

Abstract
Chromosomal translocations that generate in-frame oncogenic gene fusions are powerful examples of success of targeted cancer therapies1–3. We discovered FGFR3-TACC3 (F3-T3) gene fusions in 3% of human glioblastoma4. Subsequent studies reported similar frequencies of F3-T3 in many other cancers, thus qualifying F3-T3 as one of the most recurrent fusions across all tumor types5,6. F3-T3 fusions are potent oncogenes that confer sensitivity to FGFR inhibitors but the downstream oncogenic signaling remains largely unknown2,4–6. Here, we report that tumors harboring F3-T3 cluster within transcriptional subgroups characterized by activation of mitochondrial functions. F3-T3 activates oxidative phosphorylation and mitochondrial biogenesis and induces sensitivity to inhibitors of oxidative metabolism. We show that phosphorylation of PIN4 is the signaling intermediate for the activation of mitochondrial metabolism. The F3-T3-PIN4 axis triggers peroxisome biogenesis and new protein synthesis. The anabolic response converges on PGC1α through intracellular ROS, enabling mitochondrial respiration and tumor growth. Our analyses uncover the oncogenic circuit engaged by F3-T3, expose reliance on mitochondrial respiration as unexpected therapeutic opportunity for F3-T3-positive tumors and provide a clue to the genetic alterations that initiate the chain of metabolic responses driving mitochondrial metabolism in cancer.
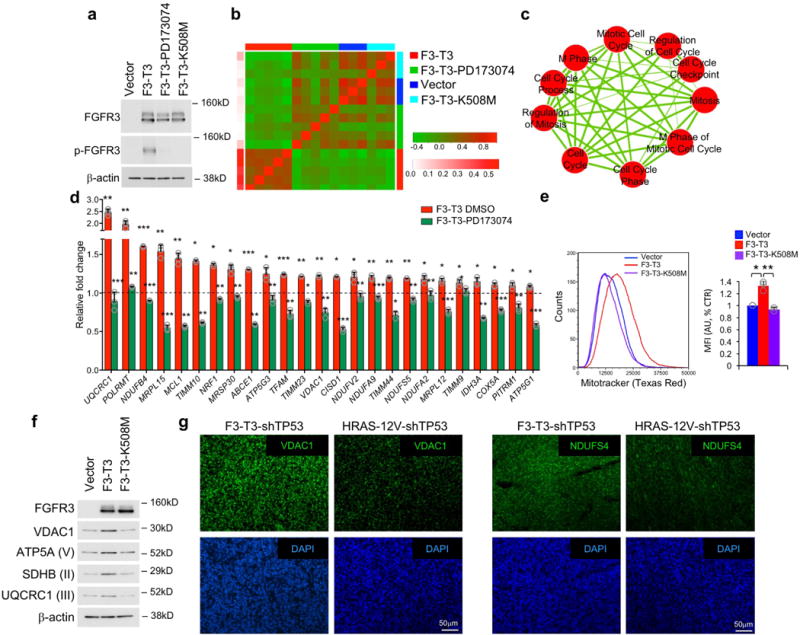
To interrogate the transcriptional changes elicited by F3-T3, we expressed F3-T3 in immortalized human astrocytes (HA) and compared gene expression profiles of cells treated with a specific inhibitor of FGFR tyrosine kinase (TK, PD173074) or vehicle. HA-F3-T3 were also compared with HA expressing the kinase-dead F3-T3 (F3-T3-K508M) and HA transduced with empty vector (Extended Data Fig. 1a). Hierarchical clustering based on genes differentially expressed between F3-T3 and PD173074-treated F3-T3 samples showed that HA-F3-T3 differed from the other three groups (Fig. 1a, Extended Data Fig. 1b). Gene Ontology-guided enrichment map demonstrated that, besides the expected enrichment for mitotic activity4, oxidative phosphorylation and mitochondrial biogenesis were the most significant categories enriched in HA-F3-T3 in each of the three independent comparisons (Fig. 1b, Extended Data Fig. 1c, Supplementary Table 1). We confirmed the expression changes of mitochondrial genes by quantitative RT-PCR (Extended Data Fig. 1d).
Figure 1. Activation of mitochondrial biogenesis and metabolism by F3-T3.
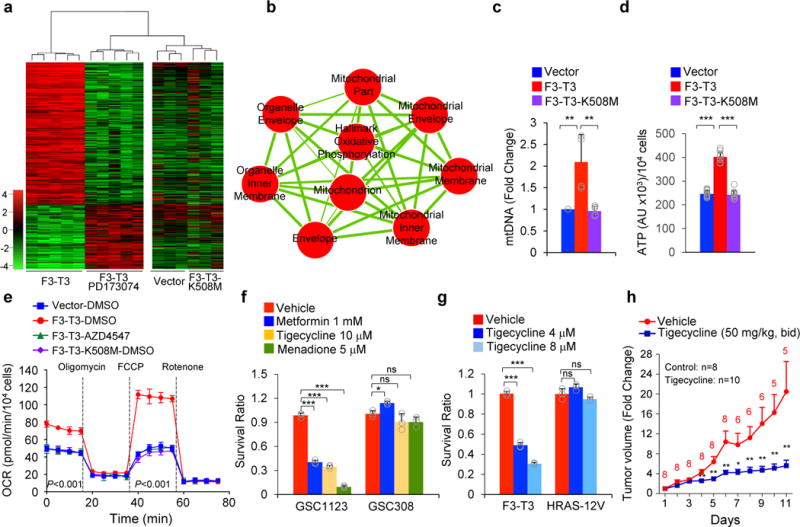
a, Hierarchical clustering of DEGs between HA-F3-T3, HA-F3-T3-PD173074 (n=5 biologically independent samples per group). HA-vector and HA-F3-T3-K508M (n=3 biologically independent samples per group) are included as controls. P-value, two-sided Mann-Whitney-Wilcoxon test. b, Enrichment map network of statistically significant GO categories (q-value 10E-6 in F3-T3 versus F3-T3-PD173074, F3-T3-K508M or vector). Nodes represent GO terms and lines their connectivity. Node size is proportional to number of genes in the category and line thickness indicates the fraction of genes shared between groups. c, qPCR of mitochondrial DNA (mtDNA) in HA-F3-T3, F3-T3-K508M or vector. d, Quantification of cellular ATP in HA as in c. e, OCR of HA-F3-T3 with or without AZD4547. f, Survival ratio of GSC1123 and GSC308 following treatment with the indicated mitochondrial inhibitors. g, Survival ratio of mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53 treated with vehicle or tigecycline. h, Tumor volume in mice treated with vehicle (n=8) or tigecycline (n=10). Data are fold change±s.e.m of controls. The number of mice remaining in the study at each time point is indicated. Data are representative of two (f, g) or three (e) independent experiments. Data are fold change±s.d. (c) or Mean±s.d. (d-g) of n=3 technical replicates (e-g); c, d n=6 and n=12 replicates, respectively, from two independent experiments. *P≤0.05, **P≤0.01, ***P≤0.001, two-tailed t-test, unequal variance.
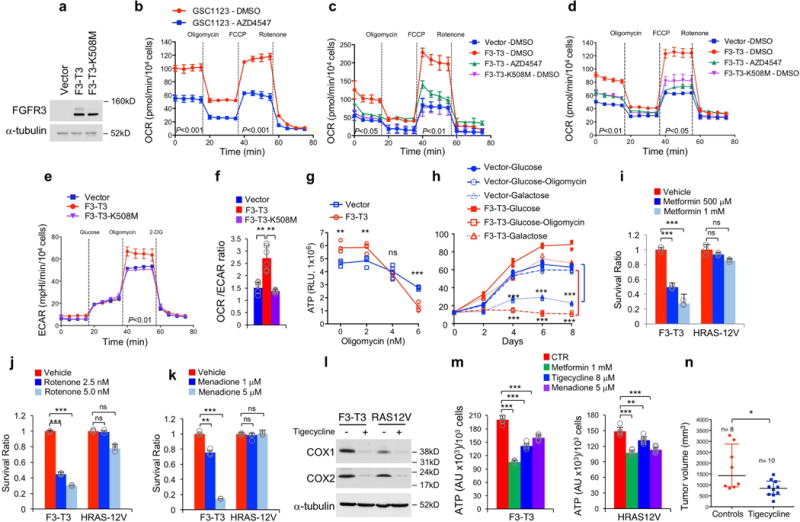
Compared with HA-vector and HA-F3-T3-K508M, HA-F3-T3 exhibited increased levels of mitochondrial DNA, mitochondrial mass (Mitotracker Red) and produced higher levels of ATP (Fig. 1c, d, Extended Data Fig. 1e). F3-T3 increased respiratory complex proteins (SDHB, UQCRC1, ATP5A1) and the mitochondrial membrane transporter VDAC1 (Extended Data Fig. 1f). We also found higher levels of VDAC1 and NDUFS4 in tumors generated by mouse Glioma Stem Cells (mGSCs) F3-T3-shTP53 than tumors formed by oncogenic HRAS-12V-shTP53 mGSCs4,7 (Extended Data Fig. 1g). Introduction of F3-T3 in HA, RPE and U251 elevated basal and maximal oxygen consumption rate (OCR) compared with cells transduced with F3-T3-K508M or empty vector and this effect was reversed by FGFR-TK inhibition with AZD4547 in cells expressing exogenous F3-T3 and human GBM-derived GSC1123 harboring endogenous F3-T34 (Fig. 1e, Extended Data Fig. 2a–d). F3-T3 elicited only a mild increase in the extracellular acidification rate (ECAR), leading to elevation of the OCR/ECAR ratio (Extended Data Fig. 2e, f). Following treatment with the inhibitor of ATP synthase oligomycin, HA-F3-T3 displayed reduced ATP levels and cell growth (>70%) but were resistant to the substitution of glucose with galactose in the culture media, a condition that imposes oxidative metabolism and markedly impacted HA-vector cell growth (Extended Data Fig. 2g, h). A 72 h treatment with the mitochondrial inhibitors metformin, menadione or tigecycline impaired growth of GSC1123 but was ineffective in GSC308 F3-T3-negative gliomaspheres4 (Fig. 1f). Similarly, mitochondrial inhibitors reduced viability of F3-T3-shTP53 mGSCs but did not affect HRAS12V-shTP53 mGSCs (Fig. 1g, Extended Data Fig. 2i–k). However, tigecycline decreased Cox1 and Cox2, two respiratory complex subunits translated by mitochondrial ribosomes8 and mitochondrial inhibitors reduced ATP production, indicating that these compounds were similarly active in both cell types (Extended Data Fig. 2l, m). We also found that treatment with tigecycline (50 mg/kg) suppressed tumor growth of F3-T3-shTP53 mGSCs glioma xenografts with >50% reduction of tumor volume after 6 days, the last day in which all controls were alive. At the end of the experiment (day 11), 3 of the 8 mice from the control group had been euthanized, whereas all the mice receiving tigecycline were alive (n=10, Fig. 1h, Extended Data Fig. 2n).
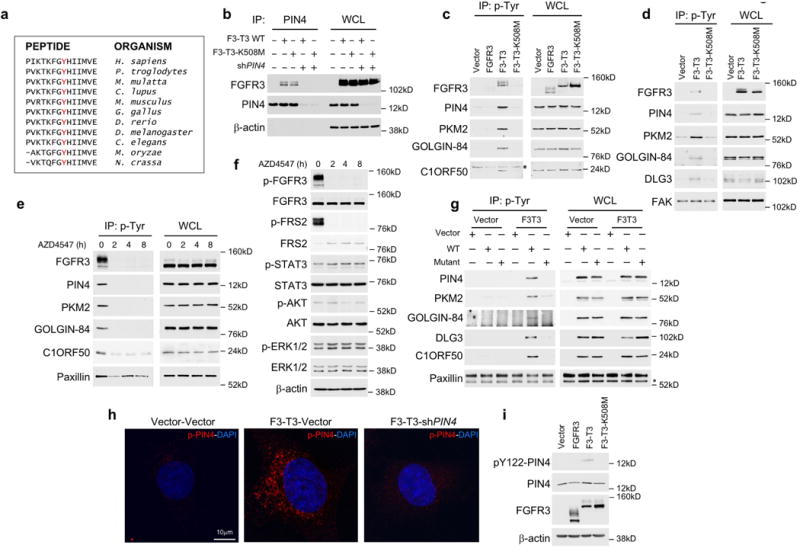
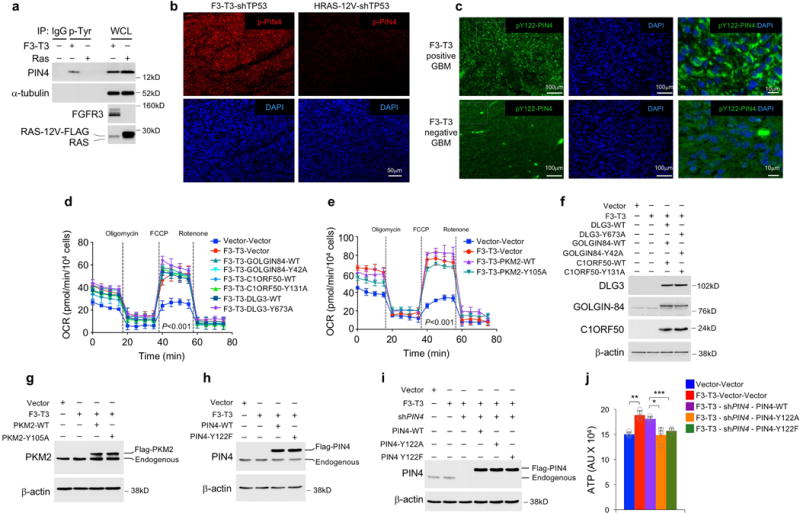
To identify F3-T3 substrates that drive oxidative metabolism, we performed anti-phosphotyrosine (anti-pY) immunoprecipitation of tryptic digests of total cellular proteins from HA-F3-T3, HA-F3-T3-K508M or HA-vector, followed by identification of phosphopeptides by LC/MS-MS (Supplementary Table 2). As expected, F3-T3 showed the largest phospho-tyrosine changes with FGFR3 Y-647 and TACC3 Y-684 scoring as #1 and #2 enriched phosphopeptides, respectively. Following F3-T3, the most enriched phosphopeptide was Y-122 of PIN4, a poorly studied homologue of the cancer driver PIN1 peptidyl-prolyl-trans-isomerase (Supplementary Table 2)9–11. Tyrosine-122 of PIN4 is conserved across evolution and we found that F3-T3 interacts with endogenous PIN4 (Extended Data Fig. 3a, b). Anti-phosphotyrosine immunoprecipitations revealed that only cells expressing active F3-T3 contained tyrosine phosphorylated PIN4, PKM2, DLG3, C1ORF50 and GOLGIN84 whereas tyrosine phosphorylated HGS was also present in FGFR3-expressing cells (Fig. 2a, Extended Data Fig. 3c, d). Treatment of GSC1123 with AZD4547 removed constitutive tyrosine phosphorylation of F3-T3, PIN4, PKM2, GOLGIN84 and C1ORF50 whereas phospho-ERK, phospho-Stat3 and phospho-AKT were not changed (Extended Data Fig. 3e, f). We confirmed F3-T3-specific tyrosine phosphorylation of exogenous wild type PIN4, PKM2, GOLGIN84, DLG3 and C1ORF50, but phosphorylation of the corresponding Y to A/F phospho-mutants was greatly reduced (Extended Data Fig. 3g). We generated and validated an anti-pY122-PIN4 phospho-specific antibody. The antibody detected PIN4 in cells expressing F3-T3 but not those transduced with vector, FGFR3 or F3-T3-K508M (Extended Data Fig. 3h, i). Tyrosine-phosphorylated PIN4 was readily detected in F3-T3-shTP53 mGSCs and xenografts but was absent in HRAS-12V-shTP53 mGSCs and corresponding tumors (Extended Data Fig. 4a, b). Immunostaining for phospho-Y122-PIN4 of primary human GBM revealed that tumors harboring F3-T3 (n=14) expressed much higher levels of pY122-PIN4 than tumors lacking F3-T3 fusions (n= 35, 15 of which harbored EGFR-Sept14, a different RTK gene fusion that signals through phospho-Stat312, Fig. 2b, Extended Data Fig. 4c).
