

Abstract
Purpose
Nicotinamide phosphoribosyltransferase (Nampt) regulates intracellular NAD+ pool and is highly expressed in a number of malignancies. FK866, a selective inhibitor of Nampt, depletes intracellular NAD+ levels, thereby blocking cellular metabolism and triggering sensitization to other drugs and cell death. Here we characterized the anti-tumor effects of Nampt inhibition in Waldenström Macroglobulinemia (WM).
Experimental Design
We investigated Nampt role in MW cells using both mRNA and protein expression analyses. We have also used loss-of-function approaches to investigate the growth and survival effects of Nampt on MW cells and further tested the anti-MW activity of dual Nampt and BTK inhibition in vitro and in vivo.
Results
We found that WM cells exhibit high levels of Nampt compared with normal B cells. Loss of function studies suggested a potential oncogenic role of Nampt in WM cells, and BTK-inhibitor ibrutinib and FK866 resulted in a significant and synergistic anti-WM cell death, regardless of MYD88 and CXCR4 mutational status. Cell death was associated with: 1) activation of caspase-3, PARP and down-regulation of Mcl-1; 2) enhanced intracellular ATP and NAD+ depletion; 3) inhibition of NF-kappa B signaling; and 4) inhibition of multiple pro-survival signaling pathways. In a murine xenograft WM model, low-dose combination FK866 and Ibrutinib is well tolerated, significantly inhibits tumor growth, and prolongs host survival.
Conclusions
our results show intracellular NAD+ level as crucial for proliferation and survival of WM cells, and provides the mechanistic preclinical rationale for targeting Nampt, either alone or with Ibrutinib, to overcome drug resistance and improve patient outcome in WM.
Keywords: BTK, NAMPT, combination therapy, Waldenstrom's Macroglobulinemia
Introduction
Cancer cells are characterized by higher NAD+ turnover than normal cells due to the increased energy required for their cell proliferation and metabolism, as well as regulation of transcription, chromatin dynamics, and DNA repair-processes.(1-6) As NAD+ is rapidly consumed and converted to nicotinamide, Nampt plays a crucial role for replenishment of the intracellular NAD+ pool in cancer cells. Thus, its aberrant activation has been reported in a number of solid and hematologic malignancies, including leukemia and multiple myeloma. (7, 8) Based on these observations, tumor cells are more susceptible to Nampt inhibition than normal cells.(8-12) Indeed, targeting Nampt with the specific inhibitor FK866 represents a novel therapeutic strategy for human cancer.(13) Clinical trials using Nampt inhibitors (CHS828, GMX1777 and APO866) as monotherapy have demonstrated tolerability (www.clinTrials.gov), and combining FK866 or other Nampt inhibitors with antineoplastic agents, chemotherapy or radiotherapy has shown promising preclinical activity.(7, 14-19)
Whole-genome sequencing has identified the MYD88L265P and CXCR4WHIM variants in 90% and 30-35% of WM patients, respectively. (20, 21) The first encodes for a mutant protein which triggers tumor growth via activation of nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) by Bruton's tyrosine kinase, whereas CXCR4WHIM mutations confer in-vitro drug-resistance.(22-24) Recently, the US Food and Drug Administration has approved the Bruton Tyrosin kinase inhibitor ibrutinib for the treatment of symptomatic WM. (25) A clinical trial in previously treated patients suggests that mutational status as affecting outcome: overall survival is lower in patients harboring MYD88WT; whereas patients with CXCR4 mutations show slower initial response which improves with prolonged (>6 months) therapy.(26, 27) The impact of mutational status on clinical response suggests that combination approaches to overcome drug-resistance may broaden therapeutic efficacy. For example, preclinical studies suggest that targeting the anti-apoptotic protein bcl-2 augments ibrutinib-mediated cytotoxicity in CXCR4WT and CXCR4S338X mutated WM cells, in vitro.(28)
Based on our previous observations regarding Nampt role in multiple myeloma,(8, 19, 29) we here investigate its impact on WM cells, both directly and in relationship to BTK pathway activation. Our data show that Nampt is constitutively active in WM patients and plays a critical role in tumor cell growth and survival; moreover, Nampt levels correlate with BTK activity status. Synergistic WM cytotoxicity was induced by combining FK866 with ibrutinib against a panel of WM cell lines and patient cells, as well as in-vivo in a WM xenograft model. Importantly, FK866 treatment restored ibrutinib-sensitivity in both MYD88 and CXCR4 mutated cells. Overall, our data provide the rationale for combining FK866 with ibrutinib as an innovative strategy to enhance sensitivity or overcome resistance to BTK inhibitors in WM.
Materials and Methods
Cell lines and patient samples
The WM cell lines (BCMW.1 and MWCL-1) and IgM-secreting low grade lymphoma cell lines (MEC-1 and RL) were used in this study. Cells were cultured at 37°C in RPMI-1640 medium containing 10% FBS (GIBCO, Life Technologies, Carlsbad, CA), 2 mM/L L-glutamine, 100 U mL-1 penicillin, and 100 μg ml–1 streptomycin (GIBCO, Life Technologies, Carlsbad, CA). Bone marrow (BM) mononuclear cells and primary WM cells from BM aspirates from WM patients were isolated using Ficoll-Hypaque density gradient sedimentation. Primary WM cells were obtained from bone marrow (BM) aspirates of patients using CD19 microbead selection (Miltenyi Biotec) with more than 90% purity, as confirmed by flow cytometric analysis. Residual CD19-negative BM mononuclear cells were cultured in Dulbecco Modified Eagle medium (DMEM) with 20% FCS for 3 to 6 weeks to generate BMSCs, as previously described.(30), (31) Peripheral blood mononuclear cells (PBMCs) were obtained from healthy subjects by Ficoll-Hypaque density gradient sedimentation, and subsequently, CD19+ selection was performed. Approval for these studies was obtained from the Dana-Farber Cancer Institute's Institutional Review Board. Informed consent was obtained from all patients and healthy volunteers according with the Declaration of Helsinki protocol.
Reagents
The Nampt inhibitor FK866 was generously provided by the NIMH Chemical Synthesis and Drug Supply Program. It was dissolved in dimethyl sulphoxide at 10 mM and stored at -80°C for in vitro study. For in vivo studies in mice, FK866 was formulated in 0.9% saline solution and stored at -20°C. BTK Inhibitor (ibrutinib) was purchased from Selleck.
Gene-expression analysis
We evaluated the expression levels of Nampt transcript (probe ID 217739_s_at) in the log10 transformed data set downloaded from NCBI's gene expression omnibus (GEO) database (series number GSE9656) of cells from WM patients and normal healthy donors. The CD19+ WM-B cells (WM-BC) were compared with normal CD19+ peripheral blood B cells (PB-BC) using the Student t-test to determine statistical significance. Also Nampt mRNA level was analyzed in GSE65816 microarray datasets including lymphoma cell lines treated with two BTK inhibitors (ibrutinib and PLS-123) in in-vitro settings. The analyses were carried out by GraphPad Prism 6.0
Immunohistochemistry (IHC)
Sections of lymph nodes samples from patients diagnosed with WM at the IRCCS AOU San Martino-IST were stained with anti-NAMPT (clone H-300; sc-67020) (Santa Cruz, CA, United States).(7) IHC was performed using the Ventana BenchMark XT automated immunostainer. Tissue sections were deparaffinized and rehydrated. After antigen retrieval, sections were incubated with primary antibodies at a dilution of 1:200 and 3,3′-diaminobenzidine (DAB) was used as a chromogen. Sections were counterstained with May-Grünwald-Giemsa and CD79α.
