

Abstract
Host cell defense against an invading pathogen depends upon various multifactorial mechanisms, several of which remain undiscovered. Here, we report a novel defense mechanism against mycobacterial infection that utilizes the histone methyltransferase, SUV39H1. Normally, a part of the host chromatin, SUV39H1, was also found to be associated with the mycobacterial bacilli during infection. Its binding to bacilli was accompanied by trimethylation of the mycobacterial histone‐like protein, HupB, which in turn reduced the cell adhesion capability of the bacilli. Importantly, SUV39H1‐mediated methylation of HupB reduced the mycobacterial survival inside the host cell. This was also true in mice infection experiments. In addition, the ability of mycobacteria to form biofilms, a survival strategy of the bacteria dependent upon cell–cell adhesion, was dramatically reduced in the presence of SUV39H1. Thus, this novel defense mechanism against mycobacteria represents a surrogate function of the epigenetic modulator, SUV39H1, and operates by interfering with their cell–cell adhesion ability.
Keywords: histone methylation, host–pathogen interaction, HupB, Mycobacterium tuberculosis, SUV39H1
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Microbiology, Virology & Host Pathogen Interaction
Introduction
The interaction of a pathogen with its host exemplifies a tussle wherein the pathogen, like Mycobacterium tuberculosis, tries to hijack the host cellular machinery and dampen the immune response for its survival, whereas the host tries to eliminate or neutralize the pathogen (Sundaramurthy & Pieters, 2007). Several proteins are released by the pathogen that interact either with the cell surface and cytoplasmic proteins involved in cell signaling (Pathak et al, 2007; Bach et al, 2008) or directly with host chromatin proteins (Pennini et al, 2010; Rolando et al, 2013; Sharma et al, 2015; Yaseen et al, 2015). Most of these proteins ultimately bring about changes in transcription of specific genes that makes the host cell amenable for survival and proliferation of the pathogen (Wang et al, 2005; Pennini et al, 2007). On the other hand, the host utilizes several proteins and miRNAs as part of its immune response to overcome the pathogen (Jayachandran et al, 2009; Holla & Balaji, 2015). A few proteins including parkin and ubiquilin1 directly bind mycobacteria and are part of non‐canonical strategies used by the host to target pathogens. Parkin, a ubiquitin ligase, directly puts ubiquitin chains on mycobacterium and helps in clearance through autophagy (Manzanillo et al, 2013). Similarly, ubiquilin1 directly interacts with mycobacteria leading to clearance of bacteria through xenophagy (Sakowski et al, 2015).
The regulation of the gene expression in the host cell is dependent upon its epigenetic circuitry consisting of DNA methylation, histone modifications, and regulatory RNAs (Schneider & Grosschedl, 2007; Holoch & Moazed, 2015). We have previously shown that mycobacterial species have devised efficient epigenetic mechanisms by which they try to directly control host cell gene expression. Rv2966c and Rv1988 help mycobacteria hijack the epigenetic circuitry by directly interacting with the host chromatin and methylating cytosines in the host DNA and a novel non‐tail arginine in histone H3, respectively (Sharma et al, 2015; Yaseen et al, 2015). Moreover, genome‐wide changes in the DNA methylation of the host have been reported during mycobacterial infection (Sharma et al, 2016). To (i) activate genes involved in immune response, (ii) prevent the mycobacteria from making changes to its epigenetic profile, or (iii) reverse the epigenetic modifications made by the mycobacterial proteins, it is conceivable that the host cell brings about changes in the expression of epigenetic effector proteins like DNA and histone methyltransferases that are involved in establishing epigenetic modifications. This study was initiated with the aim of identifying epigenetic effector proteins that play a role in host response to mycobacterial infection and also characterize the downstream changes in epigenetic modifications that ensue.
Changes in histone modifications are more dynamic than DNA methylation and hence more likely to change during an environmental insult (Stewart et al, 2015). Therefore, as an initial step, we decided to investigate whether any histone‐modifying enzyme had a role to play during mycobacterial infection. While analyzing the expression profile of the various histone methyltransferases and demethylases during mycobacterial infection, SUV39H1 (KMT1A), the histone H3 lysine 9 methyltransferase was found to be overexpressed. However, to our surprise, the increase in expression was correlated with the relocalization of SUV39H1. From being associated with the chromatin in the host nucleus, SUV39H1 was found to be predominantly associated with the mycobacterial bacilli during infection. We also show that the interaction of SUV39H1 with the intracellular mycobacteria was associated with trimethylation of the mycobacterial histone‐like protein, HupB. Importantly, SUV39H1‐mediated methylation of HupB inhibited mycobacterial survival in the host cell during infection. This inhibition was correlated with the role of HupB, in addition to being a transcriptional regulator, in mycobacterial adhesion as our results showed SUV39H1‐mediated inhibition of mycobacterial biofilm formation, a phenomenon dependent upon cell–cell adhesion. Therefore, our study has uncovered a novel mechanism, in which the host defense utilizes an epigenetic effector molecule, SUV39H1, to act against mycobacterial bacilli during infection, by interfering with their cell adhesion ability.
Results
The histone H3K9 methyltransferase SUV39H1 is overexpressed during mycobacterial infection
Expression of several host genes is altered during mycobacterial infection (Ragno et al, 2001). As change in gene expression is directly correlated with alteration in epigenetic modifications including histone modifications, it is possible that the expression levels of histone‐modifying enzymes themselves are altered during mycobacterial infection. In a preliminary experiment, where we were examining the expression profile of several histone methyltransferases and demethylases in PMA‐treated THP1 cells (THP1 macrophages) upon Mycobacterium bovis BCG infection, we found an increase in the expression of SUV39H1 (KMT1A), the histone H3K9 methyltransferase. This preliminary observation was confirmed by Western blotting using SUV39H1 antibody (Fig 1). SUV39H1 expression level, a protein that is normally expressed at very low levels in THP1 macrophages, was markedly increased during M. bovis BCG infection (Fig 1A). The increase in this expression was gradual and specific to infection by mycobacterial species [M. bovis BCG (Fig 1B); M. smegmatis and M. tuberculosis (Fig 1C)]. THP1 macrophages infected with Escherichia coli or Candida glabrata did not show any increase in SUV39H1 expression level (Fig 1B). The increase in expression of SUV39H1 was also true for infection of human peritoneal macrophages with M. bovis BCG (Fig 1D).
Figure 1. SUV39H1 is overexpressed during mycobacterial infection.

-
AUninfected (U) and Mycobacterium bovis BCG‐infected (I) THP1 macrophages were examined for the expression level of SUV39H1 (top panel) by Western blotting at different time points postinfection (indicated above the panels).
-
B, COverexpression of SUV39H1 is specific to mycobacterial species. THP1 macrophages were infected with M. bovis BCG, Escherichia coli, Candida glabrata (B), M. smegmatis (C, left panel), or M. tuberculosis H37Rv (C, right panel), and the expression level of SUV39H1 was examined at the indicated time points.
-
DOverexpression of SUV39H1 in human peritoneal macrophages. Human peritoneal macrophages were examined for the expression level of SUV39H1 (top panel) post‐M. bovis BCG infection by Western blotting (indicated above the panels).
-
ESUV39H2 is not overexpressed during mycobacterial infection. THP1 macrophages infected with M. bovis BCG were examined for SUV39H2 expression level by Western blotting using anti‐SUV39H2 antibody.
KMT1B or SUV39H2 is another human homolog of the Drosophila Su(var)3‐9 gene (O'Carroll et al, 2000). To investigate the correlation of SUV39H1 increased expression with mycobacterial infection, we examined the expression of SUV39H2 upon M. bovis BCG infection of THP1 macrophages. No change in SUV39H2 expression was observed in infected THP1 macrophages (Fig 1E).
Mycobacterial infection leads to relocalization of SUV39H1 from nucleus to cytoplasm
SUV39H1 overexpression upon M. bovis BCG infection was validated by immunostaining M. bovis BCG‐infected THP1 macrophages using SUV39H1 antibody, 48 h postinfection. To our surprise, in addition to being overexpressed in infected cells, SUV39H1 was found to be predominantly localized in the cytoplasm (Fig 2A, upper two panels) as compared to uninfected or heat‐killed M. bovis BCG‐infected THP1 macrophages where it was present in the nucleus (Fig 2A, lower two panels and Fig 2B). We noticed two different localization profiles of SUV39H1 in the cytoplasm of infected THP1 macrophages. As seen in the uppermost panel of Fig 2A, the localization of SUV39H1 in the cytoplasm was found to be speckled in most cells. However, we also observed in some fields that cells not showing the SUV39H1 speckles were stained at the cell surface for SUV39H1 (Fig 2A, second panel from top, Fig 2B, SUV39H1 staining corresponding to the cell surface is marked by red dots in the overlay image).
Figure 2. SUV39H1 is relocalized to the cytoplasm during mycobacterial infection.

-
AUninfected (second panel from below), Mycobacterium bovis BCG‐infected (upper two panels), and heat‐killed M. bovis BCG‐infected (lowermost panel) THP1 macrophages were immunostained for SUV39H1 and visualized by confocal microscopy. Note the speckled loci of SUV39H1 in the cytoplasm and on the cell surface (second panel from top) in infected THP1 macrophages in contrast to uninfected cells where the staining was predominantly in the nucleus.
-
BCell surface localization of SUV39H1. Uninfected and M. bovis BCG‐infected THP1 macrophages were immunostained for SUV39H1 and visualized by confocal microscopy. The position of the cell surface has been overlayed (white dotted line) over the SUV39H1 (green) + DAPI (blue) merge image for the infected and uninfected cells (rightmost image).
-
C, DWestern blot showing the presence of SUV39H1 in the cytoplasm (C) and plasma membrane (D) during mycobacterial infection. (C) Nuclear and cytoplasmic fractions of uninfected (lower panel) and M. bovis BCG‐infected (upper panel) THP1 macrophages at different time points after infection (indicated below the panels) were examined for the presence of SUV39H1 by Western blotting. As a control, the blots were also probed with H3 (nuclear) and tubulin (cytoplasmic) antibodies. (D) Western blot analysis for the presence of SUV39H1 in the total cell lysate and membrane fraction of THP1 macrophages infected with M. bovis BCG. As a control, the blot was also probed for the presence of SUV39H2 (SUV39H1 homolog) and TNFR1 (a known membrane protein).
