

Abstract
BACKGROUND:
West Syndrome (WS) represents as a specific epileptic encephalopathy characterised with a unique type of attacks, called infantile spasms, severe forms of abnormalities in electroencephalographic (EEG) records as a hypsarythmias and delays in the psychomotoric development. The characteristics of the disease, mostly affecting male gender, are infantile spasms and typical findings in EEG as a hypsarythmia. Infantile spasms are a consequence of many factors in the undeveloped brain.
AIM:
We aimed: (1) to see the incidence of the illness and the spreading out because of gender in rapport with other syndromes in the epileptic encephalopathies group; (2) to show principles of the treatment for the illness; and (3) to present the effects of the disease in the psycho-motoric development of affected children.
METHODS:
The study was designed as a cross-sectional study of the patients with epileptic encephalopathies, treated in Paediatric Clinic in Prishtina, from 1st of January 2013 until the 31st of December 2015.
RESULTS:
From the cohort group of 97 children diagnosed with epileptic encephalopathies, in 14 of them clinical and EEG signs of WS were noted. The earliest age of disease manifestation was 74 days (± 63.8 days). On the group of children with WS, 13 of them with Natrium Valpropat were treated, with the doses of 301.9 mg (± 64.1). From the cohort group, in 89 children (91.8%) psychomotoric retardation was documented, within the higher reoccurrence in the undifferentiated epileptic encephalopathies (96%) and the WS (78.6%).
CONCLUSION:
WS is a frequent disease of the encephalopathies with the epileptogenic framework. The resistance in anticonvulsive therapy is huge, and psychomotoric retardation follows a big percentage of children with this syndrome.
Keywords: West Syndrome, Hypsarythmia, Epileptic encephalopathy, Infantile spasm
Introduction
West Syndrome is specific epileptic encephalopathies groups characterised by a unique type of attacks, called infantile spasms, severe forms of abnormalities in the EEG - hypsarythmia and delays in the psychomotoric development. In 2004, there was a consensus by the experts in this subject, published about the West syndrome. Mostly the syndrome starts from 3 until 12 months of age, with a ratio 1:2000 - 4000 born alive, predominantly affecting male gender (60-70%) [1].
In 2001, ILEA classified the Syndrome West in the epileptic encephalopathies group.
We aimed: (1) to see the incidence of the illness and the spreading out because of gender in rapport with other syndromes in the epileptic encephalopathies group; (2) to show principles of the treatment for the illness; and (3) to present the effects of the disease in the psycho-motoric development of affected children.
Material and Methods
The study was designed as a cross-sectional of the patients with epileptic encephalopathies, treated from 1 of January 2013 until 31 of December 2015. In this presentation from the cohort group of 97 children, 14 of them with WS were diagnosed.
Diagnosis of the disease was based on the anamnestic data, clinical examination, lab analysis, and imagery examinations as a brain echosonography, computerised tomography (CT), magnetic resonance imaging (MRI), neurological examination, electroencephalographic examinations (EEG), tests on the aspect of psycho-motoric developments, therapeutic methods like physiotherapy exercises and medicaments.
For the statistical analysis, we used the program SPSS 20.0, Sigma Stat - Sigma Plot 11.0 and Microsoft Excel 2010. From the statistic parameters, we used the structure index, cumulative structure, simple arithmetic average, standard deviation, standard mistake and confidence interval with the significance level 95% (95%CI). For the testing of the differences of the nonparametric data, we used chi-cubic test (chi-test), for the exact significant level (p).
Results
The vast majority of cases were with undifferentiated epileptogenic encephalopathy (77.3%) and with Syndrome West (14.4%). With syndrome Lennox-Gastaut was diagnosed 4 cases (4.1%), 2 cases with CSWS (2) and 1 case was diagnosed with syndrome Dravet and syndrome Landau-Kleffner.
Figure 1.