Cell proliferation and viability
WM cell proliferation was measured by [3H]-thymidine (PerkinElmer, Boston, MA) incorporation assay, according to manufacturer's procedure. Dead cells were quantified by propidium iodide (PI) staining (2 mg/ml) and flow cytometry (FACS Canto II, Becton Dickinson, BD), as previously described. (8, 19)
Determination of the intracellular NAD+ and ATP levels
The intracellular content of NAD+ was assessed with a sensitive enzyme cyclic assay (BioVision, Mountain View, CA), according to the manufacturer's procedure. Intracellular ATP content was determined using Cell titer Glo Luminescent Cell Viability Assay (Promega). NAD+ and ATP values were normalized to the number of viable cells as determined using Trypan Blue (Lonza).
siRNA
RNA interference was done by using ON-TARGET PLUS SMART pool targeting NAMPT (GE Dharmacon, Lafayette, CO). Nontargeting scrambled negative control siRNA was used as negative control (GE Dharmacon, Lafayette, CO). Briefly, MWCL-1 and BCMW.1 cells were transiently transfected with NAMPT siRNA or scramble control using Amaxa technology, as per the manufacturer's instructions.
Immunoblotting
WM cells were cultured with or without stimuli, harvested, washed, and lysed using radioimmunoprecipitation assay buffer. Proteins were separated on an SDS-polyacrylamide gel and electroblotted on a polyvinylidene difluoride (PVDF) membrane (Pall Gelman Laboratory, Ann Arbor, MI). Proteins were visualized by probing the membranes with the following antibodies: anti-tubulin, -caspase-3, -PARP, p44/42 MAPK (ERK 1/2), p-p44/42 MAPK, p-AKT(Ser473), AKT, p-GSK-3α/β (Ser21/9), GSK-3α/β, Bruton's tyrosine kinase (BTK), pBTK (Tyr223), -p-p65, -p-p52, -RelB, -p-IkB, -nucleolin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signalling Technology); anti-Nampt (Bethyl Laboratories); anti-Mcl-1 (Santa Cruz Biotechnology). Standard enhanced chemiluminescence was used for protein band detection.
NF-kB luciferase reporter assay
Lentiviral NF-kB luciferase reporter and control virus (QIAGEN, Inc.) were transfected into BCMW.1 cells. After puromycin selection, cells were treated either with FK866, ibrutinib, or both. Luminescence was measured with Bright-Glo Luciferase Assay System (Promega).
Immunofluorescence
The effect of drug combination on TNF-α–induced nuclear translocation of p65 was examined by an immunocytochemical method. Briefly, BCMW.1 cells were cultured in the presence of FK866 plus ibrutinib or with either agent alone for 48 h. Cells were then stimulated with TNF-α (10 ng/mL) during the last 20 min of culture. Immunocytochemical analysis was conducted using a fluorescence microscope (Nikon Eclipse E800; Nikon) and a Photometrics Coolsnap CF color camera (Nikon), as previously described.(8)
MYD88-depletion and CXCR4 mutated WM cells generation
MYD88 RNA interference was done by using Silencer MYD88 siRNA (Life Technologies). CXCR4WT and CXCR4S338X cDNAs were subcloned into pLenti-IRES-GFP vector, and transduced using a lentiviral based strategy into BCMW.1 cells as described.(28, 31) Both transfected and transduced cells were treated with either FK866, ibrutinib, or both.
Murine xenograft model of human WM
CB17-SCID mice (28-35 days old) were purchased from Charles River Laboratories. All animal studies were conducted according to protocols approved by the Animal Ethics Committee of the Dana-Farber Cancer Institute. Mice were irradiated (200 cGy) and then inoculated subcutaneously in the right flank with 5 × 106 BCMW.1 cells in 100 μL RPMI 1640. Following detection of tumor (∼3 weeks after the injection), mice (n=6 group) were treated intraperitoneally with FK866 (30 mg/kg body weight) daily for 4 days in a week (with 3 days off), repeated for 3 weeks; ibrutinib (0.5 mg/kg/day) for 5 days, repeated weekly for 3 consecutive weeks; or the combination with the same dosing regimen used for the individual agents. The control group received the carrier alone at the same schedule as the combination group. Caliper measurements of the longest perpendicular tumor diameters were performed twice a week to estimate the tumor volume using the following formula: length × width 2 × 0.5. Tumor growth inhibition (TGI) was calculated using the formula ((Δcontrol average volume-Δ treated average volume) × 100 (Δcontrol average volume). Animals were sacrificed when tumors reached 2 cm3 or the mice appeared moribund. Survival was evaluated from the first day of treatment until death.
In situ detection of apoptosis and proliferation
Mice tumor sections were subjected to immunohistochemical (ICH) staining for caspase-3 activation to detect apoptotic cell death. Ki-67 was assessed to quantify proliferation, and tumor cell pathway activity was evaluated by IHC staining for pAKT expression, as previously described. (23) Immunostained tissues were imaged using a Canon IXY digital 700 camera.
Statistical analysis
Statistical significance of differences observed between drug-treated versus control (in both in-vitro and in-vivo experiments) was determined by Student's t-test; differences were considered significant when P≤ 0.05. Tumor growth inhibition and Kaplan-Meier survival analysis were determined using GraphPad Prism analysis software. Drug interactions were analyzed by isobologram analysis using the CalcuSyn Version 2.0 software program (Biosoft). A Combination index (CI) less than 1.0 indicates synergism; CI =1, additive effect; and CI > 1, no significant combination effect. (32)
Results
Nampt is overexpressed in WM and its targeting prevents tumor cell growth and viability
Our prior study shows higher Nampt expression in hematologic cancer cell lines and patient cells compared with healthy PBMCs, implicating its activity in tumor cell growth and function.(7, 8) Here we further investigated Nampt relevance in tumor biology by focusing on Waldenstrom's Macroglobulinema (WM). We first compared its mRNA levels in CD19+ cells obtained from WM patients with normal B-lymphocytes from healthy donors using the public available database GSE9656. As shown in Figure 1A, Nampt expression (probe ID 217739_s_at) was significantly higher in tumor than normal cells. Similar results were observed in lymph node biopsies collected from newly diagnosed WM patients (n=4, Fig. 1B), as well as in protein extracts from WM cell lines (Figure 1C, left panel). We also observed that peripheral blood derived CD19+ cells from normal donors (N=3) showed weaker Nampt levels compared with CD19- cell fraction or whole PBMCs (Figure 1C, right panel). Overall, these findings indicate that Nampt expression is greater in the malignant than normal cells. (Suppl. Fig. 1)
Figure 1.