Figure 2. Phosphorylation of PIN4 at Y122 affects mitochondrial metabolism.
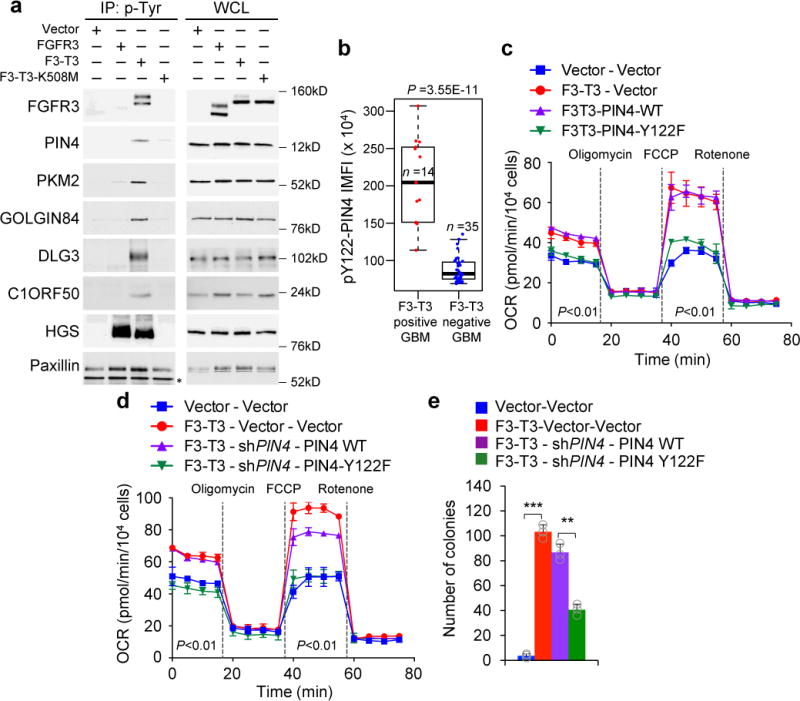
a, Immunoblot of phosphotyrosine immunoprecipitates from SF126 glioma cells (left panels). Right panels, whole cell lysates (WCL). Paxillin is loading control. b, Quantification of pY122-PIN4 integrated mean fluorescence intensity (IMFI) from F3-T3-positive and F3-T3-negative GBM. Box plot spans the first quartile to the third quartile and whiskers show the 1.5× interquartile range. P≤0.0001, two-sided Mann-Whitney test. c, OCR of HA-F3-T3 transduced with PIN4 WT, PIN4-Y122F or vector. d, OCR of HA-F3-T3 following silencing of PIN4 and reconstitution with PIN4-WT or PIN4-Y122F. e, Soft agar colony forming assay of HA treated as in d. Data in c-e are each Mean±s.d. of one representative experiment with n=3 technical replicates. Experiments were repeated three times with similar results. **P≤0.01, ***P≤0.001, two-tailed t-test, unequal variance.
Next, we expressed wild type and corresponding phospho-tyrosine mutants of PIN4, PKM2, DLG3, C1ORF50 and GOLGIN84 in HA-F3-T3 and measured oxidative metabolism. Expression of wild type and Y to A/F mutants of PKM2, DLG3, C1ORF50 and GOLGIN84 failed to impact the elevated OCR profile of HA-F3-T3 (Extended Data Fig. 4d–g). Conversely, PIN4-Y122F but not PIN4-WT reverted basal and maximum OCR of HA-F3-T3 to that of HA-vector (Fig. 2c, Extended Data Fig. 4h). We observed similar effects in HA-F3-T3 in which endogenous PIN4 had been silenced and replaced by the un-phosphorylatable Y122 mutants (Y122F, Y122A, Fig. 2d, Extended Data Fig. 4i). Reconstitution with PIN4-Y122 phospho-mutants also reversed the F3-T3-mediated increase of ATP and impaired soft agar clonogenicity (Fig. 2e, Extended Data Fig. 4j).
To identify the gene expression signature associated with F3-T3 in human tumors, we benchmarked different statistical methods for the analysis of imbalanced datasets using synthetic data and the GBM transcriptome from ATLAS-TCGA13. The combination of the EasyEnsemble (ee) undersampling technique with Mann-Whitney-Wilcoxon (MWW) test statistics (ee-MWW) exhibited the best performance for correct identification of imbalanced samples and reproducible clustering (Supplementary Information, Supplementary Table 3). We used ee-MWW to generate a ranked list of genes discriminating F3-T3-positive samples in the GBM-TCGA dataset and built a hierarchical cluster (confirmed by consensus clustering) including a small cluster of 9 F3-T3-positive samples and 9 fusion-like GBM (Fig. 3a, Extended Data Fig. 5a, Supplementary Table 4). The most significant biological processes enriched in F3-T3-positive GBM were mitochondrial categories (Fig. 3b, Extended Data Fig. 5b). Mitochondrial functions were also elevated in fusion-like GBM, which were enriched for amplification and high expression of mitochondrial RNA polymerase (POLRMT, Extended Data Fig. 5c–e). Immunostaining for oxidative phosphorylation biomarkers in an independent GBM cohort revealed that F3-T3-positive tumors expressed higher levels of mitochondrial proteins (Fig. 3c, Extended Data Fig. 5f). The ee-MWW method clustered tumors harboring other rare oncogenes (oncogenic ras in GBM and BRCA, EGFR-Sept14 gene fusion in GBM12) and identified their associated biological functions (Extended Data Fig. 5g–i, Supplementary Table 5). Using ee-MWW, we detected similarly small and homogeneous clusters of F3-T3-positive tumors enriched for mitochondrial categories in each tumor containing recurrent F3-T3 fusions in the ATLAS-TCGA dataset (pan-glioma, LUSC, HNSC, ESCA, CESC and BLCA, Extended Data Fig. 6a–k, Supplementary Table 6). The transcriptional similarity of F3-T3-positive glioma was confirmed by Topological Data Analysis14,15 (TDA, Extended Data Fig. 6l). Finally, expression of the F3-T3 fusion gene correlated with mitochondrial activities in a multi-cancer cohort (Extended Data Fig. 6m).
Figure 3. PGC1α and ESRRG are required for F3-T3-mediated mitochondrial metabolism and tumorigenesis.
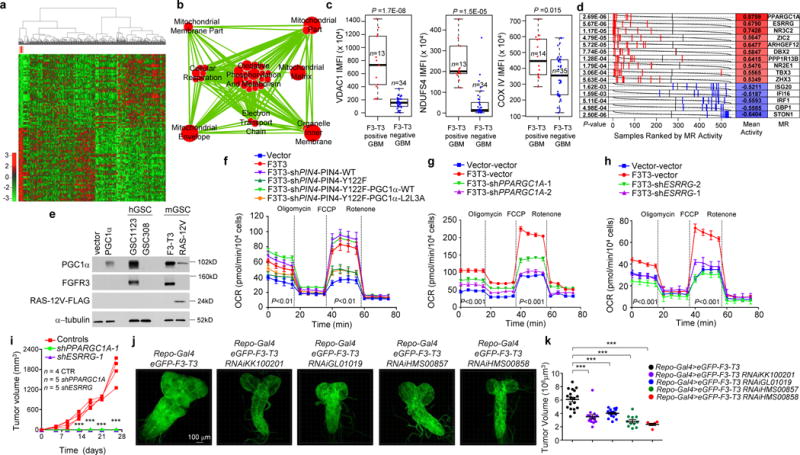
Hierarchical clustering of GBM (n=534) and normal brain (n=10) from the ATLAS-TCGA using DEGs in 9 F3-T3-positive samples (red). b, Enrichment map network of statistically significant GO categories in 9 F3-T3-positive samples (two-sided Mann-Whitney-Wilcoxon test q-value <0.001, NES>0.6). c, Quantification of IMFI of VDAC1, NDUFS4 and COXIV in F3-T3-positive and F3-T3-negative GBM. Box plot spans the first quartile to the third quartile and whiskers show the 1.5× interquartile range. P≤0.0001 (VDAC and NDUFS4); P≤0.05 (COXIV), two-sided Mann-Whitney test. d, MR activity in GBM. Grey curves represent the activity of each MR. Red and blue lines indicate individual GBM (n=534) displaying high or low MR activity, respectively. P-value, two-sided Mann-Whitney test. e, Immunoblot of PGC1α, in human (h) and mouse (m) GSCs. HA transduced with PGC1α or vector are controls. f, OCR of HA-F3-T3 following silencing of PIN4 and reconstitution with PIN4-WT or PIN4-Y122F, in the presence or the absence of PGC1α-WT or PGC1α-L2L3A. g, OCR of HA-F3-T3 transduced with shPPARGC1A. h, OCR of HA-F3-T3 transduced with shESRRG. i, Tumor growth of HA-F3-T3 transduced with vector (n=4), shPPARGC1A-1 (n=5) or shESRRG-1 (n=5). Tumor growth curves of individual mice are shown. j, Representative images of whole brain-ventral nerve cord complex optical projections from Drosophila larvae repo-Gal4>F3-T3 and repo-Gal4>F3-T3;RNAi-srl. k, Quantification of Drosophila larvae repo-Gal4>F3-T3 and repo-Gal4>F3-T3;RNAi-srl tumor volume. Data (Mean±s.d.) are from two (f, n=3 and n=5 technical replicates, respectively) or one (g, h, n=4 technical replicates; k, data are Mean±s.e.m., n=6–17 larvae) experiments. Experiments were repeated two to three times with similar results. **P≤0.01, ***P≤0.001 by two-tailed t-test, unequal variance (f-k).
To identify the transcription factors (TFs) that are causally related to the gene expression signature activated in F3-T3-positive glioma16 (master regulators, MRs), we assembled transcriptional networks from the GBM and pan-glioma datasets using the RGBM algorithm that was developed for the inference of gene regulatory networks17. In both datasets, the two most active MRs of F3-T3-positive tumors were PPARGC1A and ESRRG (coding for the PGC1α transcriptional coactivator and the nuclear receptor ERRγ, respectively; Fig. 3d, Extended Data Fig. 6n, Supplementary Table 7). Expression of PPARGC1A and ESRRG mRNAs was higher in F3-T3-positive than F3-T3-negative GBM (Extended Data Fig. 6o). As PGC1α is a coactivator of the estrogen-related receptor (ERR) subfamily of nuclear receptors and acts as MR of mitochondrial biogenesis and metabolism18,19, we asked whether PGC1α and ERRγ enable the mitochondrial functions implemented by F3-T3. Introduction of F3-T3 in HA expressing PIN4-WT increased PGC1α mRNA and protein and the expression of ROS detoxification genes20 (Extended Data Fig. 7a–d, Supplementary Table 7). Accordingly, PGC1α accumulated at higher levels in F3-T3-positive GSC1123 and F3-T3-shp53 mGSCs compared with F3-T3-negative GSC308 and HRAS-12V-shTP53 mGSCs, respectively (Fig. 3e). However, replacement of PIN4 with the un-phosphorylatable Y122F mutant blunted F3-T3-mediated induction of PGC1α (Extended Data Fig. 7a, c). Inhibition of mitochondrial metabolism and soft agar clonogenicity by PIN4-Y122F in HA-F3-T3 were both rescued by overexpression of PGC1α-WT. Conversely, PGC1α-L2L3A, a mutant in the nuclear receptor boxes L2 and L3 that are critical for binding ERRγ18 could not rescue F3-T3-mediated activation of mitochondrial metabolism in HA-F3-T3 expressing PIN4-Y122F (Fig. 3f, Extended Data Fig. 7e, f). Finally, loss of PGC1α by shRNAs and CRISPR-Cas9 gene editing reversed the activation of mitochondrial respiration by F3-T3 and depletion of ERRγ produced similar effects (Fig. 3g, h, Extended Data Fig. 7g–m). PGC1α silencing inhibited soft agar colony formation by HA-F3-T3 and impaired self-renewal of GSC1123 (Extended Data Fig. 7n–p). Silencing of either PGC1α or ERRγ extinguished tumor xenograft formation of HA-F3-T3 (Fig. 3i, Extended Data Fig. 7q) and impaired in vivo tumor growth of F3-T3-shTP53 mGSCs but not HRAS-12V-shTP53 mGSCs (Extended Data Fig. 7r–v). Next, we developed a brain tumor model in Drosophila by ectopically expressing F3-T3 from the specific glial driver repo-Gal421. Repo-Gal4-F3-T3 transgenic flies manifested glial neoplasia with enlargement and malformation of the larval brain lobe and ventral nerve cord, leading to larval lethality (Extended Data Fig. 8a, b). Cell number and proliferation of glial cells were enhanced in repo-Gal4-F3-T3 flies (Extended Data Fig. 8c, d). Cell-autonomous RNAi-mediated knockdown of spargel (srl, the Drosophila ortholog of PPARGC1A)22 in repo-Gal4-F3-T3 flies reduced glial tumor volume and decreased the number of Repo+ glial cells, without affecting repo-Gal4-driven F3-T3 protein or normal brain development (Fig. 3j, k, Extended Data Fig. 8e–g, 9a–d). Srl knockdown did not rescue Repo-Gal4-F3-T3 animals to adult viability, confirming that suppressors of glial neoplasia in Drosophila are infrequent rescuers of organismic lethality (Extended Data Fig. 9e)23. Srl knockdown in a Drosophila model of glioma driven by constitutively active EGFR (dEGFRλ) and PI3K (Dp110CAAX) in the glial lineage24 resulted in minor to no effects on tumor volume, thus highlighting the specific sensitivity of F3-T3 tumorigenesis to the perturbation of srl expression (Extended Data Fig. 9f, g). RNAi-mediated knockdown of estrogen-related receptor (ERR) also reduced F3-T3 glial tumor volume (Extended Data Fig. 9h, i).
To determine the mechanism by which pY122-PIN4 propagates F3-T3 signaling, we studied the subcellular compartmentalization of PIN4 and pY122-PIN4 and sought to uncover the set of cellular proteins interacting with PIN4. Unphosphorylated PIN4 was diffusely localized to cytoplasm and nuclear membrane whereas pY122-PIN4 concentrated on larger cytoplasmic vesicle-like structures that co-localized with F3-T3 (Extended Data Fig. 3h, 10a, b). From mass spectrometry analysis of PIN4 immunoaffinity complexes, we uncovered the peroxisomal biogenesis complex formed by Pex1 and Pex6 as the top ranking PIN4 interactor25. Other PIN4-associated proteins are implicated in vesicle formation and trafficking, nuclear and mitochondrial RNA metabolism and translation, ribosomal activity and nuclear pore/envelope functions (Supplementary Table 8, Extended Data Fig. 10c, d). Quantitative immunofluorescence revealed that Pex1 increased 2.7-fold in HA-F3-T3 without changes in PEX1 mRNA (Fig. 4a, b, Extended Data Fig. 10e, f). To ask whether F3-T3 signals through pY122-PIN4 to promote peroxisome biogenesis, we acutely transduced HA with F3-T3-expressing lentivirus, and found that both pY122-PIN4 and the total number of PMP70-positive peroxisomes were elevated after 4 days (4.3 fold increase of peroxisomes per cell, Fig. 4c, d, Extended Data Fig. 10g). The pY122-PIN4-positive cytoplasmic structures in HA-F3-T3, but not the unphosphorylated PIN4 in HA-vector, colocalized with PMP70, indicating that pY122-PIN4 trafficks to new peroxisomal membranes (Fig. 4e). The increased peroxisome biogenesis produced by acute expression of F3-T3 was annulled when F3-T3 was introduced in cells in which PIN4 had been replaced by the unphosphorylatable Y122F mutant (Fig. 4f). F3-T3 also induced a pY122-PIN4-dependent early increase of new protein synthesis (Fig. 4g, Extended Data Fig. 10h). Conversely, PGC1α and mitochondrial gene expression were unchanged 4 days after acute expression of F3-T3 but increased after 8 days (Fig. 4h, Extended Data Fig. 10i). Peroxisome biogenesis and new protein synthesis can both generate reactive oxygen species (ROS) and ROS are crucial inducers of PGC1α20,26,27. F3-T3 but not F3-T3-K508M elevated ROS at the 4-day time point and this effect required PIN4-Y122 phosphorylation (Fig. 4i, j, Extended Data Fig. 10j). Treatment of HA-F3-T3 with the ROS inhibitor N-acetyl-L-cysteine (NAC) eliminated ~70% of the increase of PGC1α by F3-T3 (Fig. 4k), thus indicating that ROS are largely responsible for elevation of PGC1α by F3-T3.