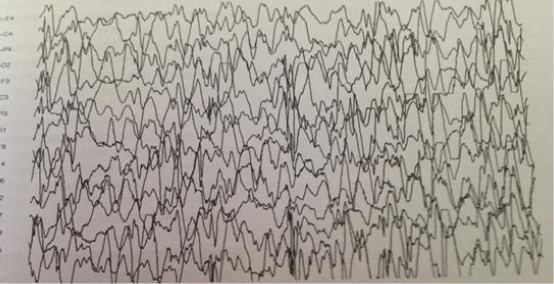
EEG records in the severe form of West Syndrome.
A similar structure was concluded analyzing the gender group; the difference of the structure in cases with undifferentiated epileptogenic encephalopathy was higher in females with (79.5% versus 75.9%). Also, with syndrome Lennox-Gastaut, we noted same relation (5.1% versus 3.4%). Moreover, in children with WS happens more often in males, with result of17.2% versus 10.3%.
The manifestation of the clinical signs approximately in our group started on the 245 days (245.5 ± 522.9), earlier on the male than the female (214.1 ± 450.9 vs 301.2 ± 634.4) (Table 2).
Table 2.
Beginning of the disease according to diagnosis
| Diagnosis | F | M | Grand Total | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | |
| CSWS | 180.0 | - | 1095.0 | - | 637.5 | 647.0 |
| Enceph.EPI | 257.7 | 638.9 | 240.9 | 501.4 | 247.6 | 556.1 |
| SD | 365.0 | - | 365.0 | - | ||
| Sy Landau-Kleffner | 913.0 | - | 913.0 | - | ||
| Sy Lennox-Gastaut | 820.0 | 905.1 | 1.0 | 0.0 | 410.5 | 704.7 |
| Sy West | 120.0 | - | 70.5 | 64.9 | 74.1 | 63.8 |
| Grand Total | 301.2 | 634.4 | 214.1 | 450.9 | 245.5 | 522.9 |
Mean = arithmetical average; SD = Standard deviation.
Table 1.
Demographic structure of the patients with diagnosis and genders
| Diagnosis | F | M | Grand Total | ||||
|---|---|---|---|---|---|---|---|
| No. | (%) | No. | (%) | No. | (%) | Report M:F | |
| CSWS | 1 | 2.6 | 1 | 1.7 | 2 | 2.1 | 1 : 1 |
| Enceph.EPI | 31 | 79.5 | 44 | 75.9 | 75 | 77.3 | 1.4 : 1 |
| Sy Dravet (SD) | 0.0 | 1 | 1.7 | 1 | 1.0 | - | |
| Sy Landau-Kleffner | 1 | 2.6 | 0.0 | 1 | 1.0 | - | |
| Sy Lennox-Gastaut | 2 | 5.1 | 2 | 3.4 | 4 | 4.1 | 1 : 1 |
| Sy West | 4 | 10.3 | 10 | 17.2 | 14 | 14.4 | 2.5 : 1 |
| Grand Total | 39 | 100 | 58 | 100 | 97 | 100 | 1.5 : 1 |
The earliest signs were in Syndrome West (74.1 ± 63.8 days). Clinical signs of the Syndrome Dravet usually are manifested after the first year, Syndrome Landau-Klenner approximately after 2.5 years (913 days), Syndrome Lennox-Gastau after 1.1 years (410.5 ± 704.7 days) and the WS after 2.5 months (74.1 ± 63.8 days).
From the group of 14 cases with WS, 13 of them with NA-Valproate were treated as a primary therapy, approximate doses of 301.9 ± 64.1 mg. In fourth children Clonazepam, as additional therapy, in an approximate dose of 0.5 mg were given. As a reason for recurrent seizures, one case with Levetiracetam was treated.
Figure 1.