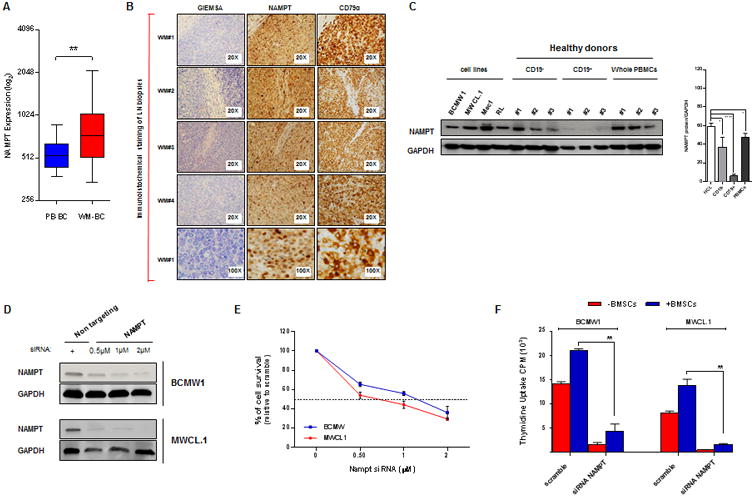
Nampt is overexpressed in WM cells and its specific knockdown impairs tumor cell growth and viability. (A) Dot plot represents Nampt (probe ID 217739_s_at) expression value from microarray analysis of WM profiles retrieved from NCBI database (GSE9656). The samples were divided into two groups: control (left panel) including normal CD19+ peripheral blood B cells (PB-BC; n=17) and tumor (right panel) including CD19+ WM cells (WM-BC; n=41). (B) Giemesa, CD79α and Nampt IHC analysis (positive cells are brown) on four representative lymph node specimens derived from WM patients (WM 1-4). Magnification: 20× (also 100× for WM 1). (C) Low grade lymphoma cell lines (BCMW.1, MWCL-1, Mec1 and RL), CD19+/-, and PBMCs from 3 healthy donors were subjected to WB analysis (one of three representative blots) using anti-Nampt Ab. Anti-GAPDH monoclonal antibody served as a loading control. Average of Nampt relative expression was calculated as the ratio of the densitometry signal for Nampt relative to GAPDH in each sample using Image J software (1.37v; National Institutes of Health, http://rsb.info.nih.gov/ij/). PBMCs, peripheral blood mononuclear cells; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HCL, human cell lines. (D) BCMW.1 and MWCL-1 cell lines were transfected using different concentrations of SMARTpool Nampt siRNA or non-targeting siRNA. Two days later, cells were subjected to immunoblotting to assess decrease in Nampt protein expression post-transfection using anti Nampt and GAPDH Abs. WB analyses confirmed reduction in Nampt protein level following transient transfection of WM cells with Nampt siRNA compared with cells transfected with scramble control. (E) The effect of Nampt knockdown on cell survival in WM cells transfected with Nampt or control siRNA were assessed by CellTiter Glo assay and presented as change relative to scramble control cells. (F) WM cell lines transfected with 2 μM of SMARTpool Nampt siRNA or control siRNA were cultured in the absence or presence of BMSCs for 24 hours. Tumor cell growth was evaluated by [3H]thymidine uptake and presented as a percentage of cell growth compared with scramble control. All data in A-F panels are shown as the mean values ± S.D. of triplicates. (one representative experiment performed in triplicate). *p=0.02, **0.001 <p< 0.003, **** p<0.0001 (Student's t test).
To further evaluate the potential oncogenic role of Nampt in WM, we examined the effect of Nampt knockdown in two WM cell lines, BCMW.1 and MWCL-1. As shown in Figure 1 D-F after 2 days of transfection Nampt depletion led to decrease viability in both cell lines, even in the presence of bone marrow stromal cells (BMSCs) derived from WM patients. By contrast, Nampt overexpression prompted cell growth and increased IgM production in the BCMW.1 cell line (Suppl. Fig. 2). These data are consistent with a previously reported role for Nampt in tumorigenesis, identifying this enzyme as crucial for WM cell growth and providing the rationale for its therapeutic targeting. We therefore next tested efficacy of FK866, a Nampt chemical inhibitor in WM. As expected, FK866 triggered cell death in WM and Ig-M secreating low grade lymphoma cell lines in a dose-dependent fashion, with IC50 ranging from 7 to 8 nmol/L. (Suppl. Fig. 3) Moreover, we also observed inhibition of cell survival in purified primary cells from WM patients, with minimal effect on mononuclear cells collected from healthy donors (Fig. 3A and data not shown).
Figure 3.
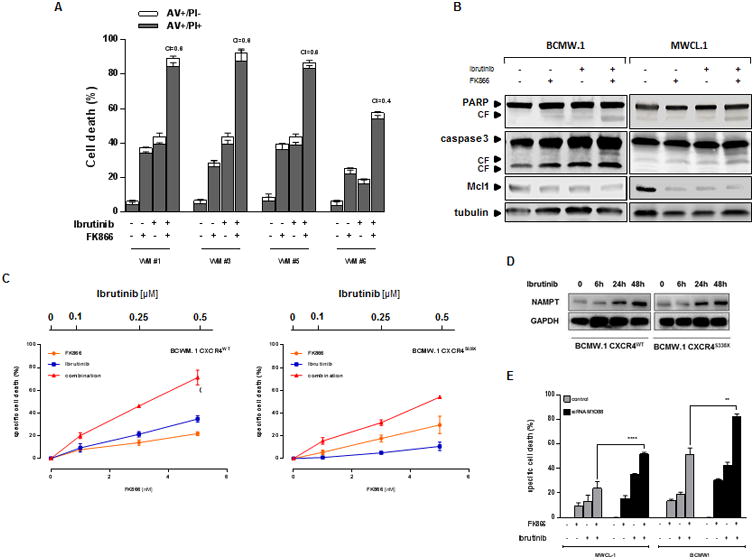
Treatment with FK866 plus ibrutinib induces apoptotic cell death in WM patients derived cells, regardless of MYD88 and CXCR4 somatic mutational status. (A) Ex vivo tumor cells (CD19+ gated) from WM patients were treated with FK866 plus ibrutinib, or either agent alone. Cell death was assessed by flow cytometry using annexin V and PI double staining after 72 hours. The percentage of early apoptotic cells (AV+/PI-) are shown as white columns and that of late apoptotic cells (AV+/PI+) are shown as solid gray columns. Data are derived from at least 3 independent experiments. (P < .05 for all patient samples). A CI less than 1 indicates synergism. (B) BCMW.1 and MWCL-1 cells were treated with either FK866 (3 nM), ibrutinib (1 μM) and the combination for 48 h. Cells were then harvested, and whole cell lysates were subjected to immunoblot analysis using anti-PARP, anti-caspase 3, anti-Mcl-1, or anti-actin antibodies. FL indicates full length and CF cleaved fragment. (C) CXCR4WT and CXCR4S338X expressing WM cells were treated with low dose FK866 (1-5 nM) plus ibrutinib (0.1-0.5 μM) or each drug alone for 72 h. Cell viability was assessed by CellTiter Glo assay and presented as a percentage of control cells (untreated cells). (D) CXCR4WT and CXCR4S338X BCMW.1 cells were treated with ibrutinb (1 μM) for 12 h, 24 h and 48 h. Cells were then harvested, and whole cell lysates were subjected to immunoblot analysis using anti-Nampt antibody. (E) BCMW.1 and MWCL-1 cells were transfected with either scramble control or MYD88-specific siRNA; 24 hours after transfection, cells were treated with either FK866, ibrutinib, or the combination, and then cultured for additional 72 hours. Cell viability was measured with specific assay and presented as a percentage of control. All data presented are a representative study set of 3. ** p=0.0032, ****p<0.0001 (Student's t test).