Figure 4. Expression of F3-T3 fusion induces peroxisome biogenesis through phosphorylation of Y122 of PIN4.
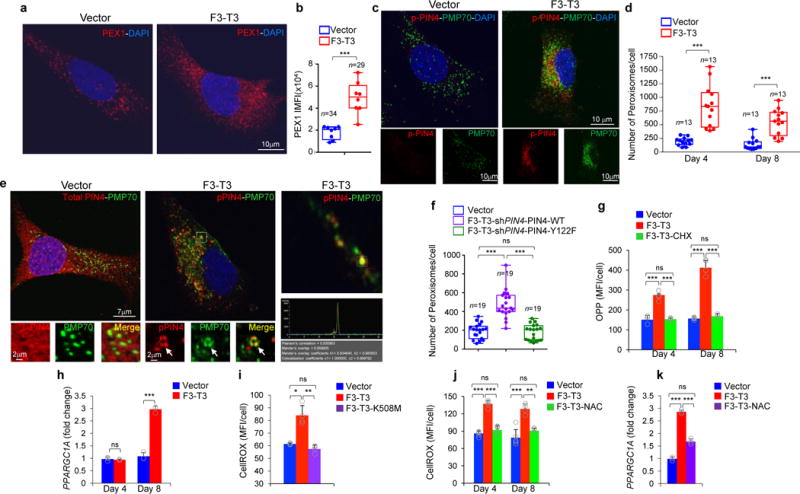
a, Representative confocal micrographs of PEX1 immunostaining (red) and DAPI (blue) in HA-vector and HA-F3-T3. b, Quantification of PEX1 IMFI in samples stained as in a (n=34 and 26 cells for vector and F3-T3, respectively). c, Representative confocal micrographs of double (upper panels) and single (lower panels) immunostaining for pY122-PIN4 (pPIN4, red) and PMP70 (green) in HA-vector and HA-F3-T3. d, Quantification of peroxisome number per cell 4 and 8 days after F3-T3 expression in HA (n=13 cells). e, Representative confocal micrographs of double immunostaining for total PIN4 and PMP70 in HA-vector (left panel) or pY122-PIN4 and PMP70 in HA-F3-T3 (middle panel). Arrowheads, pY122-PIN4-PMP70 co-localization (lower panel). Right panels, pY122-PIN4-PMP70 co-localization (upper panel) with corresponding spectral intensity profile (middle panel) and co-localization coefficient (lower panel). f, Quantification of peroxisome number per cell in HA-F3-T3 following silencing of PIN4 and expression of PIN4-WT or PIN4-Y122F (n=19 cells). g, Quantification of protein biosynthesis by OPP incorporation in HA treated as in d. Cycloheximide (CHX)-treated cultures are negative controls; (n=5 technical replicates). h, qRT-PCR for PPARGC1A in HA treated as in d (n=3 technical replicates). i, Quantification of cellular ROS in HA-vector, HA-F3-T3 and F3-T3-K508M. Bar plots from one representative experiment (n=4 technical replicates). j, Analysis of cellular ROS in HA treated as in d. Bar plots from one representative experiment (n=5 or 6 technical replicates). k, qRT-PCR for PPARGC1A in HA-vector or HA-F3-T3 treated with vehicle or NAC (n=3 technical replicates). Results in g-k are each Mean±s.d. P-values: b, d, f, two-sided Mann-Whitney-Wilcoxon test; g-k, two-tailed t-test unequal variance. Box plots span the first quartile to the third quartile and whiskers indicate the smallest and largest values. Experiment in panel i was repeated twice; all other experiments were repeated three times with similar results.
In conclusion, an integrated computational and experimental framework unraveled the chain of events propagated by F3-T3 in cancer. Signaling through pY122-PIN4 triggers vesicle trafficking to deliver building blocks for peroxisome biogenesis and new protein synthesis. The coordinated activation of these anabolic pathways results in the accumulation of ROS, which in turn elevate PGC1α-ERRγ and mitochondrial metabolism. Thus, rather than impinging exclusively on mitochondrial circuits, the oncogenic signals driving mitochondrial respiration operate within larger contexts of anabolic effectors. Dependency on mitochondrial metabolism of GBM harboring F3-T3 suggests that inhibitors of oxidative phosphorylation may be beneficial to patients carrying F3-T3-positive tumors.
METHODS
Datasets
Tumor sample data are from the Tumor Cancer Genome Atlas. Details of cohorts and samples analyzed are described in Supplementary Information.
Resampling methods for ranked list generation in imbalanced datasets
Preliminary testing and the eeMWW are reported in Supplementary Information.
Gene Ontology Networks
Gene Ontology enrichment was computed using the MWW test statistics for the genes positively regulated in tumors harboring F3-T3 or the other genetic alterations of interest (e.g. ras and EGFR-Sept14). The significant GO terms from MWW-GST analysis were further analyzed using the Enrichment Map28 application of Cytoscape29. In the network nodes represent the terms and edges represent known term interactions, and are defined by the number of shared genes between the pair of terms. Size of the nodes is proportional to statistical significance of the enrichment. The overlap between gene sets is computed according to the overlap coefficient (OC), defined as:
where and are two gene sets, and equals to the number of elements within set 30. We set a cutoff of to select the overlapping gene sets.
Correlation Analysis between GO NES and the expression of F3-T3
We selected 19 human samples harboring the F3-T3 fusions from Stransky et al.31 and the TCGA fusion gene Data Portal32. Starting from fastq data, we applied the chimeraScan pipeline33 to compute the total number of reads supporting the fusion (Supplementary Table 6m). From TCGA, we obtained the legacy level 3 RSEM counts of the samples. By using the EDASeq methodology34, we corrected the counts for GC content and applied full-quantile normalization. We transformed the normalized counts in the TPM abundance quantification, applied MWW-GST to each sample and collected the NES. We used the MDSigDB collections c5.bp, c5.mf, c5.cc and Hallmark collections of gene sets. We compared each gene set with the number of reads supporting the F3-T3 fusions by using the Spearman’s rank correlation index (Supplementary Table 6n). To test the correlation, we assumed the alternative hypothesis of the correlation greater than zero.
Assembly of the transcriptional interactomes
To identify the MRs of the gene expression signature activated in the F3-T3-positive glioma subgroup, we first assembled independent transcriptional networks from gene expression profiles (GEPs) of GBM and pan-glioma datasets using the Regularized Gradient Boosting Machine algorithm (RGBM)17 (package available on CRAN at https://cran.r-project.org/web/packages/RGBM/index.html). RGBM was used to identify regulators of the molecular subtypes of brain tumors17,35. We used GEP and a predefined list of 2,137 gene regulators/transcription factors (MRs) as input. This process was independently applied to obtain GBM and pan-glioma transcriptional interactomes comprising 430,104 (median regulon size: 203) and 300,969 (median regulon size: 141) transcriptional interactions respectively, of which 188,238 were overlapping.
Master Regulator (MR) Activity
To identify the MRs of the gene expression signature activated in F3-T3-positive glioma, we used a method that we previously described16. Briefly, the activity of a MR, defined as the index that quantifies the activation of the transcriptional program of that specific MR in each sample , is calculated as follows:
where is the expression level of the k-th positive target of the MR in the i-th sample, is the expression level of the j-th negative target of the MR in the i-th sample, the number of positive (negative) targets present in the regulon of the considered MR. If , the MR is activated in that particular sample, if , the MR is inversely activated, if it is deactivated. We used the Wilcoxon test to select MRs that showed a significant difference between the F3-T3 positive samples and all the others. In Supplementary Table 7a, b we present the list of MRs obtained by applying MR analysis ( ) and significance of differential activity <0.01.
Topological Data Analysis
Topological data analysis14,15 (TDA) of the panglioma dataset was based on the Mapper algorithm36. The topological network was built using the Ayasdi platform (http://www.ayasdi.com). Several open-source implementations of Mapper are available (https://github.com/MLWave/kepler-mapper, http://danifold.net/mapper/, https://github.com/RabadanLab/sakmapper, https://github.com/paultpearson/TDAmapper). TDA was performed using the expression matrix of the top 100 genes differentially expressed between F3-T3-positive tumors and the remaining tumors as shown in Extended Data Fig. 6a. Mapper uses a dimentionality reduction algorithm and produces a topological representation of the data that preserves locality. The projection space of the dimentional-reduction algorithm is covered with overlapping bins. The data points that fall in each bin are then clustered in the original high-dimentional space. A network is constructed by assigning a node to each cluster, and clusters that share one or more samples are connected by an edge. The result is a low-dimensional network representation of the data in which nodes represent sets of samples with similar global transcriptional profiles, and edges connect nodes that have at least one sample in common. For our analysis we used 2D Locally Linear Embedding37 as dimentionality reduction algorithm and variance normalized Euclidean metric38 as distance. Single-linkage clustering was performed in each of the pre-images of the bins using a previously described algorithm39. The number of bins (resolution) for each dimension was 20 and the degree of overlap (gain) between neighboring bins was 66%. The size of the bin was chosen such that the number of samples in each row or column of bins was the same. The open source implementations of Mapper produces results consistent with those obtained from the Ayasdi platform40.
Transcriptomic analysis of human astrocytes
We performed comparative analysis of gene expression of human astrocytes (HA) transduced with lentivirus expressing F3-T3 treated with vehicle (F3-T3-DMSO, 5 replicates), F3-T3 treated with the FGFR inhibitor PD173074 for 12 h (F3-T3-PD173074, 5 replicates), F3-T3-K508M treated with vehicle (F3-T3-K508M-DMSO, 3 replicates) and empty vector treated with vehicle (vector-DMSO, 3 replicates). Expression data were obtained using the Illumina human HT12v4 gene expression array. The list of 4,034 differentially expressed genes between the F3-T3-DMSO and F3-T3-PD173074 groups (t-test p-value < 0.01 and Wilcoxon test p-value < 0.01) was used to construct a heatmap comprising the whole dataset in which vector-DMSO and F3-T3-K508M-DMSO are control groups. Samples were clustered using the hierarchical clustering algorithm based on the Ward linkage method and Euclidean distance as implemented in R. Finally, the GO enrichment analysis was performed using the ranked list obtained from three independent comparisons: F3-T3 versus F3-T3 treated with PD173074; F3-T3 versus F3-T3-K508M; F3-T3 versus vector and the Java version of GSEA. For each comparison, statistically significant GO terms with q-value <10E-6 were selected. The statistically significant pathways common to all three comparisons were included in the construction of the visual network using the Enrichment Map application28 of Cytoscape29. The microarray data have been deposited on ArrayExpress with accession number E-MTAB-6037.
Identification of proteins phosphorylated by the F3-T3 gene fusion by mass spectrometry
Cells were lysed in buffer containing 9 M urea/20 mM HEPES pH8.0/0.1% SDS and a cocktail of phosphatase inhibitors. Six milligrams of protein were reduced with 4.5 mM DTT, alkylated with 10 mM iodoacetamide, and digested with trypsin overnight at 37°C. Samples were desalted on a C18 cartridge (Sep-Pak plus C18 cartridge, Waters). Each sample was prepared in triplicate. Phosphopeptide enrichments were performed as previously described41. An LTQ Orbitrap XL (ThermoFisher) in-line with a Paradigm MS2 HPLC (Michrom bioresources) was employed for acquiring high-resolution MS and MS/MS data. Technical duplicate data for each of the MOAC elutions and triplicate data for the phosphotyrosine immunoprecipitation samples were acquired.
RAW files were converted to mzXML using msconvert42 and searched against the Swissprot Human protein database (2013Jan09 release) appended with common proteomics contaminants and reverse sequences as decoys. Searches were performed with X!Tandem (version 2010.10.01.1) using the k-score plugin43,44. For all searches the following search parameters were used: parent monoisotopic mass error of 50 ppm; fragment ion error of 0.8 Daltons; allowing for up to 2 missed tryptic cleavages. Variable modifications were oxidation of Methionine (+15.9949@M), carbamidomethylation of Cysteine (+57.0214@C), and phosphorylation of Serine, Threonine, and Tyrosine (+79.9663@[STY]). The search results were then post-processed using PeptideProphet and ProteinProphet45–47. Spectral counts were obtained for each cell line using ABACUS48. pY-IP enrichment data were processed through ABACUS separately from the MOAC enrichment data. ABACUS results were filtered to only retain proteins with a ProteinProphet probability > 0.7. Only phosphorylated peptides with a probability > 0.8 were considered for spectral counting. For tyrosine enrichment these ABACUS parameters resulted in a protein false discovery rate (FDR) of 0.0045. This ABACUS output was used for all subsequent analysis to quantify the relative abundance of phosphorylated peptides/proteins. Phospho-site localization was performed with an in-house reimplementation of the Ascore algorithm as described49. Ascore values represent the probability of detection due to chance, with scores >19 corresponding to sites localized with >99% certainty. From four biological replicates, the application of stringent criteria selected 22 top-scoring candidate substrates of F3-T3 that exhibited at least a 1.5-fold enrichment in HA-F3-T3 compared with HA-F3-T3-KD and HA-vector (Supplementary Table 2).
Identification of PIN4 complexes by mass spectrometry
Endogenous cellular PIN4 complexes were purified from the cell line H1299 transduced with the F3-T3-expressing lentivirus. Cellular lysates were prepared in 50 mM Tris-HCl, 250 mM NaCl, 0.2% NP40, 1 mM EDTA, 10% glycerol, protease and phosphatase inhibitors. PIN4 and mock immunoprecipitates were recovered with PIN4 antibody (Abcam, ab155283) and rabbit IgG, respectively. Immunocomplexes captured on protein A-G agarose beads were washed with lysis buffer containing 300 mM NaCl and 0.3% NP40. Bound polypeptides were eluted with the PIN4 peptide used as the epitope for the PIN4 antibody (KPVFTDPPVKTKFGYH, Abcam, ab155283). Eluates were run on SDS-PAGE and four gel slices were cut from the lane containing PIN4 immunoprecipitates (columns A1, B1, C1, D1, Supplementary Table 8). Four similar gel slices were cut from the lane containing control rabbit IgG immunoprecipitates (columns A2, B2, C2, D2, Supplementary Table 8). The excised gel pieces were rehydrated and digested in 80 μl of 12.5 ng/μl Trypsin Gold/50 mM ammonium bicarbonate at 37 °C overnight. Extracted peptides were dried, reconstituted in 30 μl 0.1% TFA and stored at −20°C prior to analysis.
The concentrated peptide mix was reconstituted in a solution of 2% acetonitrile (ACN), 2% formic acid (FA) and eluted from the column using a Dionex Ultimate 3000 Nano LC system. The application of a 2.0 kV distal voltage electrosprayed the eluting peptides directly into the Thermo Fusion Tribrid mass spectrometer equipped with an EASY-Spray source (Thermo Scientific). Mass spectrometer-scanning functions and HPLC gradients were controlled by the Xcalibur data system (Thermo Finnigan, San Jose, CA). Tandem mass spectra from raw files were searched against a human protein database using the Proteome Discoverer 1.4 software (Thermo Finnigan, San Jose, CA). The peptide mass search tolerance was set to 10 ppm. A minimum sequence length of 7 amino acids residues was required. Only fully tryptic peptides were considered. Spectral counts were used for estimation of relative protein abundance between samples analyzed directly on long gradient reverse phase LC-MS/MS. A specificity score of proteins interacting with PIN4 was computed for each polypeptide as described50. Briefly, we compared the number of peptides identified from our mass spectrometry analysis to those reported in the CRAPome database that includes a list of potential contaminants from affinity purification-mass spectrometry experiments (www.crapome.org). The specificity score is computed as [(#peptide*#xcorr)/(AveSC*MaxSC*# of Expt.)], where #peptide, identified peptide count; #xcorr, the cross-correlation score for all candidate peptides queried from the database; AveSC, averaged spectral counts from CRAPome; MaxSC, maximal spectral counts from CRAPome; and # of Expt., the total found number of experiments from CRAPome.
