

Abstract
Multiple sclerosis (MS) is the most common cause of non-traumatic disability in young adults. The mechanisms underlying neurodegeneration and disease progression are poorly understood in part due to the lack of non-invasive methods to measure and monitor neurodegeneration in vivo. Sodium MRI is a topic of increasing interest in MS research as it allows the metabolic characterization of brain tissue in vivo, integrating the structural information provided by proton MRI, helping in the exploration of pathogenetic mechanisms and possibly offering insights in the disease progression and monitoring of treatment outcomes. We present an up-to-date review of the sodium MRI application in MS, organized in four main sections: 1) biological and pathogenetic role of sodium; 2) brief overview on sodium imaging techniques; 3) results of sodium MRI application in clinical studies; 4) future perspectives.
Keywords: sodium MRI, axonal degeneration, multiple sclerosis
Graphical abstract
Sodium MRI is a topic of increasing interest in multiple sclerosis (MS) research as it allows the metabolic characterization of brain tissue in vivo, helping in the exploration of pathogenic mechanisms and possibly offering insights in the disease progression and monitoring of treatment outcomes. We present an up-to-date review of the sodium MRI application in MS, organized in four main sections: biological and pathogenetic role of sodium; brief overview on sodium imaging techniques; results of sodium MRI application in clinical studies; future perspectives.
Introduction
Multiple sclerosis (MS) is the most common cause of non-traumatic disability in young adults and affects more than two million people worldwide. The disease etiology is unknown but MS prevalence increases with increasing distance north or south of the equator and the risk of developing MS correlates with the place of residence during childhood; it is therefore believed that an early exposure to an unidentified infectious agent could trigger the disease in individuals with a favorable genetic background (alleles of the MHC DR4, DR15 and DQ6) (1, 2). MS is characterized by an inflammatory component, which is responsible for acute occurrence of clinical relapses and development of focal lesions and by a degenerative component, which is responsible for accrual of progressive physical and cognitive disability (1). In about 80% of cases, the disease onset is characterized by a subacute and transient neurological deficit (clinically isolated syndrome-CIS), while, in the remaining 20%, the disease causes, from the beginning, a gradual clinical worsening over time (primary progressive MS-PP-MS). After the first episode, the presence of dissemination in time and space is required in order to confirm the diagnosis of MS (3). MS clinical course is usually characterized, during the initial stage, by unpredictable clinical and radiological relapses (relapsing-remitting MS-RR-MS); over time the recurrence of relapses tends to decrease and a gradual neurological worsening occurs (secondary progressive MS-SP-MS) (4).
The etiology of MS is still unknown but the pathogenetic process seems to start in the periphery with the priming of myelin-autoreactive T lymphocytes, which, crossing the blood brain barrier, mediate an acute autoimmune reaction against myelin and cause the activation of resident microglia and infiltrated macrophages. Auto-reactive CD4+ T cells secreting interferon-gamma and interleukin-17 are among the main mediators of the pathological process. The release of inflammatory mediators (nitric oxide, reactive oxygen species, myeloperoxidase, tumor necrosis factor-alfa) causes oligodendrocytes damage and myelin sheet disruption, and contributes to neuro-axonal damage and loss (5).
In addition to inflammation, axonal damage can be driven or amplified by a number of other pathological processes including Wallerian degeneration following axonal transection due to focal lesions (6), lack of trophic support from myelin (7), mutation of mitochondrial DNA (8, 9), astrocytes dysfunction (10), glutamate excitotoxicity (11), iron accumulation (12, 13) and sodium (23Na) ions accumulation (14).
Studies in experimental models of MS and in post-mortem samples from MS patients have provided evidence for the presence of over-expression and increased activation of persistent 23Na channels in demyelinated axons and MS plaques (15-17).
Brain 23Na MRI was introduced almost twenty years ago but poor signal to noise ratio (SNR) led to relatively long imaging times and/or poor spatial resolution compared to proton (1H) MRI and the sparse availability of MRI scanners with broadband capability limited its use. Recent technological advances in MRI hardware and software and the availability of ultra-high field magnets have prompt new developments that permit better spatial resolution with shorter imaging times and better quantitative measurements of tissue 23Na concentration (18, 19). Over time, various invasive methods have been used to measure 23Na content in animals and ex vivo human brain tissue (20-22). Non-invasive determination of brain 23Na concentration with 23Na imaging has shown to be equivalent to invasive biochemical ex vivo techniques (23).
Currently, there are eleven FDA-approved disease modifying treatments for MS with a partial efficacy in decreasing relapse rate and accumulation of white matter (WM) lesions. Since none of them is effective on the neurodegenerative component of the disease, there is an unmet need for a reliable, non-invasive technique that could help understanding the mechanisms responsible for neurodegeneration and be used for monitoring the response to new, neuroprotective therapies when they become available.
In this review we summarize the main findings obtained by the application of 23Na imaging in preclinical and clinical studies, their importance in the light of 23Na role in MS pathogenesis and their implications for disease monitoring and therapeutics development.
Biology of 23Na
23Na yields the second strongest nuclear magnetic resonance (NMR) signal among biologically relevant NMR-active nuclei. In the brain, 23Na has a bicompartimental distribution with higher concentration (140 mmol/L) in the extracellular space and a lower concentration (ranging from 10 to 15 mmol/L) in the intracellular space. 23Na has a critical role in several cellular functions such as mitosis, cellular proliferation, generation and propagation of action potentials and cell volume regulation (24-26). To ensure the maintenance of tissue homeostasis and the preservation of intracellular structures and processes, 23Na concentration is strictly controlled by the ATP-driven Na/K pump; pathological changes that determine an expansion of the extracellular space (e.g. tissue injury, edema or necrosis) or functional impairment of the Na/K pump are therefore expected to result in an increased tissue 23Na concentration (27-30).
23Na role in MS pathogenesis
Nerve fibers conduction is generated and propagated by activation of 23Na channels, which, in intact myelinated axons, are clustered in the Ranvier nodes, enabling fast saltatory conduction; in unmyelinated axons, the distribution of 23Na channels is more homogeneous along the axonal membrane and conduction is slower and continuous. 23Na ions, entering the nerve through voltage gated 23Na channels, have to be actively extruded via an energy dependent process; therefore the greater the 23Na influx, the greater the energy demand the neuron needs to fulfill (31).
When demyelination occurs, 23Na channels are redistributed from the Ranvier nodes to long segments of demyelinated membrane. Demyelinated axons express two voltage-dependent 23Na channel isoforms: Nav1.2, which is normally present along premyelinated axons, and Nav1.6, which is the predominant isoform at normal Ranvier nodes. Nav1.6 channels produce a persistent current that is able to drive reverse Na/Ca exchange even in the absence of action potentials (32).