Nampt inhibition synergistically enhances ibrutinib anti-tumor activity in WM cells
Recent reports have linked Bruton tyrosine kinase (BTK) with Nampt transcription.(33-35) To examine this relationship in WM cells, we queried the NCBI's GEO datasets: microarray analysis of lymphoma cell lines treated with BTK inhibitors for 24 hours (GSE65816) revealed that ibrutinib, as well as PLS-123, treatment significantly increased Nampt transcript values compared with untreated cells, suggesting that its expression correlates with BTK inhibition. (Suppl. Fig. 4) Given this correlation, we next investigated dual Nampt and BTK inhibition (via ibrutinib and FK866, respectively) as an innovative therapeutic approach on WM cells. Minimally toxic concentrations of both drugs were used for combined cytotoxicity studies. WM (BCMW.1 and MWCL-1) and IgM-secreting low-grade lymphoma cell lines (MEC-1 and RL) were treated with increasing doses of FK866 (1-3 nM); ibrutinib, over a range of 0.5-5 μM, was then added followed by assessment of cell viability after 72 h. As shown in Fig. 2 and Supp. Fig. 5-6, combined therapy decreased survival and DNA synthesis in all cell lines. Chou and Talalay analysis confirmed synergistic activity with combination index (CI) < 1.0. Next we tested efficacy of combined therapies on purified tumor cells derived from WM patients both sensitive (WM #1-5) or resistant to ibrutinib (WM #6). Surprisingly, a slight heterogeneity was observed among tested samples: 4/6 demonstrated synergy (CI < 1) whilst 2/6 showed additivity (CI=1; Fig. 3A and Suppl. Fig. 7). Such variability was most likely due to different previous therapies that made tumor cells collected from pts #2 and 4 less sensitive to this co-treatment. Several studies demonstrate that FK866-based combination therapies induce apoptosis in hematological malignancies.(7, 19) As shown in Figures 3B and Suppl. Fig. 8, combined low doses of both drugs increased proteolytic cleavage of caspase-3, poly-(ADP-ribose) polymerase (PARP), and Mcl-1 in BCMW.1 and MWCL-1 cell lines. Overall our findings show that apoptotic cell death is triggered by dual Nampt and BTK inhibition.
Figure 2.
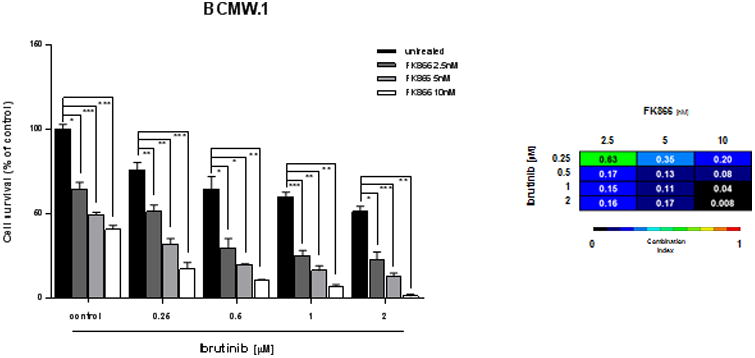
Dual inhibition of Nampt and BTK leads to synergistic killing in WM cells. (A) BCMW.1 cells were treated with an inhibitor of Nampt (FK866), BTK (ibrutinib), or both. Cell death was measured by cell viability assay and presented as a percentage of control cells (untreated). Synergism was calculated by CI analysis with heat maps depicting the CI values at increasing doses of FK866 and ibrutinib. Data are presented as mean ± S.D (n=3). CIs were generated with CalcuSyn software for each set of combination. CI<1, =1 and >1 denote synergism, additive effect, and antagonism, respectively. ns, not significant, *0.04<p<0.01, **0.009<p<0.001, ***0.001<p< 0.0001 (Student's t test).
Increased anti-WM activity of combination therapy occurs regardless of MYD88 and CXCR4 somatic mutational status
Whole-genome sequencing studies have revealed activating somatic mutations MYD88L265P and CXCR4WHIM in 90% and 30% of WM patients, respectively.(21, 26, 31) Both variants play important roles not only in WM pathogenesis, but also in clinical presentation and overall survival. Indeed, a recent clinical trial showed an influence of mutational status on response to ibrutinib treatment: increased overall and major response rate are observed in patients harboring MYD88L265P and CXCR4WT variants (100% and 91.7%, respectively).(26) By contrast, CXCR4WHIM mutation confers both in-vitro and in-vivo resistance to ibrutinib treatment.(Fig. 3C) (22-24) We therefore next examined whether anti-WM activity of this drug combination is affected by the somatic mutational status. We treated CXCR4WT and CXCR4S338X BCMW.1 cells with increasing doses of FK866 (1-3 nM); ibrutinib, over a range of concentrations depending on its IC50 value, was then added followed by assessment of cell viability by PI staining and flow cytometry analysis after 72 h. Synergistic cytotoxicity (CI <1.0) was evident at nearly all doses in both WM cell lines tested. Since Nampt/BTK dual inhibition is cytotoxic even in WHIM-like mutated CXCR4 WM cells, our data suggest that this combination may overcome ibrutinib-resistance. Indeed although CXCR4WT BCMW.1 cells show a slight increase of Nampt protein level relative to BCMW.1 CXCR4S338X cells at baseline, ibrutinib treatment triggers marked and similar upregulation of this protein in both cell types, confirming that FK866 can overcome ibrutinib resistance regardless of CXCR4 mutational status.
Next, we investigated the efficacy of combined treatment in MYD88-silenced and parental expressing MYD88L265P WM cells. As shown in Fig. 3E, addition of FK866 to ibrutinib enhanced cytotoxicity even in MYD88-depleted cells.(20, 31, 36, 37) Finally this synergistic effect was also observed in the MYD88WT-expressing MEC.1 and Ramos cells, (Fig. 2) further supporting its ability to overcome ibrutinib-resistance in WM.