Cell culture
Human cell lines
h-TERT-immortalized HA51, SF126 cells52, U87 (ATCC HTB-14), h-TERT-RPE-1 (ATCC CRL-4000), HEK293T (ATCC CRL-11268), U251 (Sigma # 09063001). Cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (FBS, Sigma). Cells were transfected using Lipofectamine 2000 (Invitrogen) or the calcium phosphate method.
Murine glioma stem cells
mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53 were isolated from the brains of mice that had received injection of lentivirus containing a bi-cistronic expression cassette including F3-T3 or HRAS-12V and TP53-shRNA into the dentate gyrus as described4,7. 2–4 months after intracranial injection mice showing neurological symptoms were sacrificed, brain tumors were identified macroscopically, dissected and cultured in DMEM:F12 containing 1× N2 and B27 supplements (Invitrogen) and human recombinant FGF-2 and EGF (20 ng ml−1 each; Peprotech). Studies were approved by the IACUC at Columbia University (#AAAL7600).
Human glioma stem cells
the GBM-derived glioma stem cells (GSCs) used in this study have been described4,12. GBM-derived GSCs were grown in DMEM:F12 containing 1× N2 and B27 supplements (Invitrogen) and human recombinant FGF-2 and EGF (20 ng ml−1 each; Peprotech). Cells were transduced using lentiviral particles in medium containing 4 μg ml−1 of polybrene (Sigma). Cells were routinely tested for mycoplasma contamination using the Mycoplasma Plus PCR Primer Set (Agilent Technologies) and were found to be negative. Cell authentication was performed using short tandem repeats (STR) at the ATCC facility.
Limiting dilution assay (LDA) for human GSCs was performed as described previously50. Briefly, spheres were dissociated into single cells and plated into 96-well plates in 0.2 ml of medium containing growth factors at increasing densities (1–100 cells per well) in triplicate. Cultures were left undisturbed for 14 days, and then the percent of wells not containing spheres for each cell dilution was calculated and plotted against the number of cells per well. Linear regression lines were plotted, and we estimated the minimal frequency of glioma cells endowed with stem cell capacity (the number of cells required to generate at least one sphere in every well = the stem cell frequency) based on the Poisson distribution and the intersection at the 37% level using Prism 6.0 software. Data represent the means of three independent experiments performed on different days.
The soft agar colony assay was performed by seeding HA at a density of 10,000 cells/well into 6-well plates in 0.3% agar in DMEM 10% FBS. The number of colonies/well was determined using an Olympus 1×70 microscope equipped with a digital camera.
Subcutaneous xenograft glioma models
Mice were housed in a pathogen-free animal facility. All animal studies were approved by the IACUC at Columbia University (#AAAQ2459; AAAL7600). Mice were 4–6 week old male and female athymic nude (Nu/Nu, Charles River Laboratories). No statistical method was used to pre-determine sample size. No method of randomization was used to allocate animals to experimental groups. Mice in the same cage were generally part of the same treatment. The investigators were not blinded during outcome assessment. In none of the experiments did tumors exceed the maximum volume allowed according to our IACUC protocol, specifically 20 mm in the maximum diameter. 5×105 HA-F3-T3 transduced with a lentivirus expressing shRNA sequence against PPARGC1α of ESRRG or the empty vector were injected subcutaneously in the right flank in 150 μl of saline solution (5 mice per each group). 0.5×105 mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53 transduced with lentivirus expressing two independent shRNA sequences against PPARGC1α were injected subcutaneously in the right flank in 100 μl of saline solution (5 mice per each group). Treatment with tigecycline (10 mice) or vehicle (8 mice) was performed in mice injected with 1×105 mGSC-F3-T3-shTP53 when tumors reached 150–270 mm3 (10 days after injection). Tigecycline was diluted in saline pH 7 and administered at dose of 50 mg/Kg body weight by intraperitoneal injection b.i.d. Tumor diameters were measured daily with a caliper and tumor volumes estimated using the formula: (width2 × length)/2 = V (mm3). Mice were euthanized when tumor size reached the maximum diameter allowed by our IACUC protocol (20 mm in the maximum diameter) or when mice displayed body weight loss equal to or greater than 20% of total body mass, or showed signs of compromised health or distress.
Plasmids, cloning, and lentivirus production
cDNAs for FGFR3, PIN4, PKM2, GOLGIN84, DLG3, C1ORF50 and PGC1A were amplified by PCR and cloned into the pLOC vector in frame with FLAG or V5 tag. F3-T3, F3-T3-K508M and FLAG-tagged PEX1 were cloned into pLVX-puro vector (Clontech). To generate PIN4-Y122A, PIN4-Y122F, PKM2-Y105A, GOLGIN84-Y42A, DLG3-Y673A and C1ORF50-Y131A a site-directed mutagenesis was performed using the QuickChange Site-Directed mutagenesis kit (Agilent) and the resulting plasmids were sequence verified. Lentivirus was produced by co-transfection of the lentiviral vectors with pCMV-ΔR8.1 and pCMV-MD2.G plasmids into HEK293T cells as previously described7,16. shRNA sequences are:
shPIN4: GTCAGACACATTCTATGTGAACTCGAGTTCACATAGAATGTGTCTGAC;
shPPARGC1A-Hs-1: GCAGAGTATGACGATGGTATTCTCGAGAATACCATCGTCATACTCTGC;
shPPARGC1A-Hs-2: CCGTTATACCTGTGATGCTTTCTCGAGAAAGCATCACAGGTATAACGG;
shPpargc1a-Ms-1: CCAGAACAAGAACAACGGTTTCTCGAGAAACCGTTGTTCTTGTTCTGG;
shPpargc1a-Ms-2: CCCATTTGAGAACAAGACTATCTCGAGATAGTCTTGTTCTCAAATGGG;
shESRRG-1: CAAACAAAGATCGACACATTGCTCGAGCAATGTGTCGATCTTTGTTTG;
shESRRG-2: CATGAAGCGCTGCAGGATTATCTCGAGATAATCCTGCAGCGCTTCATG;
For the generation of PPARGC1A knockout HA-F3-T3 cells with CRISPR/Cas9, guide RNA (gRNA) sequences were designed to target PPARGC1A coding sequence as described (http://crispr.mit.edu/). We designed two gRNAs against exon 1 of the PPARGC1A gene and validated three clones for loss of PGC1α expression. sgRNA sequence #1 (GGCGTGGGACATGTGCAACC) and #2 (ACCAGGACTCTGAGTCTGTA) were then inserted by linker cloning in the lentiviral vector pLCiG253,54. HA expressing the empty vector or F3-T3 were infected either with pLCiG2/Control, pLCiG2/PPARGC1A-gRNA #1 or pLCiG2/PPARGC1A-gRNA #2. 72 h after infection, cells were seeded in a 96 well plate at a density of 0.6 cell/well. Two weeks later, colonies were isolated and PPARGC1A deletion was analyzed by qRT-PCR and western blot. For acute expression of F3-T3 in HA, cells were first transduced with pLOC expressing FLAG-PIN4 WT or the Y122F mutant. Subsequently, cells were transduced with pLKO.1-puro vector encoding shRNA targeting PIN4. The levels of endogenous and ectopically expressed proteins were then verified by immunoblotting. Finally, cells were transduced with pLVX vector expressing F3–T3.
Generation of phospho-Y122-PIN4 antibody
The anti-phospho-PIN4 antibody was generated by immunizing rabbits with a short synthetic peptide containing phosphorylated Y122 (PVKTKFG–pY–HIIMVE) (Yenzym Antibodies, LLC). A two-step purification process was applied. First, antiserum was cross-absorbed against the phospho-peptide matrix to purify antibodies that recognize the phosphorylated peptide. Next, the anti-serum was purified against the un-phosphorylated peptide matrix to remove non-specific antibodies. Antibodies were validated using lysates from cells transfected with PIN4 WT or the phospho-mutant PIN4-Y122A.
Immunoblot and immunoprecipitation
For western blot, cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP40, 0.5% sodium dexoycholate, 0.1% sodium dodecyl sulfate, 1.5 mM Na3VO4, 10 mM sodium fluoride, 10 mM sodium pyrophosphate, 10 mM β-glycerolphosphate and EDTA-free protease inhibitor cocktail, Roche). Lysates were cleared by centrifugation at 15,000 r.p.m. for 15 min at 4 °C. Phospho-Tyrosine immunoprecipitation was performed on cells freshly harvested in cold PBS containing Na3VO4 and lysed in RIPA buffer. 800 μg of protein extracts were incubated with 30 μl of phospho-Tyrosine sepharose beads (P-Tyr-100, Cell Signaling Technology, #9419) in a final volume of 800 μl overnight at 4°C. Beads were washed 5 times with cold RIPA buffer and eluted by 2× SDS sample buffer. Immunoprecipitates were separated by SDS–PAGE and transferred to nitrocellulose membrane. Membranes were blocked in TBS with 5% non-fat milk and 0.1% Tween20, and probed with primary antibodies overnight at 4°C. PIN4 and FLAG-PEX1 immunoprecipitation was performed on cells freshly harvested in cold PBS and lysed in 50 mM Tris pH 8.0, 150mM NACl, 0.5% NP40, 1mM EDTA, 10% Glycerol, protease and phosphatase inhibitors. 2,000 μg of protein extracts were incubated with PIN4 antibody (Abcam, ab155283) at concentration of 0.6 μg/mg cell lysates or FLAG-M2 agarose beads in a final volume of 1,000 μl overnight at 4°C. For PIN4 immunoprecipitation, Protein A/G Plus agarose beads (Santa Cruz Biotechnology) were added for 2 h at 4°C. Beads were washed 5 times with cold lysis buffer including 300 mM NaCl, and immunocomplexes were eluted with PIN4 or FLAG-M2 peptide at room temperature for 4 h or 45 min, respectively. Immunoprecipitates were separated by SDS–PAGE and transferred to nitrocellulose membrane. Membranes were blocked in TBS with 5% non-fat milk and 0.1% Tween20, and probed with primary antibodies overnight at 4°C.
Antibodies and concentrations are: FGFR3 1:1000 (Santa Cruz, B-9, sc-13121), PIN4 1:1000 (Abcam, ab155283), PKM2 1:1000 (Cell Signaling, #3198), DLG3/SAP102 1:1000 (Cell Signaling, #3733), GOLGIN84 1:2000 (Santa Cruz, H-283, sc-134704); C1ORF50 1:1000 (Novus Biologicals, NBP1-81053), HGS 1:1000 (Abcam, ab72053), FAK 1:1000 (Cell Signaling, 3285), Paxillin 1:1000 (BD Transduction, 610051), PGC1α 1:500 (Santa Cruz, H300, sc-13067), PGC1α 1:1000 (Novus Biological, NBP104676), ESRRG 1:500 (Abcam, ab128930), ESRRG 1:500 (R7D, PP-H6812000) p-FRS2 1:1000 (Cell Signaling, 3861), FRS2 1:1000 (Santa Cruz, sc-8318), p-STAT3 1:1000 (Cell Signaling, #9131), STAT3 1:1000 (Santa Cruz, C-20 sc-482,), p-AKT 1:1000 (Cell Signaling, #4060), AKT 1:1000 (Cell Signaling, #9272), p-ERK1/2 1:1000 (Cell Signaling, #4370), ERK1/2 1:1000 (Cell Signaling, #9102), β-actin 1:2000 (Sigma, A5441), PEX1 1:500 (BD Biosciences #611719), PEX6 1:500 (Stress Marq, #SMC-470), NUP214 1:500 (Abcam #ab70497), SEC16A 1:500 (Abcam #ab70722), DHX30 1:500 (Novus Biologicals, NBP1-26203), SUN-2 1:500 (Abcam #ab124916), FLAG 1:1000 (Abcam ab1162), Retinoblastoma 1:1000 (BD Pharmingen 554136), α-tubulin 1:2000 (Sigma, T5168), total OXPHOS 1:1000 (Abcam, #ab110411), MTCO1 1:1000 (Abcam, #ab14705). Secondary antibodies horseradish-peroxidase-conjugated were purchased from Pierce and Enhanced ChemiLuminescence (Amersham) or Super Signal West Femto (Thermo Scientific) was used for detection.
Quantitative RT–PCR
Total RNA was prepared using the Trizol reagent (Invitrogen) and cDNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen) as described16,55. Quantitative RT–PCR (qRT-PCR) was performed with a Roche 480 thermal cycler, using SYBR Green PCR Master Mix (Applied Biosystems). Human primers used in qRT–PCR are: UQCRC1 Fw 5′_CACCGTGATGATGCTCTACC_3′ and Rv 5′_CCACCACCATAAGTGCAGTC_3′; POLRMT Fw 5′_TATTCATGGTGAAGGATGCC_3′ and Rv 5′_TCTGTTCCAGACACCTTTCG_3′; NDUFB4 Fw 5′ _TGCTTCAGTACAACGATCCC _3′ and Rv 5′_CACACAGAGCTCCCATGAGT_3′; MRPL15 Fw 5′ _TGCTTCCACCAGAAGAACTG_3′ and Rv 5′_ACTTCCTGGCGAGTTCAAGT_3′; MCL1 Fw 5′_GCATCGAACCATTAGCAGAA_3′ and Rv 5′_TGCCACCTTCTAGGTCCTCT_3′; MRSP30 Fw 5′_TATTCCTCGTGGTCATCGAA_3′ and Rv 5′_CTCTGCGAGTTGCTTGGATA_3′; TIMM10 Fw 5′ _CCTGGACCGATGTGTCTCTA_3′ and Rv 5′ _GCACCCTCTTCATCAGCTCT_3′; NRF1 Fw 5′_GGAAACGGCCTCATGTATTT_3′ and Rv 5′_TCATCTAACGTGGCTCGAAG_3′; ATP5G3 Fw 5′_CCCAGAATGGTGTGTCTCAG_3′ and Rv 5′_TTCCAATACCAGCACCAGAA_3′; ABCE1 Fw 5′_TCATTGATCAAGAGGTGCAGA_3′ and Rv 5′_TAGACATCAGCAGGTTTGCC_3′; TIMM23 Fw 5′_GGATTGAAGGAAACCCAGAA_3′ and Rv 5′_CCCTTGCCTAGTCACCATATT_3′; TFAM Fw 5′_GCTCAGAACCCAGATGCAA _3′ and Rv 5′_CACTCCGCCCTATAAGCATC_3′; TIMM9 Fw 5′_TGAAGAGACCACCTGTTCAGA_3′ and Rv 5′_AAGGAGTCCTGCTTTGGCT_3′; TIMM44 Fw 5′ _TCCAAGACAGAGATGTCGGA_3′ and Rv 5′_GATGTCGTTCTCGCACTGTT_3′; VDAC1 Fw 5′_AATGTGAATGACGGGACAGA_3′ and Rv 5′_ACAGCGGTCTCCAACTTCTT_3′; NDUFA9 Fw 5′_TATGCATCGGTTTGGTCCTA_3′ and Rv 5′_GACCAACGAAAGCAAAGGAT_3′; NDUFV2 Fw 5′_AAAGGCAGAATGGGTGGTT_3′ and Rv 5′_CTTTCCAACTGGCTTTCGAT_3′; COX5A Fw 5′_GATGCTCGCTGGGTAACATA_3′ and Rv 5′_GGGCTCTGGAACCATATCAT_3′; NDUFS5 Fw 5′_TGGTGAACAGCCCTACAAGA_3′ and Rv 5′_TCTGCCCGAGTATAACCGAT_3′; NDUFA2 Fw 5′_CGTCAGGGACTTCATTGAGA _3′ and Rv 5′_AAGGGACATTCGTCTCTTGG_3′; PITRM1 Fw 5′_TCTCGGATGAGATGAAGCAG_3′ and Rv 5′_CCCAGTGCCGAGGTATCTAT_3′; CISD1 Fw 5′_TGACTTCCAGTTCCAGCGTA_3′ and Rv 5′_GATAACCAATTGCAGCTGTCC_3′; ATP5G1 Fw 5′_AGCTCTGATCCGCTGTTGTA_3′ and Rv 5′_GGAAGTTGCTGTAGGAAGGC_3′; MRPL12 Fw 5′_TCAACGAGCTCCTGAAGAAA_3′ and Rv 5′_GTGTCCGTTCTTTCGCTATG_3′; IDH3A Fw 5′_CCGACCATGTGTCTCTATCG_3′ and Rv 5′_GCACGACTCCATCAACAATC_3′; 18S Fw 5′_C G C C G C T A G A G G T G A A A T T C_3′ and Rv 5′_C T T T C G C T C T G G T C C G T C T T_3′; PPARGC1A Fw 5′_CTCACACCAAACCCACAGAG_3′ and Rv 5′_GTGTTGTGACTGCGACTGTG_3′; PEX1 Fw 5′_AGTCACCAGCCTGCATTCTT_3′ and Rv 5′_ATGGGAACATGGCTTGAGAA_3′. Mouse primers used in qRT-PCR are: Ppargc1a Fw 5′_GACAGCTTTCTGGGTGGATT_3′ and Rv 5′_CGCAGGCTCATTGTTGTACT_3′; β-actin Fw 5′_GATGACGATATCGCTGCGCTG_3′ and Rv 5′_GTACGACCAGAGGCATACAGG_3′. qRT–PCR results were analyzed by the ΔΔCT method56 using 18S or β-actin as the housekeeping gene.