While channels re-distribution represents an adaptive mechanism to preserve action potential conduction and facilitate recovery of neurological deficits, it imposes a huge burden on the axonal metabolism thus increasing the risk of axonal damage secondary to energy deprivation (16, 33). In MS, the state of virtual hypoxia secondary to mitochondrial dysfunction (34, 35) determines a decrease in ATP production, which, associated to the increased energy request needed to guarantee conduction along demyelinated axons, causes neuronal energy failure (8). Since the maintenance of 23Na balance is an active process controlled by the Na/K pump, the ATP deficit induces intracellular 23Na accumulation and reverse activation of Na/Ca exchanger; the activation of the 20Ca dependent proteases and the cytoskeleton disruption represent the final step leading to cellular death (Fig. 1) (5, 12, 31, 36, 37). Increased concentrations of intracellular 23Na stimulate further 20Ca accumulation by release from the endoplasmic reticulum, triggered by inositol 1,4,5-trisphosphate receptors and ryanodine receptors (Fig. 1) (38).
Fig. 1.

Role of 23Na channels in the axon degeneration cascade. Mitochondrial damage determines energy failure, with ATP deprivation and loss of function of Na/K ATPase. Consequent loss of ionic transmembrane gradient activates Nav1.6 channels producing a sustained 23Na influx and reversing the operation of the Na/Ca exchanger. Further 20Ca release into the axoplasm occurs from injured mitochondria and intracellular stores, triggered by inositol 1,4,5-trisphosphate receptors and ryanodine receptors, stimulate by increased intracellular 23Na concentrations. Elevated intracellular levels of 20Ca activate downstream proteolytic cascade, which produce axonal injury. Reproduced from Waxman 2006 by permission of Elsevier Ltd.
In summary, the two key factors leading to abnormal 23Na influx in MS are (i) the defective mitochondrial function and (ii) the 23Na influx via Nav1.6 channels (32, 39, 40); however, their relative contribution to axonal injury is still unclear.
Supporting this hypothesis, over expression of 23Na channels along demyelinated axons (15, 16) and upregulation of 23Na channels in activated macrophages, microglia and astrocytes (41, 42) have been reported in MS plaques (Fig. 2). Moreover, it has been shown that in the animal models of experimental autoimmune encephalomyelitis (EAE), the mutation of the 23Na channel subunit, which controls the expression of 23Na channels on the cells surfaces, determines reduced axonal degeneration and neurological disability (17). Adaptation to the increased energy demand has been reported not only in lesions and normal appearing white matter (NAWM) but also in the normal appearing grey matter (GM), where pathological studies have shown an increased mitochondrial density (43). Moreover, the 23Na related damage in the GM could be linked to the presence of cortical demyelinating lesions and to the abnormal neuronal expression of 23Na channels with atypical properties, as exemplified by the expression of Nav1.8 channels, resistant to inactivation, in the Purkinje neurons of animals with EAE and patients with progressive MS (44).
Fig. 2.
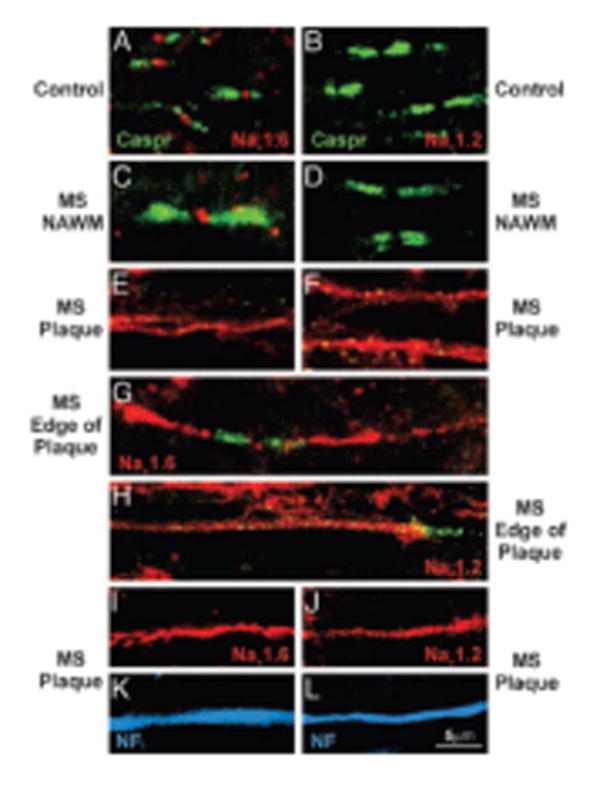
Altered axonal expression of 23Na channels in MS. Sections of postmortem spinal cord white matter from control (A and B) and MS (C–L) patients, immunostained to show Nav1.6 (red), Nav1.2 (red), Caspr (integral constituent of paranodal junctions-green), and neurofilaments (blue). In control white matter (A) and in normal-appearing white matter in MS tissue (C), Nav1.6 is localized at nodes of Ranvier whereas Nav1.2 is not detectable (B and D). Within MS plaques, continuous Nav1.6 (E) and Nav1.2 (F) immunostaining are present; in some instances bounded by Caspr (G-H). Colocalization of Nav1.6 (I) and Nav1.2 (J) with neurofilament immunostaining (K and L; blue) confirms the axonal identity of these profiles. Reproduced from Craner et al.2004 Copyright (2004) National Academy of Sciences, U.S.A.
Since 23Na channels upregulation is responsible for axonal degeneration, 23Na channels blockers are expected to exert neuroprotective effects. Indeed, state-dependent 23Na channels blockers (e.g. class I anti-arrhythmic or anticonvulsants) are able to protect axons from anoxic-ischemic injury in vitro (40, 45-48) and in animal models of MS (49-53), at concentrations that do not compromise the conduction of action potentials. This is further supported by the demonstration that the abrupt withdrawal of phenytoin and carbamazepine seems to induce disease exacerbation and increase of the inflammatory markers in EAE (51).