Nampt and its outputs, NAD+ and ATP intracellular levels, are crucial in enhancing ibrutinib-sensitivity of WM cells
To assess the role of Nampt in the observed synergism, we treated Nampt-depleted WM cells with increasing concentrations of ibrutinib. As shown in Fig. 4A, Nampt knockdown (KD) cells were more sensitive to BTK-inhibition than scramble control transfectants. By contrast, no differences were observed in WM cells ectopically overexpressing Nampt compared with control (data not shown).
Figure 4.
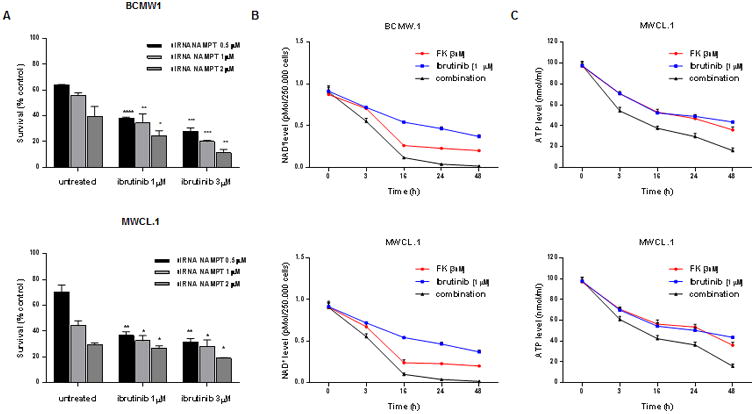
Nampt and its outputs, NAD+ and ATP intracellular levels, are crucial in enhancing ibrutinib-sensitivity of WM cells. (A) BCMW.1 (up) and MWCL-1 (low) cell lines were transfected using different concentrations of Nampt siRNA or non-targeting siRNA. 48 hours later, cells were treated with increased doses of ibrutinib (1-3 μM). After an additional 48 hours, cell viability was measured using a specific assay. Viability of WM cells Nampt depleted was compared with WM cells transfected with scramble control. (B-C) BCMW.1 and MWCL-1 cell lines were treated with FK866 (3 nM), ibrutinib (1 μM) or combined therapy for 3 h. Cells were then harvested, and intracellular NAD+ (B) and ATP (C) levels were measured using an enzyme cyclic and luminescent assay, respectively. All data presented are mean ± SD of 3 independent experiments. *0.04<p<0.01, **0.005<p<0.001, ***0.0009<p<0.0005, ****p<0.0001 (Student's t test).
FK866 mediates its anti-tumor effect by depleting intracellular NAD+ stores, followed by ATP depletion.(7, 38, 39) Utilizing a cycling enzymatic assay, we analyzed these activity markers following exposure to FK866, ibrutinib, and the combination. As expected, combined treatment reduced both NAD+ and ATP cellular content to a greater extent than either single agent therapy. Fig. 4B-C shows a time-course experiment where intracellular NAD+ and ATP level were monitored simultaneously after single or combined agents treatment: a significant NAD+ decrease was detected as early as 3 h after addition of both drugs, with complete depletion at day 2. Marked intracellular ATP shortage was evident at 24 and 48 h. The observation that ibrutinib enhances NAD+ and ATP depletion triggered by Nampt inhibition suggests that depletion of the energy pool contributes functionally to the anti-WM effect of FK866 plus ibrutinib. Exogenous NAD+ completely rescued combined treatment-induced cell death, confirming that the effect on WM viability is a consequence of depleted energy stores.(Suppl. Fig. 9)
Combined treatment targets WM cells in the context of BM microenvironment via robust inhibition of pro-survival signaling pathways
Interaction of tumor cells with BM microenvironment confers growth-promoting effects to malignant cells.(8, 22, 40) We therefore next evaluated the anti-proliferative effect of such combination in the context of BM milieu. WM cells were co-cultured with WM-BMSCs, and then treated with FK866, ibrutinib or both drugs for 72 h; proliferation was then measured by thymidine uptake assay (Fig 5A). Importantly, co-treatment suppressed WM cell growth also in the context of the BM microenvironment without effect on viability of BMSCs. Therefore, low doses of FK866 enhance the anti-WM effect of ibrutinib even in the context of the BM microenvironment. Exposure of WM cells (BCMW.1 and MWCL-1) to either single agent or the combination triggered drug-dependent inhibition of proliferative signaling triggered in the BM microenvironment, including p44/42 MAPK, pGSK3, and pAKT. (Fig. 5B) Western blot analysis showed that TNF-α–induced nuclear translocation of p65, p52 and RelB in WM cells was more significantly inhibited by combination treatment than with either agent alone. (Fig. 5C) p-p65 immunofluorescence staining confirmed block of nuclear translocation of canonical NF-kB pathway markers following combination versus single agent therapy. (Fig. 5D) To further confirm these results, studies with WM cells expressing NF-kB luciferase reporter construct were next performed. As shown in Fig. 5E the increased anti-WM effect triggered by BTK/Nampt dual inhibition was associated with significant inhibition of NF-kB activity. Overall our findings demonstrate inhibition of pro-survival signaling pathways induced by combined treatment effect even in the presence of BM.
Figure 5.
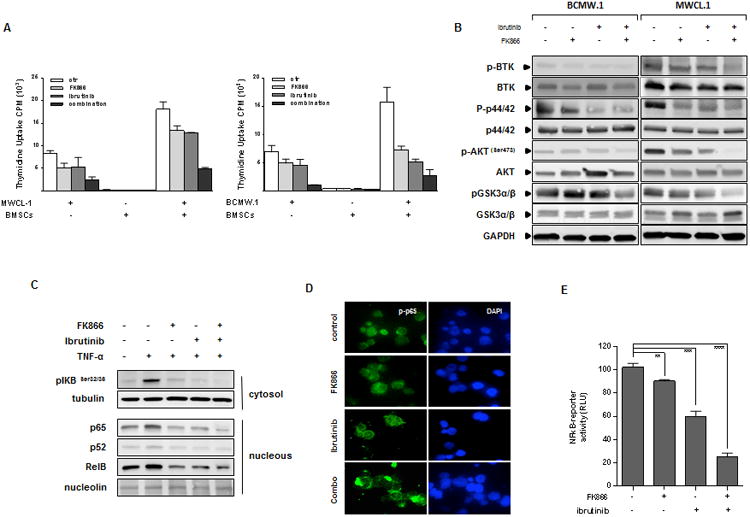
Combined treatment decreases WM cell growth in the context of the BM microenvironment via inhibition of pro-survival signaling pathways (A) MWCL-1 and BCMW.1 cells were cultured in the absence (-) or presence (+) of BMSCs from WM patients with FK866, ibrutinib, or the combination. Cell proliferation was assessed by [3H]-thymidine incorporation assay. Data are mean ± SD of triplicate samples. Error bars represent SD. (B) BCMW.1 and MWCL-1 cells were treated with FK866, ibrutinib or combined therapy for 24 h. Whole-cell lysates were then immunoblotted using with the indicated antibodies. Blots shown are representative of 3 independent experiments. (C) Inhibition of NF-kB activity was assessed using western blot analysis of BCMW.1 cells treated with either FK866, ibrutinib, or their combination for 6 h, and then exposed to TNF-α (10 ng/mL) during the last 20 min. Cytoplasmic and nuclear extracts were subjected to Western blotting using specific antibody for analysis of NF-kB canonical (anti–p-NF-κBp65 and -p-IkB) and non-canonical (-NF-κBp52, -RelB) activity. (D) BCMW.1 cells were cultured with FK866, ibrutinib, or the combination for 6 h, with TNF-α (10 ng/mL) added for the last 20 min. Immunocytochemical analysis was performed using anti-pospho-NF-κBp65 antibody. DAPI (4′,6-diamidino-2-phenylindole) was used to stain nuclei. (E) p65-NF-κB activity was also assessed by a luciferase promoter assay at 6 hours in BCMW.1 cells following treatment with FK866 (3 nM), ibrutinib (0.5 μM),, or the combination. Experiments were performed in triplicate. A representative experimental set is shown. **p=0.0038, ***p= 0.002, ****p<0.0001 (Student's t test).