Quantification of mitochondrial DNA content
Total DNA was isolated using the Puregene Blood Core kit (Qiagen) according to the manufacturer’s instructions. Mitochondrial DNA content was measured using real time quantitative PCR (qPCR) as previously described57. Briefly, relative quantification of mitochondrial DNA content for each sample was determined using a set of mitochondrial specific primers: mt-Mito: Fw 5′-CACTTTCCACACAGACATCA -3′, Rv 5′-TGGTTAGGCTGGTGTTAGGG -3′; and a set of nuclear-specific primers: B2M Fw 5′-TGTTCCTGCTGGGTAGCTCT -3′ and Rv 5′-CCTCCATGATGCTGCTTACA -3′. QPCR was performed with a Roche 480 thermal cycler, using SYBR Green PCR Master Mix (Applied Biosystems). QPCR conditions used were: 95 °C for 10 min followed by 40 cycles 95 °C for 15 s, 72 °C for 15 s followed by a melting cycle going up to 95 °C. Primer specificity was determined by melt curve analysis and agarose gel electrophoresis, confirming a single band of the amplification product. The relative mitochondrial DNA content was calculated using the 2−ΔΔCt method where ΔCt is Ctmt-Mito-CtB2M.
Metabolic assays
Measurement of oxygen consumption rate and extracellular acidification rate
The functional status of mitochondria in cells expressing F3-T3 was determined by analizying multiple parameters of oxidative metabolism using the XF96 Extracellular Flux Analyzer (Agilent, Santa Clara, CA, USA) that measures the extracellular flux changes of oxygen and protons. Cells were plated in XF96-well microplates (6,000–7,000 cells/well) in a final volume of 80 μl of DMEM medium (25 mM glucose, 2 mM glutamine) supplemented with 10% FBS, 48 h prior to the assay. For experiments requiring AZD4547 treatment, cells were plated as previously described in the presence of 150 nM of AZD4547. For the Mito stress test, cells were washed twice with 200 μl of XF Assay Medium Modified DMEM (Agilent, Santa Clara, CA, USA), supplemented with 25 mM glucose, 2 mM glutamine (XF-Mito-MEM) and incubated at 37°C in the absence of CO2 for 1 h prior to the assay in 180 μl/well of XF-Mito-MEM. The ports of the sensor cartridge were sequentially loaded with 20 μl/well of the appropriate compound: the ATP coupler oligomycin (Sigma, O4876), the uncoupling agent carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, Sigma C2920) and the Complex I inhibitor Rotenone (Sigma, R8875). Compound concentration used for the different cell lines are indicated in Supplementary Table 9a. For the Glycolysis stress test, cells were washed twice with 200 μl of XF Assay Medium Modified DMEM supplemented with 2 mM glutamine (XF-Glyco-MEM) and incubated at 37°C in the absence of CO2 for 1 h prior to the assay in 180 μl/well of XF-Mito-MEM. The ports of the sensor cartridge were sequentially loaded with 20 μl/well of the appropriate compound: glucose [10 mM final] (Sigma, 8769), oligomycin [2 μM final] and 2-DG [100 mM final]. Oxygen consumption rate (OCR) and extra-cellular acidification rate (ECAR) were measured through 16 rates: 4 rates under basal conditions, 4 rates after oligomycin or glucose injection, 4 rates after FCCP or oligomycin injection, and 4 rates after Rotenone or 2-DG injection, for OCR or ECAR evaluation, respectively. The protocol was: mix (2 min), wait (1 min) and measure (2 min). OCR and ECAR values were normalized to the number of cells per well. The ratio OCR/ECAR was determined by dividing the normalized value of rate 4 of the OCR (basal condition) with the normalized value of rate 8 of the ECAR (after glucose injection).
ATP Assay
Cells were cultured in DMEM medium containing 25 mM glucose, 2 mM glutamine and 10% FBS. Twenty-four hours later, 5,000 cells/130 μl were plated in opaque white 96 well plates in 5 mM glucose, 2 mM glutamine and 0.2% FBS DMEM medium. CellTiterGlo assay reagent (Promega, G7570) was added 12–16 h later according to the manufacturer’s instructions and luminescence was measured using GloMax-Multi+ Microplate Multimode Reader (Promega). For experiments testing the effect of oligomycin, cells were cultured in glucose-free DMEM medium for 24 h and ATP levels were determined as described above.
Cell growth assay
Time course analysis of cellular growth of HA expressing F3-T3 or the empty vector was performed by plating 12,500 cells/well in triplicate in 6-well plate in DMEM containing 10% FBS. 24 h later, cells were washed and cultured in glucose-free DMEM medium containing 0.2% FBS and supplemented with 25 mM glucose or 25 mM galactose in the presence or absence of 100 nM of oligomycin. Viable cells were scored every 2 days by trypan blue exclusion. Survival assays of human and mouse GSCs treated with mitochondrial inhibitors were performed by plating 25,000 cells/well in 12 well plates in triplicate. Cells were counted after 72 h.
Mitochondria analysis by flow cytometry
Cells were seeded in 60 mm dishes and cultured in DMEM containing 10% FBS. Cells were washed once with FBS free medium. Mitotracker Red (LifeTecnologies, M7212) was added to a final concentration of 20–40 nM and incubated for 20–30 min at 37°C. Cells were then quickly washed with PBS, trypsinized, collected in phenol red free medium and incubated for 10 min at 37°C in the dark prior to analysis. Unstained cells were negative control. Acquisition was performed on LSR II Flow Cytometer (BD Biosciences, San Jose, CA) on the basis of Forward and Sideward Scatter parameters and texas red fluorescence using BD FACSDiva software. Eight to ten thousand events from each sample were evaluated. Data were analyzed using the FCS Express 6 Flow software (De Novo Software).
Gene Set Enrichment Analysis (GSEA) for ROS detoxification genes
We generated a gene set of 46 genes participating in ROS detoxification programs by combining genes extracted from specific references20,58 and the Detoxification of Reactive Oxygen Species Reactome Pathway R-HSA-3299685 (http://reactome.org/pages/download-data/). The full list of genes is reported in Supplementary Table 7c. The GSEA analysis was performed comparing the gene expression of HA-F3-T3 and HA transduced with vector control using default settings59.
Analysis of protein biosynthesis and ROS by high content microscopy
HA transduced as indicated in figure legends were plated at density of 6,000 cells/well in 96-well clear bottom black plates (Greiner) 18 h before the analysis in preparation for the evaluation of both protein biosynthesis and cellular ROS. Protein biosynthesis was detected by the Click-iT Plus OPP Alexa Fluor 594 protein synthesis assay kit (Molecular Probes, #C10457). Cells were incubated in the dark with O-propargyl-puromycin (OPP) reagent at concentration of 10 μM for 30 min. Identical samples were treated with CHX at concentration of 30 μM for 30 min before addition of OPP reagent and used as negative controls. Samples were washed with Click-iT rinse buffer, fixed in 3.7% formaldehyde for 15 min followed by permeabilization in 0.5% Triton X-100 for 15 min. Click-iT OPP reaction cocktail was then added for 30 min followed by one wash in Click-iT rinse buffer, nuclear counterstaining with DAPI. Acquisition of fluorescence intensity was performed using IN Cell Analyzer 2000 (GE Healthcare) equipped with a 2048 × 2048 CCD camera. Assay conditions were: two-color assay (DAPI and Cy3), 20× objective, exposure time 0.5 msec for Cy3 and 0.1 msec for DAPI. Four fields around the center of each well including 2,000 to 6,000 cells were imaged. Data were analyzed using IN Cell Investigator software (GE Healthcare). Fluorescence intensity was normalized for the number of cells as determined by the number of DAPI positive objects in each well.
For determination of cellular ROS, cells were incubated in the dark with CellROX Deep Red reagent at concentration of 2.5 μM for 30 min at 37°C. Identical samples were treated with NAC at concentration of 5 μM for 2 h before addition of CellROX Deep Red reagent and used as negative controls. Samples were washed once with PBS, fixed in 3.7% formaldehyde for 15 min followed by two additional washes in PBS and nuclear counterstaining with DAPI. Acquisition of fluorescence intensity was performed as described above. Assay conditions were: two-color assay (DAPI and Cy5), 20× objective, exposure time 0.8 msec for Cy5 and 0.1 msec for DAPI. Four fields around the center of each well including 2,000 to 6,000 cells were imaged. Data were analyzed using IN Cell Investigator software (GE Healthcare). Fluorescence intensity was normalized for the number of cells as determined by the number of DAPI positive objects in each well.
Immunofluorescence of cultured cells and primary tissue
Cells were fixed with 4% paraformaldehyde containing 4% sucrose, permeabilized with 0.1–1% Triton X-100, 0.1% BSA in TBS for 4 min at 4°C, and blocked with 3% BSA, 0.05% Triton X-100 in TBS. The primary antibodies used were as follows: phospho-PIN4 (1:100); PMP70 (Sigma, #SAB420018, 1:20); PEX1 (BD Bioscience, #611719, 1:100); FGFR3 (Santa Cruz, sc-13121 B9, 1:1000). Secondary antibodies were anti mouse Alexa Fluor-647, anti-rabbit Alexa Fluor-568, or Cy3-conjugated (Molecular Probes, Invitrogen). Nuclei were stained with DAPI (Sigma).
Fluorescence microscopy was performed on a Nikon A1R MP microscope using a 100×, 1.45 Plan Apo Lambda lens. Images were recorded with a Z-optical spacing of 0.15 μm and analyzed using the NIS Elements Advanced Research software (Nikon Instruments Inc.). The number of peroxisomes per cell was scored as the average of PMP70 positive objects in 5 Z-optical sections (one at the equatorial plane, 2 above, and 2 below the equatorial plane). Quantification of PEX1 fluorescence intensity was performed on maximum intensity images of Z-optical sections. After calibration and thresholding, the integrated density (product of the area and the mean intensity value, IMFI) was averaged between 30 cells in at least 6 representative pictures per each sample.
Tissue preparation and immunostaining on mouse and human tissues were performed as previously described7,50,60. The human GBM samples analyzed by immunostaining had been stored in the Onconeurotek tumorbank (certified NF S96 900), and received the authorization for analysis from ethical comittee (CPP Ile de France VI, ref A39II), and French Ministry for research (AC 2013-1962). Briefly, tumor sections were deparaffinized in xylene and rehydrated in a graded series of ethyl alcohol. Antigen retrieval was performed in Citrate Solution pH = 6.0 using a decloaking chamber (10 min for phosho-PIN4 and 7 min for COXIV, VDAC, NDUFS4 and FGFR3). Primary antibodies were incubated at 4 °C overnight: COXIV (Cell Signaling # 4850, 1:1,500), VDAC1 (Abcam ab14734, 1:700), NDUFS4 (Abcam ab55540, 1:700), phosho-Y122-PIN4 (1:200) and FGFR3 (Santa Cruz, B9, sc-13121, 1:500). Sections were incubated in biotinylated secondary antibody for 1 h, followed by 30 min of streptavidin-HRP conjugated (Vector Laboratories) for phospho-Y122-PIN4 and FGFR3, VDAC1, and NDUFS4 or HRP-conjugated anti-rabbit secondary antibody (Dako) for COXIV and TSA-Cy3 or TSA-Fluorescein (Perkin-Elmer). Nuclei were counterstained with DAPI (Sigma). Images were acquired under 20× magnification using an Olympus 1×70 microscope equipped with digital camera. Quantification of fluorescence intensity was performed using NIH ImageJ software. After calibrating and standardizing the 8-bit grayscale images, the integrated density (product of the area and the mean intensity value, IMFI) was averaged between three 20× representative pictures per each sample section.
Drosophila studies
The UAS-F3-T3 flies were generated by inserting the human F3-T3 fusion gene into the pACU2 plasmid followed by embryo injection of the plasmid and selection of the correct transgenic fly. All other genotypes were established through standard genetics. repo-Gal4 was used to drive gene expression in the glial lineage. UAS-eGFP or UAS-mRFP were introduced to visualize and quantify tumor volume. repo-Gal4; UAS-dEGFRλ; UAS-Dp110CAAX (as previously described24) and repo-Gal4; UAS-F3-T3 stocks were balanced over the CyoWeeP and TM6B balancers. srl RNAi lines were obtained from the Bloomington Drosophila Stock Center (BDSC) and the Vienna Drosophila Resource Center (VDRC): P{KK100201}VIE-260B (VDRC v103355), y1 sc* v1; P{TRiP.GL01019}attP40 (BDSC 57043), y1 sc* v1; P{TRiP.HMS00857}attP2 (BDSC 33914), and y1 sc* v1; P{TRiP.HMS00858}attP2 (BDSC 33915). The following ERR RNAi lines were used: y1 v1; P{TRiP.JF02431}attP2 (BDSC 27085), y1 v1; P{TRiP.HMC03087}attP2 (BDSC 50686), and P{KK108422}VIE-260B (VDRC v108349). y1 v1; P{UAS-GFP.VALIUM10}attP2 (BDSC 35786) was used as a control from RNAi experiments.
Fly culture, immunohistochemistry and imaging
Flies were mated and maintained at 29 °C. Fly larvae were retrieved at late third instar stage for brain dissections followed by fixation and immunohistochemical analysis. Larval brains were dissected, fixed, and stained as previously described61. Briefly, third instar larval brains were dissected in PBS, fixed in 4% paraformaldehyde solution for 20 min at room temperature, and incubated with primary antibodies, including: rat anti-phospho-Histone-H3 (Abcam ab10543, 1:300) and mouse anti-repo (Developmental Study Hybridoma Bank, 1:60) overnight at 4 °C and secondary antibody for 2 h at room temperature. Fluorescence images were acquired using a Leica SP8 confocal microscope.
Image analysis
To determine tumor volume, we acquired image stacks using a Leica SP8 confocal microscope with a Z-step size at 5.0 μm per optical slice using a 20× objective throughout the entire thickness of the brain and ventral nerve cord. The confocal LIF files were converted into Imaris file through ImarisFileConverter 6.4.2. All subsequent image processing was conducted with Imaris 5.5 software. Z-series stacks were used to make three-dimensional reconstructions. A smooth level of 1.0 was given to every measurement for consistency. Brain tumor volumes were quantified using three-dimensional reconstructions.
Statistical analysis
In general, two to four independent experiments were performed. Comparisons between groups were analyzed by t-test with Welch correction (two-tailed, unequal variance) and/or the Mann-Whitney-Wilcoxon non-parametric test when appropriate. Results in graphs are expressed as Mean ± s.d. or Mean ± s.e.m. for the indicated number of observations. All statistical analyses were performed and P-values were obtained using the GraphPad Prism software 6.0 or the R software (https://www.r-project.org) and are reported in Source Data.
