1 mg/kg), PSNCBAM-1 (10 mg/kg), RTICBM-15 (10 mg/kg), and RTICBM-28 (10 mg/kg) tested in combination with vehicle (unfilled bars) or THC (filled bars) on spontaneous activity (4A), anti-nociception (4B), rectal temperature (4C), and catalepsy (4D). Values represent the mean (± SEM) of 6 mice per group. Asterisks (*) and number symbols (#) indicate a significant post-hoc confirmation of difference between compound and the vehicle/vehicle condition or the vehicle/THC condition (left side of each panel), respectively following significant (allosteric modulator/antagonist X agonist) interaction. VEH=vehicle; THC=delta-9-tetrahydrocannabinol; PSN=PSNCBAM-1; Rim=Rimonabant/SR141716; 28=RTICBM-28; 15=RTICBM-15
3.4 Drug Discrimination
In the first group of mice trained to discriminate THC (5.6 mg/kg) from vehicle, THC substituted for itself with an ED50 of 1.2 mg/kg (CL: 0.82 – 1.7) (Figure 5A). Pretreatment with RTICBM-15 (3 mg/kg) or pregnenolone (3–20 mg/kg) did not alter the dose-response relationship of THC for aperture selection (Figure 5A) [ED50 for THC & RTICBM-15 = 0.99 mg/kg (CL: 0.79 – 1.23)] or response rate (Figure 5B). When administered in combination with vehicle, neither compound produced substitution or response rate effects. A higher dose (10 mg/kg) of RTICBM-15 was also without effect when tested against the THC training dose (5.6 mg/kg). Similarly, THC also dose dependently substituted for the 5.6 mg/kg training dose in the second group of mice, with ED50 values of 0.88 mg/kg (CL: 0.64 – 1.21) and 0.85 mg/kg (CL: 0.56 – 1.28) for the first (Figure 6A) and second (Figure 6B) determinations, respectively. Whereas PSNCBAM-1 (10 mg/kg) did not alter THC’s discriminative stimulus effects [ED50 value for THC & PSNCBAM-1 = 0.95 mg/kg (CL: 0.77 – 1.17)] (Figure 6A), RTICBM-28 (10 mg/kg) lowered THC potency [ED50 value for THC & RTICBM-28 = 2.25 mg/kg (CL: 1.67 – 3.05)], but did not alter its efficacy (Figure 6B). Neither compound decreased response rates (Figure 6D–E). By comparison, the two CB1 receptor antagonists, otenabant and rimonabant, blocked THC’s discriminative stimulus effects without substituting themselves when administered alone (Figure 6C). Each antagonist significantly decreased response rates (Figure 6F).
Figure 5.
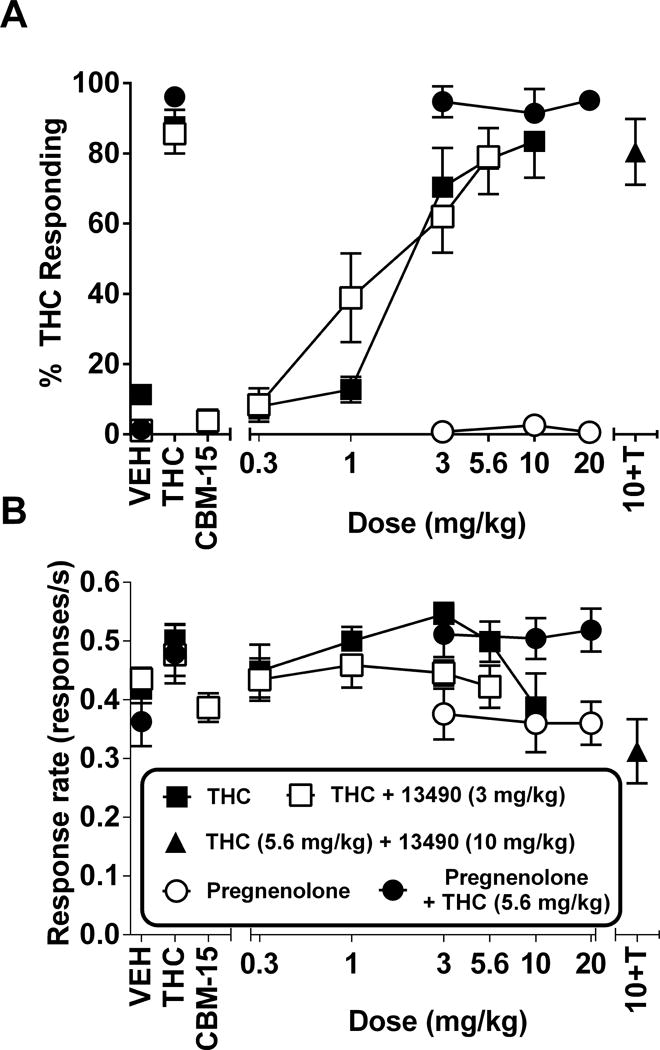