These findings have prompt clinical trials to investigate the neuroprotective effect of voltage-gated 23Na channel blockers in patients with MS. Unfortunately, the first clinical trial assessing the neuroprotective effect of lamotrigine in MS patients failed to show an effect on brain atrophy accrual. In particular, cerebral volume of patients treated with lamotrigine did not differ from that of placebo over 24 months; moreover, lamotrigine seemed to cause early volume loss that reversed partially on discontinuation of treatment. In contrast with the pseudoatrophy described over the first few months of therapy with other immunomodulatory agents, the decrease in cerebral volume during lamotrigine treatment occurred slowly over 6-12 months and was not associated with reduction in relapse rate and MRI activity; it is therefore possible that it reflected the development of actual axonal loss (54). Although the treatment failure may in part be explained by the high rate of non-adherence to therapy in the lamotrigine group, it is also possible that the decrease of cells volume induced by reduced entry of 23Na ions and water caused by 23Na channel blockade and the lamotrigine anti-inflammatory activity within normal-appearing tissue may have contributed to the results.
Even if the direct blockage of 23Na voltage channel has not produced the expected results in the lamotrigine trial (54), the systemic administration of amiloride, and the consequent blockage of 23Na and 20Ca influx through the proton-gated acid-sensing ion channel 1, has proven a neuroprotective effect not only in acute and chronic experimental models of MS (55, 56), but also in progressive MS patients (57).
There are a few ongoing trials testing the efficacy of 23Na channel blockers in different MS phenotypes (see ClinicalTrials.gov for details) and, therefore, once validated in longitudinal studies, 23Na imaging might prove useful in providing and additional measure of cellular and metabolic brain changes during treatment with 23Na blockers administration.
23Na imaging
Single quantum (SQ) 23Na MRI is an imaging technique that exploits the magnetic resonance properties of 23Na atomic nuclei, allowing the metabolic characterization of brain tissue in vivo. Unlike other metabolic imaging techniques (e.g. MR spectroscopy) it allows exploration and quantitative assessment of brain metabolism both at a global and regional level. Unfortunately, since the concentration of 23Na ions in the human body is much lower than 1H concentration, 23Na MRI presents a poor SNR, which is responsible for the longer acquisition time and the poor spatial resolution of 23Na MRI in comparison to standard 1H MRI. In addition, in most biologic tissues, 23Na interactions with macromolecules determines a bi-exponential transverse relaxation time (T2) with the signal main component (up to 60%) hardly detectable due to its short echo time (58).
These technical limitations have been partially overcome by the development of ultra-short TE sequences (18) and the availability of ultra-high field magnets (19) leading to a rekindled interest and application of brain 23Na imaging in neurological diseases such as ischemic stroke, brain tumors and Alzheimer's disease (19, 59, 60).
23Na MRI quantifies the tissue total sodium concentration (TSC), which represents the weighted average of intracellular and extracellular 23Na (respectively 10-15 mmol/L and 140 mmol/L). TSC is sensitive to changes in both extra- and intra-cellular space, being affected by cellular death, swelling, proliferation (27, 30) as well as by metabolic changes that affect 23Na exchange across the cell membrane (28, 29). In the CNS, we may therefore assume that TSC increase is related to intra-axonal accumulation of 23Na ions, determined by Na/K pump dysfunction, as well as to enlargement of extra-axonal space consequent to neuronal degeneration.
Clinical studies: evidence of 23Na accumulation in MS
The first application of 23Na MRI in patients with MS has been reported by Inglese et al. (61) and has demonstrated that patients with RR-MS show higher NAWM TSC in comparison with healthy controls; such increase in23Na concentration is even higher in acute and chronic lesions compared to areas of NAWM. In addition, TSC levels in lesions, NAWM and GM showed a direct correlation with T2-weighted and T1-weighted lesion load while NAGM TSC was found to be negatively associated with GM volume. In the same study the EDSS (Expanded Disability Status Scale) (62) showed a mild, positive association with the mean TSC value in chronic lesions, NAWM and GM. These results suggest that the abnormal increase of TSC in MS patients might reflect changes in cellular and metabolic integrity of WM lesions as well as normal appearing brain tissue. These findings have been reproduced in different laboratories around the world and the application of the method has been extended to patients with clinical phenotypes other than RR-MS (63-65). In MS patients at early disease stage 23Na increase seems to be limited to macroscopic lesions (63) while in patients with longer disease duration (>5 years) TSC appears to be increased not only in lesions, but also in NAWM, cortical and deep GM (61, 63, 64) with higher concentration reported in more destructive lesions (Fig. 3) (61, 64) and in patients with progressive phenotypes (Fig. 4) (64, 65). While TSC increase in lesions might be explained by gliosis, tissue disruption and replacement with extracellular fluid, TSC increase in normal appearing brain tissue might be related not only to increased extracellular space, caused by demyelination and axonal loss, but also to intra-axonal 23Na increase.
Fig. 3.
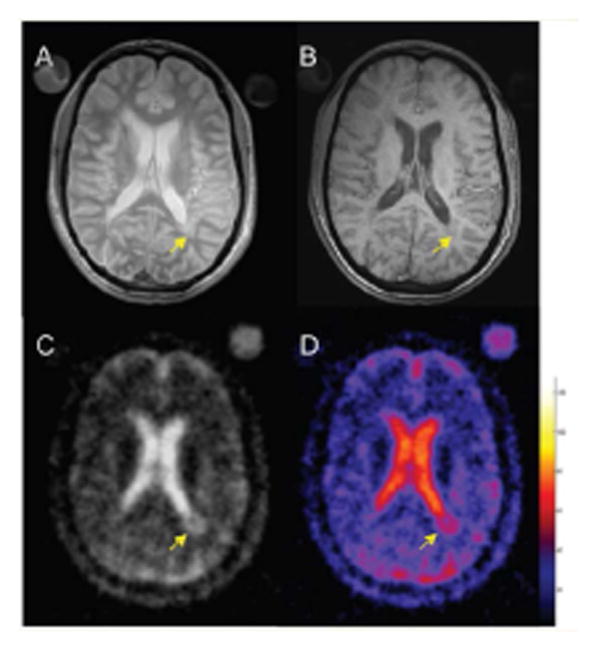
Selected brain axial proton density (A), T1-weighted (B), 23Na images (C) and corresponding TSC map (D) from an MS patient. The arrow indicates a hypointense periventricular lesion (B) that shows a higher TSC value. The color bar represents the TSC values (mM). Reproduced by Inglese et al. 2010 by permission of Oxford University Press.
Fig. 4.