Code and data availability
A collection of the R procedures to perform ee-MWW is available at http://github.com/miccec/yaGST. The RGBM package is available on CRAN at https://cran.r-project.org/web/packages/RGBM/index.html. Transcriptomic microarray gene expression data have been deposited on ArrayExpress with accession number E-MTAB-6037. Source data for western blot are provided in Supplementary Figure 1. Source data underlying the graphical representations used in the figures are provided as Excel spreadsheets corresponding to each individual figure.
Extended Data
Extended Data Figure 1. Activation of mitosis and mitochondria by F3-T3.
a, Immunoblot analysis of FGFR3 and p-FGFR3 in HA-F3-T3 treated with DMSO or PD173074, HA-F3-T3-K508M and HA-vector. β-actin is shown as loading control. Experiment was repeated at least five times with similar results. b, Heatmap of correlations among HA-F3-T3, HA-F3-T3 treated with PD173074, HA-vector and HA-F3-T3-K508M. Upper and right track colors represent sample type; left track color scale represents correlation between each sample and the F3-T3 group. HA-F3-T3, HA-F3-T3-PD173074 (n=5 biologically independent samples per group). HA-vector and HA-F3-T3-K508M (n=3 biologically independent samples per group). c, Enrichment map network of GO categories scoring as significant (q-value <10E-6 in each comparison, NES >0.6, two-sided Mann-Whitney-Wilcoxon test) from three independent gene set enrichment analyses (F3-T3 versus F3-T3-PD173074; F3-T3 versus F3-T3-K508M; F3-T3 versus vector). Nodes represent GO terms and lines indicate their connectivity. Size of nodes is proportional to enrichment significance and thickness of lines indicates the fraction of genes shared between the groups. d, qRT–PCR from HA-vector or HA-F3-T3 treated with vehicle (DMSO) or PD173074 for 12 h. Data are fold changes relative to vector (dotted line) of one representative experiment out of two independent experiments (data are Mean ± s.d., n=3 technical replicates). P-values were calculated using a two-tailed t-test, unequal variance. P: * <0.05; **<0.01; ***<0.001. For complete list of P-values refer to Excel source data. e, Analysis of mitochondrial mass by MitoTracker FACS analysis in HA-F3-T3, F3-T3-K508M or vector (left panel). Right panel, quantification of MFI. Data are Mean±s.d. of three (vector and F3-T3) and two (F3-T3-K508M) independent experiments; P: * <0.05; **<0.01, two-tailed t-test, unequal variance. f, Immunoblot analysis of mitochondrial proteins in HA-F3-T3, HA-F3-T3-K508M and HA-vector. Experiment was repeated independently three times with similar results. g, Representative microphotographs of VDAC1 and NDUFS4 immunofluorescence (upper panels, green) in mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53. DAPI staining of nuclei is shown as an indication of cellular density (lower panels, blue). Experiment was repeated independently two times with similar results. Molecular weights are indicated on all immunoblots.
Extended Data Figure 2. F3-T3 induces sensitivity to inhibitors of mitochondrial metabolism.
a, Immunoblot analysis using the FGFR3 antibody in HA-vector, HA-F3-T3 or HA-F3-T3-K508M. α-tubulin is shown as loading control. Experiment was repeated five times with similar results. b, OCR of GSC1123 harboring F3-T3 in the presence or absence of AZD4547. Data are Mean±s.d. (n=6 technical replicates) of one representative experiment out of two independent experiments. P: <0.001 for rate 1–4 and 9–12, two-tailed t-test, unequal variance. c, OCR of RPE cells expressing F3-T3 in the presence or absence of AZD4547, F3-T3-K508M or the empty vector. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment out of three independent experiments performed in triplicate with similar results. P: <0.05 for rate 1–4; P: <0.001 for rate 9–12, two-tailed t-test, unequal variance. d, OCR of U251 expressing F3-T3, F3-T3-K508M or the empty vector in the presence or absence of AZD4547. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment out of two independent experiments performed in triplicate with similar results. P: <0.01 for rate 1–4; P: <0.001 for rate 9–12, two-tailed t-test, unequal variance. e, ECAR of HA-F3-T3, F3-T3-K508M or the empty vector. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment out of two independent experiments performed in triplicate with similar results. P: <0.01 for rate 9–12, two-tailed t-test, unequal variance. f, Ratio between OCR (rate 4) and ECAR (rate 8) in HA-F3-T3, HA-F3-T3-K508M or HA-vector; Data are Mean±s.d. (n=6 replicates) of two independent experiments each performed in triplicate. P: <0.01, two-tailed t-test, unequal variance. g, Quantification of ATP production in HA-F3-T3 or HA-vector following treatment with the indicated concentrations of oligomycin for 72 h. Data are independent technical replicates (n=4) and Means (connecting lines) of one representative experiment out of two independent experiments performed with similar results. P: **<0.01; ***<0.001; two tailed t-test unequal variance. h, Time course analysis of cellular growth of HA-F3-T3 or HA-vector cultured in the presence of glucose (25 mM) or galactose (25 mM) with or without oligomycin (100 nM). Data are independent technical replicates (n=3) of one representative experiment out of two independent experiments performed with similar results. P: ***<0.001; two-tailed t-test, unequal variance. i, Survival ratio of mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53 treated for 72 h with vehicle or metformin, j, rotenone, k, menadione at the indicated concentrations. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment out of two independent experiments performed with similar results. P: **<0.01; ***<0.001; two-tailed t-test, unequal variance. l, Western blot analysis of COX1 and COX2 proteins in mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53 treated with vehicle or tigecycline at concentration of 8 μM for 72 h; α-tubulin is shown as loading control. Experiment was independently repeated twice with similar results. m, Quantification of cellular ATP in mGSC-F3-T3-shTP53 (bar graph on the left) and mGSC-HRAS-12V-shTP53 (bar graph on the right) treated with vehicle or metformin (1mM), tigecycline (8 μM), or menadione (5 μM) for 16 h. Data are Mean±s.d. of one experiment (n=6 technical replicates). P: **<0.01; ***<0.001; two-tailed t-test, unequal variance. n, Quantification of mGSC-F3-T3-shTP53 tumor volume in control and tigecycline treated mice. Data are tumor volumes (Median with interquartile range) at day 6 of treatment, a time when all mice were still in the study; n=8 for control (median=1,427mm3) and n=10 for tigecycline treated mice (median=843.4mm3); P: *<0.05; two-sided Mann-Whitney test. Molecular weights are indicated in immunoblots.
Extended Data Figure 3. Phosphorylation of Tyr-122 of PIN4 by F3-T3.
a, Amino acid sequence flanking Tyr-122 of PIN4 (in red) is evolutionarily conserved. b, Immunoprecipitation-western blot analysis of HA-F3-T3 or F3-T3-K508M with or without silencing of endogenous PIN4. β-actin is shown as loading control. WCL, whole cell lysates. c, Immunoblot analysis of phosphotyrosine immunoprecipitates from U87 glioma cells expressing empty vector, FGFR3, F3-T3 or F3-T3-K508M using the indicated antibodies (left panels). WCL, right panels. The asterisk indicates a non-specific band. d, Immunoblot analysis of phosphotyrosine immunoprecipitates from HA expressing empty vector, F3-T3 or F3-T3-K508M using the indicated antibodies (left panels); WCL, right panels. FAK is shown as loading control. e, Immunoblot analysis of phosphotyrosine immunoprecipitates from GSC1123 harboring endogenous F3-T3 shows a decreased phosphorylation of F3-T3 substrates following treatment with AZD4547 for the indicated times (left panels); WCL, right panels. Paxillin is shown as loading control. f, Immunoblot analysis of canonical FGFR signaling proteins in GSC1123 treated with AZD4547 for the indicated time. β-actin is shown as loading control. g, Immunoblot analysis of phosphotyrosine immunoprecipitates from HA-F3-T3 or HA-vector transduced with WT or the unphosphorylable (Y/A) F3-T3 kinase substrate mutants. Paxillin is shown as loading control. The asterisk indicates a non-specific band. h, Confocal images of immunofluorescence staining using the pY122-PIN4 specific antibody (red) in HA transduced with vector or F3-T3 without or with silencing of endogenous PIN4. Nuclei were stained with DAPI (blue). i, Immunoblot analysis of pY122-PIN4, total PIN4 and FGFR3 in SF126 cells transduced with FGFR3, F3-T3, F3-T3-K508M or the empty vector. β-actin is shown as loading control. Molecular weights are indicated in all panels. Experiments in b-g, and i were repeated independently three times with similar results. Experiment in h was repeated independently four times with similar results.
Extended Data Figure 4. Functional analysis of tyrosine phosphorylation of F3-T3 kinase substrates.
a, Western blot analysis of phosphotyrosine immunoprecipitation of mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53 using PIN4 antibody. F3-T3 and RAS-12V expression are shown. α-tubulin is shown as loading control b, Microphotographs of immunofluorescence using the pY122-PIN4 specific antibody (red, upper panels) in tumors from mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53. Nuclei were counterstained with DAPI (blue, lower panels). Experiment was repeated independently two times with similar results. c, Representative microphotographs of pY122-PIN4 immunofluorescence in F3-T3-positive (upper panels) and F3-T3-negative (lower panels) GBM (green). Right panels show higher magnification of pY122-PIN4-DAPI co-staining depicting cytoplasmic localization of pY122-PIN4. DAPI staining of nuclei is shown as an indication of cellular density (middle panels). d, Analysis of OCR of HA-F3-T3 transduced with WT, or the unphosphorylable mutant (Y/A) of GOLGIN84, C1ORF50 and DLG3. HA-vector are included as control. Data are Mean±s.d. (n=5 technical replicates) of one representative experiment out of two independent experiments performed in triplicate with similar results. P <0.001, rate 9–12 for vector versus every F3-T3 combination, two-tailed t-test, unequal variance. e, Analysis of OCR of HA-F3-T3 transduced with PKM2-WT, PKM2-Y105A or the empty vector. HA-vector is included as control. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment out of three independent experiments; P <0.001, rate 9–12 for vector versus every F3-T3 combination, two-tailed t-test, unequal variance. f, Immunoblot analysis of GOLGIN84, C1ORF50 and DLG3 WT or Y/A mutants in HA-F3-T3 or HA- vector. g, Immunoblot analysis of HA transduced with F3-T3 or the empty vector for the expression of PKM2 WT or PKM2-Y105A. h, Immunoblot analysis of HA transduced with F3-T3 or the empty vector for the expression of PIN4 WT or PIN4-Y122F. i, Immunoblot analysis of PIN4 proteins in HA expressing F3-T3 following silencing of endogenous PIN4 and reconstitution with PIN4 WT, PIN4-Y122A or PIN4-Y122F. In f-i, β-actin is shown as loading control. Molecular weights are indicated on all immunoblots. j, Quantification of ATP levels in HA treated as in i. Data are Mean±s.d. (n=4 technical replicates) of one out of two independent experiments. P: *<0.05; **<0.01; ***<0.001; two-tailed t-test, unequal variance. Experiments in a, f-i were repeated independently three times with similar results.
Extended Data Figure 5. Transcriptomic analysis of F3-T3 fusion-positive and fusion-like GBM and validation of eeMWW using different cancer driving alterations.
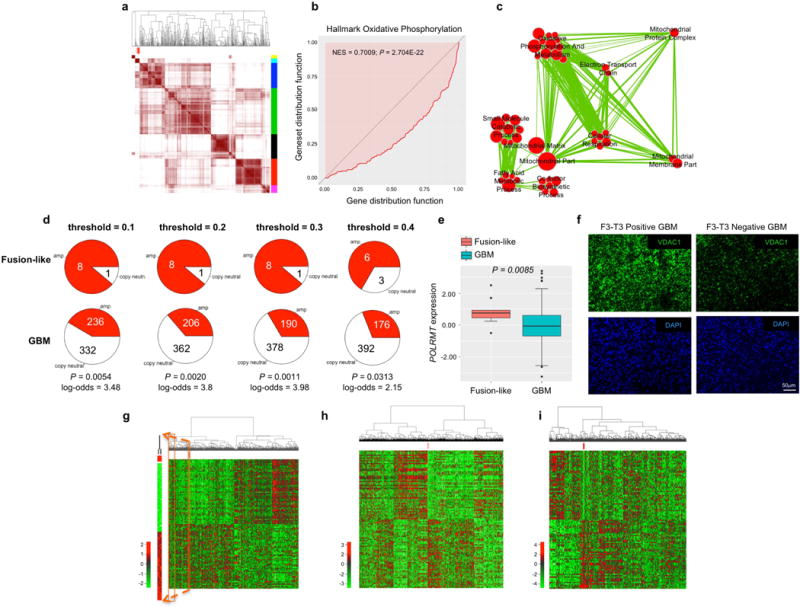
a, Consensus clustering on the Euclidean distance matrix based on the top and bottom 50 genes having the highest and lowest probability to be up- or down-regulated, respectively, in the 9 F3-T3 fusion-positive GBM. The consensus matrix is obtained from 10,000 random samplings using 70% of the 544 samples. The 9 F3-T3 positive GBM (in red) fall in one cluster (cyan). b, Mann-Whitney-Wilcoxon enrichment plot of the “Hallmark Oxidative Phosphorylation” GO category in F3-T3-positive GBM. NES and P-value are indicated. c, Enrichment map network of statistically significant GO categories (q-value <0.001, NES >0.6, two-sided Mann-Whitney-Wilcoxon test) in 9 fusion-like GBM. Nodes represent GO terms and lines demonstrate their connectivity. Size of nodes is proportional to number of gene in the category and thickness of lines indicates the fraction of genes shared between the groups. d, Analysis of copy number amplification of the POLRMT gene comparing fusion-like GBM with all other GBM at different thresholds for amplification detection on log-R ratio from SNP arrays. P-value and log-odds at different thresholds are indicated (Fisher’s Exact test). e, POLRMT gene expression in fusion-like GBM (n=9) and the remaining samples (n=535). Box plot spans the first quartile to the third quartile and whiskers show the 1.5× interquartile range. P-value, two-sided Mann-Whitney-Wilcoxon test. f, Representative microphotographs of VDAC1 immunofluorescence in F3-T3-positive (green, left panels) and F3-T3-negative (right panels) GBM. DAPI staining of nuclei is shown as an indication of cellular density (blue, lower panels). g, Hierarchical clustering for 2 GBM harboring a KRAS mutation (red) out of 544 samples. Heatmap of the 2 KRAS mutant samples is enlarged to the left. h, Hierarchical clustering for 5 KRAS-mutated samples (red) in the BRCA cohort (n=1,093). i, Hierarchical clustering for 6 EGFR-SEPT14-positive GBM (red) out of 544 samples. Data in g-i, were obtained using Euclidean distance and Ward linkage method and are based on the top and bottom 50 genes having the highest and lowest probability to be up- or down-regulated, respectively.
Extended Data Figure 6. Pan-glioma and multi-cancer analysis of F3-T3 fusion-positive samples.