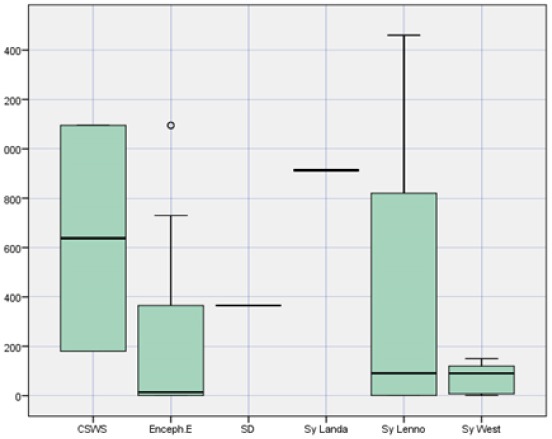
Approximate age of the disease manifestation
Figure 2.
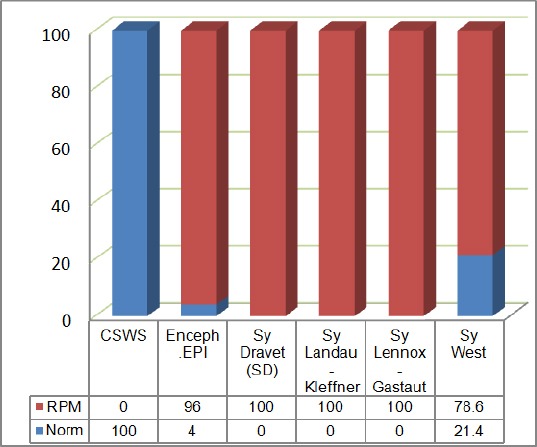
Prevalence of psychomotoric awkwardness according to the diagnosis
Topiramate was given in two cases; in the approximate dose of 50 mg. Clinical signs of psychomotoric retardation were concluded in 89 patients (91.8%), with a higher reoccurrence of undifferentiated epileptic encephalopathies (96 %) and the with WS in (78.6%).
Figure 3.
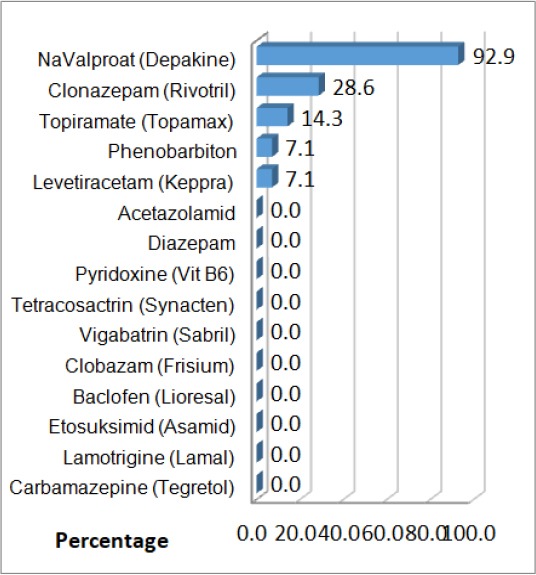
Structure of cases with West Syndrome according to the medical therapy
Discussion
According to the publication of Primec ZR from Slovenia during the 11 years, there were diagnosed 47 cases with infantile spasms. The cumulative incidence was 2.06 in 10.000 births. The age of first attacks was from 2 to 10 months [2].
However, Brna PM with colleague’s reported the average incidence of infantile spasms in Canada with the incidence of 30.7/100.00 at births [3]. Boys were affected more than girls (60%).
In the presentation from Salonga, Filipine the ratio male-female was about 1:1. In the research done by Chiemc Hanya S with bp. in the paediatric department in Bangkok, Thailand, from 628 patients with epilepsy, 31 patients (4.9%) were diagnosed with Syndrome West. The report of gender was close 1:07:1, whereas the approximate age of the manifestation of disease is 9.7 months (range 4-32 months), while the seizures are from 5.7 months (range 2-11 months) [4].
According to the Wong V et all. in Hong Kong, from 1970 to 2000, 105 cases were diagnosed with WS. The number of new cases is 108 per year. The average incidence of WS to the new epileptic cases is approximately 0.8- 4.8% [5].