Its cytoplasmic localization was confirmed by Western blotting proteins corresponding to cytoplasmic and nuclear fractions of M. bovis BCG‐infected THP1 macrophages and probing for the presence of SUV39H1. The purity of the subcellular fractions (Fig 2C) was confirmed by localization of histone H3 (nucleus) and tubulin (cytoplasmic). As compared to uninfected THP1 macrophages where it was detected only in the nuclear fraction (Fig 2C, bottom panel), SUV39H1 was detected in both nuclear and cytoplasmic fractions of M. bovis BCG‐infected THP1 macrophages (Fig 2C, upper panel). While the level of SUV39H1 increased in both fractions, there was a substantial increase in its level in the cytoplasmic fraction with increasing time, postinfection. Interestingly, in the cytoplasmic fraction, a second band of higher molecular weight corresponding to SUV39H1 was observed. This could indicate post‐translational modification of SUV39H1 upon mycobacterial infection, which could be a trigger for relocalization of SUV39H1 from nucleus to cytoplasm.
To confirm the cell surface localization of SUV39H1 in some of the infected THP1 macrophages, total membrane fraction isolated from the uninfected or M. bovis BCG‐infected THP1 macrophages was examined for the presence of SUV39H1 by Western blotting. SUV39H1 was found to be absent in the membrane fraction of the uninfected cells (Fig EV1A). On the other hand, SUV39H1 was present in the membrane fraction of the M. bovis BCG‐infected THP1 macrophages (Fig 2D, uppermost panel). The purity of the membrane fraction was checked by probing for TNFR1 (Figs 2D and EV1A, lower panel), a known plasma membrane protein (Sedger & McDermott, 2014). As a control, the Western blot was also probed for the SUV39H2 protein. As can be seen in Fig 2D (middle panel), SUV39H2 was not found to be enriched in the membrane fraction, indicating that localization to the cell surface was specific to SUV39H1 during mycobacterial infection.
Figure EV1. Relocalisation to the cytoplasm during mycobacterial infection is specific to SUV39H1.

-
AWestern blot analysis for the presence of SUV39H1 in the total cell lysate and membrane fraction of uninfected THP1 macrophages. As a control, the blot was also probed for the presence of TNFR1 (a known membrane protein).
-
B, CSubcellular localization of EZH2 during mycobacterial infection. Uninfected and Mycobacterium bovis BCG‐infected THP1 macrophages were immunostained for EZH2 (B) and SUV39H2 (C) and visualized by confocal microscopy. Scale bar: 10 μm. Nuclei were counterstained with DAPI.
Source data are available online for this figure.
To confirm that the change in localization of a histone methyltransferase from nucleus to cytoplasm was specific to SUV39H1, we also performed immunostaining for SUV39H2 as well as for another histone methyltransferase, EZH2, in M. bovis BCG‐infected THP1 macrophages. As shown in Fig EV1B and C, neither was an increase in SUV39H2 and EZH2 expression observed nor were these proteins relocalized to another subcellular region in the infected THP1 macrophages.
SUV39H1 associates with mycobacterial bacilli in the phagolysosomes during infection
As can be seen in the uppermost panel of Fig 2A, the SUV39H1 speckles in the cytoplasm appeared rod‐shaped and seemed to resemble bacilli. Since internalized mycobacterial bacilli are normally present within the phagolysosomes, we decided to investigate whether SUV39H1 was localized in the phagolysosomes in these infected macrophages. To test this, phagosomal fraction was isolated from uninfected and M. bovis BCG‐infected THP1 macrophages by sucrose gradient (see Materials and Methods) and the presence of SUV39H1 was examined by Western blotting. As can be seen in Fig 3A, SUV39H1 was indeed present in the phagosome fraction of the infected macrophages. The phagosomal fraction was confirmed by examining the presence of the known phagosomal protein LAMP1 (Fig 3A, middle panel). To rule out contamination of cytoplasmic proteins, the same blot was also probed with GAPDH antibodies (Fig 3A, lower panel).
Figure 3. SUV39H1 colocalizes with the mycobacterial bacilli in the phagolysosomes.

-
ASUV39H1 is present in the phagolysosomal fraction of infected THP1 macrophages. The phagolysosomal fraction isolated from the cell lysates of uninfected and Mycobacterium bovis BCG‐infected THP1 macrophages was probed for SUV39H1. As a control, the blot was also probed for LAMP1 (phagosomal marker) and GAPDH (cytosolic protein).
-
BSUV39H1 colocalizes with the intracellular mycobacterial bacilli. GFP::M. bovis BCG (GFP::BCG, upper panel)‐ or RFP::M. bovis BCG (RFP::BCG, lower panel)‐infected THP1 macrophages were immunostained with SUV39H1 antibody and examined by confocal microscopy. Nuclei were counterstained with DAPI. Scale bar: 2 μm (upper panels) and 5 μm (lower panels).
-
C, DM. bovis BCG bacilli were isolated from the phagolysosomal fraction of infected THP1 macrophages and examined for SUV39H1 by Western blotting (C) or confocal microscopy (D). To rule out contamination by cytoplasmic and phagosomal host proteins, the Western blot (C) was also probed for LAMP1 (phagosomal marker) and GAPDH (cytoplasmic marker). The blot was also probed for GroEL1, a mycobacterial protein. (D) To show colocalization of SUV39H1 and the mycobacterial bacilli, a portion of the confocal image in the top panel (within the upper right hand quandrant). Scale bar: 5 μm (upper panel) and 1 μm (lower zoomed panel).
Source data are available online for this figure.
Since the staining for SUV39H1 in phagolysosomes resembled the shape of bacilli, we next asked whether SUV39H1 was directly associated with the M. bovis BCG bacilli rather than being in their vicinity in the phagolysosomes. To test this, THP1 macrophages infected with GFP::M. bovis BCG (expressing GFP) or RFP::M. bovis BCG (expressing RFP) were immunostained for SUV39H1, 48 h postinfection. SUV39H1 signal was found to colocalize with the GFP/RFP signal (Fig 3B). To confirm that this colocalization indicated the association of SUV39H1 with M. bovis BCG bacilli, phagosomal fraction isolated from M. bovis BCG‐infected THP1 macrophages was treated with 0.5% NP‐40 to remove the phagolysosomal membrane and the other soluble host proteins from the phagolysosomal fraction (Healy & O'Connor, 2009). The bacilli residing in this host cell compartment were pelleted and probed for SUV39H1. SUV39H1 was found to be present in the isolated mycobacterial fraction (Fig 3C). As a control, the same blot was probed with GAPDH antibody (to confirm the absence of cytoplasmic contamination), with LAMP1 antibody (to confirm the phagolysosomal fraction), and GROEL1 (to confirm mycobacterial fraction; Fig 3C). When these isolated bacilli were immunostained in vitro for SUV39H1, the SUV39H1 antibody stained the M. bovis BCG bacilli confirming the association of SUV39H1 with mycobacterial bacilli isolated from the phagolysosomal fraction (Fig 3D).
THP1 macrophages without the speckled SUV39H1 staining in the second panel from the top of Fig 2A would indicate that these cells were devoid of internalized M. bovis BCG bacilli. Since SUV39H1 staining in these cells was on the cell surface, this could indicate that THP1 macrophages had probably localized SUV39H1 on its cell surface for interaction with the bacilli and the staining of bacilli in the phagosomes was the end result of this interaction followed by phagocytosis.
SUV39H1 can bind to mycobacteria in vitro
To examine whether the association of SUV39H1 with M. bovis BCG during infection indicated its binding to M. bovis BCG, we performed bacterial binding assay (Materials and Methods; Sakowski et al, 2015). In this assay, lysate of HEK293 cells transfected with SFB‐tagged SUV39H1 was incubated with M. bovis BCG in vitro. After five washes with PBS containing 0.5% Triton X‐100, to remove non‐specific binding proteins, proteins bound to M. bovis BCG were analyzed by Western analysis. The FLAG antibody detected SFB‐SUV39H1 in the M. bovis BCG‐bound fraction (Fig 4A). The blot was also probed with anti‐MTB antibody [detects primarily lipoarabinomannan (LAM) on mycobacterial cell surface] to confirm the presence of mycobacteria. As a control, HEK293 cell lysate transfected with SFB vector alone was also incubated with M. bovis BCG cells (Fig 4A). The interaction of SUV39H1 with M. bovis BCG was quite strong as even an increase in concentration of Triton X‐100 in wash buffer to 1.5% did not disrupt the interaction (Fig 4B). Direct interaction of SUV39H1 with M. bovis BCG was also confirmed by (i) in vitro incubation of recombinant MBP‐SUV39H1 (expressed in E. coli) with M. bovis BCG followed by Western analysis (Fig 4C), or (ii) SFB‐SUV39H1 (expressed in HEK293 cells) with GFP::M. bovis BCG followed by immunostaining (Fig 4D).
Figure 4. SUV39H1 binds to mycobacterial bacilli in vitro .

- Bacterial binding assay. SFB‐SUV39H1 pulled down from HEK293 cells was incubated with Mycobacterium bovis BCG bacilli. The bacilli were pelleted and non‐specific proteins were removed by giving washes with buffer containing 0.5% Triton X‐100. As a control, bacilli were also incubated with SFB‐tag alone pulled down from HEK293 cells.
- SUV39H1 associates very strongly with mycobacterial cells. The bacterial binding assay (as in A) was repeated but with wash buffer containing increasing concentration of Triton X‐100 (indicated below the panel).
- Bacterial binding assay performed with recombinant MBP‐SUV39H1 protein from Escherichia coli. FLAG antibody (A and B) and MBP antibody (C) detected SUV39H1, while an antibody that detects LAM present on the surface of mycobacterial cells (MTB) detected M. bovis BCG.
- SFB‐SUV39H1 was incubated with GFP::M. bovis BCG, immunostained for SUV39H1, and examined by confocal microscopy. Nuclei were counterstained with DAPI. Scale bar: 5 μm.
Source data are available online for this figure.
SUV39H1 methylates the mycobacterial protein, HupB
SUV39H1 is a histone methyltransferase that specifically methylates histone H3 lysine 9 in the host nucleus. We were therefore intrigued with its colocalization and binding with M. bovis BCG in the phagosomes. A preliminary mass spectrometry experiment, wherein host cytoplasmic proteins interacting with SUV39H1 in cytoplasm were analyzed, we identified alpha‐laminin 2 as one of the interacting partners (Fig EV2A and B). In the literature, it is known that mycobacterium binds to host cells through alpha‐laminin (Lefrançois et al, 2011). In this interaction, mycobacterial protein HupB binds to alpha‐laminin (HupB or Rv2986c is also known as laminin binding protein, LBP). Interestingly, HupB is histone‐like protein from mycobacteria and has been shown to be present both in the mycobacterial cytosol and on the cell wall (Yeruva et al, 2006). Since SUV39H1 is a protein (histone) methyltransferase, we decided to examine whether SUV39H1 could methylate HupB present on the cell wall surface of the mycobacteria during infection.