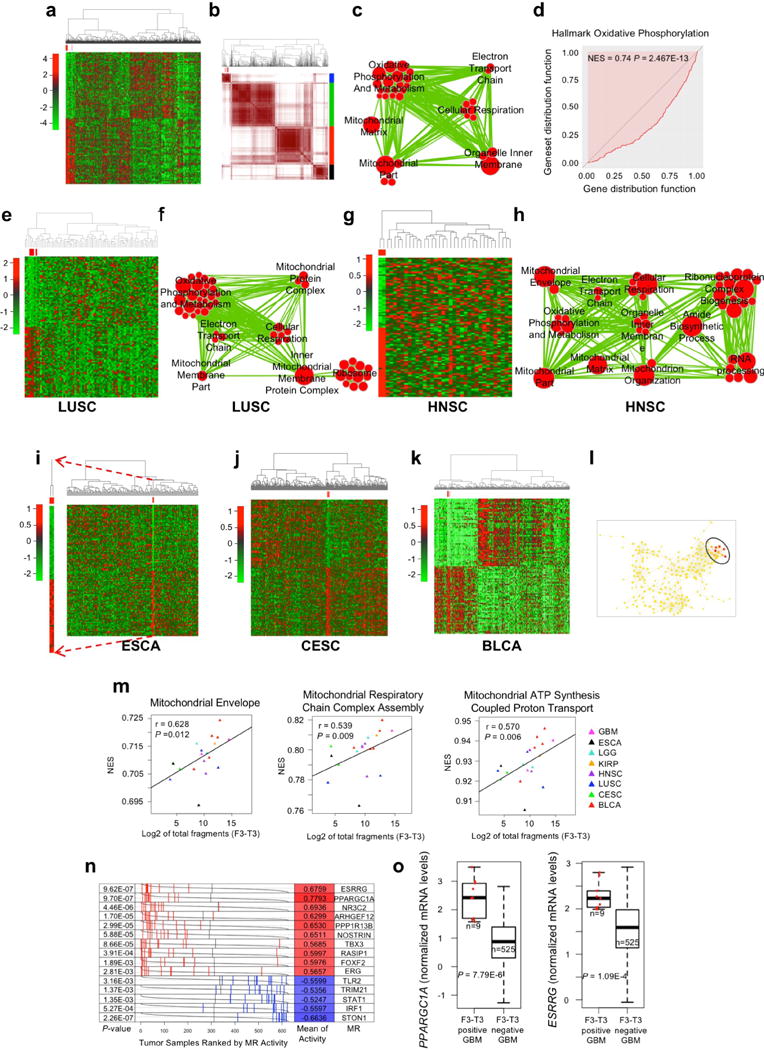
a, Hierarchical and b, consensus clustering for 11 F3-T3-positive (red) out of 627 pan-glioma samples. The 11 F3-T3-positive samples (red) in b fall in one cluster (blue). c, Enrichment map network of statistically significant GO categories (q-value <0.001, NES >0.6, two-sided Mann-Whitney-Wilcoxon test) in the 11 F3-T3 fusion positive pan-glioma samples. Nodes represent GO terms and lines demonstrate their connectivity. Size of nodes is proportional to number of genes in the GO category and thickness of lines indicates the fraction of genes shared between the groups. d, Mann-Whitney-Wilcoxon enrichment plot of the “Hallmark Oxidative Phosphorylation” GO category in F3-T3-positive samples in the pan-glioma cohort. e, Hierarchical clustering for 4 F3-T3-positive (red) out of 86 LUSC samples. f, Enrichment map network of statistically significant GO categories (q-value <0.001, NES >0.6, two-sided Mann-Whitney-Wilcoxon test) in 4 F3-T3-positive LUSC. g, Hierarchical clustering for 2 F3-T3-positive, HPV-positive HNSC (in red) out of 36 samples. h, Enrichment map network of statistically significant GO categories (q-value <0.001, NES >0.6, two-sided Mann-Whitney-Wilcoxon test) in 2 F3-T3-positive HNSC. i, Hierarchical clustering for 2 F3-T3-positive (red) out of 184 ESCA samples. Heatmap of the 2 F3-T3-positive samples is enlarged to the left. j, Hierarchical clustering for 4 F3-T3-positive (red) out of 305 CESC samples. k, Hierarchical clustering for 5 F3-T3-positive (red) out of 408 BLCA samples. l, TDA network of pan-glioma samples (n=627) reconstructed using variance normalized Euclidean distance and locally linear embedding as filter function. The nodes containing F3-T3-positive samples are highlighted in red. m, Correlation between the expression of F3-T3 (Log2 of total fragment, x-axis) and NES (y-axis) of three top ranking mitochondrial functional categories in a multi-cancer cohort including F3-T3-positive samples (n=19) from 8 tumor types (r and P-values are indicated, two-sided Mann-Whitney-Wilcoxon test). n, Analysis of the activity of MRs in the pan-glioma cohort (n=627 glioma). Grey curves represent the activity of each MR with tumor samples ranked according to MR activity. Red and blue lines indicate individual GBM samples displaying high and low MR activity, respectively. P-values, two-sided Mann-Whitney-Wilcoxon test, for differential activity (left column) and mean of the activity of the MR (right column) in F3-T3-positive versus F3-T3-negative samples are indicated. o, Gene expression analysis of PPARGC1A and ESRRG genes in F3-T3-positive and F3-T3-negative GBM; n=9 F3-T3-positive tumors; n=525 F3-T3-negative tumors. Box plot spans the first quartile to the third quartile and whiskers show the 1.5× interquartile range. P-value, two-sided Mann-Whitney-Wilcoxon test. Data in a, e, g, i-k, were obtained using Euclidean distance and Ward linkage method and are based on the top and bottom 50 genes having the highest and lowest probability to be up- or down-regulated, respectively.
Extended Data Figure 7. PGC1α and ESRRG are required for mitochondrial metabolism and tumorigenesis of cells transformed by F3-T3.
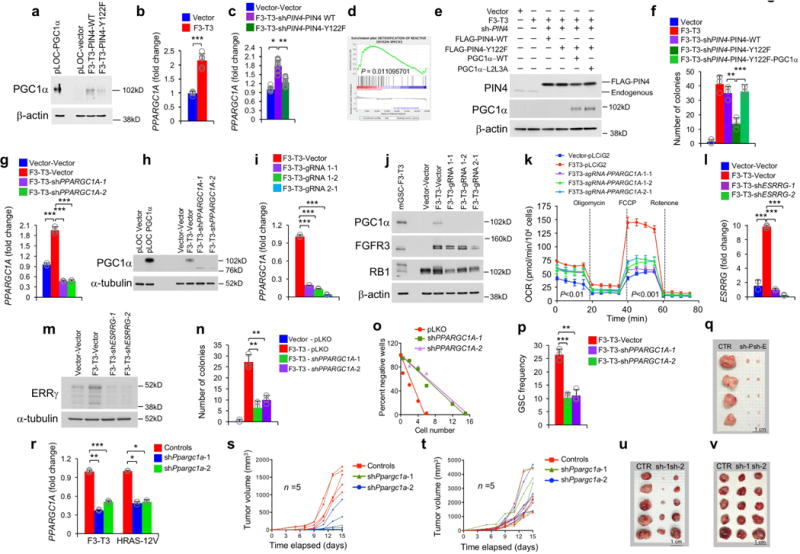
a, Immunoblot of endogenous PGC1α in HA-F3-T3 following silencing of PIN4 and reconstitution with WT or Y122F PIN4. Exogenous expression of PGC1α in HA is included as positive control. Experiment was independently repeated three times with similar results. b, qRT-PCR for PPARGC1A in HA-F3-T3 or HA-vector. Data are Mean±s.d. (n=6 replicates) from two independent experiments each performed in triplicate. c, qRT-PCR of PPARGC1A in HA-F3-T3 treated as in a. Data are Mean±s.d. (n=4 biological replicates) from four independent experiments. d, Gene Set Enrichment Analysis (GSEA) shows upregulation of ROS detoxification genes in HA-F3-T3 (n=5 biological replicates) compared with HA-vector (n=3 biological replicates). Nominal P-value is indicated. e, Immunoblot of FLAG-PIN4 (WT and Y122F) and PGC1α (WT and L2L3A) in HA-F3-T3 after silencing of the PIN4. Experiment was repeated two times independently with similar results. f, Soft agar colony forming assay of HA-F3-T3 following silencing of PIN4 and reconstitution with WT or Y122F FLAG-PIN4 in the presence or the absence of PGC1α. Data are Mean±s.d. (n=3 technical replicates) from one representative experiment out of two independent experiments. g, qRT-PCR for PPARGC1A in HA-vector or HA-F3-T3 transduced with shPPARGC1A-1 or shPPARGC1A-2 lentivirus. Data are Mean±s.d. (n=3 technical replicates) from one representative experiment. h, Immunoblot analysis of PGC1α in HA-F3-T3 treated as in g. Experiment was repeated two times independently with similar results. Exogenous expression of PGC1α is included as positive control. Experiment was repeated two times independently with similar results. i, qRT-PCR for PPARGC1A in HA-F3-T3 expressing two independent gRNAs for PPARGC1A (gRNA-PPARGC1A-1, 2 clones, gRNA- PPARGC1A-2, 1 clone) or the empty vector. Data are Mean±s.d. (n=3 technical replicates) from one representative experiment. j, Western blot of cells treated as in I using the indicated antibodies. Experiment was repeated two times independently with similar results. k, OCR of HA-vector or HA-F3-T3 transduced with PPARGC1A gRNA-1 or gRNA-2. Data are Mean±s.d. (n=5 technical replicates) from one representative experiment out of two independent experiments. P: <0.001 for rate 1–4 and 9–12, two-tailed t-test, unequal variance. l, qRT-PCR for ESRRG in HA-vector or HA-F3-T3 infected with shESRRG-1 or shESRRG-2 lentivirus. Data are Mean±s.d. (n=3 technical replicates) from one representative experiment. m, Immunoblot analysis of ESRRG in HA-F3-T3 treated as in l. Experiment was repeated two times independently with similar results. n, Soft agar colony forming assay of HA treated as in Figure 3g. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment out of two independent experiments performed in triplicate. o, GSC1123 cells were transduced with shPPARGC1A lentiviruses or the empty vector. Cells were analyzed by in vitro LDA. Representative regression plot used to calculate the frequency of gliomaspheres in 96-well cultures from three independent infections. p, Bar graph shows the frequency of gliomaspheres from three independent infections analyzed by LDA as shown in o. Data are Mean±s.d. (n=3 biological replicates). q, Photograph shows tumors generated by HA-F3-T3 transduced with shPPARGC1A-1, shESRRG-1 or vector lentivirus in Figure 3i at the time of mouse euthanasia. sh-P, shPPARGC1A-1; sh-E, shESRRG-1. r, qRT-PCR for Ppargc1a in mGSC-F3-T3-shTP53 and mGSC-HRAS-12V-shTP53 transduced with shPpargc1a-1 or shPpargc1a-2 lentivirus. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment. s, Tumor volume of mGSC-F3T3-shTP53 expressing pLKO-vector (n=5), shPpargc1a-1 (n=5) or shPpargc1a-2 (n=5). Data are tumor growth curve of individual mice. t, Tumor volume of mice injected subcutaneously with mGSC-HRAS-12V-shTP53 expressing pLKO-vector (n=5) or shPpargc1a-1 (n=5) or shPpargc1a-2 (n=5). Data are tumor growth curve of individual mice; P: ns, two-tailed t-test, unequal variance (time points 1–7). u, Photograph shows tumors generated by mGSC-F3-T3-shTP53 transduced with shPpargc1a-1 or shPpargc1a-2 or vector lentivirus in s at the time of mouse euthanasia. v, Photograph shows tumors generated by mGSC-HRAS-12V-shTP53 transduced with shPpargc1a-1 or shPpargc1a-2 or vector lentivirus in t at the time of mouse euthanasia. Molecular weights are indicated and β-actin or α-tubulin is shown as loading control in all immunoblots. P: *<0.05, **<0.01, ***<0.001, two-tailed t-test, unequal variance.
Extended Data Figure 8. Drosophila PGC1α homolog spargel (srl) mediates F3-T3-induced tumor growth.
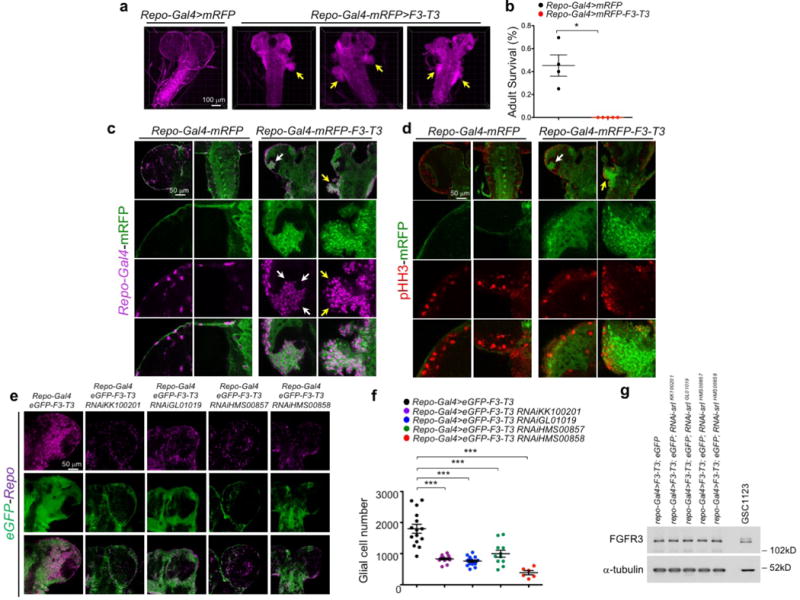
a, Optical projections of whole brain-ventral nerve cord complexes from Drosophila larvae. Expression of the F3-T3 fusion oncogene using the repo-Gal4 (repo-Gal4>F3-T3) pan-glial driver induced pathological changes in brain and ventral nerve cord with ectopic tissue protrusions (yellow arrows) due to excessive glial cell proliferation and accumulation. b, Survival of larvae bearing F3-T3-driven glial tumors. Larvae bearing F3-T3-driven glial tumors die before developing into adulthood (n=100 biologically independent samples). Data are shown as Mean±s.e.m. P: *<0.05, two-tailed t-test, unequal variance. Individual dots represent the fraction of surviving animals. c, Glial expression of F3-T3 resulted in increased total glial cell number (Repo+; mRFP+ cells) compared to controls. Note the excessive accumulation of glial cells in the brain lobe (white arrows) and ventral nerve cord (yellow arrows). d, Glial expression of F3-T3 increases glial cell proliferation (mRFP+;pHH3+ cells) compared to control. Note the excessive accumulation of glial cells in the brain lobe (white arrows) and ventral nerve cord (yellow arrows). e, Glia-specific srl knockdown in F3-T3-induced glial tumors resulted in decreased total glial number (Repo+; eGFP+ cells) compared to controls. f, Quantification of glia number in control and srl-deficient tumors; (n=15 for repo-Gal4>F3-T3; n = 15 for repo-Gal4>F3-TACC3;RNAiKK100201; n=16 for repo-Gal4>F3-T3;RNAiGL01019; n=11, for repo-Gal4>F3-T3;RNAiHMS00857; n=6 for repo-Gal4>F3-T3;RNAiHMS00858. Data are shown as Mean±s.e.m. P: ***<0.001, two-tailed t-test, unequal variance. g, Western blot analysis of the F3-T3 protein in repo-Gal4>F3-T3 and repo-Gal4>F3-T3;RNAi-srl Drosophila brains. The expression of F3-T3 in human GSC1123 is shown as positive control for F3-T3 and α-tubulin is shown as loading control. Experiments in c–e, g were performed twice.
Extended Data Figure 9. Glia-specific knockdown of srl has little to no effect on EGFR-PI3K-induced tumor growth but glia-specific knockdown ERR inhibits F3-T3-induced tumor growth.