Combination FK866 plus ibrutinib treatment induces synergistic inhibition of human WM cell growth in vivo
Based on our in vitro findings, we next evaluated the effect of this drug combination in vivo, using a xenograft mouse model in which BCMW.1 cells were injected s.c. in SCID mice. After development of tumor (approximately 3 weeks from cells injection), mice were treated either with low doses of either ibrutinib (0.5 mg/kg) or FK866 (30 mg/kg) daily for 4 days a week administered intraperitoneally. As shown in Fig. 6A, low doses of either agent alone had minimal effect on tumor growth relative to control mice. Importantly, when ibrutinib was combined with FK866, there was a significant reduction in tumor growth relative to untreated mice noted as early as week one and maintained at the end of therapy (p=0.002 and p<0.0001, respectively). Furthermore, combined treatment was well tolerated, with the single daily FK866 better tolerated than twice a day schedule; no significant weight loss, neurological changes or bleeding events were observed. (Suppl. Fig. 10) The median overall survival of FK866- and low dose ibrutinib-treated mice was significantly longer than vehicle-treated mice (44 vs 20 days; P=0.02) or mice treated with either drug alone (22.5 days for ibrutinib and 28 days for FK866) (Fig. 6B). Overall, these findings suggest that combining FK866 with ibrutinib markedly reduces tumor growth and is well tolerated in vivo.
Figure 6.
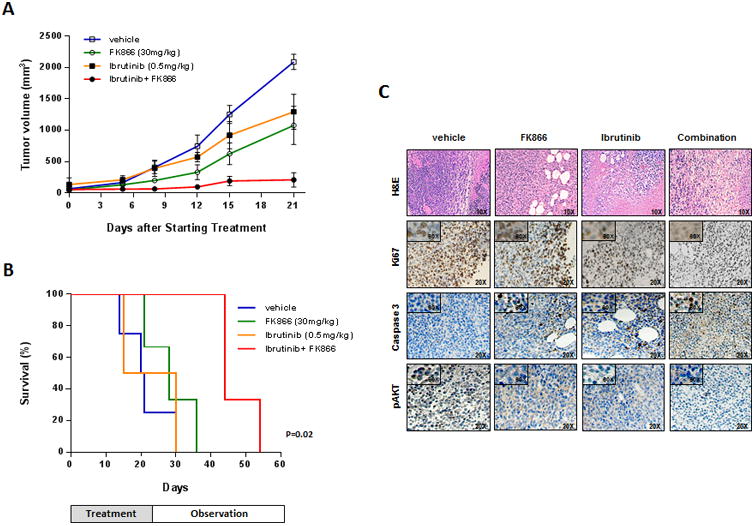
FK866 plus ibrutinib trigger synergistic inhibition of human WM cell growth in vivo. (A) Average and SD of tumor volume (mm3) from groups of mice (n=6/group) versus time (days) when tumor was measured. BCMW.1 cells (5 × 106 in 100 μL of serum free RPMI-1640 medium) were implanted in the flank of CB17-SCID mice. After tumor detection, mice were randomized to IP treatment with vehicle, FK866, ibrutinib, or the combination at the indicated doses for three weeks. A significant decrease in tumor growth was noted in combination-treated mice versus vehicle-treated mice (p=0.002 after the first week and p<0.0001 at the end of treatment). Data are mean tumor volume ± SD. (B) Kaplan-Meier survival plot showing survival for mice treated with vehicle, FK866, ibrutinib or their combination at the indicated concentrations. FK866 plus ibrutinib-treated mice show significantly increased survival (p=0.02) compared with vehicle-treated mice. The mean overall survival (OS) was 20 days in the vehicle- versus 44 days in combination-treated cohorts. Error bars represent mean ± SD. (C) Hematoxylin Eosin (10×) and immunohistochemical staining for Ki-67, caspase 3 and pAKT (20×; 60× in insets) in tumors sectioned on day 30 (endpoint) from vehicle-, FK866- (30 mg/kg), ibrutinib- (0.5 mg/kg) and combination-treated mice. Photographs are representative of 2 mice receiving each treatment.
We also examined the effect of drugs combination in vivo by caspase 3 staining of xenografts tumors harvested from treated mice. Combination therapy, but not treatment with either agent alone, dramatically increased the number of cleaved caspase-3 positive WM cells. (Fig. 6C) A significant decrease in the proliferation marker Ki67 was also noted in tumor sections from combination-treated mice relative to mice receiving single agent or vehicle. Finally, consistent with our vitro data, a significant reduction of pAKT protein level was also observed in tumors harvested from mice treated with combination versus single agent or vehicle control.
These results confirm the potent in vivo anti-WM activity of this combination at doses that are well tolerated, providing the framework for its clinical evaluation in WM.
Discussion
Whole genome sequencing studies revealed somatic mutations affecting MYD88 and CXCR4 genes in 90% and 30-35% of WM patients, respectively. The newly US Food and Drug Administration-approved drug ibrutinib is highly active in previously treated WM patients with an overall response rate of 90.5%, and 2-year progression-free and overall survival rates of 69.1% and 95.2%, respectively.(26) However, treatment responses are influenced by MYD88 and CXCR4 mutation status, with higher major response observed in MYD88L265P and CXCR4WT harboring patients. Therefore, novel single agent and combination therapeutic approaches are needed to improve outcome in WM.
We have previously demonstrated that Nampt, the rate-limiting enzyme in NAD+ synthesis, plays a critical role in maintaining viability of hematologic cancer cells.(3, 7, 8) Moreover, the selective Nampt inhibitor FK866 induces potent cell death in broad range of tumors in vitro. To date, clinical use of FK866 has been less promising, suggesting that drug combinations may increase the utility of Nampt-targeting therapy.