Figure EV2. SUV39H1 trimethylates M. tuberculosis protein HupB.

-
A, BMass spectrometric analysis of SUV39H1 interacting proteins. (A) MS spectra of the identified protein. (B) Spectrum data analysis by MASCOT identified nine unique peptides of laminin alpha 2 that was identified as an interacting partner of SUV39H1 from the cytoplasmic fraction of Mycobacterium bovis BCG‐infected THP1 cells.
-
CHupB is the only protein trimethylated by SUV39H1. Recombinant MBP‐SUV39H1 and M. bovis BCG lysates were incubated in the presence of SAM. As a control, the lysate was incubated with BSA. Shown here is the full gel pictures of the Western blot shown in Fig 6A probed with trimethyl lysine and HupB‐specific antibodies The trimethyl lysine antibody (left panel) picked up only one single band, which corresponds to exactly the size of HupB (right panel).
-
DRecombinant SUV39H1 cannot trimethylate recombinant mycobacterial protein HupB expressed in Escherichia coli. Recombinant MBP‐SUV39H1 or BSA (control) was incubated with recombinant mycobacterial protein HupB that was expressed in E. coli followed by Western blotting with mono/dimethyl lysine (middle panel)‐ or trimethyl lysine (upper panel)‐specific antibodies. The blot was also reprobed with HupB antibody.
-
EPurification of recombinant MBP, MBP‐SUV39H1, 6XHis‐HupB. Recombinant M. tuberculosis HupB (6XHis‐Rv2986c) was overexpressed and purified from M. smegmatis. MBP and MBP‐SUV39H1 were expressed and purified from E. coli.
Source data are available online for this figure.
Methyltransferase assay was set up with recombinant MBP‐SUV39H1 or BSA (as control) and M. bovis BCG lysate in the presence of SAM as a methyl group donor, and the reaction mix was analyzed by Western blotting using either mono/dimethyl lysine (middle panel)‐ or trimethyl lysine (upper panel)‐specific antibodies. Mono/dimethyl lysine antibody detected HupB in both BSA and MBP‐SUV39H1 lanes (Fig 5A), indicating that HupB from M. bovis was already mono‐ or dimethylated by an endogenous mycobacterial methyltransferase. When the same blot was probed with trimethyl lysine antibody (Fig 5A, upper panel), signal was significantly more in M. bovis BCG lysate incubated with SUV39H1 as compared to BSA (additional data on replicate experiments for this figure is provided as source data). Only one band, which corresponded to HupB, was detected in the whole blot when the trimethyl antibody was used as a probe (Fig EV2C). The level of HupB in the lysate was same for both control and SUV39H1 lanes antibody (Fig 5A, lower panel). This suggested that SUV39H1 trimethylates the mycobacterial protein, HupB. Interestingly, when the same experiment was performed with recombinant MBP‐SUV39H1 and 6XHis‐HupB (M. tuberculosis protein) purified from E. coli, signal was detected with mono/dimethyl lysine antibody, but not with trimethyl lysine antibody (Fig EV2D). This would indicate that the HupB lysine residues acquiring mono/dimethylation modifications in E. coli were different from the ones in mycobacterial species. Therefore, to confirm SUV39H1‐mediated trimethylation of HupB by in vitro methylation assay, recombinant M. tuberculosis HupB (6XHis‐Rv2986c) was overexpressed and purified from M. smegmatis (Fig EV2E) and incubated with E. coli purified recombinant MBP‐SUV39H1 (Fig EV2E) and SAM. As can be seen in Fig 5B, trimethyl lysine antibody detected HupB that was incubated with MBP‐SUV39H1, but not with MBP‐LacZ.
Figure 5. SUV39H1 trimethylates HupB at lysine 138.

-
A, BSUV39H1 trimethylates HupB. Recombinant MBP‐SUV39H1 or BSA (control) and Mycobacterium bovis BCG lysate were incubated in the presence of SAM followed by Western blotting with mono/dimethyl lysine (middle panel)‐ or trimethyl lysine (upper panel)‐specific antibodies. The blot was also reprobed with HupB antibody. The ratio of the normalized signal for trimethyl lysine antibody and HupB antibody is given below the panels. (B) SUV39H1 trimethylates HupB in vitro. Recombinant MBP‐SUV39H1 purified from Escherichia coli was incubated with recombinant M. tuberculosis HupB (6XHis‐HupB) expressed and purified from M. smegmatis (lowermost panel shows Coomassie Blue‐stained gel for the proteins present in the reaction mix) in the presence of cold SAM followed by Western blotting and probing with trimethyl lysine antibody and control HupB antibody.
-
CSUV39H1 trimethylates HupB on the mycobacterial cell surface. Recombinant M. bovis BCG was incubated with MBP‐SUV39H1 in the presence of SAM followed by fractionation of the M. bovis BCG into cell wall and cytosolic fractions, Western‐blotted, and probed with trimethyl lysine (upper panel)‐specific antibodies. As a control, the blot was also probed with GroEL1 (for cytosolic fraction) and HupB antibodies.
-
DIn vivo interaction of SUV39H1 with HupB during infection. THP1 macrophages infected with wild‐type M. tuberculosis H37Rv or MtbΔhupB strains were examined for the localization of SUV39H1 protein. The outline of some of the internalized mycobacterial bacilli as observed in DIC is marked in white. The cells were counterstained with DAPI. Scale bar: 5 μm.
-
EHupB contains the H3K9 motif. Comparison of H3 and M. tuberculosis H37Rv HupB protein sequence showing the presence of H3K9 motif in HupB centered around K138. The rectangles indicate regions of similarity.
-
F, GSUV39H1 trimethylates HupB at K138. (F) Recombinant MBP‐SUV39H1 and lysate from either MtbΔhupB strain (ΔHupB) or MtbΔhupB transformed with 6XHis‐HupB (HupB‐WT) or 6XHis‐hupB K138A ((HupB‐K138A) constructs were incubated in the presence of SAM followed by Western blotting and probing with trimethyl lysine (upper panel)‐specific antibodies. As a control, the blot was also reprobed with HupB antibody and an antibody that detects LAM present on the surface of mycobacterial cells (MTB). (G) Recombinant 6XHis‐HupB (Hup‐WT) or 6XHis‐hupB K138A (HupB‐K138A) proteins expressed and purified from M. smegmatis (lowermost panel) were incubated with recombinant MBP‐SUV39H1 (second panel from bottom) in the presence of SAM, Western‐blotted, and probed with trimethyl lysine and HupB (control) antibodies.
Source data are available online for this figure.
HupB is present both on the cell surface and in the cytosol of a mycobacterial cell (Yeruva et al, 2006). To examine whether the trimethylation by SUV39H1 was on the HupB protein present on the cell surface or in the cytosol of the mycobacteria, the experiment in Fig 5A was repeated on M. bovis BCG followed by fractionation into cell wall and cytosolic fractions. As shown before (Yeruva et al, 2006), HupB was found to be present in both the fractions. However, the trimethylated form of HupB was present only in the cell wall fraction. The fractionation was confirmed by probing the Western blot for GroEL1, a protein known to be present only in the cytosol (Fig 5C).
SUV39H1 was found associated with mycobacterial bacilli in the phagosomes (Fig 3). To examine whether this association was through interaction of SUV39H1 with HupB, THP1 macrophages were infected with wild‐type and HupB mutant (in which hupB gene had been deleted, MtbΔhupB) M. tuberculosis H37Rv strains. The infected cells were immunostained for SUV39H1. As can be seen in Fig 5D, SUV39H1 antibody stained mycobacterial bacilli (marked by white outline) in the wild‐type M. tuberculosis H37Rv strain, but not in MtbΔhupB strain.
SUV39H1 trimethylates HupB at lysine 138
SUV39H1 methylates lysine at position 9 in the histone H3 (Rathert et al, 2008). This would indicate that the amino acid sequence around H3K9 could be the target sequence for SUV39H1 action. Using the amino acid sequence motif (TK–ARK—KAP) that encompasses H3K9, we tested whether similar motif was present in HupB or any other mycobacterial protein using the pattern search program on the Tuberculist server. While the central part of the motif “ARK” was present in several proteins, the TK–ARK—KAP motif was present only in the M. tuberculosis HupB protein (amino acids 132–144, Figs 5E and EV3A). In addition, a recent study has identified the specificity profile of SUV39H1 and characterized non‐histone protein targets of SUV39H1 (Kudithipudi et al, 2017). The amino acid profile of M. tuberculosis HupB around K138 was also found to be similar to the SUV39H1 profile with an invariant arginine at −1 position and a lysine at −4 position (from K138; Kudithipudi et al, 2017).
Figure EV3. SUV39H1 trimethylates HupB at K138.
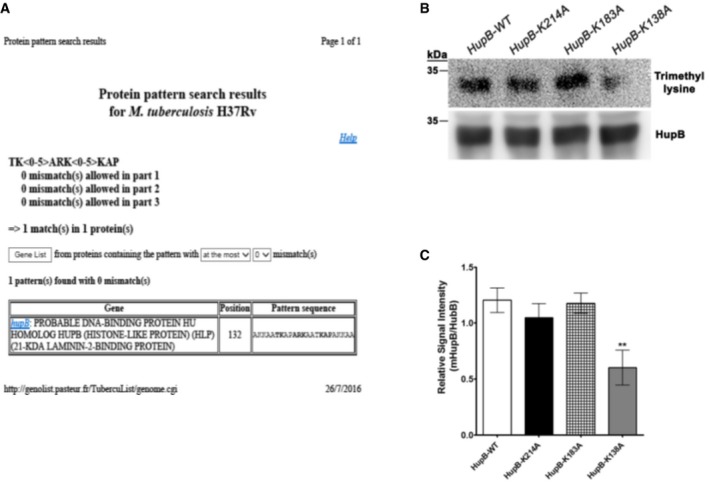
-
AThe sequence motif encompassing H3K9 in histone H3 is present in HupB. Pattern search for a motif containing the following amino acids, TK<0–5>ARK<0–5>KAP, was carried out on the mycobacterial protein database using the software available on the Tuberculist website (genolist.pasteur.fr). In this search, only HupB matched with the above‐mentioned pattern.