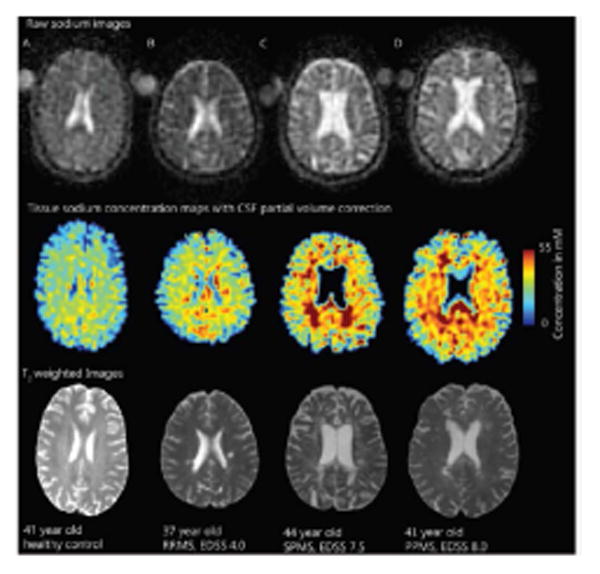
Global 23Na concentration across MS phenotypes. Raw 23Na images in 23Na space (top), tissue 23Na maps with CSF partial volume correction (middle) and T2-weighted images (bottom) registered to the T1 volumetric scan in controls (A) and patients with MS (B-C-D). Increased 23Na is seen in relapsing remitting- MS patients lesions (B) and, more extensively, in lesions and normal appearing white matter of secondary- (C) and primary- (D) progressive MS patients. Reproduced from Paling et al.2013 by permission of Oxford University Press.
Brain regional analysis of TSC distribution has shown a limited involvement of the NAWM (brainstem, cerebellum and temporal poles) in the early stage of the disease, and a widespread TSC increase, involving the entire brain, in more advanced MS (63). In particular, while in PP patients TSC increase seems to be restricted to the motor system, in SP patients it is more diffuse, involving also frontal, limbic and visual cortex, deep GM and cerebellum (Fig. 5) (65).
Fig. 5.
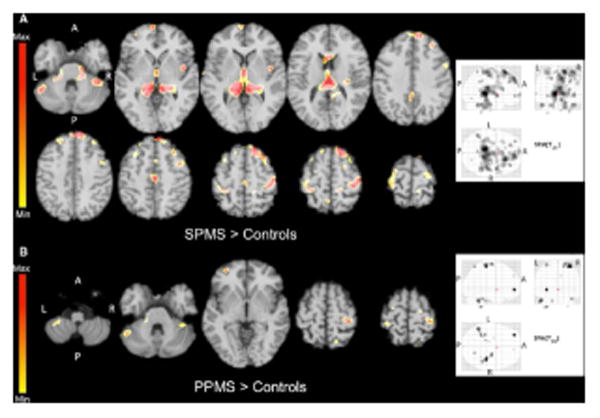
Statistical mapping of TSC increases in secondary progressive MS patients relative to controls (A) and in primary progressive MS patients relative to controls (B). In order to reduce CSF contamination grey matter, normal appearing white matter and T2 lesion masks were applied onto the co-registered quantitative sodium concentration maps to obtain TSC distribution maps of each compartment for each patient. Reproduced from Maarouf et al. 2013 by permission of Springer Ltd.
In both relapsing and progressive patients, TSC shows only a modest correlation with clinical disability (61, 64) and a weak correlation with lesion load and GM atrophy (61, 63). The correlation between TSC increase, clinical disability and MRI parameters of tissue loss, although present and consistently replicated across studies, is only modest; this could indicate that TSC reflects not only the irreversible neuronal loss responsible for clinical disability, but also the potentially reversible neuronal functional damage and could therefore be especially useful as predictive factor of clinical outcome. Supporting this hypothesis, only a small overlap has been identified between local brain atrophy and regions showing TSC increase (65); moreover, disability seems to correlate with NAWM TSC but not with WM fraction (64).
Future perspectives
23Na MRI allows direct visualization, in vivo, of ongoing cellular metabolic dysfunction and death. Unfortunately, the impossibility to determine if TSC increase is linked to an accumulation of intracellular 23Na or an increase in extracellular volume represents a major limitation. TSC can be helpful in monitoring the occurrence of tissue injury and disability, but it is not useful in discriminating the metabolic dysfunction from the irreversible cellular damage. Metabolic changes that affect 23Na exchange across cells membrane influence the intracellular sodium concentration (ISC) (27) that could therefore be considered as a pure functional marker. Currently there are three MRI techniques that allow the in vivo measurement of ISC (shift reagents–SRs; inversion recovery-IR pulses and multiple quantum filters-MQFs) but, considering the toxicity of SRs, only two of them are applicable in human studies (66-71). The IR technique is based on the assumption of a different longitudinal relaxation time of the 23Na nuclei in the intra- and extra-cellular compartments. Unfortunately, sodium IR imaging of the human brain in vivo is complicated by specific absorption rate limitations.
The MQF technique is based on the different transverse relaxation properties of the 23Na nuclei in different compartments and it allows, in biological tissues, the detection of the signal coming primarily from the intracellular 23Na (72-75). Because of the weak nature of the multi-quantum sodium MR signal, the application of high and ultra-high fields is particularly suited for MQF 23Na MRI. Others limitations of the MQF technique are: (i) the low SNR, which might be improved developing specific multichannel receive arrays, obtaining a reduction of imaging time and an increased spatial resolution; (ii) the sensitivity to B0– and B1– field inhomogeneities, that can be effectively minimized applying a B0–inhomogeneity insensitive TQF acquisition (66), and correcting in post–processing with the help of auxiliary B1–maps (67).
Combining SQ and triple-quantum filtered (TQF) 23Na MRI, it is possible to quantify TSC and intracellular sodium molar fraction (ISMF); from these measures it is possible to derive ISC and intracellular 23Na volume fraction (ISVF), an indirect measure of extracellular 23Na concentration (59) (Fig. 6). In these experiments, it is recommended to choose the TE in order to optimize the TQF signal, which is much weaker than the SQ and to acquire both TQF and SQ with the same TE, to keep the same distortions in TQF and SQ data; unfortunately, such a choice, while optimizing TQF acquisition, leads to an unavoidable signal loss of 40% in SQ images. Finally, it is important to remember that TQF signal is mainly, but not exclusively, generated by intracellular 23Na, therefore, possible contribution to TQF signal from extracellular 23Na has to be taken into account when interpreting the results. Nevertheless, as long as the intracellular 23Na values experimentally obtained are within expected physiological range, this bias can be considered small enough not to significantly affect the results. Over time, the development of new tissue models and acquisition schemes might offer a better solution to this problem.
Fig. 6.