In Malaysia, during the period 1997-1999, WS was reported in 3% of new cases diagnosed with epilepsy [6].
Depending on the age of children with WS, the attacks usually start between 3 and 12 months, reaching the peak of attacks at the age of 5th month in 90% of the cases. In 70% of the cases, a first convulsive attack starts on the age of 6 months; it is very rare for WS to start in the middle of the 1st and 3rd year (around 5%). The location of the focal cortical lesion can impact the age of epileptic spasms at the beginning [7].
In the results of our research, the male is represented in a higher percentage than girls (10/4), ratio M: F is 2:5:1. Average of the presentations of WS is 74 days, (around 2.5 months). In our group, the incidence WS is 4.05 in 10000 live births. Mental and development signs were seen in 13 cases or 78.6% of cases with WS.
There are many causes of WS. Infantile spasms are as a consequence of many factors in the undeveloped brain. WS can occur in the children with cognitive delays or a healthy child up till then. The symptomatic form is known in 80% of cases while the idiopathic form remains in 20 % of the cases.
Idiopathic forms appear in families with hereditary character, whereby family members suffer from various forms of epilepsy or febrile convulsions.
Symptomatic forms happen more often, wherein 80% of the cases are connected alongside with other problems such are tuberous sclerosis, pre and perinatal damages, causing hypoxia, ischemia, premature birth, infections or disability of migration in the neurons in case of hemimegalencephalia, agenesis of corpus callosum and lissencephaly.
Clinical manifestations: WS usually starts with an infantile spasm of lights that occur two or three times in a row, which come being added in the coming weeks with many cases of stroke in various attacks per day from 20 to 150 times. The attacks are short tonic or clonic and they happen before getting up or during, which follows crying that doesn’t present any ictal signs. Usually, attacks are symmetric, rapid, presenting with mix forms of the flexor or extensor type [8].
Diagnosis: A correct anamnesis data and clinical neurological evaluation is a big step in the diagnosing of disease. The lab analyses usually are within the normal limits. Neurometabolic examinations have an important role in eliminating metabolic diseases. Neuroradiological examinations as a CT and brain MRI shows atrophy of the brain mass. Characteristic changes in the EEG between two attacks presented as a hypsarythmia, with predominate of a slow elementary activity with a high voltage which is characterised with multiple peaks, steep waves and complex SW with a lot of hearth’s focal. The EEG changes can be symmetric and asymmetric. During the sleep, EEG is discontinued, and it reminds us of the suppression of the basic brain activity. In some cases, all EEG manifestations of hypsarythmia can be absent, and the findings of the EEG are characterised from a fragmental and modified hypsarythmia.
Prognosis and treatment: Prognosis of WS is variable. In most cases, it comes to motoric and mental regression, more often as an axial hypotonic. The continuous epileptic activity often is the cause of a cognitive deficit, loss of visual contact is a bad prognostic sign [9]. The attacks and hypsarythmia are stopped before the age of 3, in 40 to 50% of cases. The other cases evolve in the syndrome Lennox-Gastaut. 5 to 10% of children have a normal psychometric development. Children with idiopathic or cryptogenic form have a better prognosis. The children with Syndrome West have a fatal ending in 5 to 20% of cases. The deaths can happen because of the basic reason for the illness or the treatment with ACTH and corticosteroids.
Treatment: There is no consensus on the effective long-term treatment of WS. Treatment of choice is ACTH and Vigabatrin, however. As a reason for the multiple side effects, we used Valproate as a therapy of choice. In the absent of good control, we used on of the next medications as Lamotrigine, Levetiracetam, Nitrazepam, Pyridoxine, Sulthiam, Topiramate or Zonisamide. Vigabatrin was used relatively successfully in the children with WS, but, because of side effects in the damaging of the retina, the usage is limited. For the children with the Syndrome West and tuberous sclerosis, the use of Vigabatrin is more effective than the ACTH. In some cases, imunoglobin I.V was successful used. Also, the high doses of Pyridoxine 100-300 mg/kg during the first and second were given, and good results arrived. The ketogenic diet was successful in some cases of symptomatic/cryptogenic forms. In some cases when the attacks were very resistant calosototmia was applied but without satisfactory results.