Effects of THC (filled squares), RTICBM-15 (3 mg/kg) + THC (unfilled squares), and pregnenolone in combination with vehicle (unfilled circles) or 5.6 mg/kg THC (filled circles) on percentage of THC-aperture responding (5A) and response rates (5B) in mice trained to discriminate 5.6 mg/kg THC from vehicle. Results of a combination test with 10 mg/kg RTICBM-15 and 5.6 mg/kg THC (filled triangle) are shown at the right side of each panel. Points above V, T and CBM-15 represent the results of control tests with vehicle, 5.6 mg/kg THC, and 3 mg/kg RTICBM-15 alone, respectively, conducted before the dose-effect determinations. Each point represents the mean (± SEM) of 7–9 mice. VEH=vehicle; THC,T=delta-9-tetrahydrocannabinol; CBM-15=RTICBM-15
Figure 6.
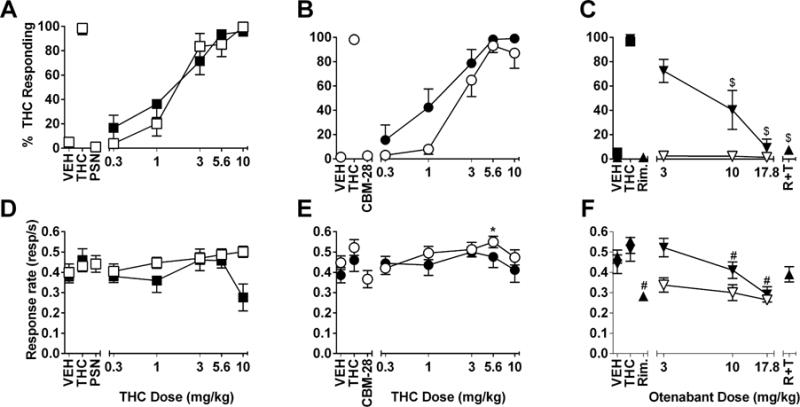
Effects of combinations of THC (0.3–10 mg/kg; filled symbols in 6A and 6B) and 10 mg/kg PSNCBAM-1 (unfilled squares; 6A) or 10 mg/kg RTICBM-28 (unfilled circles; 6B) on percentage of THC-aperture responding in mice trained to discriminate 5.6 mg/kg THC from vehicle. The same mice were also assessed with the CB1 receptor antagonists, otenabant (with vehicle, unfilled inverted triangles and with 5.6 mg/kg THC, filled inverted triangles; 6C) and 1 mg/kg rimonabant and 5.6 mg/kg THC (filled triangle; right side of 6C). Response rates for each dose-effect curve are shown in the respective bottom panels (6D–F). Points above V and T represent the results of control tests with vehicle and 5.6 mg/kg THC. Points above PSN (6A, 6D), 136 (6B, 6E), and Antag (6C, 6F) represent the results of control tests with combinations of vehicle and PSNCBAM-1 (10 mg/kg), RTICBM-28 (10 mg/kg), and rimonabant (1 mg/kg), respectively. Each point represents the mean (± SEM) of 7–9 mice. VEH=vehicle; THC,T=delta-9-tetrahydrocannabinol; PSN=PSNCBAM-1; Rim,R=Rimonabant/SR141716; CBM-28=RTICBM-28
4. Discussion
Theoretically, allosteric modulation can provide numerous benefits over orthosteric agonists or antagonists including greater receptor subtype selectivity, permissive augmentation of endogenous signaling, stabilization of novel receptor conformations that may bias signaling towards preferred pathways, and dissociation of duration and magnitude of effect (Kenakin, 2009). The obvious caveat is that these molecular changes must extend from the test tube to the whole animal for any therapeutic benefit to be realized. To fulfill this requirement, we sought to characterize the CB1 allosteric modulator PSNCBAM-1 as well as two structural analogs, RTICBM-15 and RTICBM-28, both in vitro and in vivo.