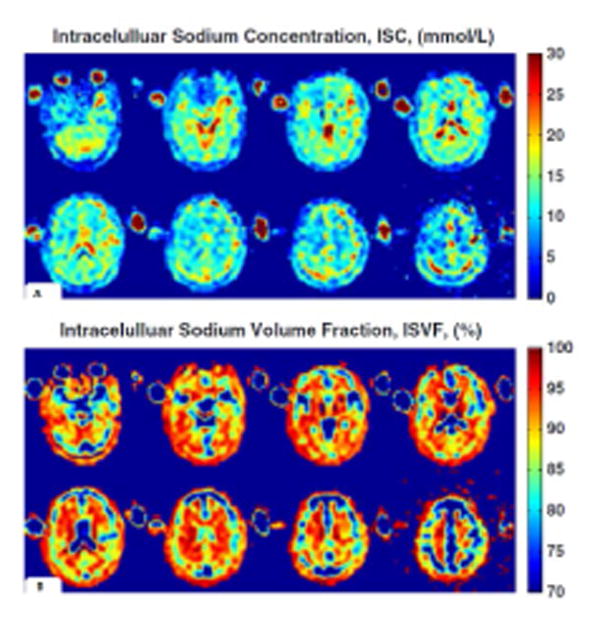
ISC and ISVF quantification. ISC map (a) and ISVF map (b) derived from MRI measurements of a healthy young 27-year-old male. ISCs of the grey matter and white matter regions are relatively uniform, while ISVF for white matter is higher than for grey matter, consistently with previous findings obtained with invasive methods in animals or ex vivo human brain tissue. Reproduced from Fleysher et al. 2013 by permission of John Wiley & Sons, Ltd.
In the future, technical improvements of 23Na MRI imaging should focus on the distinction of intra- from extra-cellular component of 23Na increase (67), while clinical applications should consider the combined use of 23Na MRI with different MRI modalities sensitive to neuroaxonal loss (76), not only in cross-sectional but also in longitudinal studies.
In MS patients, TSC and ISC increase might indicate axonal dysfunction, offering insights in axonal metabolism before the generation of stable, irreversible, axonal damage they could be a putative target for therapeutic interventions (77). TSC and the more technically challenging ISC, might enable in vivo assessment of the metabolic state on the brain and identification of an ‘intervention window’, providing a better tool to investigate the neuroprotective effects of experimental therapies and to monitor the response to putative neuroprotective agents and 23Na blockers in clinical trials. 23Na imaging, as well as the combined application of different MRI modalities such as MR spectroscopy and diffusion tensor imaging, could also be helpful in studying and understanding the role of energy failure, clarifying MS pathophysiology in comparison with others neuroinflammatory conditions (e.g. neuromyelitis optica, and acute disseminated encephalomyelitis).
Finally, the associations between TSC increase, disability and progressive course, identified in cross-sectional studies, need further confirmation from longitudinal evaluations. Analyzing the presence and degree of 23Na accumulation over time, would be important to clarify the role of 23Na increase as predictive marker of disease course. If confirmed and validated in longitudinal studies, 23Na concentration could therefore be utilized for identification of patients at higher risk of progression, candidate to more aggressive therapeutic approaches.
Acknowledgments
This study was supported in part by NIH grant R56 NS079116-01A1 and by the Noto Foundation (M.I.). M.P. is supported by a research fellowship grant from Fondazione Italiana Sclerosi Multipla (FISM-2013/B/7).
List of abbreviations (excluding standard abbreviations)
- MS
multiple sclerosis
- CIS
clinically isolated syndrome
- PP
primary progressive
- RR
relapsing remitting
- SP
secondary progressive
- 23Na
sodium
- SNR
signal to noise ratio
- 1H
proton
- WM
white matter
- NMR
nuclear magnetic resonance
- 20Ca
calcium
- EAE
experimental autoimmune encephalomyelitis
- NAWM
normal appearing white matter
- GM
grey matter
- TSC
total sodium concentration
- EDSS
Expanded Disability Status Scale
- ISC
intracellular sodium concentration
- SRs
shift reagents–SRs
- IR
inversion recovery
- MQFs
multiple quantum filters
- SQ
single quantum
- TQF
triple-quantum filtered
- ISMF
intracellular sodium molar fraction
- ISVF
intracellular 23Na volume fraction
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.International Multiple Sclerosis Genetics C, Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, Shah TS, Spencer C, Booth D, Goris A, Oturai A, Saarela J, Fontaine B, Hemmer B, Martin C, Zipp F, D'Alfonso S, Martinelli-Boneschi F, Taylor B, Harbo HF, Kockum I, Hillert J, Olsson T, Ban M, Oksenberg JR, Hintzen R, Barcellos LF, Wellcome Trust Case Control C, International IBDGC. Agliardi C, Alfredsson L, Alizadeh M, Anderson C, Andrews R, Sondergaard HB, Baker A, Band G, Baranzini SE, Barizzone N, Barrett J, Bellenguez C, Bergamaschi L, Bernardinelli L, Berthele A, Biberacher V, Binder TM, Blackburn H, Bomfim IL, Brambilla P, Broadley S, Brochet B, Brundin L, Buck D, Butzkueven H, Caillier SJ, Camu W, Carpentier W, Cavalla P, Celius EG, Coman I, Comi G, Corrado L, Cosemans L, Cournu-Rebeix I, Cree BA, Cusi D, Damotte V, Defer G, Delgado SR, Deloukas P, di Sapio A, Dilthey AT, Donnelly P, Dubois B, Duddy M, Edkins S, Elovaara I, Esposito F, Evangelou N, Fiddes B, Field J, Franke A, Freeman C, Frohlich IY, Galimberti D, Gieger C, Gourraud PA, Graetz C, Graham A, Grummel V, Guaschino C, Hadjixenofontos A, Hakonarson H, Halfpenny C, Hall G, Hall P, Hamsten A, Harley J, Harrower T, Hawkins C, Hellenthal G, Hillier C, Hobart J, Hoshi M, Hunt SE, Jagodic M, Jelcic I, Jochim A, Kendall B, Kermode A, Kilpatrick T, Koivisto K, Konidari I, Korn T, Kronsbein H, Langford C, Larsson M, Lathrop M, Lebrun-Frenay C, Lechner-Scott J, Lee MH, Leone MA, Leppa V, Liberatore G, Lie BA, Lill CM, Linden M, Link J, Luessi F, Lycke J, Macciardi F, Mannisto S, Manrique CP, Martin R, Martinelli V, Mason D, Mazibrada G, McCabe C, Mero IL, Mescheriakova J, Moutsianas L, Myhr KM, Nagels G, Nicholas R, Nilsson P, Piehl F, Pirinen M, Price SE, Quach H, Reunanen M, Robberecht W, Robertson NP, Rodegher M, Rog D, Salvetti M, Schnetz-Boutaud NC, Sellebjerg F, Selter RC, Schaefer C, Shaunak S, Shen L, Shields S, Siffrin V, Slee M, Sorensen PS, Sorosina M, Sospedra M, Spurkland A, Strange A, Sundqvist E, Thijs V, Thorpe J, Ticca A, Tienari P, van Duijn C, Visser EM, Vucic S, Westerlind H, Wiley JS, Wilkins A, Wilson JF, Winkelmann J, Zajicek J, Zindler E, Haines JL, Pericak-Vance MA, Ivinson AJ, Stewart G, Hafler D, Hauser SL, Compston A, McVean G, De Jager P, Sawcer SJ, McCauley JL. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nature genetics. 2013;45(11):1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O'Connor P, Sandberg-Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Annals of neurology. 2011;69(2):292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, Wolinsky JS, Balcer LJ, Banwell B, Barkhof F, Bebo B, Jr, Calabresi PA, Clanet M, Comi G, Fox RJ, Freedman MS, Goodman AD, Inglese M, Kappos L, Kieseier BC, Lincoln JA, Lubetzki C, Miller AE, Montalban X, O'Connor PW, Petkau J, Pozzilli C, Rudick RA, Sormani MP, Stuve O, Waubant E, Polman CH. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–286. doi: 10.1212/WNL.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waxman SG. Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis-current status. Nature clinical practice Neurology. 2008;4(3):159–169. doi: 10.1038/ncpneuro0735. [DOI] [PubMed] [Google Scholar]
