

Abstract
Accurate early diagnosis of Parkinson's disease is essential. Using data available from the Parkinson's Progression Markers Initiative study, we identified a multivariate logistic regression model including cerebrospinal fluid α‐synuclein, olfactory function, age, and gender that achieved a high degree of discrimination between patients with Parkinson's disease and healthy control or scan without evidence of dopaminergic deficit participants. Additionally, the model could predict the conversion of scan without evidence of dopaminergic deficit to Parkinson's disease, as well as discriminate between normal and impaired subjects with leucine‐rich repeat kinase 2 mutations. Although further validation is needed, this model may serve as an alternative method to neuroimaging screening in Parkinson's disease studies.
Introduction
Parkinson's disease (PD) features neurodegeneration in key brain regions and pathological accumulations of α‐synuclein (α‐syn). Diagnosis is based on clinical symptoms, and its accuracy varies according to disease duration (lower at early stages), patient age, and the expertise of the clinician.1 Functional neuroimaging aids PD diagnosis, but cost and access restrict its usage. Promising ancillary diagnostic tests, including olfactory loss, have demonstrated ≥80% specificity in relatively large‐scale independent studies,1 but the diagnostic accuracy needs further improvement. A previous study has shown the integration of several key clinical factors as well as genetic risks for PD diagnosis yielded good sensitivity and specificity for PD diagnosis.2 Genetic risk or clinical features based prediction models have been studied in the prediction of PD falls or cognitive impairment.3, 4, 5 Body fluid biomarkers, particularly in the cerebrospinal fluid (CSF), have also been widely tested, including α‐syn, total and phosphorylated tau (t‐tau and p‐tau), and amyloid beta 1‐42 (Aβ 1‐42), but no single biomarker to date has been capable of accurate diagnosis. In one study, clinical and body fluid markers were combined to successfully predict cognitive impairment in PD.5 However, to our knowledge, no study has used a similar combined model to predict PD diagnosis.
The aim of this report was to explore the possibility of combining clinical measures with CSF biomarkers to increase PD diagnostic accuracy, particularly early diagnosis. To accomplish this, we examined data available from the Parkinson's Progression Markers Initiative (PPMI), an international, multicenter, longitudinal study designed to identify biomarkers of PD progression. The PPMI study has recruited a large cohort of patients with early PD and controls and collected a rich dataset including demographics, olfactory function, biomarker measurements, and clinical and behavioral assessments, which we combined to generate a diagnostic model for PD. The PPMI study also includes a cohort of scan without evidence of dopaminergic deficit (SWEDD) subjects, who meet all PD inclusion criteria except for a confirmation of screening dopamine transporter (DAT) single‐photon emission computed tomography (SPECT) scan showing no evidence of DAT deficit. SWEDD is considered as a heterogeneous population consisting of misdiagnosed different disorders of PD lookalikes, a phase of PD that precedes imaging‐detectable degeneration of dopaminergic neurons, and PD patients with incorrect imaging findings. To test whether our model could be used to predict premotor or preclinical PD, we then applied this model to SWEDD patients and a second cohort of subjects carrying a genetic mutation that puts them at high risk of PD.
Methods
Participants and sample selection
From the 423 patients with PD and 196 healthy controls (HC) enrolled in PPMI (ppmi‐info.org/study‐design), 290 PD and 138 HC subjects with low CSF blood contamination (hemoglobin < 200 ng/mL) at baseline and no missing values of University of Pennsylvania Smell Identification Test (UPSIT) and CSF α‐syn were included (data downloaded at May 4th, 2017). From the 55 subjects described as SWEDD at baseline in the same cohort, 51 subjects, who had baseline CSF samples with low hemoglobin, were included in the current analysis.
A second cohort, consisting of 14 leucine‐rich repeat kinase 2 (LRRK2) mutation carriers with available CSF α‐syn, UPSIT, and positron emission tomography (PET) imaging data, has been previously described.6, 7, 8 All subjects were scanned using 11C‐(±)‐α‐dihydrotetrabenazine (TBZ), a ligand of vesicular monoamine transporter 2.