-
B, CSUV39H1 trimethylates HupB at K138. Recombinant MBP‐SUV39H1 and lysate from MtbΔhupB strain transfected with HupB, 6XHis‐hupBK138A, 6XHis‐hupBK183A, and 6XHis‐hupBK214A constructs were incubated in the presence of SAM followed by Western blotting with trimethyl lysine (upper panel, B)‐specific antibodies. The blot was also reprobed with HupB antibody. The signal was quantified for each band after densitometric scanning from three independent experiments and represented as ratio of methylated HupB to HupB in (C). Error bars represent standard deviation (SD). Asterisks indicate significant difference by Student's t‐test: **P < 0.005.
Source data are available online for this figure.
To test whether SUV39H1 methylates HupB protein at the lysine present within the central ARK motif (K138), lysine at 138 position in recombinant 6XHis‐HupB was mutated to alanine (6XHis‐HupBK138A) by site‐directed mutagenesis. The wild‐type and mutant HupB constructs (6XHis‐HupB, 6XHis‐HupBK138A) were transformed into the MtbΔhupB strain. Thus, the expression of hupB in this strain transformed by the above constructs would correspond to the appropriate wild‐type or mutant 6XHis‐HupB fusion protein. The mycobacterial cell lysate from this strain with or without the various constructs was incubated with SUV39H1 and SAM and examined by Western blotting for HupB trimethylation. A significant decrease in the level of HupB methylation was observed for 6XHis‐HupBK138A mutant as compared to 6XHis‐HupB (Fig 5F). No signal was detected in MtbΔhupB protein lysate lane with trimethyl lysine antibody.
Lysines present in the “RK” submotif at two other positions (182–183 and 214) were also mutated to create 6XHis‐HupBK183A and 6XHis‐HupBK214A mutants of HupB and examined for methylation by SUV39H1. Cell lysate from MtbΔhupB strain transformed with the various constructs was incubated with SUV39H1 and SAM and examined by Western blotting for HupB trimethylation. The level of HupB trimethylation in 6XHis‐HupBK183A mutant was similar to the wild type. Slight reduction in HupB methylation was observed in 6XHis‐HupBK214A mutant but was not statistically significant in contrast to the significant decrease in the level of HupB methylation observed for 6XHis‐HupBK138A mutant (Fig EV3B and C). This indicated that SUV39H1 was methylating HupB at lysine present at 138 position and within the motif similar to the one present around H3K9 in histone H3 and the defined SUV39H1 specificity profile (Kudithipudi et al, 2017).
The methylation of HupB protein at K138 was also confirmed by in vitro methylation assay. Recombinant 6XHis‐HupB and 6XHis‐HupBK138A proteins were overexpressed and purified from M. smegmatis and incubated with recombinant MBP‐SUV39H1 and SAM. The trimethylation of 6XHis‐HupBK138A was found to be significantly less than 6XHis‐HupB (Fig 5G), indicating that SUV39H1 methylates specifically at K138 (additional data on replicate experiments for this figure is provided as source data). The residual HupB trimethylation observed in 6XHis‐HupBK138A could be due to another lysine in HupB being a minor secondary target of SUV39H1.
SUV39H1 downregulation makes THP1 macrophages more susceptible to mycobacterial infection
To investigate whether the association of SUV39H1 with mycobacterial bacilli reflected its role during mycobacterial infection, stable SUV39H1‐knockdown THP1 cell lines were derived using SUV39H1 shRNA (see Materials and Methods). A THP1 cell line transfected with scrambled shRNA was used as a control. The repression of SUV39H1 expression was confirmed by Western blot analysis. As THP1 cell lines with more than 90% decrease in SUV39H1 (SUV39H1‐KD3) expression showed very limited growth, we used the THP1‐SUV39H1‐KD1 cell line with ~60% decrease in SUV39H1 expression for further experimentation (Fig 6A). THP1 macrophages stably transfected with scrambled (control) or SUV39H1 (SUV39H1‐KD) shRNA were infected with GFP::M. bovis BCG and the number of intracellular bacteria inside the macrophages was determined either visually by confocal microscope or by sorting GFP‐positive cells by FACS (Ranjbar et al, 2015). The number of intracellular mycobacteria 24 and 48 h postinfection was found to be significantly higher in SUV39H1‐KD as compared to control in both the assays (Fig 6B and C, confocal; Fig EV4A, FACS).
Figure 6. Downregulation of SUV39H1 expression makes THP1 macrophages more susceptible to mycobacterial infection.

-
AshRNA‐mediated knockdown of SUV39H1 expression. Western blot showing SUV39H1 expression in THP1 cells stably transfected with SUV39H1 (SUV39H1‐KD1/2/3) or scrambled shRNA. Tubulin was used as a control.
-
B–EIncreased number of intracellular bacilli are present in THP1 macrophages with decreased SUV39H1 expression levels. THP1 macrophages stably transfected with scrambled or SUV39H1 (SUV39H1‐KD) shRNA were infected with GFP::M. bovis BCG and examined by confocal microscopy (B) at different time points, and the number of bacilli present was quantified (i) visually in least eight randomly chosen fields from three independent experiments (C), and (ii) by Alamar blue assay (D). (E) The same experiment was repeated with M. tuberculosis H37Rv and the number of intracellular bacilli was quantified by CFU assay. Values in (C–E) have been obtained from at least three biological replicates.
Figure EV4. Downregulation of SUV39H1 expression makes THP1 macrophages more susceptible to mycobacterial infection.

-
ATHP1 macrophages stably transfected with scrambled (scrambled) or SUV39H1 (SUV39H1) shRNA were infected with GFP::M. bovis BCG and subjected to FACS (for GFP‐positive cells). At 24‐ and 48‐h time points, cell counts were normalized with 0‐h cell count.
-
B, CSUV39H1 prevents mycobacterial infection through HupB methylation. SUV39H1‐KD1 or control THP1 macrophages were infected with M. smegmatis expressing either wild‐type (6XHis‐HupB) or mutant forms (6XHis‐HupBK138A or 6XHis‐HupBK183A) of M. tuberculosis hupB gene. The number of intracellular bacilli was quantified by CFU assay at the indicated time points postinfection.
-
DSurvival of M. tuberculosis in the presence of SUV39H1 inhibitor. THP1 macrophages infected with M. tuberculosis H37Rv were cultured in the presence of DMSO (control group) or chaetocin (SUV39H1 inhibitor). The number of intracellular bacilli was quantified by CFU assay at the indicated time points postinfection.
This was also confirmed by examining the number of viable intracellular bacteria as quantified by Alamar assay (Ganji et al, 2016) counting. Significantly more numbers of GFP::M. bovis BCG bacilli were found in infected SUV39H1‐KD as compared to control macrophages (Fig 6D). The observed increase in the number of surviving bacilli was small although significant probably as the expression of SUV39H1 was only partially reduced in the THP1‐SUV39H1‐KD1 macrophages.
The same was also true for M. tuberculosis infection. The number of viable bacteria was calculated by CFU quantification for bacteria isolated from SUV39H1‐KD and control macrophages infected with H37Rv strain of M. tuberculosis. As can be seen in Fig 6E, significantly more number of bacteria were present in SUV39H1‐KD THP1 macrophages as compared to control cells. All these observations indicate that THP1 macrophages were more susceptible to mycobacterial infections in the absence of SUV39H1.
SUV39H1 prevents mycobacterial infection through trimethylation of HupB
HupB, apart from being a transcriptional regulator, is also present on the mycobacterial cell surface and involved in cell–cell adhesion (Shimoji et al, 1999). As mentioned above, HupB is also a part of the mechanism by which mycobacteria bind to host cells during infection (Lefrançois et al, 2011). Since SUV39H1 gets localized to the host cell surface during infection, it was possible that the role of SUV39H1 in prevention of mycobacterial infection was dependent upon trimethylation of HupB by SUV39H1. To examine whether the role of SUV39H1 during mycobacterial infection was indeed through HupB methylation, we performed infection of SUV39H1‐KD or control (stably transfected with scrambled shRNA) THP1 macrophages with M. smegmatis transformed with either wild type (6XHis‐HupB::M. smegmatis) or mutant forms (6XHis‐HupBK138A::M. smegmatis, 6XHis‐HupBK183A::M. smegmatis) of M. tuberculosis hupB gene. The survival of the bacilli was checked 24 h after infection by CFU counting. As shown in Fig EV4B and C, increased survival of bacteria was observed in case of SUV39H1‐KD infected with 6XHis‐HupB::M. smegmatis 6XHis‐HupBK183A::M. smegmatis. However, in case of M. smegmatis transformed with 6XHis‐HupBK138A (mutant that cannot be methylated by SUV39H1), negligible increase in CFU was observed for SUV39H1‐KD as compared to control knockdown cells.
The same was also true for M. tuberculosis H37Rv infection. To investigate the effect of HupB methylation on infection, a M. tuberculosis H37Rv strain in which the hupB gene was deleted (MtbΔhupB) was used. SUV39H1‐KD or control (stably transfected with scrambled shRNA) THP1 macrophages were infected with MtbΔhupB strain complemented with either wild‐type (6XHis‐HupB) or mutant HupB (6XHis‐HupBK138A and 6XHis‐HupBK183A) constructs. Since MtbΔhupB strain transformed with 6XHis‐HupBK214A showed increased growth rate, this mutant strain was excluded from any further assay to draw unbiased inference from our results. The survival of MtbΔhupB strain inside macrophages during infection is known to be significantly reduced (Pandey et al, 2014). We also observed that the number of MtbΔhupB bacilli gaining entry (446,700 ± 49,100 vs. 1,927,000 ± 95,280) and survival (13,400 ± 1,217 vs. 51,570 ± 1,484) within the macrophages was significantly less than the HupB‐complemented MtbΔhupB strain (MtbΔhupB + 6XHis‐HupB, P < 0.001, Fig 7A and B). But no difference was observed in the survivability of MtbΔhupB strain in wild‐type or SUV39H1‐KD THP1 macrophages during infection. Importantly, while MtbΔhupB + 6XHis‐HupB and MtbΔhupB + 6XHis‐HupBK183A strains showed a significant increase in bacterial survivability in SUV39H1‐KD as compared to control, no survival advantage was observed for the MtbΔhupB + 6XHis‐HupBK138A strain in the SUV39H1‐KD THP1 macrophages.