- 6.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. The New England journal of medicine. 1998;338(5):278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 7.Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annual review of neuroscience. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- 8.Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, Gudz T, Macklin WB, Lewis DA, Fox RJ, Rudick R, Mirnics K, Trapp BD. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Annals of neurology. 2006;59(3):478–489. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- 9.Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM, Mahad DJ. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Annals of neurology. 2011;69(3):481–492. doi: 10.1002/ana.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cambron M, D'Haeseleer M, Laureys G, Clinckers R, Debruyne J, De Keyser J. White-matter astrocytes, axonal energy metabolism, and axonal degeneration in multiple sclerosis. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32(3):413–424. doi: 10.1038/jcbfm.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nature medicine. 2000;6(1):67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 12.Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nature reviews Neurology. 2012;8(11):647–656. doi: 10.1038/nrneurol.2012.168. [DOI] [PubMed] [Google Scholar]
- 13.Lassmann H. Mechanisms of white matter damage in multiple sclerosis. Glia. 2014;62(11):1816–1830. doi: 10.1002/glia.22597. [DOI] [PubMed] [Google Scholar]
- 14.Smith KJ. Sodium channels and multiple sclerosis: roles in symptom production, damage and therapy. Brain pathology. 2007;17(2):230–242. doi: 10.1111/j.1750-3639.2007.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moll C, Mourre C, Lazdunski M, Ulrich J. Increase of sodium channels in demyelinated lesions of multiple sclerosis. Brain research. 1991;556(2):311–316. doi: 10.1016/0006-8993(91)90321-l. [DOI] [PubMed] [Google Scholar]
- 16.Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(21):8168–8173. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Malley HA, Shreiner AB, Chen GH, Huffnagle GB, Isom LL. Loss of Na+ channel beta2 subunits is neuroprotective in a mouse model of multiple sclerosis. Molecular and cellular neurosciences. 2009;40(2):143–155. doi: 10.1016/j.mcn.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boada FE, G J, Shen GX, Chang SY, Thulborn KR. Fast three dimensional sodium imaging. Magn Reson Med. 1997:37. doi: 10.1002/mrm.1910370512. [DOI] [PubMed] [Google Scholar]
- 19.Thulborn KR, Gindin TS, Davis D, Erb P. Comprehensive MR imaging protocol for stroke management: tissue sodium concentration as a measure of tissue viability in nonhuman primate studies and in clinical studies. Radiology. 1999;213(1):156–166. doi: 10.1148/radiology.213.1.r99se15156. [DOI] [PubMed] [Google Scholar]
- 20.Winter PM, Bansal N. Triple-quantum-filtered (23)Na NMR spectroscopy of subcutaneously implanted 9l gliosarcoma in the rat in the presence of TmDOTP(5-1) Journal of magnetic resonance. 2001;152(1):70–78. doi: 10.1006/jmre.2001.2390. [DOI] [PubMed] [Google Scholar]
- 21.Winter PM, Seshan V, Makos JD, Sherry AD, Malloy CR, Bansal N. Quantitation of intracellular [Na+] in vivo by using TmDOTP5- as an NMR shift reagent and extracellular marker. Journal of applied physiology. 1998;85(5):1806–1812. doi: 10.1152/jappl.1998.85.5.1806. [DOI] [PubMed] [Google Scholar]
- 22.Woodward DL, Reed DJ, Woodbury DM. Extracellular space of rat cerebral cortex. The American journal of physiology. 1967;212(2):367–370. doi: 10.1152/ajplegacy.1967.212.2.367. [DOI] [PubMed] [Google Scholar]
- 23.Thulborn KR, Davis D, Adams H, Gindin T, Zhou J. Quantitative tissue sodium concentration mapping of the growth of focal cerebral tumors with sodium magnetic resonance imaging. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine/Society of Magnetic Resonance in Medicine. 1999;41(2):351–359. doi: 10.1002/(sici)1522-2594(199902)41:2<351::aid-mrm20>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 24.Koch KS, Leffert HL. Increased sodium ion influx is necessary to initiate rat hepatocyte proliferation. Cell. 1979;18(1):153–163. doi: 10.1016/0092-8674(79)90364-7. [DOI] [PubMed] [Google Scholar]
- 25.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. The Journal of physiology. 1952;117(4):500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lang F. Mechanisms and significance of cell volume regulation. Journal of the American College of Nutrition. 2007;26(5 Suppl):613S–623S. doi: 10.1080/07315724.2007.10719667. [DOI] [PubMed] [Google Scholar]
- 27.Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer research. 1987;47(12):3039–3051. [PubMed] [Google Scholar]
- 28.Cameron IL, Smith NK, Pool TB, Sparks RL. Intracellular concentration of sodium and other elements as related to mitogenesis and oncogenesis in vivo. Cancer research. 1980;40(5):1493–1500. [PubMed] [Google Scholar]
- 29.Nagy I, Lustyik G, Lukacs G, Nagy V, Balazs G. Correlation of malignancy with the intracellular Na+:K+ ratio in human thyroid tumors. Cancer research. 1983;43(11):5395–5402. [PubMed] [Google Scholar]