CSF assays and clinical assessments
CSF levels of α‐syn (the measurement in 2013), Aβ 1‐42, t‐tau and p‐tau, and hemoglobin have been previously reported for the PPMI cohort,9, 10 as have α‐syn and PET for our LRRK2 cohort.6 Due to the difference of platform (Luminex for LRRK2 and ELISA for PPMI) used in α‐syn measurement between PPMI and LRRK2 cohorts, the α‐syn concentration in the LRRK2 cohort was significantly lower. The average α‐syn concentration of healthy controls in PPMI and baseline asymptomatic carriers in LRRK2 cohorts were 2243.8 pg/mL and 388.3 pg/mL, and a conversion factor of 5.78 (2243.8/388.3 = 5.78) was used for normalization between platforms when applying α‐syn concentration to the equation for integrated probability calculation. Clinical measures included scores for Movement Disorder Society – Unified Parkinson's Disease Rating Scale (MDS‐UPDRS) III, Montreal Cognitive Assessment (MoCA), and UPSIT, as previously described.11
Statistical analysis
Statistical analyses were performed using IBMS SPSS version 23 and GraphPad Prism 6. Normality of CSF biomarkers was tested using the Kolmogorov–Smirnov test, and groups compared using the Mann–Whitney test. Stepwise multivariate logistic regression was used to identify models differentiating PD from HC in the PPMI cohort, controlling for age and sex of participants. The initial model included UPSIT, RBD Questionnaire, Geriatric Depression Scale and other nonmotor test scores, self‐report of constipation, caffeine intake, smoking status, as well as CSF α‐syn, Aβ 1‐42, tau and p‐tau, or their ratios. The top contributors, including CSF α‐syn and UPSIT, were retained in the final integrated model, along with age and sex of the subject, while the clinical and biochemical markers that were less significant were excluded. The diagnostic utility of biomarkers was determined using a receiver operating characteristic (ROC) analysis with the cut points providing the highest Youden index (i.e., maximized sum of sensitivity and specificity), but considering balance of sensitivity and specificity. P values <0.05 were regarded as significant.
Results
Demographics, clinical evaluations, and CSF biomarkers in PPMI
No significant differences in age or sex were observed between the two groups. As expected, MoCA, UPSIT, and UPDRS III motor scores differed between groups (P < 0.05; Table 1). CSF α‐syn, total tau, and p‐tau181 concentrations were reduced in patients with PD compared to HC (Mann–Whitney U test, P < 0.05), with no significant difference observed in Aβ 1‐42 (Table 1), as previously published.12, 13 The ratios of α‐syn/Aβ 1‐42 and t‐tau/Aβ 1‐42 were significantly lower (P < 0.05; Table 1) in patients with PD compared to HC.
Table 1.
Demographic information and cerebrospinal fluid measures
| HC N = 138 | PD N = 290 | Significance | ROC AUC | |
|---|---|---|---|---|
| Sex (% male) | 65.2% | 66.7% | 0.093b | |
| Age (mean, SD) | 60.7 (11.9) | 61.6 (9.7) | 0.8197a | |
| UPDRS III (mean, range) | 1.2 (0–10) | 22.5 (5–48) | <0.0001a | |
| MoCA (mean, range) | 28.2 (26–30) | 27.0 (17–30) | <0.0001a | |
| UPSIT score (mean, range) | 34.7 (15–40) | 22.2 (5–40) | <0.0001a | 0.9180 |
| CSF α‐syn (pg/mL) (mean, SD) | 2243.8 (1162.9) | 1823.7 (726.5) | 0.0010a | 0.5982 |
| CSF Aβ 1‐42 (pg/mL) (mean, SD) | 379.8 (111.2) | 374.5 (103.0) | 0.4079a | 0.5248 |
| CSF total tau (pg/mL) (mean, SD) | 54.1 (29.1) | 44.9 (18.0) | 0.0029a | 0.5890 |
| CSF p‐tau181 (pg/mL) (mean, SD) | 18.7 (12.1) | 15.9 (9.7) | 0.0087a | 0.5784 |
| CSF α‐syn/Aβ 1‐42 (mean, SD) | 6.7 (6.1) | 5.1 (2.2) | 0.0145a | 0.5731 |
| CSF total tau/Aβ 1‐42 (mean, SD) | 170.2 (211.3) | 126.7 (64.7) | 0.0357a | 0.5628 |
| CSF p‐tau181/Aβ 1‐42 (mean, SD) | 56.3 (65.0) | 43.5 (26.6) | 0.0694a | 0.5543 |
| CSF p‐tau181/total tau (mean, SD) | 0.37 (0.19) | 0.37 (0.20) | 0.9347a | 0.5025 |
CSF, cerebrospinal fluid; PD, Parkinson's disease; HC, healthy control.
Based on Mann–Whitney U test.
Based on chi‐square test.