In the current study, we observed that Nampt is highly expressed in WM cells compared with normal CD19+ B cells, and that BTK is crucial for its activity, since upregulation of Nampt mRNA is triggered by ibrutinib treatment. This new oncogenic significance of Nampt in WM prompted us to assess its role in conferring ibrutinib-sensitivity in WM. Dual Nampt and BTK inhibition resulted in significant dose-dependent cytotoxicity in a panel of low-grade lymphoma cell lines. Importantly, this effect persisted even in the presence of WHIM-like mutated CXCR4 and in MYD88-silenced cells, suggesting that FK866 treatment may overcome ibrutinib-resistance. Moreover, the anti-WM effect of dual Nampt and BTK targeting was via interference with pro-survival pathways, overcoming the WM-cell growth advantage conferred by BM microenvironment. Similar responses were observed in WM patient cells and in a human WM xenograft model, without effects on PBMCs, suggesting its tumor-specificity and a favorable therapeutic index. Mechanistically, synergistic anti-WM cytotoxicity is associated with caspase activation and Mcl-1 down-regulation, resulting in apoptotic cell death. Importantly, there is an absence of autophagic features (data not shown), as in other combination FK866-based regimens.(7, 8, 19, 41, 42) Moreover, combined treatment induces energy depletion, which further downregulates pro-survival signaling activity leading to WM cell death both in vitro and in vivo. Finally, exogenous NAD+ fully rescued FK866-induced WM cell death, both as single agent and in combination with ibrutinib, confirming the role of NAD+ depletion in the observed synergism.
Despite the great therapeutic benefit of ibrutinib, drug-resistance develops and there remains a need for innovative therapeutic regimes. (22, 28) Mechanisms of drug-resistance may be multifactorial, including oncogene mutations and up-regulation of anti-apoptotic proteins such as Bcl-2 or Mcl-1.(43-46)
Here we postulated that Mcl-1 downregulation following Nampt inhibition may contribute to the synergistic anti-WM activity of FK 866 with ibrutinib. Indeed, combination Nampt and BTK treatment markedly decreases Mcl-1 protein expression, associated with WM cell death.
In summary, our data show that adding low doses of NAD+-depleting agent FK866 to ibrutinib both enhances sensitivity and overcomes resistance to BTK-inhibitor, suggesting that targeting NAD+ salvage pathway may enhance efficacy of ibrutinib-based therapies in WM. Importantly, our study establishes that dual BTK/Nampt targeted therapies may enhance cytotoxicity regardless of MYD88 and CXCR4 mutational status, providing the framework for clinical evaluation of combination therapy to improve patient outcome in WM.
Supplementary Material
Translational Relevance.
An upregulated NAD+ biosynthesis, which is needed to face increased proliferation and metabolic processes, represents an important feature distinguishing cancer cells from their normal counterparts. As a result, the NAD+ biosynthetic pathway emerges as highly promising therapeutic target for tumors, as suggested by the use of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors in a number of malignancies, including multiple myeloma and leukemia. Here, by using different approaches, we found that Waldenstrӧm Macroglobulinemia (WM) cells exhibit high levels of Nampt compared with normal B cells, indicating its potential oncogenic role. As a result, dual Nampt and BTK inhibition leads to significant and synergistic WM cell death, regardless of MYD88 and CXCR4 mutational status. Overall, our data provide the mechanistic preclinical rationale for targeting Nampt, either alone or with Btk inhibitor Ibrutinib, to overcome drug resistance and improve patient outcome in WM.
Acknowledgments
Grant Support: This work was supported by National Institutes of Health (grants RO-1 50947, RO-1 73878), DF/HCC SPORE in Multiple Myeloma (P-50100707); International Multiple Myeloma Foundation (A.C.); American Italian Cancer Foundation (M.C.). K.C.A. is an American Cancer Society Clinical Research Professor.
Footnotes
Authorship: M.C. and A.C. designed and performed research, analyzed data, and wrote the paper; T.C. contributed to perform animal work; C. A., P. A., Y.-T.T., C. Y., D.L., A.B., D.S., M.M., T.H., D.C., and A.N. provided reagents, analytic tools and input to studies; G.F.O. and L.M. performed IHC analysis; N. M., R. L. and M.G., provided WM patient samples and critically evaluated the manuscript; S. T. and K.C.A. critically evaluated and edited the manuscript.
Conflict-of-interest disclosure: All the authors declare no competing financial interests
References
- 1.Berger F, Ramirez-Hernandez MH, Ziegler M. The new life of a centenarian: signalling functions of NAD(P) Trends in biochemical sciences. 2004;29:111–8. doi: 10.1016/j.tibs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature reviews Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 3.Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome--a key determinant of cancer cell biology. Nature reviews Cancer. 2012;12:741–52. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocrine reviews. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cagnetta A, Soncini D, Caffa I, Acharya C, Acharya P, Adamia S, et al. Apo866 Increases Anti-Tumor Activity of Cyclosporin-a by Inducing Mitochondrial and Endoplasmic Reticulum Stress in Leukemia Cells. Clinical cancer research. 2015 doi: 10.1158/1078-0432.CCR-14-3023. [DOI] [PubMed] [Google Scholar]
- 8.Cea M, Cagnetta A, Fulciniti M, Tai YT, Hideshima T, Chauhan D, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. 2012;120:3519–29. doi: 10.1182/blood-2012-03-416776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bi TQ, Che XM, Liao XH, Zhang DJ, Long HL, Li HJ, et al. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncology reports. 2011;26:1251–7. doi: 10.3892/or.2011.1378. [DOI] [PubMed] [Google Scholar]
- 10.Gehrke I, Bouchard ED, Beiggi S, Poeppl AG, Johnston JB, Gibson SB, et al. On-target effect of FK866, a nicotinamide phosphoribosyl transferase inhibitor, by apoptosis-mediated death in chronic lymphocytic leukemia cells. Clinical cancer research. 2014;20:4861–72. doi: 10.1158/1078-0432.CCR-14-0624. [DOI] [PubMed] [Google Scholar]
- 11.Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, et al. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood. 2009;113:3276–86. doi: 10.1182/blood-2008-08-173369. [DOI] [PubMed] [Google Scholar]
- 12.Thakur BK, Dittrich T, Chandra P, Becker A, Kuehnau W, Klusmann JH, et al. Involvement of p53 in the cytotoxic activity of the NAMPT inhibitor FK866 in myeloid leukemic cells. International journal of cancer Journal international du cancer. 2013;132:766–74. doi: 10.1002/ijc.27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galli U, Travelli C, Massarotti A, Fakhfouri G, Rahimian R, Tron GC, et al. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. Journal of medicinal chemistry. 2013;56:6279–96. doi: 10.1021/jm4001049. [DOI] [PubMed] [Google Scholar]