Figure 7. SUV39H1 prevents mycobacterial infection through HupB methylation.

-
A, BSUV39H1‐KD1 or control THP1 macrophages were infected with HupB‐deleted Mycobacterium tuberculosis H37Rv strain (MtbΔhupB) complemented with either wild‐type (6XHis‐HupB) or mutant (6XHis‐HupBK138A or 6XHis‐HupBK183A) forms of M. tuberculosis hupB gene.
-
C–FMouse (C and D) or human (E and F) peritoneal macrophages infected with M. smegmatis expressing either wild‐type (6XHis‐HupB) or mutant forms (6XHis‐HupBK138A or 6XHis‐HupBK183A) were cultured in the presence of DMSO (control group) or chaetocin (SUV39H1 inhibitor). The number of intracellular bacilli was quantified by CFU assay at the indicated time points postinfection. As CFU counts of HupB and HupBK183A were similar in mouse peritoneal macrophages, HupBK183A mutant was not examined in human peritoneal macrophages.
-
GSurvival advantage for 6XHis‐HupBK183A:: M. smegmatis in infected mice. CFU counting was performed 7 days postinfection for the indicated organs in BALB/c mice infected intravenously with M. smegmatis expressing either wild type (6XHis‐HupB) or mutant forms (6XHis‐HupBK138A or 6XHis‐HupBK183A).
-
HBody weights of infected mice were measured on 0 and 7th day postinfection. n = 5 for all the groups. % body weight for each animal was calculated with respect to their weight on the day of infection.
The role of SUV39H1‐mediated methylation of HupB during mycobacterial infection was also confirmed by the use of a SUV39H1 inhibitor, chaetocin (Greiner et al, 2005). THP1 macrophages were infected with M. tuberculosis H37Rv in the presence and absence of the inhibitor and the survivability of the internalized bacilli 48 h after infection was quantified by CFU counting. The survival of the M. tuberculosis H37Rv was significantly better in the presence of chaetocin (Fig EV4D). We also examined the effect of HupB on the survivability of mycobacterial cells during infection in mouse and human peritoneal macrophages. Mouse peritoneal macrophages were infected with either wild type (HupB::M. smegmatis) or mutant forms (6XHis‐HupBK138A::M. smegmatis, 6XHis‐HupBK183A::M. smegmatis) of M. tuberculosis hupB gene in the presence or absence of chaetocin. Forty‐eight hours after infection, the survival of HupB::M. smegmatis and 6XHis‐HupBK183A::M. smegmatis in mouse peritoneal macrophages was significantly better in the presence of the inhibitor (Fig 7C and D). On the other hand, no difference was observed in the survival of 6XHis‐HupBK138A::M. smegmatis strain in the presence or absence of chaetocin. This was also true for infection of human peritoneal macrophages with wild type (HupB::M. smegmatis) or mutant form (6XHis‐HupBK138A::M. smegmatis) of M. tuberculosis hupB gene in the presence or absence of chaetocin. Twenty‐four hours after infection, the survival of HupB::M. smegmatis was significantly better in the presence of the inhibitor but the survival of 6XHis‐HupBK138A::M. smegmatis strain was same in the presence and absence of chaetocin (Fig 7E and F). Interestingly, the survival of the 6XHis‐HupBK138A::M. smegmatis strain in both mouse and human peritoneal macrophages was significantly better than that of the HupB::M. smegmatis strain even in the absence of SUV39H1 inhibitor, chaetocin (CFU/ml: 1,490 ± 215.0 vs. 810.0 ± 110.2, P < 0.05 and 3,197,000 ± 254,400 vs. 1,583,000 ± 110,500, P < 0.01 in mouse and human peritoneal macrophages, respectively).
To further confirm the role of SUV39H1‐mediated methylation of HupB on mycobacterial infection, BALB/c mice were injected intravenously with the wild type (6XHis‐HupB::M. smegmatis) or mutant form (6XHis‐HupBK138A::M. smegmatis) of M. tuberculosis hupB gene. As a control, infection was also carried out with M. smegmatis transformed with the vector alone (pVV16::M. smegmatis). The bacterial load was analyzed in liver, lung, and spleen of the infected mice 7 days after infection by CFU counting. The bacterial load was found to be significantly more in all tissues for 6XHis‐HupBK138A::M. smegmatis as compared to HupB::M. smegmatis and pVV16::M. smegmatis (Fig 7G). Infection with M. smegmatis affected the health of the mice as they showed weight loss after infection. While the weight loss during infection was observed for all infected mice as compared to uninfected, the weight loss was significantly more in 6XHis‐HupBK138A::M. smegmatis as compared to infection with 6XHis‐HupB::M. smegmatis and pVV16::M. smegmatis (Fig 7H).
The above experiments confirmed that the inhibitory effect of SUV39H1 on mycobacterial infection was mediated through SUV39H1 methylation of HupB at lysine 138.
SUV39H1 inhibits mycobacterial biofilm formation
The mycobacterial protein, HupB, has similarity with histone proteins, and it plays an important role in regulation of transcription in mycobacteria (Pandey et al, 2014). As discussed above, HupB has also been found to be tethered to the mycobacterial cell wall (Yeruva et al, 2006). While the exact role of the HupB protein fraction that is present on the cell wall has not been elucidated, a few reports have suggested its role in adhesion (in M. leprae, Shimoji et al, 1999) and biofilm formation (in uropathogenic E. coli, Devaraj et al, 2015). Biofilm formation, an important part of the bacterial infection process, is dependent upon cell–cell adhesion (Joo & Otto, 2012). Therefore, to examine whether SUV39H1 could affect mycobacterial adhesion properties, we decided to investigate the effect of SUV39H1 on biofilm formation and mycobacterial cell adhesion. To test the effect of SUV39H1 on mycobacterial cell adhesion, we performed in vitro biofilm assay with M. bovis BCG cultured in 7H9 medium in the presence of recombinant MBP‐SUV39H1 or BSA (as control) and SAM for about 25 days. While M. bovis BCG was able to form biofilm in the presence of BSA, biofilm formation was severely inhibited in the presence of SUV39H1 (Fig 8A). We quantified the biofilm formation by crystal violet blue assay, which confirmed a significant defect in biofilm formation in the presence of SUV39H1 (Fig 8B).
Figure 8. SUV39H1 inhibits mycobacterial biofilm formation through methylation of HupB.

-
A–FSUV39H1 inhibits Mycobacterium bovis BCG (A–D) and M. tuberculosis H37Rv (E and F) from forming biofilms. MBP‐SUV39H1 or BSA (control) and SAM were added to M. bovis BCG (A and B) or M. tuberculosis H37Rv, MtbΔhupB (HupB deleted) or MtbΔhupB + pMS101 (hupB complemented) strains (E and F) culture medium and the bacilli were allowed to form biofilm over a period of 25 days. The biofilm formation was either imaged (A and E) or quantified by crystal violet staining performed for three independent experiments (B, F). (C) Western blot showing the presence of trimethylated HupB in the lysate of M. bovis BCG bacilli cultured in the presence of MBP‐SUV39H1, but not with BSA. (D) Quantification of HupB trimethylation after the biofilm assay as shown in (C) and performed for three independent experiments. (F) Quantification of HupB trimethylation after the biofilm assay as shown in (E) and performed for three independent experiments.
To examine whether the inhibition of biofilm formation was related to methylation of HupB, the mycobacterial bacilli in the above‐mentioned in vitro assay (Fig 8A) were lysed and subjected to Western blot analysis using trimethyl lysine antibody. HupB was significantly trimethylated in M. bovis BCG bacilli incubated with SUV39H1 as compared to BSA (Fig 8C, upper panel and 8D, see also additional data on replicate experiments for this figure that is provided as source data).
This was also true for M. tuberculosis. The same in vitro experiment was repeated with M. tuberculosis H37Rv. The effect of SUV39H1 on HupB methylation and biofilm formation was examined in the wild‐type M. tuberculosis H37Rv and MtbΔhupB (M. tuberculosis H37Rv strain in which hupB gene was deleted) strains. HupB‐complemented MtbΔhupB strains were also used in the experiment. SUV39H1 was able to inhibit biofilm formation by the wild‐type M. tuberculosis H37Rv strain (Fig 8E). A previous report had indicated the role of HupB in biofilm formation in uropathogenic E. coli (Devaraj et al, 2015). As MtbΔhupB was found to be deficient in biofilm formation (no biofilm formation observed in BSA panel, Fig 8E), it indicated that HupB had a role in biofilm formation even in M. tuberculosis H37Rv. In the hupB‐complemented MtbΔhupB strain, the biofilm formation was observed in BSA, but not in SUV39H1 panel. The biofilm formation was quantified by crystal violet blue assay (Fig 8E, lower panel and Fig 8F). This not only confirmed involvement of HupB in biofilm formation but also showed that SUV39H1‐mediated HupB methylation was involved in preventing biofilm formation.
SUV39H1 methylation of M. tuberculosis HupB at K138 inhibits biofilm formation
To investigate whether methylation of HupB K138 by SUV39H1 interfered in biofilm formation by mycobacterial bacilli, MtbΔhupB strain transformed with wild‐type (6XHis‐HupB) or mutant HupB (6XHis‐HupBK138A and 6XHis‐HupBK183A) constructs was cultured in the presence of BSA + SAM or SUV39H1 + SAM. While biofilm formation was inhibited in the 6XHis‐hupB‐ and 6XHis‐hupBK183A‐transformed MtbΔhupB strains, inhibition of biofilm formation was significantly compromised in the MtbΔhupB + 6XHis‐HupBK138A mutant strain (Fig 9A and B). Since K138 methylation was significantly reduced in 6XHis‐HupBK138A, this not only indicated the involvement of K138 in biofilm formation but once again confirmed that SUV39H1 prevents mycobacterial biofilm formation by trimethylating HupB.
Figure 9. SUV39H1 inhibits mycobacterial biofilm formation through methylation of lysine 138 of HupB.

-
A, BMBP‐SUV39H1 or BSA (control) and SAM were added to the culture medium of MtbΔhupB + HupB, MtbΔhupB + 6XHis‐hupBK138A and MtbΔhupB + 6XHis‐hupBK183A strains and the bacilli were allowed to form biofilm over a period of around 25 days. The biofilm formation after crystal violet staining was either imaged (A) or quantified for three independent experiments (B).