In the ROC analysis, each single CSF marker or ratio discriminated PD from HC poorly to moderately due to high overlap between groups (Table 1). The UPSIT score alone, reflecting olfactory loss, showed an area under curve (AUC) of 0.918 (sensitivity 82.6%, specificity 89.3%). When a stepwise logistic regression model was used to control for age and sex and eliminate markers with low independent contribution, the final diagnostic model included CSF α‐syn, UPSIT score, age, and sex. The AUC of this integrated four‐factor model was 0.927 with a sensitivity and specificity of 89.7% and 80.4%, respectively, and a cutoff value of 0.495 in the ROC analysis (Fig. 1).
Figure 1.
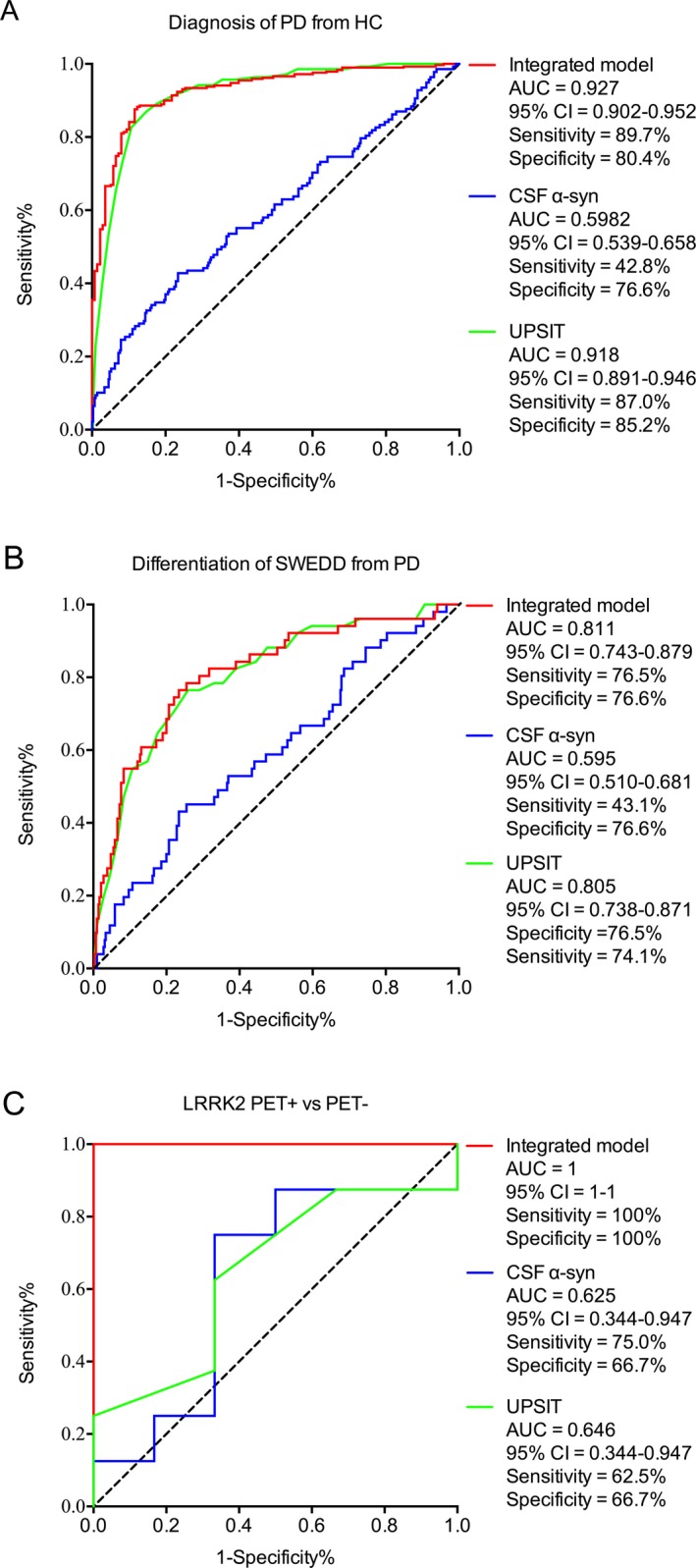
Figure 1 A. Receiver operating characteristic curves for the integrated model (red line), ?‐syn (blue line) and UPSIT (green line) to discriminate PD from HC in PPMI cohort. B. Receiver operating characteristic curve for the integrated model (red line), α‐syn (blue line) and UPSIT (green line) to differentiate SWEDD from PD. C. Receiver operating characteristic curves for the integrated model (red line), α‐syn (blue line) and UPSIT (green line) to discriminate PET+ from PET‐ subjects in LRRK2 mutation cohort. Integrated model included α‐syn, UPSIT score, age and sex. AUC = area under curve.