PSNCBAM-1, RTICBM-15 and RTICBM-28 non-competitively antagonized CP55,940-and THC-stimulated [35S]GTPγS binding. Consistent with our data, PSNCBAM-1 has previously been reported to act as a non-competitive antagonist of CP55,940-stimulated [35S]GTPγS binding (Baillie et al., 2013; Horswill et al., 2007). The IC50 value for PSNCBAM-1 reported here (49.2 nM at 100 nM CP55,940), was similar to that reported by the Horswill study (42.1 nM at 200 nM CP55,940) whereas Baillie et al reported PSNCBAM-1 as slightly more potent (2.72 nM).
Fitting of the operational model of allosterism allowed for determination of the binding cooperativity (alpha) and efficacy modifier (beta) and revealed greater binding cooperativity for RTICBM-28 as compared with PSNCBAM-1. In addition, we constructed inhibition curves for PSNCBAM-1, RTICBM-15 and RTICBM-28 at multiple CP55,940 concentrations to determine if PSNCBAM-1’s potency changed depending upon the concentration of CP55,940 tested. Indeed, all three compounds exhibited significantly greater potency at higher concentrations of CP55,940 as determined by non-overlapping 95% confidence intervals. This was to be expected as allosteric vectors are bi-directional (Kenakin, 2014) and positive binding cooperativity with [3H]CP55,940 was observed for these compounds in the present study and was reported previously (German et al., 2014; Horswill et al., 2007). Notably, RTICBM-28 exhibited greater potency at inhibiting CP55,940 than PSNCBAM-1 or RTICBM-15 and a produced a larger maximal increase in specific [3H]CP55,940 binding. These results combined with the greater calculated alpha for RTICBM-28 in the [35S]GTPγS binding suggest that the cyano substitution at the 4-chlorophenyl position enhances the binding cooperativity between the allosteric modulator and the probe.
The calculated KB values for these analogs did not significantly differ in binding studies with [3H]SR141716, further suggesting that differences in potency between RTICBM-28 and PSNCBAM-1 are due to binding cooperativity and not greater affinity of the modulator for the receptor. Inclusion of 30 μM GDP to reflect the conditions in which the allosteric modulators are binding with the receptor in the functional assays reduced the apparent affinity of all three compounds but did not reveal differences between KB values suggesting that while GDP appears to reduce the affinity of the allosteric modulators for the receptor, it does not account for differences in inhibitory potency. It was previously reported that azide and isothiocyanate substitutions at the 4-chlorophenyl position of PSNCBAM-1 reduced potency to inhibit CP55,940’s effects on cAMP accumulation and β-arrestin recruitment (Kulkarni et al., 2016), further supporting the importance of this position on the molecule. For the most part, our [35S]GTPγS data correspond with the calcium data from a previous study showing insurmountable antagonism of CP55,940 with IC50 values within the same range (German et al., 2014). However, while we observed differences in potencies between all three structures, the previous study observed relatively similar potencies in their calcium mobilization functional assay. This could be due to 1) differences in sensitivity of the assays, 2) differences between the mouse and human CB1 receptors, or 3) differences in receptor/G-protein coupling as the cell line used in the previous study contains recombinant Gαq with the 5 carboxy-teminal amino acids replaced with those of Gαi which effectively facilitates Gαi/o GPCRs to couple to Gαq, allowing for assessment of receptor activity via calcium imaging. Here we also observed that the two structural modifications to PSNCBAM-1, either a cyano substitution at the 4-chlorophenyl position (RTICBM-28) or a dimethyl substitution at the 2-pyrrolidinyl position (RTICBM-15), do not appear to affect the negative binding cooperativity with [3H]SR141716. After fitting binding data to the allosteric ternary complex model, derived apparent affinity (KB) and cooperativity (α) factors for RTICBM-15 and RTICBM-28 did not significantly differ from PSNCBAM-1.