- 30.Thulborn KR, Davis D, Snyder J, Yonas H, Kassam A. Sodium MR imaging of acute and subacute stroke for assessment of tissue viability. Neuroimaging clinics of North America. 2005;15(3):639–653. xi–xii. doi: 10.1016/j.nic.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. The Lancet Neurology. 2009;8(3):280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- 32.Rush AM, Dib-Hajj SD, Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. The Journal of physiology. 2005;564(Pt 3):803–815. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.England JD, Gamboni F, Levinson SR. Increased numbers of sodium channels form along demyelinated axons. Brain research. 1991;548(1-2):334–337. doi: 10.1016/0006-8993(91)91144-p. [DOI] [PubMed] [Google Scholar]
- 34.Waxman SG. Ions, energy and axonal injury: towards a molecular neurology of multiple sclerosis. Trends in molecular medicine. 2006;12(5):192–195. doi: 10.1016/j.molmed.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Lassmann H. Multiple sclerosis: is there neurodegeneration independent from inflammation? Journal of the neurological sciences. 2007;259(1-2):3–6. doi: 10.1016/j.jns.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain: a journal of neurology. 2009;132(Pt 5):1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stys PK. General mechanisms of axonal damage and its prevention. Journal of the neurological sciences. 2005;233(1-2):3–13. doi: 10.1016/j.jns.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 38.Nikolaeva MA, Mukherjee B, Stys PK. Na+-dependent sources of intra-axonal Ca2+ release in rat optic nerve during in vitro chemical ischemia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25(43):9960–9967. doi: 10.1523/JNEUROSCI.2003-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waxman SG. Conduction in Myelinated, Unmyelinated, and Demyelinated Fibers. Arch Neurol. 1977:34. doi: 10.1001/archneur.1977.00500220019003. [DOI] [PubMed] [Google Scholar]
- 40.Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1992;12(2):430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Craner MJ, Damarjian TG, Liu S, Hains BC, Lo AC, Black JA, Newcombe J, Cuzner ML, Waxman SG. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia. 2005;49(2):220–229. doi: 10.1002/glia.20112. [DOI] [PubMed] [Google Scholar]
- 42.Black JA, Newcombe J, Waxman SG. Astrocytes within multiple sclerosis lesions upregulate sodium channel Nav1.5. Brain : a journal of neurology. 2010;133(Pt 3):835–846. doi: 10.1093/brain/awq003. [DOI] [PubMed] [Google Scholar]
- 43.Blokhin A, Vyshkina T, Komoly S, Kalman B. Variations in mitochondrial DNA copy numbers in MS brains. Journal of molecular neuroscience : MN. 2008;35(3):283–287. doi: 10.1007/s12031-008-9115-1. [DOI] [PubMed] [Google Scholar]
- 44.Black JA, Dib-Hajj S, Baker D, Newcombe J, Cuzner ML, Waxman SG. Sensory neuron-specific sodium channel SNS is abnormally expressed in the brains of mice with experimental allergic encephalomyelitis and humans with multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(21):11598–11602. doi: 10.1073/pnas.97.21.11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stys PK, Ransom BR, Waxman SG. Tertiary and quaternary local anesthetics protect CNS white matter from anoxic injury at concentrations that do not block excitability. Journal of neurophysiology. 1992;67(1):236–240. doi: 10.1152/jn.1992.67.1.236. [DOI] [PubMed] [Google Scholar]
- 46.Stys PK, Lesiuk H. Correlation between electrophysiological effects of mexiletine and ischemic protection in central nervous system white matter. Neuroscience. 1996;71(1):27–36. doi: 10.1016/0306-4522(95)00430-0. [DOI] [PubMed] [Google Scholar]
- 47.Fern R, Ransom BR, Stys PK, Waxman SG. Pharmacological protection of CNS white matter during anoxia: actions of phenytoin, carbamazepine and diazepam. The Journal of pharmacology and experimental therapeutics. 1993;266(3):1549–1555. [PubMed] [Google Scholar]
- 48.Stys PK. Protective effects of antiarrhythmic agents against anoxic injury in CNS white matter. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1995;15(3):425–432. doi: 10.1038/jcbfm.1995.53. [DOI] [PubMed] [Google Scholar]
- 49.Bechtold DA, Kapoor R, Smith KJ. Axonal protection using flecainide in experimental autoimmune encephalomyelitis. Annals of neurology. 2004;55(5):607–616. doi: 10.1002/ana.20045. [DOI] [PubMed] [Google Scholar]
- 50.Lo AC, Saab CY, Black JA, Waxman SG. Phenytoin protects spinal cord axons and preserves axonal conduction and neurological function in a model of neuroinflammation in vivo. Journal of neurophysiology. 2003;90(5):3566–3571. doi: 10.1152/jn.00434.2003. [DOI] [PubMed] [Google Scholar]
- 51.Black JA, Liu S, Carrithers M, Carrithers LM, Waxman SG. Exacerbation of experimental autoimmune encephalomyelitis after withdrawal of phenytoin and carbamazepine. Annals of neurology. 2007;62(1):21–33. doi: 10.1002/ana.21172. [DOI] [PubMed] [Google Scholar]
- 52.Bechtold DA, Miller SJ, Dawson AC, Sun Y, Kapoor R, Berry D, Smith KJ. Axonal protection achieved in a model of multiple sclerosis using lamotrigine. Journal of neurology. 2006;253(12):1542–1551. doi: 10.1007/s00415-006-0204-1. [DOI] [PubMed] [Google Scholar]
- 53.Al-Izki S, Pryce G, Hankey DJ, Lidster K, von Kutzleben SM, Browne L, Clutterbuck L, Posada C, Edith Chan AW, Amor S, Perkins V, Gerritsen WH, Ummenthum K, Peferoen-Baert R, van der Valk P, Montoya A, Joel SP, Garthwaite J, Giovannoni G, Selwood DL, Baker D. Lesional-targeting of neuroprotection to the inflammatory penumbra in experimental multiple sclerosis. Brain : a journal of neurology. 2014;137(Pt 1):92–108. doi: 10.1093/brain/awt324. [DOI] [PubMed] [Google Scholar]
- 54.Kapoor R, Furby J, Hayton T, Smith KJ, Altmann DR, Brenner R, Chataway J, Hughes RA, Miller DH. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: a randomised, double-blind, placebo-controlled, parallel-group trial. The Lancet Neurology. 2010;9(7):681–688. doi: 10.1016/S1474-4422(10)70131-9. [DOI] [PubMed] [Google Scholar]