SWEDD and conversion of SWEDD to PD
Subjects with PD‐like clinical symptoms but no dopaminergic dysfunction on DAT scan received the SWEDD designation. When the integrated model was applied to SWEDD subjects, an AUC of 0.811 was achieved for SWEDD versus PD (sensitivity: 76.5%; specificity: 76.6%) (Fig. 1B), indicating that the SWEDD population is distinct from PD. Remarkably, when the cutoff value determined for the PPMI PD versus HC was applied to the 31 subjects classified as SWEDD with longitudinal DAT scan results, 15 were predicted to have a PD profile. Notably, using the 1‐ to 2‐year follow‐up data available at the time of analysis, four of the 15 subjects predicted to have a PD profile by our model showed dopaminergic dysfunction evidenced by DAT scan (DAT+), whereas none of the 16 subjects who had a HC profile in the model developed abnormal DAT scan results (Table 2), a separation with an AUC of 0.815 (DAT+ vs. DAT− at follow‐up; sensitivity: 100%; specificity: 59.3% for conversion into PD).
Table 2.
Predictive value of integrated model
| PPMI SWEDD | LRRK2 | ||||
|---|---|---|---|---|---|
| Predicted outcome (cutoff = 0.495) | Normal (N = 16) | PD (N = 15) | Normal (N = 2) | PD (N = 12) | |
| Age (mean, SD) | 59.7 (11.3) | 61.6 (10.9) | 28.0 (2.82) | 53.5 (12.09) | |
| Sex (male%) | 43.8% | 60% | 100% | 42% | |
| UPSIT (mean, range) | 35.5 (30–39) | 25.8 (12–34) | 35.5 (34–37) | 27.4 (13–31) | |
| CSF α‐syn (ng/mL) (mean, SD) | 2.4 (0.89) | 1.7 (0.48) | 3.52 (0.70) | 1.91 (0.62) | |
| Number of subjects converted to PD after 1‐ to 2‐year follow‐up | 0 | 4 | Number of subjects with verified PET abnormality | 0 | 6 |
| DAT scan (mean, SD) | 2.5 (0.49) | 1.9 (0.55) | TBZ score mean % of normal control (range) | 122 (119–125) | 79 (17–140) |
Sensitivity = 100%; specificity = 59.3% for SWEDD cohort (DAT+ vs. DAT− at follow‐ups). Sensitivity = 100%; specificity = 25% for LRRK2 cohort (PET+ vs. PET−).
LRRK2 and PET abnormality
We next sought to determine the utility of the model in subjects without overt clinical symptoms, by testing it in a cohort of genetic mutation carriers. Most (12/14) LRRK2 subjects were asymptomatic at the time of CSF collection. When the model was applied to this cohort, those with PET abnormality, defined as a deficit of ≥20% compared to age‐matched control,6, 7, 8 were identified with 100% sensitivity and specificity, and an AUC of 1 (Fig. 1C). We then applied the cutoff value determined in the PPMI cohort to predict conversion from normal (−) to impaired (+) PET. Both cases categorized as “control” were PET‐; the “PD” group consisted of 12 subjects, including all six PET+ subjects (PET+ vs. PET− AUC of 0.708; 100% sensitivity and 25% specificity) (Table 2). The single subject who converted from asymptomatic to symptomatic and two subjects who converted from PET‐ to PET+ during follow‐up were sorted into the PD group based on baseline marker values.