We also examined PSNCBAM-1, RTICBM-15 and RTICBM-28 using THC as a probe in [35S]GTPγS binding. All three compounds insurmountably antagonized THC-stimulated [35S]GTPγS binding. To our knowledge, effects of PSNCBAM-1 have not been reported using THC as a probe. However, the CB1 allosteric modulator Org27569—which exhibits a very similar pharmacology to that of PSNCBAM-1—was previously shown to antagonize CP55,940- and THC-stimulated ERK activation in hCB1 transfected HEK293 cells (Gamage et al., 2016). Others have reported that Org27569 fully antagonizes CP55,940-stimulated ERK activation but does not affect THC-stimulated ERK activation (Khajehali et al., 2015); however, differences between those two studies may be due to methods used to assess ERK phosphorylation.
In contrast to the in vitro findings that PSNCBAM-1, RTICBM-15 and RTICBM-28 insurmountably antagonize THC in G protein signaling, their effects in vivo were not as clear. All three allosteric modulators were largely ineffective at attenuating the pharmacological in vivo effects of XLR-11 and THC including hypomotility, hypothermia and catalepsy. However, a small but significant reduction in the anti-nociceptive effect of THC was observed for PSNCBAM-1. While PSNCBAM-1 and RTICBM-15 did not affect THC’s discriminative stimulus, RTICBM-28 did produce a modest rightward shift in THC’s potency (Figure 6). These data suggest that allosteric modulators can affect CB1 receptor signaling in vivo as it relates to the anti-nociceptive and discriminative stimulus effects of THC; however, their effects are small.
These disparate findings underscore the difficulty in observing effects of CB1 allosteric modulators in behavioral models. While PSNCBAM-1 was reported to reduce feeding in rats (Horswill et al., 2007), we did not observe an effect of PSNCBAM-1 (30 mg/kg) on feeding in mice (unpublished results). Aside from the Horswill study, no other investigations into the effects of PSNCBAM-1 in vivo have been reported. Of the few behavioral studies of CB1 allosteric modulators, Org27569 has been the primary modulator examined but has largely been ineffective. Org27569 did not reduce CP55,940 or THC’s effects in any measure of cannabinoid tetrad whether administered systemically or centrally. Org27569 also did not affect anandamide’s discriminative stimulus in drug discrimination when tested in mice (Gamage et al., 2014). While it did produce non-CB1 mediated reductions in food intake (Gamage et al., 2014), this behavior is highly sensitive and reductions could be non-specific. However, two other studies reported promising results for Org27569. Despite no observed effects on CP55,940 induced catalepsy or anti-nociception, Org27569 attenuated the development of hypothermia over time following a single dose of CP55,940 (Ding et al., 2014). Notably, the time at which SR141716’s effects became apparent was sooner than that observed for Org27569 (i.e. 30 min vs 45 min) and the temperature then plateaued for approximately two hours. In contrast, Org27569’s (10 mg/kg) effects became apparent later and the temperature did not plateau but slowly recovered to control levels. This would be consistent with slower association kinetics which are a common feature of allosteric modulators (Kenakin and Miller, 2010).