-
CMycobacteria are present in groups in SUV39H1‐KD THP1 macrophages. The number of loci where GFP::M. bovis BCG bacilli were present in groups of three or more was quantified in eight or more randomly selected fields from three independent experiments after infection into THP1 macrophages transfected with scrambled or SUV39H1 (SUV39H1‐KD) shRNAs.
-
DSUV39H1‐mediated HupB trimethylation inhibits mycobacterial infection. Upon localization to the cell surface, SUV39H1 interacts with the mycobacterial bacilli and trimethylates the mycobacterial cell surface protein, HupB. The trimethylation of HupB inhibits the interaction of mycobacteria with the host cell and consequently inhibits infection.
The same was true for M. smegmatis strains transformed with wild type (HupB::M. smegmatis) or mutant forms (6XHis‐HupBK138A::M. smegmatis, 6XHis‐HupBK183A::M. smegmatis) of M. tuberculosis hupB gene. As can be seen in Fig EV5A and B, SUV39H1 was able to inhibit biofilm formation of wild‐type (HupB::M. smegmatis) and 6XHis‐HupBK183A::M. smegmatis strains, and it was unable to inhibit biofilm formation in 6XHis‐HupBK138A::M. smegmatis strain.
Figure EV5. SUV39H1 inhibits mycobacterial biofilm formation.

-
A, BSUV39H1 inhibits mycobacterial biofilm formation through methylation of lysine 138 of HupB. Biofilm formation by M. smegmatis + HupBK138A mutant is not inhibited by SUV39H1. MBP‐SUV39H1 or BSA (control) and SAM were added to the culture medium of HupB::M. smegmatis, 6XHis‐HupBK138A::M. smegmatis, and 6XHis‐hupBK183A::M. smegmatis strains and the bacilli were allowed to form biofilm. The biofilm formation after crystal violet staining was either imaged (A) or quantified (B). Error bars represent standard deviation (SD). Asterisks indicate significant difference by Student's t‐test: ***P < 0.001.
-
CCharacterization of SUV39H1 antibody specificity by Western blotting. Protein lysate of M. bovis BCG bacilli and uninfected THP1 macrophages was Western‐blotted and probed with SUV39H1 antibody. The blot was also probed with GroEL1 antibody as a control. SUV39H1 antibody does not cross‐react with any mycobacterial protein.
-
DCharacterization of SUV39H1 antibody specificity by immunostaining. M. bovis BCG bacilli were immunostained with SUV39H1 antibody and visualized by confocal microscopy. DIC—brightfield image of the bacilli. Scale bar: 2 μm.
Source data are available online for this figure.
That the inhibition of mycobacterial infection by SUV39H1 was through its action on mycobacterial cell adhesion was further supported by the observation that during infection of THP1 macrophages stably transfected with SUV39H1 (SUV39H1‐KD) shRNA with GFP::M. bovis BCG the mycobacteria bacilli were present in groups (three or more per locus) at significantly higher number of loci in SUV39H1‐KD as compared to control THP1 macrophages [Fig 9C, see also Fig 6B (48‐h panel)], suggesting that the presence of SUV39H1 in macrophages was probably inhibiting adherence between mycobacterial cells.
Discussion
The present study has not only identified a novel host defense mechanism against mycobacterial infection but also revealed the capability of a mammalian cell in utilizing its protein repertoire to perform surrogate functions when challenged by an environmental insult.
SUV39H1 acts in a surrogate function during mycobacterial infection
A homolog of the Drosophila protein, suppressor of variegation 3(9) (first identified as a gene whose product could suppress the effect of position effect variegation), SUV39H1 is an evolutionary conserved protein (Aagaard et al, 1999). In addition to its role during mammalian development (Peters et al, 2001), several studies have shown correlation of altered SUV39H1 expression with cancers (Watson et al, 2014; Chiba et al, 2015). In almost all the previous studies that have examined suppressor of variegation 3(9) or its homologs, this protein has been shown to function by modulating chromatin organization, through its role as a histone H3K9 methyltransferase in the nucleus (Rea et al, 2000; Peters et al, 2002). In this study, we show that during infection, SUV39H1 methylates the mycobacterial cell surface protein HupB. HupB is a histone‐like protein from mycobacteria. Moreover, K138, the lysine in HupB that was found to be methylated by SUV39H1, is present within a sequence motif that is almost same as the one that encompasses H3K9 and is present in a sequence motif that is similar to specificity profile of SUV39H1 (Kudithipudi et al, 2017). Interestingly, mycobacteria have two more histone‐like protein, HNS and HBHA. But both of them do not have this motif and as our result showed only one mycobacterial protein is trimethylated by SUV39H1 (Fig EV2C). Therefore, our finding that SUV39H1 methylates the mycobacterial histone‐like protein HupB, at a motif that is almost same as one that encompasses H3K9, confirms the substrate specificity of this protein. Moreover, the region containing the K138 lysine present in the C‐terminal portion of the mycobacterial HupB is absent from known histone‐like proteins (including homologs of mycobacterial HupB) present in other bacterial species. This would suggest that the action of SUV39H1 could be specific to infection by mycobacterial species. Testing the role of SUV39H1 during other bacterial infections would be able to answer this hypothesis.
Based on our observations, we hypothesize that during mycobacterial infection, the macrophage cell upregulates SUV39H1 gene expression and the resultant protein is localized to the cell surface where it interacts with the mycobacterial bacilli and trimethylates the HupB protein present on their surface. Inhibition of biofilm formation by SUV39H1‐mediated HupB trimethylation indicates that the adhesion capabilities of the mycobacterial bacilli including their interaction with the host cell is hindered by SUV39H1 during infection, which in turn decreases the ability of mycobacteria to infect and survive within the host cell (Fig 9D). It is possible that the methylation of HupB at K138 prevents its interaction with host proteins including laminin, thereby inhibiting the adhesion of mycobacterial bacilli to the host cell. This, in turn, could result in decreased mycobacterial infection efficiency. Further work is required to delineate the HupB‐host cell protein(s) interaction that is affected by HupB methylation. Moreover, the mechanism and cues that prompt the cellular machinery to relocalize SUV39H1 from the nucleus to the cell surface remain unclear. Since post‐translational modifications of proteins are known to be involved in shuttling of protein across various subcellular loci, further work needs to be done to test the possibility that the mechanism of SUV39H1 relocalization is dependent upon its post‐translational modification and relevant associated effector molecules. Furthermore, to understand the role of SUV39H1 during mycobacterial infection, it would be important to dissect out the cascade of events that eventually triggers SUV39H1 relocalization.
Importantly, our finding that SUV39H1 acts to prevent mycobacterial infections, both in macrophages (THP1 as well as peritoneal) cells in culture and in mice infection studies, through its surrogate role as a methyltransferase of the mycobacterial protein HupB, adds a novel epigenetic paradigm to the duel between the host cell and the infecting pathogen. Our laboratory had previously shown that mycobacteria utilize a subset of its repertoire of secretory proteins to modulate the host cell by directly interacting with the epigenetic circuitry (Sharma et al, 2015; Yaseen et al, 2015). Here, we show that the host cells have also developed the ability to utilize components of its epigenetic circuitry to directly interact with the mycobacterial cells. The next step in our effort to unravel this novel defense mechanism would be to understand whether these two sets of proteins, one from the host and other from the pathogen, interact with or counteract each other and which other proteins are involved in this network.
SUV39H1 inhibits mycobacterial biofilm formation
Formation of biofilms by various species of bacteria is being recognized as an important reason for antibiotic resistance (Costerton et al, 1999; Cos et al, 2010). In several cases, infection by commensal bacteria species has also been attributed as a reason for their ability to form biofilms (Reisner et al, 2006; Fey & Olson, 2010). Only a limited number of studies have examined the response of a mammalian host cell against bacteria biofilms (Hänsch, 2012; Rabin et al, 2015). Some of these studies have focused on the role of antimicrobial peptides (AMPs) like LL‐37 in combating biofilm formation (Overhage et al, 2008). A few studies have investigated the correlation between expression level of the protein, lactoferrin (part of host innate immune response), and biofilm formation (Hänsch, 2012). Moreover, biopeptides from various plant and animal sources have been investigated for their ability in the treatment of biofilm formation by various bacterial species (Rabin et al, 2015). Our finding that SUV39H1, which functions as a histone methyltransferase in normal circumstances, inhibits mycobacterial adhesion not only adds a new paradigm to the host response against bacterial infection but also identifies a protein, which is a natural component of the host defense against infection, could be a good candidate with likely lesser side effects for therapeutic use in the treatment for mycobacterial infections. Furthermore, understanding how trimethylation of HupB by SUV39H1 alters mycobacterial cell adhesion and virulence would help in understanding this novel mechanism.
In summary, our study has unlocked not only novel facets of SUV39H1 function but also new paradigms of host defense mechanism. It appears highly unlikely that the use of an evolutionary conserved protein like SUV39H1 by the cellular machinery in a non‐canonical surrogate function during an environmental insult was a chance event. Further investigations that examine the validity of this possibility would provide a novel perspective to research on evolution of host–pathogen interactions.
Materials and Methods
Subcellular fractionation
Subcellular fractionation was performed as per Healy and O'Connor (2009). Briefly, cells were lysed in hypotonic buffer (20 mM Tris, pH 8.0, 10 mM KCl, 1 mM MgCl2, 0.1% v/v TX‐100, and 20% v/v glycerol) for 15 min, with vortexing for 30 s after every 5 min and subjected to centrifugation at 2,400 g for 2 min, supernatants were collected as cytoplasmic fraction, and pellets were dissolved in hypertonic buffer (20 mM Tris pH 8, 400 mM NaCl, 1 mM EDTA, 1% TX‐100, and 20% glycerol) and sonicated for 5 min. After sonication, samples were centrifuged at 10,000 g for 5 min and supernatants were collected as the nuclear fraction.