Discussion
The accurate diagnosis of PD is crucial, especially in early disease stages. Recently, a model including olfactory function (UPSIT), genetic risk, family history of PD, age, and gender was developed to facilitate an accurate diagnostic model, which achieved an AUC of 0.923 (sensitivity = 83.4%, specificity = 90.3%) in the PPMI cohort and validated in several independent cohorts.2 Whether this model can identify individuals with prodromal or preclinical PD has not been tested. CSF biochemical markers were also analyzed using multivariate logistic regression for PD diagnosis, with only relatively low AUCs achieved.13 As far as we know, there have been no attempts to combine body fluid biomarkers with clinical measures in a multivariate diagnosis model for PD diagnosis or prediction. This investigation found that integrating four independent parameters (CSF α‐syn, UPSIT score, age, and sex) yielded an excellent AUC (0.927; Fig. 1A) for PD versus HC in the PPMI cohort. The four‐factor model could also differentiate SWEDD from PD, and likely predicted the conversion of SWEDD to PD. Of 31 SWEDD subjects, four converted to PD over the next 1–2 years, consistent with previous studies showing a conversion rate between 2 and 12%.14, 15, 16 All four were classified as “PD‐like” by the model, while none of those classified as “normal” converted to PD. Although the reasons for the appearance of parkinsonism without apparent dopamine deficiency are not fully understood, several hypotheses, including abnormal scan/faulty interpretation, distinct pathophysiology, or prodromal PD, have been proposed. While these questions remain unanswered, our findings support the idea that some SWEDD subjects may be in a phase of PD that precedes imaging‐detectable degeneration of dopaminergic neurons, or undergoing atypical disease development. Their relationship to typical PD remains to be elucidated, but the model may provide clues in how to identify these cases and guide their treatment.
Because all PD subjects in the PPMI cohort have clinical symptoms, we sought to determine if the model could identify subjects at risk in a cohort of asymptomatic LRRK2 mutation carriers. To apply the PPMI model to the LRRK2 cohort, the α‐syn concentrations acquired in the LRRK2 cohort were normalized to the levels obtained in the PPMI cohort because CSF α‐syn was measured using two different platforms in the two cohorts (see Methods for details). The model performed with very high sensitivity, identifying all subjects with PET abnormality. As a majority of those without PET deficit were also categorized as PD‐like using the PPMI cohort cutoff, the specificity appears to be low (PET+ vs. PET−); however, these subjects are all at an elevated risk of PD, and two of these PET‐ subjects converted to PET+ at follow‐up. Because no more sensitive diagnostic tool for very early PD pathology exists, determining whether or not the subjects identified as PD‐like will progress requires further follow‐up.
Intriguingly, the model is mostly driven by UPSIT in PD versus HC, with minimal contribution from CSF α‐syn in the PPMI cohort. However, while UPSIT alone distinguished PD from HC in this carefully selected cohort, the nonspecific nature of olfactory dysfunction17, 18, 19, 20 and the low prevalence of PD (2% of the population above 60 years old)21 make it insufficient to diagnose PD in general populations. It is possible that CSF α‐syn indicates underlying PD‐type pathophysiology and might increase the specificity of the panel for PD in the general population that includes those with other neurodegenerative diseases. This hypothesis is supported by the finding that α‐syn contributes more to improving the AUC of the model in the LRRK2 cohort than it does when examining patients with symptomatic PD in the PPMI cohort. To further test this idea, it is critical to investigate the four‐factor model in asymptomatic subjects with long‐term follow‐up. One limitation of the current study is that the group sizes of SWEDD subjects and LRRK2 mutation carriers are low. Further, the uneven distribution of symptomatic and asymptomatic LRRK2 subjects by sex, and possible differences in the markers by sex in this cohort may artificially increase the sensitivity and specificity in differentiating PET+ and PET− subjects. Notably, the prediction efficiency of the model is similar with or without including sex as a parameter (data not shown). Our findings need to be further validated in larger cohorts such as the cohort in the ongoing Michael J. Fox Foundation LRRK2 study, when appropriate samples and imaging data become available for replication. However, the clinical application of a model that is capable of diagnosing PD at preclinical stage robustly is unquestionably important.
Author Contributions
Z.Y., T.S., M.S., and J.Z. contributed to study concept and design; Z.Y. and J.A. contributed to data acquisition and analysis; Z.Y., T.S., M.S., and J.Z. contributed to drafting the manuscript and figures.
Conflict of Interests
Nothing to report.
Supporting information
Table S1. Characteristics of LRRK2 mutation carriers.
Acknowledgments
Data used in the preparation of this article were obtained from the Parkinson's Progression Markers Initiative (PPMI) database (www.ppmi-info.org/data). PPMI – a public–private partnership – is funded by the Michael J. Fox Foundation for Parkinson's Research funding partners AbbVie, Avid Radiopharmaceuticals, Biogen Idec, BioLegend, Bristol‐Myers Squibb, Eli Lilly & Co., F. Hoffman‐La Roche, Ltd., GE Healthcare, Genentech, GlaxoSmithKline, Lundbeck, Merck, Meso Scale Discovery, Piramal, Pfizer, Sanofi Genzyme, Servier, Takeda, Teva, and UCB. For up‐to‐date information on the study, visit www.ppmi-info.org. Funding was provided by NIH grants U01 NS082137 and U01 NS091272 to JZ.