- 14.Zoppoli G, Cea M, Soncini D, Fruscione F, Rudner J, Moran E, et al. Potent synergistic interaction between the Nampt inhibitor APO866 and the apoptosis activator TRAIL in human leukemia cells. Experimental hematology. 2010;38:979–88. doi: 10.1016/j.exphem.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 15.Pogrebniak A, Schemainda I, Azzam K, Pelka-Fleischer R, Nussler V, Hasmann M. Chemopotentiating effects of a novel NAD biosynthesis inhibitor, FK866, in combination with antineoplastic agents. European journal of medical research. 2006;11:313–21. [PubMed] [Google Scholar]
- 16.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Travelli C, Drago V, Maldi E, Kaludercic N, Galli U, Boldorini R, et al. Reciprocal potentiation of the antitumoral activities of FK866, an inhibitor of nicotinamide phosphoribosyltransferase, and etoposide or cisplatin in neuroblastoma cells. The Journal of pharmacology and experimental therapeutics. 2011;338:829–40. doi: 10.1124/jpet.111.184630. [DOI] [PubMed] [Google Scholar]
- 18.Muruganandham M, Alfieri AA, Matei C, Chen Y, Sukenick G, Schemainda I, et al. Metabolic signatures associated with a NAD synthesis inhibitor-induced tumor apoptosis identified by 1H-decoupled-31P magnetic resonance spectroscopy. Clinical cancer research. 2005;11:3503–13. doi: 10.1158/1078-0432.CCR-04-1399. [DOI] [PubMed] [Google Scholar]
- 19.Cagnetta A, Cea M, Calimeri T, Acharya C, Fulciniti M, Tai YT, et al. Intracellular NAD(+) depletion enhances bortezomib-induced anti-myeloma activity. Blood. 2013;122:1243–55. doi: 10.1182/blood-2013-02-483511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. The New England journal of medicine. 2012;367:826–33. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 21.Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–46. doi: 10.1182/blood-2013-09-525808. [DOI] [PubMed] [Google Scholar]
- 22.Roccaro AM, Sacco A, Jimenez C, Maiso P, Moschetta M, Mishima Y, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123:4120–31. doi: 10.1182/blood-2014-03-564583. [DOI] [PubMed] [Google Scholar]
- 23.Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom's Macroglobulinemia. Leukemia. 2015;29:169–76. doi: 10.1038/leu.2014.187. [DOI] [PubMed] [Google Scholar]
- 24.Treon SP, Hunter ZR. A new era for Waldenstrom macroglobulinemia: MYD88 L265P. Blood. 2013;121:4434–6. doi: 10.1182/blood-2013-04-494849. [DOI] [PubMed] [Google Scholar]
- 25.Anderson KC, Alsina M, Bensinger W, Biermann JS, Cohen AD, Devine S, et al. Waldenstrom's macroglobulinemia/lymphoplasmacytic lymphoma, version 2.2013. Journal of the National Comprehensive Cancer Network : JNCCN. 2012;10:1211–9. doi: 10.6004/jnccn.2012.0128. [DOI] [PubMed] [Google Scholar]
- 26.Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. The New England journal of medicine. 2015;372:1430–40. doi: 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- 27.Treon SP, Xu L, Hunter Z. MYD88 Mutations and Response to Ibrutinib in Waldenstrom's Macroglobulinemia. The New England journal of medicine. 2015;373:584–6. doi: 10.1056/NEJMc1506192. [DOI] [PubMed] [Google Scholar]
- 28.Cao Y, Yang G, Hunter ZR, Liu X, Xu L, Chen J, et al. The BCL2 antagonist ABT-199 triggers apoptosis, and augments ibrutinib and idelalisib mediated cytotoxicity in CXCR4 and CXCR4 mutated Waldenstrom macroglobulinaemia cells. British journal of haematology. 2015 doi: 10.1111/bjh.13278. [DOI] [PubMed] [Google Scholar]
- 29.Cea M, Cagnetta A, Patrone F, Nencioni A, Gobbi M, Anderson KC. Intracellular NAD(+) depletion induces autophagic death in multiple myeloma cells. Autophagy. 2013;9:410–2. doi: 10.4161/auto.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fulciniti M, Amodio N, Bandi RL, Munshi M, Yang G, Xu L, et al. MYD88-independent growth and survival effects of Sp1 transactivation in Waldenstrom macroglobulinemia. Blood. 2014;123:2673–81. doi: 10.1182/blood-2014-01-550509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood. 2013;122:1222–32. doi: 10.1182/blood-2012-12-475111. [DOI] [PubMed] [Google Scholar]
- 32.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 33.Audrito V, Serra S, Brusa D, Mazzola F, Arruga F, Vaisitti T, et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood. 2015;125:111–23. doi: 10.1182/blood-2014-07-589069. [DOI] [PubMed] [Google Scholar]
- 34.Kenny EF, Quinn SR, Doyle SL, Vink PM, van Eenennaam H, O'Neill LA. Bruton's tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PloS one. 2013;8:e74103. doi: 10.1371/journal.pone.0074103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y, Liu X, et al. MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121:2051–8. doi: 10.1182/blood-2012-09-454355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulain S, Roumier C, Decambron A, Renneville A, Herbaux C, Bertrand E, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121:4504–11. doi: 10.1182/blood-2012-06-436329. [DOI] [PubMed] [Google Scholar]
- 38.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer research. 2003;63:7436–42. [PubMed] [Google Scholar]
- 39.Bruzzone S, Fruscione F, Morando S, Ferrando T, Poggi A, Garuti A, et al. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PloS one. 2009;4:e7897. doi: 10.1371/journal.pone.0007897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roccaro AM, Sacco A, Husu EN, Pitsillides C, Vesole S, Azab AK, et al. Dual targeting of the PI3K/Akt/mTOR pathway as an antitumor strategy in Waldenstrom macroglobulinemia. Blood. 2010;115:559–69. doi: 10.1182/blood-2009-07-235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.David E, Kaufman JL, Flowers CR, Schafer-Hales K, Torre C, Chen J, et al. Tipifarnib sensitizes cells to proteasome inhibition by blocking degradation of bortezomib-induced aggresomes. Blood. 2010;116:5285–8. doi: 10.1182/blood-2010-03-272393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cea M, Cagnetta A, Patrone F, Nencioni A, Gobbi M, Anderson KC. Intracellular NAD (+) depletion induces autophagic death in multiple myeloma cells. Autophagy. 2012;9 doi: 10.4161/auto.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–4. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 44.Michels J, Obrist F, Vitale I, Lissa D, Garcia P, Behnam-Motlagh P, et al. MCL-1 dependency of cisplatin-resistant cancer cells. Biochemical pharmacology. 2014;92:55–61. doi: 10.1016/j.bcp.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 45.Rooswinkel RW, van de Kooij B, de Vries E, Paauwe M, Braster R, Verheij M, et al. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood. 2014;123:2806–15. doi: 10.1182/blood-2013-08-519470. [DOI] [PubMed] [Google Scholar]
- 46.Hermanson DL, Das SG, Li Y, Xing C. Overexpression of Mcl-1 confers multidrug resistance, whereas topoisomerase IIbeta downregulation introduces mitoxantrone-specific drug resistance in acute myeloid leukemia. Molecular pharmacology. 2013;84:236–43. doi: 10.1124/mol.113.086140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.