Phagosome isolation
The phagosome isolation was performed as described elsewhere (Dietrich et al, 2000) with slight modifications. M. bovis BCG‐infected THP1 macrophages were washed twice with PBS and resuspended in the lysis buffer (20 mM HEPES pH 6.5 and 8.55% sucrose). The cells were passed 15–20 times through a 28‐gauge syringe, and after the 15th passage, lysis was monitored under a microscope. The procedure was stopped when nine out of 10 cells were found to have been lysed. The lysate was subjected to centrifugation at 120 g for 8 min; the supernatants were collected and again recentrifuged twice under same conditions. Final supernatant was layered on top of a 12%/50% (2 ml each) sucrose gradient and centrifuged for 45 min at 800 g. After centrifugation, 1 ml interface was recovered using syringe. The recovered fraction was diluted 10 times with 20 mM HEPES buffer and subjected to centrifugation at 18,000 g for 10 min and the pellet collected as the phagosomal fraction. For isolating intracellular mycobacteria, phagosomal fraction was treated with the same lysis buffer that also contained 0.5% NP‐40 and subjected to centrifugation at 18,000 g for 5 min. The pellet contained the bacteria, and supernatant contained the phagosomal matrix.
Immunofluorescence
Mycobacterium bovis BCG‐infected or uninfected THP1 macrophages were washed twice with PBS; cells were fixed with 3.7% formaldehyde for 10 min followed by permeabilization with 0.1% Triton X‐100 for 5 min. Cells were incubated with SUV39H1 antibody at the dilution of 1:200 overnight at 4°C. After incubation, cells were washed three times with PBS, followed by staining with Alexa Flour‐conjugated secondary antibody at the dilution of 1:2,000 for 1 h at room temperature. Finally, cells were mounted using DAPI‐containing Vectashield and images were taken using confocal microscope.
Bacterial binding assay
Bacterial binding assay was followed as described elsewhere (Sakowski et al, 2015). Briefly, M. bovis BCG was incubated with recombinant SUV39H1 or HEK lysate overexpressing SFB‐SUV39H1 in PBS containing 0.5% Triton X‐100 for 4 h. After incubation, M. bovis BCG cells were washed five times with the same buffer and resuspended in SDS–PAGE gel loading dye.
Biofilm assay
We followed the protocol described by Kulka et al (2012) for biofilm assay with modifications. The biofilm assay was set up in 7H9‐OADC media supplemented with methyltransferase buffer (50 mM Tris pH 8.8, 20 μM SAM, 4 mM DTT, 5 mM MgCl2, and 1 μg of SUV39H1 or BSA). 2% inoculum of M. bovis BCG or different strains of M. tuberculosis H37Rv having OD 0.7–1 was added to the medium and cultured for 4 weeks to examine biofilm formation. Biofilm was estimated by crystal violet staining followed by fluorescence reading at 570 nm.
Infection of THP1 and peritoneal macrophages
As described previously (Yaseen et al, 2015), THP cells treated with 10 ng of PMA for 12 h were cultured further for 24 h in PMA‐free RPMI media followed by infection with M. bovis BCG or different strains of M. tuberculosis at an MOI of 10:1 for 6 h in antibiotic‐free media. The viability of infected THP1 macrophages is not significantly different at MOI 1:1 vs. 10:1 (Sharma et al, 2016; the THP1 cell line used in the present study is the same as used in Sharma et al, 2016). Thereafter, cells were washed twice with PBS and recultured in antibiotic‐containing media for 48 h. Before infection, mycobacterial culture was passed 10 times through 27.5‐G needle to make a single cell suspension. Peritoneal exudate cells (PEC) were collected from BALB/c mice as per approved IEAC guidelines (PCD/CDFD/15). The infections were done as described above for THP1 macrophages.
MOI for E. coli was 1:10, and for C. glabrata, it was 1:5. The remaining infection conditions were same as those for mycobacterial species.
Mouse infection assay
Animal experimentation was done in accordance with the guidelines of CPCSEA (Government of India) at VIMTA Labs Limited, Hyderabad. The protocol was approved by the Institutional Animal Ethics Committee (IAEC) of the Vimta Labs Ltd. (IAEC Protocol Approval Number: PCD/OS/17). 4‐ to 6‐week‐old male BALB/c mice were injected intravenously (tail vein) with different strains of M. smegmatis (using ~2 × 107 bacteria) as per approved IEAC guidelines (PCD/CDFD/17). Seven days postinfection, mice were sacrificed. To examine the bacterial load in lung, liver, and spleen, the organs were homogenized in 1 ml PBS. Body weight of the individual mice was also taken at different times after infection as indicated.
CFU and Alamar assay on intracellular bacteria
For CFU counting of intracellular bacteria, infected THP1 macrophages were washed twice with PBS followed by incubation in PBS containing 0.2% Triton X‐100 for 10 min for cell lysis. After cell lysis, serial dilutions were made and 100 μl from each dilution was plated on 7H10 agar plates.
For Alamar assay, we followed the protocol mentioned in Ganji et al (2016). Briefly, cells in 24‐well dish were washed twice with PBS and lysed in 200 μl of sterile water for 15 min at 37°C. 50 μl of 1:1 mixture of Tween‐80 and 10% Alamar blue solution was added to the lysate and incubated at 37°C overnight. The fluorescence was measured in a fluorescence plate reader with excitation and emission at 530 and 590 nm, respectively.
Methyltransferase assay
One to two microgram of MBP‐SUV39H1 was incubated with 1 μg of HupB or 50 μg of mycobacteria lysate in buffer containing 50 mM Tris pH 8.8, 5 mM MgCl2, 4 mM DTT, and 20 μM S‐adenosylmethionine (SAM). The reaction was incubated at 30°C for 2 h. After incubation, reaction was stopped by adding the SDS electrophoresis dye.
Generation of knockdown stable cell lines
For the generation of knockdown THP1 cell line, THP1 cells were treated with polybrene (8 μg/ml) for 2 h, followed by the addition of SUV39H1 or scrambled shRNA lentiviral transduction particles (Sigma) and incubated for 48 h. After lentiviral infection, cells were selected on puromycin for 1 week and the selected cells were examined for SUV39H1 expression by Western blotting.
Site‐directed mutagenesis
Mycobacterium tuberculosis HupB was cloned in the pVV16 vector having 6XHis tag at the C‐terminus. PCR‐mediated SDMs were generated for the following lysines (138, 183, and 214) which were converted to alanine.
Mycobacterial cell fractionation
The subcellular fractionation of mycobacteria was performed by the protocol mentioned elsewhere (Ghosh et al, 2013). Briefly, mycobacteria cells were sonicated in lysis buffer (10 mM Tris–HCl pH 7.5, 100 mM NaCl, and 0.1 mM EDTA). The lysate was subjected to centrifugation at 27,000 g for 1 h, and the supernatant was collected as cytosolic fraction and pellet as cell wall fraction.
Total membrane isolation
107 THP1 cells infected with BCG were sonicated in 200 μl of buffer containing 100 mM Tris–HCl pH 10.7, 5 mM EDTA, and 2 mM DTT. After sonication, the sample was diluted to 1 ml with buffer containing 100 mM Tris–HCl pH 8, 0.33 M sucrose, 5 mM EDTA, and 2 mM DTT. The sample was subjected to centrifugation for 3 min at 1,000 g. The supernatant was collected and again subjected to centrifugation for 5 min at 3,000 g. Resulting supernatant was subjected to centrifugation at 19,000 g for 45 min. The resulting pellet corresponding to total membrane fraction was dissolved in buffer containing 10 mM Tris–HCl pH 7.5, 0.1 mM DTT, and 20% glycerol.
Collection of human PBMCs
Blood was collected from three healthy volunteers; 15 ml from each was kept for 15 min at room temperature and diluted with equal volume of PBS. 3 ml of Ficoll‐paque was taken in 15‐ml tubes and 7 ml of blood mixture was added slowly from the top to avoid mixing. The samples were subjected to centrifugation at 800 g for 45 min. The whitish layer above the Ficoll layer containing the PBMCs was collected and washed with PBS. The PBMCs were seeded and infected for 2 h with various mycobacterium strains. The samples were collected at various time points either for use in CFU counting or for Western blotting.
Specificity of SUV39H1 antibodies
To confirm that the anti‐SUV39H1 antibodies did not cross‐react with any mycobacterial proteins, protein lysate from M. bovis BCG‐infected and uninfected THP1 cells was electrophoresed, Western‐blotted, and probed with anti‐SUV39H1 antibodies (Fig EV5C). SUV39H1 antibodies did not recognize any mycobacterial proteins and detected only the SUV39H1 protein in the THP1 cell lysate (the upper band corresponds to post‐translationally modified SUV39H1 protein). This was also true for immunofluorescence experiment as no signal was detected for M. bovis BCG bacilli immunostained with anti‐SUV39H1 antibodies (Fig EV5D).
Source of antibodies
Antibodies against SUV39H1 were purchased from both Millipore and Abcam (07‐550 & ab155164). Antibodies against trimethylated lysine (ab76118), di/monomethylated lysine (ab23366), SUV39H2 (ab5264), histone H3 (ab1791) and H4 (ab10158), mycobacterium (ab20832), tubulin (ab125267), GAPDH (ab22555), β‐actin (ab8227), and LAMP1 (ab24170) were from Abcam. MBP antibody (AB3596) was purchased from Millipore. TNFR1 (sc7895) antibody was from Santa Cruz. HupB antibody was generated in Dr. Sritharan's laboratory (University of Hyderabad). GroEL1 antibody was a kind gift from Dr. Shekhar Mande NCCS, Pune, India.
Author contributions
SK and IY designed the experiments. IY performed the experiments. Experimental work related to M.tb H37Rv and MtbΔhupB strains was planned by MS and performed by MC and IY in MS laboratory. SK wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 8
Acknowledgements
The authors thank Dr. Prakruti Singh and Mr. Ambey Prasad Dwivedi for help in a few of the experiments, Mr Sridhar for help in animal experimentation, and Dr. Vinay K Nandicoori for his suggestions and comments on the manuscript. IY and MC are recipients of Senior Research Fellowship of the Council of Scientific and Industrial Research (CSIR) and University Grants Commission (UGC), India, toward the pursuit of a PhD degree of the Manipal University, Manipal, and University of Hyderabad, Hyderabad, respectively. This study was supported by funds from DBT and CDFD core funds to SK. MS acknowledges the financial assistance under “UPE‐II” Program of the University of Hyderabad.