- 55.Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, Vincent A, Fugger L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nature medicine. 2007;13(12):1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- 56.Vergo S, Craner MJ, Etzensperger R, Attfield K, Friese MA, Newcombe J, Esiri M, Fugger L. Acid-sensing ion channel 1 is involved in both axonal injury and demyelination in multiple sclerosis and its animal model. Brain : a journal of neurology. 2011;134(Pt 2):571–584. doi: 10.1093/brain/awq337. [DOI] [PubMed] [Google Scholar]
- 57.Arun T, Tomassini V, Sbardella E, de Ruiter MB, Matthews L, Leite MI, Gelineau-Morel R, Cavey A, Vergo S, Craner M, Fugger L, Rovira A, Jenkinson M, Palace J. Targeting ASIC1 in primary progressive multiple sclerosis: evidence of neuroprotection with amiloride. Brain : a journal of neurology. 2013;136(Pt 1):106–115. doi: 10.1093/brain/aws325. [DOI] [PubMed] [Google Scholar]
- 58.Maudsley AA, Hilal SK. Biological aspects of sodium-23 imaging. British medical bulletin. 1984;40(2):165–166. doi: 10.1093/oxfordjournals.bmb.a071964. [DOI] [PubMed] [Google Scholar]
- 59.Ouwerkerk R, Bleich KB, Gillen JS, Pomper MG, Bottomley PA. Tissue sodium concentration in human brain tumors as measured with 23Na MR imaging. Radiology. 2003;227(2):529–537. doi: 10.1148/radiol.2272020483. [DOI] [PubMed] [Google Scholar]
- 60.Mellon EA, Pilkinton DT, Clark CM, Elliott MA, Witschey WR, 2nd, Borthakur A, Reddy R. Sodium MR imaging detection of mild Alzheimer disease: preliminary study. AJNR American journal of neuroradiology. 2009;30(5):978–984. doi: 10.3174/ajnr.A1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inglese M, Madelin G, Oesingmann N, Babb JS, Wu W, Stoeckel B, Herbert J, Johnson G. Brain tissue sodium concentration in multiple sclerosis: a sodium imaging study at 3 tesla. Brain : a journal of neurology. 2010;133(Pt 3):847–857. doi: 10.1093/brain/awp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33(11):1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 63.Zaaraoui W, Konstandin S, Audoin B, Nagel AM, Rico A, Malikova I, Soulier E, Viout P, Confort-Gouny S, Cozzone PJ, Pelletier J, Schad LR, Ranjeva JP. Distribution of brain sodium accumulation correlates with disability in multiple sclerosis: a cross-sectional 23Na MR imaging study. Radiology. 2012;264(3):859–867. doi: 10.1148/radiol.12112680. [DOI] [PubMed] [Google Scholar]
- 64.Paling D, Solanky BS, Riemer F, Tozer DJ, Wheeler-Kingshott CA, Kapoor R, Golay X, Miller DH. Sodium accumulation is associated with disability and a progressive course in multiple sclerosis. Brain : a journal of neurology. 2013;136(Pt 7):2305–2317. doi: 10.1093/brain/awt149. [DOI] [PubMed] [Google Scholar]
- 65.Maarouf A, Audoin B, Konstandin S, Rico A, Soulier E, Reuter F, Le Troter A, Confort-Gouny S, Cozzone PJ, Guye M, Schad LR, Pelletier J, Ranjeva JP, Zaaraoui W. Topography of brain sodium accumulation in progressive multiple sclerosis. Magma. 2014;27(1):53–62. doi: 10.1007/s10334-013-0396-1. [DOI] [PubMed] [Google Scholar]
- 66.Fleysher L, Oesingmann N, Inglese M. B(0) inhomogeneity-insensitive triple-quantum-filtered sodium imaging using a 12-step phase-cycling scheme. NMR in biomedicine. 2010;23(10):1191–1198. doi: 10.1002/nbm.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fleysher L, Oesingmann N, Brown R, Sodickson DK, Wiggins GC, Inglese M. Noninvasive quantification of intracellular sodium in human brain using ultrahigh-field MRI. NMR in biomedicine. 2013;26(1):9–19. doi: 10.1002/nbm.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Madelin G, Kline R, Walvick R, Regatte RR. A method for estimating intracellular sodium concentration and extracellular volume fraction in brain in vivo using sodium magnetic resonance imaging. Scientific reports. 2014;4:4763. doi: 10.1038/srep04763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stobbe R, Beaulieu C. In vivo sodium magnetic resonance imaging of the human brain using soft inversion recovery fluid attenuation. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine/Society of Magnetic Resonance in Medicine. 2005;54(5):1305–1310. doi: 10.1002/mrm.20696. [DOI] [PubMed] [Google Scholar]
- 70.Tsang A, Stobbe RW, Beaulieu C. Triple-quantum-filtered sodium imaging of the human brain at 4.7 T. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine/Society of Magnetic Resonance in Medicine. 2012;67(6):1633–1643. doi: 10.1002/mrm.23147. [DOI] [PubMed] [Google Scholar]
- 71.Tsang A, Stobbe RW, Beaulieu C. In vivo double quantum filtered sodium magnetic resonance imaging of human brain. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine/Society of Magnetic Resonance in Medicine. 2014 [Google Scholar]
- 72.Navon G. Complete elimination of the extracellular 23Na NMR signal in triple quantum filtered spectra of rat hearts in the presence of shift reagents. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine/Society of Magnetic Resonance in Medicine. 1993;30(4):503–506. doi: 10.1002/mrm.1910300415. [DOI] [PubMed] [Google Scholar]
- 73.Eliav U, Shinar H, Navon G. The Formation of a 2nd-Rank Tensor in Na-23 Double-Quantum-Filtered Nmr as an Indicator for Order in a Biological Tissue. Journal of magnetic resonance. 1992;98(1):223–229. [Google Scholar]
- 74.Pekar J, Leigh JS. Detection of Biexponential Relaxation in Na-23 Facilitated by Double-Quantum Filtering. Journal of magnetic resonance. 1986;69(3):582–584. [Google Scholar]
- 75.Pekar J, Renshaw PF, Leigh JS. Selective Detection of Intracellular Sodium by Coherence-Transfer Nmr. Journal of magnetic resonance. 1987;72(1):159–161. [Google Scholar]
- 76.Paling D, Golay X, Wheeler-Kingshott C, Kapoor R, Miller D. Energy failure in multiple sclerosis and its investigation using MR techniques. Journal of neurology. 2011;258(12):2113–2127. doi: 10.1007/s00415-011-6117-7. [DOI] [PubMed] [Google Scholar]
- 77.Nikic I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, Bruck W, Bishop D, Misgeld T, Kerschensteiner M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nature medicine. 2011;17(4):495–499. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]