Funding Statement
This work was funded by NIH grants U01 NS082137 and U01 NS091272; Zhang and National Key Technology Research and Development Program of China grant 2016YFC1306500.
References
- 1. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30:1591–1601. [DOI] [PubMed] [Google Scholar]
- 2. Nalls MA, McLean CY, Rick J, et al. Diagnosis of Parkinson's disease on the basis of clinical and genetic classification: a population‐based modelling study. Lancet Neurol 2015;14:1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paul SS, Canning CG, Sherrington C, et al. Three simple clinical tests to accurately predict falls in people with Parkinson's disease. Mov Disord 2013;28:655–662. [DOI] [PubMed] [Google Scholar]
- 4. Skogseth RE, Bronnick K, Pereira JB, et al. Associations between cerebrospinal fluid biomarkers and cognition in early untreated Parkinson's disease. J Parkinsons Dis 2015;5:783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schrag A, Siddiqui UF, Anastasiou Z, et al. Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson's disease: a cohort study. Lancet Neurol 2017;16:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi M, Furay AR, Sossi V, et al. DJ‐1 and alphaSYN in LRRK2 CSF do not correlate with striatal dopaminergic function. Neurobiol Aging 2012;33(836):e835–e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aasly JO, Shi M, Sossi V, et al. Cerebrospinal fluid amyloid beta and tau in LRRK2 mutation carriers. Neurology 2012;78:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stewart T, Sossi V, Aasly JO, et al. Phosphorylated inverted question mark‐synuclein in Parkinson inverted question marks disease: correlation depends on disease severity. Acta Neuropathol Commun 2015;3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mollenhauer B, Locascio JJ, Schulz‐Schaeffer W, et al. alpha‐Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 2011;10:230–240. [DOI] [PubMed] [Google Scholar]
- 10. Mollenhauer B, Trautmann E, Taylor P, et al. Total CSF alpha‐synuclein is lower in de novo Parkinson patients than in healthy subjects. Neurosci Lett 2013;532:44–48. [DOI] [PubMed] [Google Scholar]
- 11. Lawton M, Hu MT, Baig F, et al. Equating scores of the University of Pennsylvania Smell Identification Test and Sniffin' Sticks test in patients with Parkinson's disease. Parkinsonism Relat Disord 2016;33:96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kang JH, Mollenhauer B, Coffey CS, et al. CSF biomarkers associated with disease heterogeneity in early Parkinson's disease: the Parkinson's Progression Markers Initiative study. Acta Neuropathol 2016;131:935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kang JH, Irwin DJ, Chen‐Plotkin AS, et al. Association of cerebrospinal fluid beta‐amyloid 1‐42, T‐tau, P‐tau181, and alpha‐synuclein levels with clinical features of drug‐naive patients with early Parkinson disease. JAMA Neurol 2013;70:1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Batla A, Erro R, Stamelou M, et al. Patients with scans without evidence of dopaminergic deficit: a long‐term follow‐up study. Mov Disord 2014;29:1820–1825. [DOI] [PubMed] [Google Scholar]
- 15. Marek K, Seibyl J, Eberly S, et al. Longitudinal follow‐up of SWEDD subjects in the PRECEPT Study. Neurology 2014;82:1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marshall VL, Patterson J, Hadley DM, et al. Two‐year follow‐up in 150 consecutive cases with normal dopamine transporter imaging. Nucl Med Commun 2006;27:933–937. [DOI] [PubMed] [Google Scholar]
- 17. Schofield PW, Ebrahimi H, Jones AL, et al. An olfactory ‘stress test’ may detect preclinical Alzheimer's disease. BMC Neurol 2012;12:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doty RL, Li C, Mannon LJ, Yousem DM. Olfactory dysfunction in multiple sclerosis. N Engl J Med 1997;336:1918–1919. [DOI] [PubMed] [Google Scholar]
- 19. Mahlknecht P, Iranzo A, Hogl B, et al. Olfactory dysfunction predicts early transition to a Lewy body disease in idiopathic RBD. Neurology 2015;84:654–658. [DOI] [PubMed] [Google Scholar]
- 20. Abele M, Riet A, Hummel T, et al. Olfactory dysfunction in cerebellar ataxia and multiple system atrophy. J Neurol 2003;250:1453–1455. [DOI] [PubMed] [Google Scholar]
- 21. Gasser T. Genetics of Parkinson's disease. Curr Opin Neurol 2005;18:363–369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of LRRK2 mutation carriers.