In conclusion, West Syndrome is a frequent disease at the encephalopathic epileptogenic framework. The disease often involves syndrome Lennox-Gastaut. The resistance in anticonvulsive therapy is huge, and psychomotoric retardation follows a big percentage of children with this syndrome. The characteristics of the illness are infantile spasms and characteristics differences in EEG like hypsarythmia. This disease happens more often to the boys than the girls.
Footnotes
Funding: This research did not receive any financial support.
Competing Interests: The authors have declared that no competing interests exist.
References
- 1.Chugani HT. Pathophysiology of infantile spasms. Adv Exp Med Biol. 2002;497:111–121. doi: 10.1007/978-1-4615-1335-3_13. https://doi.org/10.1007/978-1-4615-1335-3_13 PMid:11993727. [DOI] [PubMed] [Google Scholar]
- 2.Primec ZR, Kopac S, Neubauer D. Epidemiologic features of infantile spasms in Slovenia. Epilepsia. 2002;43(2):183–7. doi: 10.1046/j.1528-1157.2002.26201.x. https://doi.org/10.1046/j.1528-1157.2002.26201.x PMid:11903466. [DOI] [PubMed] [Google Scholar]
- 3.Brna PM, Gordon KE, Dooley JM, Wood EP. The epidemiology of infantile spasms. Can J Neurol Sci. 2001;28(4):309–12. doi: 10.1017/s0317167100001517. https://doi.org/10.1017/S0317167100001517 PMid:11766774. [DOI] [PubMed] [Google Scholar]
- 4.Chiemchanya S, Visudtibhan A, Visudhiphan P. West syndrome in Thailand:a hospital-based survey. Brain Dev. 2001;23(7):605–8. doi: 10.1016/s0387-7604(01)00294-7. https://doi.org/10.1016/S0387-7604(01)00294-7. [DOI] [PubMed] [Google Scholar]
- 5.Wong V. West syndrome – The University of Hong Kong experience (1970-2000) Brain Dev. 2001;23(7):609–15. doi: 10.1016/s0387-7604(01)00296-0. https://doi.org/10.1016/S0387-7604(01)00296-0. [DOI] [PubMed] [Google Scholar]
- 6.Thambyayah M. Early epileptic encephalopathies including West syndrome:a 3-year retrospective study from Klang Hospital, Malaysia. Brain Dev. 2001;23(7):603–4. doi: 10.1016/s0387-7604(01)00293-5. https://doi.org/10.1016/S0387-7604(01)00293-5. [DOI] [PubMed] [Google Scholar]
- 7.Hrachovy RA, Frost JD. Severe Encephalopathic Epilepsy in Infants:Infantile Spasms (West Syndrome) In: Pellock JM, ourgeois BFD, Dodson WE, editors. Undefined. Pediatric Epilepsy. Third Edition. New York, NY: Demos Medical Publishing; 2008. p. 16. [Google Scholar]
- 8.Lux AL, Osborne JP. A proposal for case definitions and outcome measures in studies of infantile spasms and west syndrome:consensus statement of the west Delphi group. Epilepsia. 2004;45:1416–28. doi: 10.1111/j.0013-9580.2004.02404.x. https://doi.org/10.1111/j.0013-9580.2004.02404.x PMid:15509243. [DOI] [PubMed] [Google Scholar]
- 9.Pavone P, Striano P, Falsaperla R, Pavone L, Ruggieri M. Infantile spasms syndrome, West syndrome and related phenotypes:what we know in 2013. Brain Dev. 2014;36(9):739–51. doi: 10.1016/j.braindev.2013.10.008. https://doi.org/10.1016/j.braindev.2013.10.008 PMid:24268986. [DOI] [PubMed] [Google Scholar]