We also examined the steroid hormone pregnenolone in [35S]GTPγS and drug discrimination. Pregnenolone has been reported to act as an endogenous CB1 allosteric modulator, with THC administration increasing circulating pregnenolone levels and pregnenolone antagonizing THC’s effects in several in vitro and in vivo assays including ERK activation. We did not observe an effect of pregnenolone on THC’s efficacy or potency in stimulating [35S]GTPγS binding nor did we observe augmentation of THC’s discriminative stimulus effects in mice. Previously, pregnenolone was shown to act as an antagonist in vitro in assays of THC-induced ERK activation but pregnenolone did not affect THC’s ability to attenuate forskolin-stimulated cAMP production (Vallee et al., 2014). While CB1-mediated inhibition of adenylyl cyclase is Gi/o dependent (Bouaboula et al., 1995a; Felder et al., 1995; Howlett et al., 1988; Howlett et al., 1986), CB1 receptors have been reported to activate ERK independently of G-proteins via beta arrestin (Ahn et al., 2013). It is possible that pregnenolone, in the presence of THC, is producing a receptor conformation that permits canonical G-protein signaling but inhibits G-protein independent ERK activation. However, ERK activation by CB1 receptors has also been shown to be pertussis toxin sensitive (Bouaboula et al., 1995b; Chen et al., 2010; Gamage et al., 2016), which raises interesting questions regarding how pregnenolone may inhibit ERK but not G-protein activation. Other groups also have had difficulty in observing an effect of pregnenolone. While Vallee et al. (2014) reported antagonism of THC induced ERK activation assay, another group reported no effect of pregnenolone on THC’s effects on ERK (Khajehali et al., 2015). Interestingly, Khajehali et al. (2015) did observe slight displacement of [3H]SR141716 binding at 10 μM whereas Vallee et al. (2014) reported no effects of pregnenolone on specific binding of [3H]SR141716 or [3H]CP55,940. Another group examined pregnenolone in slice electrophysiology measuring depolarization-induced suppression of excitation (DSE), a CB1 mediated effect that results from on-demand synthesis of 2-AG, and found that while Org27569 and PSNCBAM-1 attenuated DSE, pregnenolone was without effect (Straiker et al., 2015). A hallmark of allosteric modulators is probe-dependence (Kenakin and Miller, 2010), whereby the probe ligand that typically binds to the presumed orthosteric site of the receptor can affect the allosteric compound’s affinity and ability to alter the receptor to modulate signaling. While pregnenolone was shown to antagonize THC’s and WIN55,212-2’s effects, it was not tested against other CB1 ligands. It is possible that it has a high degree of probe dependence and does not affect endocannabinoids in the case of DSE. However, our data suggest that pregnenolone does not appreciably alter THC-stimulated G-protein signaling. More data are needed to examine the effects of pregnenolone in other assays using multiple pharmacological probes.
In behavioral studies, pregnenolone was previously reported to inhibit hypotility, hypothermia, catalepsy and anti-nociception induced by THC (Vallee et al., 2014). Moreover, pregnenolone was shown to inhibit THC-induced increases in food consumption in rats and mice as well as attenuate self-administration of the cannabinoid WIN55,212-2. We examined pregnenolone in THC discrimination in mice, as it has good predictive validity for the abuse-related effects of established abused-drugs and high degree of pharmacological specificity (Balster and Prescott, 1992). Mice were trained to discriminate THC (5.6 mg/kg) from VEH and then substitution tests for THC were conducted (3, 10 and 20 mg/kg) in the presence of pregnenolone (20 mg/kg s.c.) or VEH. We observed no effect of pregnenolone alone or when administered prior to THC. Doses of THC used in the substitution tests included those within the range previously tested (3 – 15 mg/kg) in the cannabinoid tetrad (Vallee et al., 2014). In the present study and the Vallee study, pregnenolone was administered both subcutaneously and 30 min prior to THC injection, however our dose of pregnenolone was higher (20 mg/kg vs 6 mg/kg). Our data suggest that pregnenolone does not affect the discriminative stimulus effects of THC which are highly predictive of the subjective effects of cannabis intoxication.