See also: C Gutierrez et al (January 2018)
References
- Aagaard L, Laible G, Selenko P, Schmid M, Dorn R, Schotta G, Kuhfittig S, Wolf A, Lebersorger A, Singh PB, Reuter G, Jenuwein T (1999) Functional mammalian homologues of the Drosophila PEV‐modifier Su(var)3‐9 encode centromere‐associated proteins which complex with the heterochromatin component M31. EMBO J 18: 1923–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av‐Gay Y (2008) Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe 3: 316–322 [DOI] [PubMed] [Google Scholar]
- Chiba T, Saito T, Yuki K, Zen Y, Koide S, Kanogawa N, Motoyama T, Ogasawara S, Suzuki E, Ooka Y, Tawada A, Otsuka M, Miyazaki M, Iwama A, Yokosuka O (2015) Histone lysine methyltransferase SUV39H1 is a potent target for epigenetic therapy of hepatocellular carcinoma. Int J Cancer 136: 289–298 [DOI] [PubMed] [Google Scholar]
- Cos P, Toté K, Horemans T, Maes L (2010) Biofilms: an extra hurdle for effective antimicrobial therapy. Curr Pharm Des 16: 2279–2295 [DOI] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284: 1318–1322 [DOI] [PubMed] [Google Scholar]
- Devaraj A, Justice SS, Bakaletz LO, Goodman SD (2015) DNABII proteins play a central role in UPEC biofilm structure. Mol Microbiol 96: 1119–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich G, Schaible UE, Diehl KD, Mollenkopf H, Wiek S, Hess J, Hagens K, Kaufmann SH, Knapp B (2000) Isolation of RNA from mycobacteria grown under in vitro and in vivo conditions. FEMS Microbiol Lett 186: 177–180 [DOI] [PubMed] [Google Scholar]
- Fey PD, Olson ME (2010) Current concepts in biofilm formation of Staphylococcus epidermidis . Future Microbiol 5: 917–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganji R, Dhali S, Rizvi A, Sankati S, Vemula MH, Mahajan G, Rapole S, Banerjee S (2016) Proteomics approach to understand reduced clearance of mycobacteria and high viral titers during HIV‐mycobacteria co‐infection. Cell Microbiol 18: 355–368 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Indi SS, Nagaraja V (2013) Regulation of lipid biosynthesis, sliding motility, and biofilm formation by a membrane‐anchored nucleoid‐associated protein of Mycobacterium tuberculosis . J Bacteriol 195: 1769–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A (2005) Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3‐9. Nat Chem Biol 1: 143–145 [DOI] [PubMed] [Google Scholar]
- Hänsch GM (2012) Host defence against bacterial biofilms: “Mission impossible”? ISRN Immunol 2012: 853123 [Google Scholar]
- Healy NC, O'Connor R (2009) Sequestration of PDLIM2 in the cytoplasm of monocytic/macrophage cells is associated with adhesion and increased nuclear activity of NF‐kappaB. J Leukoc Biol 85: 481–490 [DOI] [PubMed] [Google Scholar]
- Holla S, Balaji KN (2015) Epigenetics and miRNA during bacteria‐induced host immune responses. Epigenomics 7: 1197–1212 [DOI] [PubMed] [Google Scholar]
- Holoch D, Moazed D (2015) RNA‐mediated epigenetic regulation of gene expression. Nat Rev Genet 16: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayachandran R, Scherr N, Pieters J (2009) Analyzing the interaction of pathogens with the host immune system. Immunol Lett 122: 112–114 [DOI] [PubMed] [Google Scholar]
- Joo HS, Otto M (2012) Molecular basis of in vivo biofilm formation by bacterial pathogens. Chem Biol 19: 1503–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudithipudi S, Schuhmacher MK, Kebede AF, Jeltsch A (2017) The SUV39H1 protein lysine methyltransferase methylates chromatin proteins involved in heterochromatin formation and VDJ recombination. ACS Chem Biol 12: 958–968 [DOI] [PubMed] [Google Scholar]
- Kulka K, Hatfull G, Ojha AK (2012) Growth of Mycobacterium tuberculosis biofilms. J Vis Exp 15: 3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrançois LH, Pujol C, Bodier CC, Teixeira‐Gomez AP, Drobecq H, Rosso ML, Raze D, Dias AA, Hugot JP, Chacon O, Barletta RG, Locht C, Vidal Pessolani MC, Biet F (2011) Characterization of the Mycobacterium avium subsp. Paratuberculosis laminin‐binding/histone‐like protein (Lbp/Hlp) which reacts with sera from patients with Crohn's disease. Microbes Infect 13: 585–594 [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS (2013) The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501: 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Carroll D, Scherthan H, Peters AH, Opravil S, Haynes AR, Laible G, Rea S, Schmid M, Lebersorger A, Jerratsch M, Sattler L, Mattei MG, Denny P, Brown SD, Schweizer D, Jenuwein T (2000) Isolation and characterization of Suv39 h2, a second histone H3 methyltransferase gene that displays testis‐specific expression. Mol Cell Biol 20: 9423–9433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overhage J, Campisano A, Bains M, Torfs EC, Rehm BH, Hancock RE (2008) Human host defense peptide ll‐37 prevents bacterial biofilm formation. Infect Immun 76: 4176–4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SD, Choudhury M, Yousuf S, Wheeler PR, Gordon SV, Ranjan A, Sritharan M (2014) Iron‐regulated protein HupB of Mycobacterium tuberculosis positively regulates siderophore biosynthesis and is essential for growth in macrophages. J Bacteriol 196: 1853–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, Kaisho T, Kundu M, Basu J (2007) Direct extracellular interaction between the early secreted antigen ESAT‐6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol 8: 610–618 [DOI] [PubMed] [Google Scholar]
- Pennini ME, Liu Y, Yang J, Croniger CM, Boom WH, Harding CV (2007) CCAAT/enhancer‐binding protein beta and delta binding to CIITA promoters is associated with the inhibition of CIITA expression in response to Mycobacterium tuberculosis 19‐kDa lipoprotein. J Immunol 179: 6910–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennini ME, Perrinet S, Dautry‐Varsat A, Subtil A (2010) Histone methylation by NUE, a novel nuclear effector of the intracellular pathogen Chlamydia trachomatis . PLoS Pathog 6: e1000995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schöfer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T (2001) Loss of the Suv39 h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323–337 [DOI] [PubMed] [Google Scholar]
- Peters AH, Mermoud JE, O'Carroll D, Pagani M, Schweizer D, Brockdorff N, Jenuwein T (2002) Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet 30: 77–80 [DOI] [PubMed] [Google Scholar]
- Rabin N, Zheng Y, Opoku‐Temeng C, Du Y, Bonsu E, Sintim HO (2015) Agents that inhibit bacterial biofilm formation. Future Med Chem 7: 647–671 [DOI] [PubMed] [Google Scholar]
- Ragno S, Romano M, Howell S, Pappin DJ, Jenner PJ, Colston MJ (2001) Changes in gene expression in macrophages infected with Mycobacterium tuberculosis: a combined transcriptomic and proteomic approach. Immunology 104: 99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjbar S, Haridas V, Jasenosky LD, Falvo JV, Goldfeld AE (2015) A role for IFITM proteins in restriction of Mycobacterium tuberculosis infection. Cell Rep 13: 874–8783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng X, Jeltsch A (2008) Protein lysine methyltransferase G9a acts on non‐histone targets. Nat Chem Biol 4: 344–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T (2000) Regulation of chromatin structure by site‐specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Reisner A, Krogfelt KA, Klein BM, Zechner EL, Molin S (2006) In vitro biofilm formation of commensal and pathogenic Escherichia coli strains: impact of environmental and genetic factors. J Bacteriol 188: 3572–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolando M, Sanulli S, Rusniok C, Gomez‐Valero L, Bertholet C, Sahr T, Margueron R, Buchrieser C (2013) Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 13: 395–405 [DOI] [PubMed] [Google Scholar]
- Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, Maurer K, Cadwell K, Philips JA (2015) Ubiquilin 1 promotes IFN‐γ‐induced xenophagy of Mycobacterium tuberculosis . PLoS Pathog 11: e1005076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Grosschedl R (2007) Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev 21: 3027–3043 [DOI] [PubMed] [Google Scholar]
- Sedger LM, McDermott MF (2014) TNF and TNF‐receptors: from mediators of cell death and inflammation to therapeutic giants ‐ past, present and future. Cytokine Growth Factor Rev 25: 453–472 [DOI] [PubMed] [Google Scholar]
- Sharma G, Upadhyay S, Srilalitha M, Nandicoori VK, Khosla S (2015) The interaction of mycobacterial protein Rv2966c with host chromatin is mediated through non‐CpG methylation and histone H3/H4 binding. Nucleic Acids Res 43: 3922–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Sowpati DT, Singh P, Khan MZ, Ganji R, Upadhyay S, Banerjee S, Nandicoori VK, Khosla S (2016) Genome‐wide non‐CpG methylation of the host genome during M. tuberculosis infection. Sci Rep 6: 25006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoji Y, Ng V, Matsumura K, Fischetti VA, Rambukkana A (1999) A 21‐kDa surface protein of Mycobacterium leprae binds peripheral nerve laminin‐2 and mediates Schwann cell invasion. Proc Natl Acad Sci USA 96: 9857–9862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart KR, Veselovska L, Kim J, Huang J, Saadeh H, Tomizawa S, Smallwood SA, Chen T, Kelsey G (2015) Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev 29: 2449–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramurthy V, Pieters J (2007) Interactions of pathogenic mycobacteria with host macrophages. Microbes Infect 9: 1671–1679 [DOI] [PubMed] [Google Scholar]
- Wang Y, Curry HM, Zwilling BS, Lafuse WP (2005) Mycobacteria inhibition of IFN‐gamma induced HLA‐DR gene expression by up‐regulating histone deacetylation at the promoter region in human THP‐1 monocytic cells. J Immunol 174: 5687–5694 [DOI] [PubMed] [Google Scholar]
- Watson GW, Wickramasekara S, Palomera‐Sanchez Z, Black C, Maier CS, Williams DE, Dashwood RH, Ho E (2014) SUV39H1/H3K9me3 attenuates sulforaphane‐induced apoptotic signaling in PC3 prostate cancer cells. Oncogenesis 3: e131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaseen I, Kaur P, Nandicoori VK, Khosla S (2015) Mycobacteria modulate host epigenetic machinery by Rv1988 methylation of a non‐tail arginine of histone H3. Nat Commun 6: 8922 [DOI] [PubMed] [Google Scholar]
- Yeruva VC, Duggirala S, Lakshmi V, Kolarich D, Altmann F, Sritharan M (2006) Identification and characterization of a major cell wall‐associated iron‐regulated envelope protein (Irep‐28) in Mycobacterium tuberculosis . Clin Vaccine Immunol 13: 1137–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 8