5. Conclusions
In this study we sought to characterize the CB1 allosteric modulator PSNCBAM-1, two of its structural analogs, and pregnenolone in assays of receptor function, physiology and behavior. PSNCBAM-1 and its analogs exhibited insurmountable antagonism of CP55,940- and THC- stimulated [35S]GTPγS binding and exhibited greater inhibitory potency when tested at higher concentrations of CP55,940, consistent with positive binding cooperativity and an allosteric mechanism. Furthermore, compound RTICBM-28 exhibited greater potency than PSNCBAM-1, suggesting that modifications to the parent structure can yield enhanced pharmacological activity. Fitting of the allosteric operational model revealed a greater alpha value for RTICBM-28, suggesting this structural modification enhances binding cooperativity with CP55,940. Pregnenolone did not produce appreciable effects on receptor function when tested against THC. In contrast to their effects in receptor signaling, PSNCBAM-1, RTICBM-15 and RTICBM-28 produced minimal augmentation of the physiological and behavioral effects of THC. While no effects on hypothermia, catalepsy or locomotor activity were observed for any of the modulators tested, PSNCBAM-1 moderately attenuated THC induced anti-nociception and RTICBM-28 produced a modest rightward shift of THC’s ED50 in drug discrimination. While small, these effects were significant and demonstrate that pharmacological effects of CB1 allosteric modulators can be observed in vivo. These findings further underscore the difficulty in observing effects for CB1 allosteric modulators in vivo. Further studies exploring the pharmacokinetics and timing of these and similar compounds as well as their effects on other probe agonists are needed.
Highlights.
PSNCBAM-1 and two structural analogs exhibit insurmountable antagonism of CP55,940 and THC-stimulated [35S]GTPγS binding
Cyano substitution of 4-chlorophenyl position of PSNCBAM-1 enhances binding cooperativity with CP55,940
PSNCBAM-1 partially attenuates anti-nociceptive response to THC in mice
Structural analog to PSNCBAM-1 reduced THC’s potency in drug discrimination in mice
Acknowledgments
Research supported by National Institute on Drug Abuse grants DA-03672, DA-040693 and DA-040460; NIDA had no further role in the writing of the review or in the decision to submit the paper for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abood ME. Allosteric Modulators: A Side Door. J Med Chem. 2016;59:42–43. doi: 10.1021/acs.jmedchem.5b01824. [DOI] [PubMed] [Google Scholar]
- Adamczyk P, Miszkiel J, McCreary AC, Filip M, Papp M, Przegalinski E. The effects of cannabinoid CB1, CB2 and vanilloid TRPV1 receptor antagonists on cocaine addictive behavior in rats. Brain Res. 2012;1444:45–54. doi: 10.1016/j.brainres.2012.01.030. [DOI] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of beta-arrestin 1 and beta-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1) J Biol Chem. 2013;288:9790–9800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggadiredja K, Nakamichi M, Hiranita T, Tanaka H, Shoyama Y, Watanabe S, Yamamoto T. Endocannabinoid system modulates relapse to methamphetamine seeking: possible mediation by the arachidonic acid cascade. Neuropsychopharmacology. 2004;29:1470–1478. doi: 10.1038/sj.npp.1300454. [DOI] [PubMed] [Google Scholar]
- Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, McAllister S, Strange PG, Stephens GJ, Pertwee RG, Ross RA. CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol. 2013;83:322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balster RL, Prescott WR. Delta 9-tetrahydrocannabinol discrimination in rats as a model for cannabis intoxication. Neurosci Biobehav Rev. 1992;16:55–62. doi: 10.1016/s0149-7634(05)80051-x. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Bourrie B, Rinaldi-Carmona M, Shire D, Le Fur G, Casellas P. Stimulation of cannabinoid receptor CB1 induces krox-24 expression in human astrocytoma cells. J Biol Chem. 1995a;270:13973–13980. doi: 10.1074/jbc.270.23.13973. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G, Casellas P. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995b;312(Pt 2):637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XP, Yang W, Fan Y, Luo JS, Hong K, Wang Z, Yan JF, Chen X, Lu JX, Benovic JL, Zhou NM. Structural determinants in the second intracellular loop of the human cannabinoid CB1 receptor mediate selective coupling to G(s) and G(i) Br J Pharmacol. 2010;161:1817–1834. doi: 10.1111/j.1476-5381.2010.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet. 2007;370:1706–1713. doi: 10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Ding Y, Qiu Y, Jing L, Thorn DA, Zhang Y, Li JX. Behavioral effects of the cannabinoid CB1 receptor allosteric modulator ORG27569 in rats. Pharmacol Res Perspect. 2014;2:e00069. doi: 10.1002/prp2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- Gamage TF, Anderson JC, Abood ME. CB1 allosteric modulator Org27569 is an antagonist/inverse agonist of ERK1/2 signaling. Cannabis and Cannabinoid Research. 2016;1:272–280. doi: 10.1089/can.2016.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage TF, Ignatowska-Jankowska BM, Wiley JL, Abdelrahman M, Trembleau L, Greig IR, Thakur GA, Tichkule R, Poklis J, Ross RA, Pertwee RG, Lichtman AH. In-vivo pharmacological evaluation of the CB1-receptor allosteric modulator Org-27569. Behav Pharmacol. 2014;25:182–185. doi: 10.1097/FBP.0000000000000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German N, Decker AM, Gilmour BP, Gay EA, Wiley JL, Thomas BF, Zhang Y. Diarylureas as allosteric modulators of the cannabinoid CB1 receptor: structure-activity relationship studies on 1-(4-chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl)pyridin-2-yl]phenyl}urea (PSNCBAM-1) J Med Chem. 2014;57:7758–7769. doi: 10.1021/jm501042u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C, Wong Kai In P. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Johnson MR, Melvin LS, Milne GM. Nonclassical cannabinoid analgetics inhibit adenylate cyclase: development of a cannabinoid receptor model. Mol Pharmacol. 1988;33:297–302. [PubMed] [Google Scholar]
- Howlett AC, Qualy JM, Khachatrian LL. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol Pharmacol. 1986;29:307–313. [PubMed] [Google Scholar]
- Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, Damaj IM, Poklis J, Wiley JL, Zanda M, Zanato C, Greig IR, Lichtman AH, Ross RA. A Cannabinoid CB1 Receptor-Positive Allosteric Modulator Reduces Neuropathic Pain in the Mouse with No Psychoactive Effects. Neuropsychopharmacology. 2015;40:2948–2959. doi: 10.1038/npp.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa MA, Narang S, Jamison RN, Michna E, Edwards RR, Penetar DM, Wasan AD. The subjective psychoactive effects of oral dronabinol studied in a randomized, controlled crossover clinical trial for pain. Clin J Pain. 2014;30:472–478. doi: 10.1097/AJP.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, Qiu Y, Zhang Y, Li JX. Effects of the cannabinoid CB(1) receptor allosteric modulator ORG 27569 on reinstatement of cocaine- and methamphetamine-seeking behavior in rats. Drug Alcohol Depend. 2014;143:251–256. doi: 10.1016/j.drugalcdep.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. A pharmacology primer: theory, application and methods. fourth. Academic Press; San Diego: 2009. [Google Scholar]
- Kenakin T. What is pharmacological ‘affinity’? Relevance to biased agonism and antagonism. Trends Pharmacol Sci. 2014;35:434–441. doi: 10.1016/j.tips.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K. Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor. Mol Pharmacol. 2015;88:368–379. doi: 10.1124/mol.115.099192. [DOI] [PubMed] [Google Scholar]
- Kulkarni PM, Kulkarni AR, Korde A, Tichkule RB, Laprairie RB, Denovan-Wright EM, Zhou H, Janero DR, Zvonok N, Makriyannis A, Cascio MG, Pertwee RG, Thakur GA. Novel Electrophilic and Photoaffinity Covalent Probes for Mapping the Cannabinoid 1 Receptor Allosteric Site(s) J Med Chem. 2016;59:44–60. doi: 10.1021/acs.jmedchem.5b01303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Kulkarni AR, Kulkarni PM, Hurst DP, Lynch D, Reggio PH, Janero DR, Pertwee RG, Stevenson LA, Kelly ME, Denovan-Wright EM, Thakur GA. Mapping Cannabinoid 1 Receptor Allosteric Site(s): Critical Molecular Determinant and Signaling Profile of GAT100, a Novel, Potent, and Irreversibly Binding Probe. ACS Chem Neurosci. 2016;7:776–798. doi: 10.1021/acschemneuro.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein–coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- Morales P, Goya P, Jagerovic N, Hernandez-Folgado L. Allosteric Modulators of the CB1 Cannabinoid Receptor: A Structural Update Review. Cannabis and Cannabinoid Research. 2016;1:22–30. doi: 10.1089/can.2015.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Li JX, Thomas BF, Wiley JL, Kenakin TP, Zhang Y. Allosteric Modulation: An Alternate Approach Targeting the Cannabinoid CB1 Receptor. Med Res Rev. 2016;37:441–474. doi: 10.1002/med.21418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Ross RA. Allosterism and cannabinoid CB(1) receptors: the shape of things to come. Trends Pharmacol Sci. 2007;28:567–572. doi: 10.1016/j.tips.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Schindler CW, Panlilio LV, Gilman JP, Justinova Z, Vemuri VK, Makriyannis A, Goldberg SR. Effects of cannabinoid receptor antagonists on maintenance and reinstatement of methamphetamine self-administration in rhesus monkeys. Eur J Pharmacol. 2010;633:44–49. doi: 10.1016/j.ejphar.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore DM, Baillie GL, Hurst DH, Navas F, 3rd, Seltzman HH, Marcu JP, Abood ME, Ross RA, Reggio PH. Allosteric modulation of a cannabinoid G protein-coupled receptor: binding site elucidation and relationship to G protein signaling. J Biol Chem. 2014;289:5828–5845. doi: 10.1074/jbc.M113.478495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mitjavila J, Yin D, Gibson A, Mackie K. Aiming for allosterism: Evaluation of allosteric modulators of CB1 in a neuronal model. Pharmacol Res. 2015;99:370–376. doi: 10.1016/j.phrs.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Thomas BF, Lefever TW, Cortes RA, Kovach AL, Cox AO, Patel PR, Pollard GT, Marusich JA, Kevin R, Gamage TF, Wiley JL. Thermolytic degradation of synthetic cannabinoids: chemical exposures and pharmacological consequences. J Pharmacol Exp Ther. 2017;361:162–171. doi: 10.1124/jpet.116.238717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee M, Vitiello S, Bellocchio L, Hebert-Chatelain E, Monlezun S, Martin-Garcia E, Kasanetz F, Baillie GL, Panin F, Cathala A, Roullot-Lacarriere V, Fabre S, Hurst DP, Lynch DL, Shore DM, Deroche-Gamonet V, Spampinato U, Revest JM, Maldonado R, Reggio PH, Ross RA, Marsicano G, Piazza PV. Pregnenolone can protect the brain from cannabis intoxication. Science. 2014;343:94–98. doi: 10.1126/science.1243985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vann RE, Warner JA, Bushell K, Huffman JW, Martin BR, Wiley JL. Discriminative stimulus properties of delta9-tetrahydrocannabinol (THC) in C57Bl/6J mice. Eur J Pharmacol. 2009;615:102–107. doi: 10.1016/j.ejphar.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]