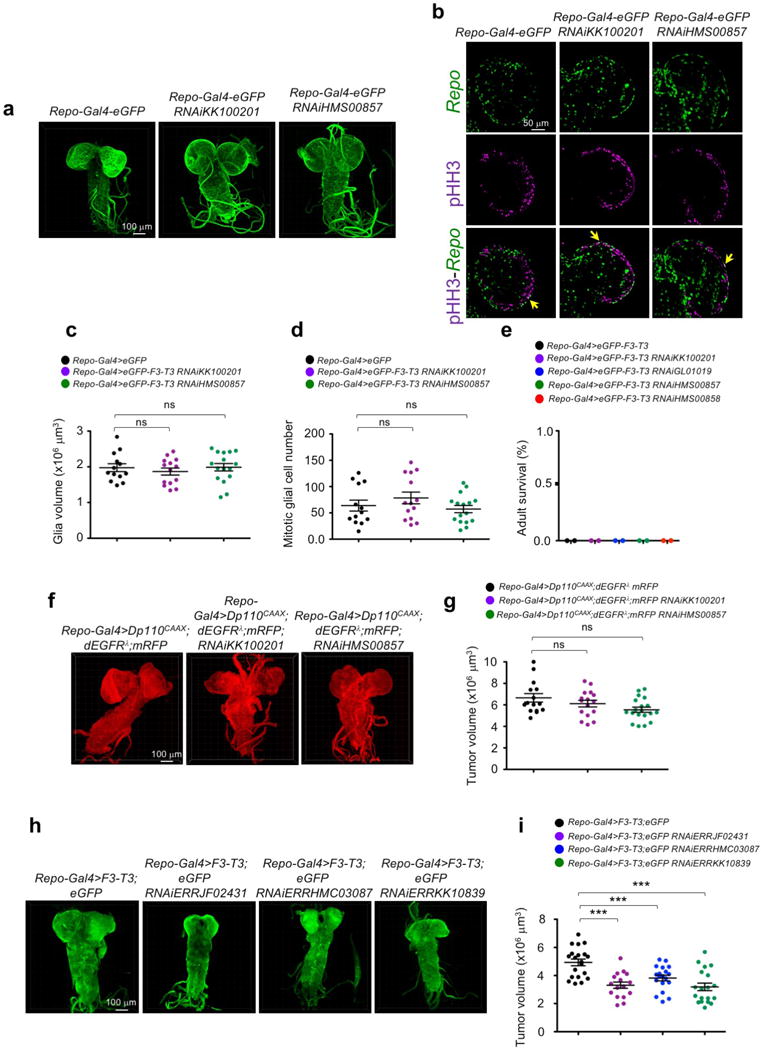
a, Optical projections of whole brain-ventral nerve cord complexes from larvae with control and srl-deficient glia. b, Glia-specific srl knockdown in larval brains did not significantly impact the overall glial population (Repo+ cells) nor the mitotic index of glial cells (Repo+; pHH3+ cells, yellow arrows). c, Quantification of glia volume in larval brains with control and srl-deficient glia; n=13 for repo-Gal4>eGFP; n=14 for repo-Gal4>eGFP;RNAiKK100201; and n=16, for repo-Gal4>eGFP;RNAiHMS00857. Data are Mean±s.d. P: ns, two-tailed t-test, unequal variance. d, Quantification of proliferating glia number (Repo+; pHH3+ cells) in larval brains with control and srl-deficient glia; n=13 for repo-Gal4>eGFP; n=14 for repo-Gal4>eGFP;RNAiKK100201; and n=16 for repo-Gal4>eGFP;RNAiHMS00857. Data are Mean±s.d. P: ns, two-tailed t-test, unequal variance. e, Adult lethality in repo-Gal4>F3-T3 and repo-Gal4>F3-T3;RNAi-srl larvae (n=100). f, Optical projections of control and srl-deficient brain tumors from repo-Gal4>Dp110CAAX;dEGFRλ;mRFP larvae. g, Quantification of tumor volume in control and srl-deficient tumors; n=15 for repo-Gal4>Dp110CAAX;dEGFRλ;mRFP; n=16 for repo-Gal4>Dp110CAAX;dEGFRλ;mRFP;RNAiKK100201; n=19 for repo-Gal4>Dp110CAAX;dEGFRλ;mRFP;RNAiHMS00857. Data are Mean±s.d. P: ns, two-tailed t-test, unequal variance. h, Optical projections of brain tumors from Drosophila larvae repo-Gal4>F3-T3 and repo-Gal4>F3-T3;RNAi-ERR. RNAi-mediated knockdown of ERR reduces the volume of F3-T3-induced glial tumors. i, Quantification of tumor volume in the control and ERR-deficient tumors; n=20 for repo-Gal4>F3-T3; n=16 for repo-Gal4>F3-T3;RNAiJF02431; n=19, for repo-Gal4>F3-T3;RNAiHMC03087; n=19, for repo-Gal4>F3-T3;RNAiKK10839). P: ***<0.001, two-tailed t-test, unequal variance. In all experiments n are biologically independent animals. Experiments in a, b, f, h were repeated twice with similar results.
Extended Data Figure 10. Acute expression of F3-T3 fusion induces peroxisome biogenesis through phosphorylation of Y122 of PIN4.
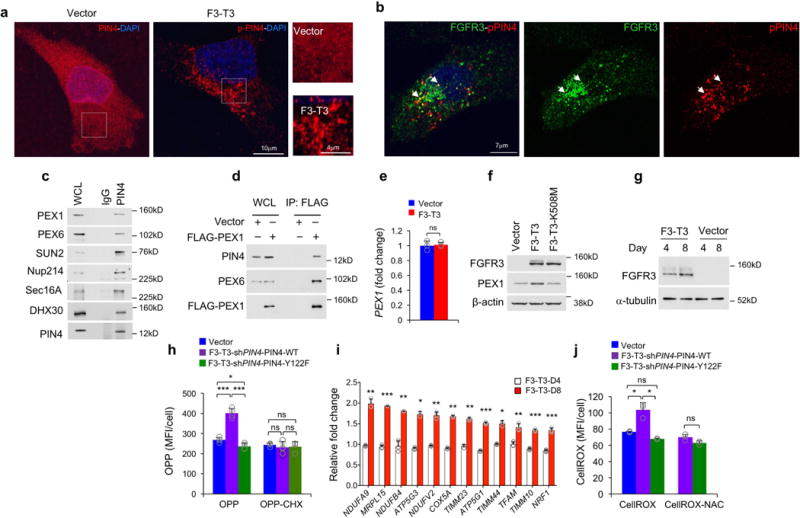
a, Representative confocal microphotographs (maximum intensity) of immunofluorescence staining for total PIN4 (PIN4, red, left panel) and pY122-PIN4 (pPIN4, red, middle panel) in vector and HA expressing F3-T3. Right panels show higher magnification of dotted boxes. Nuclei were counterstained with DAPI (blue). Experiment was repeated independently two times with similar results. b, Maximum intensity of confocal images of double immunofluorescence staining for FGFR3 (green, middle panel) and pY122-PIN4 (red, right panel) in HA-F3-T3. Arrows indicate protein co-localization. Experiment was repeated independently two times with similar results. c, Co-immunoprecipitation from H1299 cells using PIN4 antibody. Endogenous PIN4 immunocomplexes and input (WCL) were analyzed by western blot using the indicated antibodies. Input is 10% for PEX1, PEX6, SUN2 and NUP214; 5% for SEC16A and DHX30; 2% for PIN4. d, Western blot analysis of co-immunoprecipitation of exogenous FLAG-PEX1 in HA-F3-T3. WCL: 1% for PIN4 and 10% for PEX1 and PEX6. Experiment was repeated independently four times with similar results. e, qRT-PCR for PEX1 in HA-F3-T3 and HA-vector. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment out of three independent experiments performed in triplicate. f, Western blot analysis of PEX1 expression in HA transduced with F3-T3, F3-T3-K508M or the empty vector. β-actin is shown as loading control. Experiment was repeated independently three times with similar results. g, Time course analysis of F3-T3 expression in HA by western blot. α-tubulin is shown as loading control. Experiment was repeated independently two times with similar results. h, Quantification of protein biosynthesis by OPP incorporation measured by high-content fluorescent microscopy in HA reconstituted with PIN4-WT or PIN4-Y122F after silencing of the endogenous PIN4 and acutely transduced with F3-T3 or vector. Representative bar plots (n=4 technical replicates) from one out of three independent experiments. P: *<0.05, ***<0.001, two-tailed t-test, unequal variance. Cycloheximide (CHX)-treated cultures were used as negative controls. i, Time course expression analysis by qRT-PCR for the indicated mitochondrial genes in HA expressing F3-T3 or empty vector. Data are Mean±s.d. (n=3 technical replicates) of one representative experiment out of two independent experiments performed in triplicate. Values were normalized to vector (dotted line). P: *<0.05, **<0.01, ***<0.001, two-tailed t-test, unequal variance). j, Quantification of cellular ROS (measured by high-content microscopy) in HA reconstituted with PIN4-WT or PIN4-Y122F after silencing of the endogenous PIN4 and acutely transduced with F3-T3 or vector. Representative bar plots from one out of three independent experiments. Data are Mean±s.d. (n=3 technical replicates). P: *<0.05, (two-tailed t-test, unequal variance). NAC-treated cultures were used as negative controls. Molecular weights are indicated in all immunoblots.
Supplementary Material
Acknowledgments
We thank Claudio Scuoppo for donation of pLCiG2 plasmid and support with gRNA design, Emily Chen for PIN4 immunocomplexes identification and Hai Li for high content microscopy. This work was supported by NIH R01CA101644, U54CA193313 and R01CA131126 to A.L.; R01CA178546, U54CA193313, R01CA179044, R01CA190891, R01NS061776 and The Chemotherapy Foundation to A.I.; SickKids Garron Family Cancer Centre Pitblado Discovery and Ontario Institute for Cancer Research (OICR) Brain Translational Research Initiative to X.H.; American Brain Tumor Association (ABTA) and Cancer Biology Taining Grant (T32CA009503) fellowship to V.F.; NRF-2013R1A6A3A03063888 fellowship to S.B.L.; Italian Association for Cancer Research (AIRC) fellowship to M.V.R.
Footnotes
Author Contributions
A.I. and A.L. conceived and coordinated the studies and provided overall supervision. M.C. and S.M.P. developed and performed bioinformatics analyses and wrote the computational sections. V.F. performed cell, molecular biology and metabolic assays, with collaboration of T., M.V.R., A.M.C. and S.B.L. J.J.F. and X.H. developed and analyzed the Drosophila F3-T3 model. K.S.J.E-J and D.M.C.R. conducted the phosphoproteomics experiments. M.S. and K.M. provided GBM tissues and assisted in immunostaining. G.L., T., Va.F. and H.S. performed immunostaining and protein analyses. T. and P.S. performed mouse experiments. L.G., J.Z., L.C. and R.M. conducted gene expression and bioinformatics analyses. A.I. and A.L. wrote the manuscript with input from all authors.
References
- 1.Ali MA. Chronic Myeloid Leukemia in the Era of Tyrosine Kinase Inhibitors: An Evolving Paradigm of Molecularly Targeted Therapy. Mol Diagn Ther. 2016;4:315–333. doi: 10.1007/s40291-016-0208-1. [DOI] [PubMed] [Google Scholar]
- 2.Di Stefano AL, et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin Cancer Res. 2015;21:3307–3317. doi: 10.1158/1078-0432.CCR-14-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerber DE, Minna JD. ALK inhibition for non-small cell lung cancer: from discovery to therapy in record time. Cancer Cell. 2010;18:548–551. doi: 10.1016/j.ccr.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh D, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costa R, et al. FGFR3-TACC3 fusion in solid tumors: mini review. Oncotarget. 2016;7:55924–55938. doi: 10.18632/oncotarget.10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lasorella A, Sanson M, Iavarone A. FGFR-TACC gene fusions in human glioma. Neuro-Oncol. 2016;19:475–483. doi: 10.1093/neuonc/now240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niola F, et al. Mesenchymal high-grade glioma is maintained by the ID-RAP1 axis. J Clin Invest. 2013;123:405–417. doi: 10.1172/JCI63811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skrtic M, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uchida T, Fujimori F, Tradler T, Fischer G, Rahfeld JU. Identification and characterization of a 14 kDa human protein as a novel parvulin-like peptidyl prolyl cis/trans isomerase. FEBS Lett. 1999;446:278–282. doi: 10.1016/s0014-5793(99)00239-2. [DOI] [PubMed] [Google Scholar]
- 10.Uchida T, et al. Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem Biol. 2003;10:15–24. doi: 10.1016/s1074-5521(02)00310-1. [DOI] [PubMed] [Google Scholar]
- 11.Yeh ES, Means AR. PIN1, the cell cycle and cancer. Nat Rev Cancer. 2007;7:381–388. doi: 10.1038/nrc2107. [DOI] [PubMed] [Google Scholar]
- 12.Frattini V, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He HB, Garcia EA. Learning from Imbalanced Data. IEEE Trans Knowl Data Eng. 2009;21:1263–1284. [Google Scholar]
- 14.Carlsson G. Topology and Data. Bull Amer Math Soc. 2009;46:255–308. [Google Scholar]
- 15.Nicolau M, Levine AJ, Carlsson G. Topology based data analysis identifies a subgroup of breast cancers with a unique mutational profile and excellent survival. Proc Natl Acad Sci USA. 2011;108:7265–7270. doi: 10.1073/pnas.1102826108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carro MS, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mall R, et al. RGBM: Regularized Gradient Boosting Machines For The Identification of Transcriptional Regulators Of Discrete Glioma Subtypes. BoiRxiv. 2017 doi: 10.1093/nar/gky015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devarakonda S, et al. Disorder-to-order transition underlies the structural basis for the assembly of a transcriptionally active PGC-1alpha/ERRgamma complex. Proc Natl Acad Sci USA. 2011;108:18678–18683. doi: 10.1073/pnas.1113813108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 20.St-Pierre J, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 21.Xiong WC, Okano H, Patel NH, Blendy JA, Montell C. repo encodes a glial-specific homeo domain protein required in the Drosophila nervous system. Genes Dev. 1994;8:981–994. doi: 10.1101/gad.8.8.981. [DOI] [PubMed] [Google Scholar]
- 22.Tiefenbock SK, Baltzer C, Egli NA, Frei C. The Drosophila PGC-1 homologue Spargel coordinates mitochondrial activity to insulin signalling. EMBO J. 2010;29:171–183. doi: 10.1038/emboj.2009.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Read RD, et al. A kinome-wide RNAi screen in Drosophila Glia reveals that the RIO kinases mediate cell proliferation and survival through TORC2-Akt signaling in glioblastoma. PLoS Genet. 2013;9:e1003253. doi: 10.1371/journal.pgen.1003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Read RD, Cavenee WK, Furnari FB, Thomas JB. A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet. 2009;5:e1000374. doi: 10.1371/journal.pgen.1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith JJ, Aitchison JD. Peroxisomes take shape. Nat Rev Mol Cell Biol. 2013;14:803–817. doi: 10.1038/nrm3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isserlin R, Merico D, Voisin V, Bader GD. Enrichment Map - a Cytoscape app to visualize and explore OMICs pathway enrichment results. F1000Res. 2014;3:141. doi: 10.12688/f1000research.4536.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshihara K, et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015;34:4845–4854. doi: 10.1038/onc.2014.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27:2903–2904. doi: 10.1093/bioinformatics/btr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ceccarelli M, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lum PY, et al. Extracting insights from the shape of complex data using topology. Sci Rep. 2013;3:1236. doi: 10.1038/srep01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roweis ST, Saul LK. Nonlinear dimensionality reduction by locally linear embedding. Science. 2000;290:2323–2326. doi: 10.1126/science.290.5500.2323. [DOI] [PubMed] [Google Scholar]
- 38.Nielson JL, et al. Topological data analysis for discovery in preclinical spinal cord injury and traumatic brain injury. Nat Commun. 2015;6:8581. doi: 10.1038/ncomms9581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh G, Memoli F, Carlsson GE. Topological methods for the analysis of high dimensional data sets and 3D object recognition. In: Botsch M, Pajarola R, editors. SPBG. Citeseer; 2007. pp. 91–100. [Google Scholar]
- 40.Rizvi AH, et al. Single-cell topological RNA-seq analysis reveals insights into cellular differentiation and development. Nat Biotechnol. 2017;35:551–560. doi: 10.1038/nbt.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rolland D, et al. Global phosphoproteomic profiling reveals distinct signatures in B-cell non-Hodgkin lymphomas. Am J Pathol. 2014;184:1331–1342. doi: 10.1016/j.ajpath.2014.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fenyo D, Beavis RC. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal Chem. 2003;75:768–774. doi: 10.1021/ac0258709. [DOI] [PubMed] [Google Scholar]
- 44.MacLean B, Eng JK, Beavis RC, McIntosh M. General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics. 2006;22:2830–2832. doi: 10.1093/bioinformatics/btl379. [DOI] [PubMed] [Google Scholar]
- 45.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 46.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 47.Pedrioli PG. Trans-proteomic pipeline: a pipeline for proteomic analysis. Methods Mol Biol. 2010;604:213–238. doi: 10.1007/978-1-60761-444-9_15. [DOI] [PubMed] [Google Scholar]
- 48.Fermin D, Basrur V, Yocum AK, Nesvizhskii AI. Abacus: a computational tool for extracting and pre-processing spectral count data for label-free quantitative proteomic analysis. Proteomics. 2011;11:1340–1345. doi: 10.1002/pmic.201000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 50.Lee SB, et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature. 2016;529:172–177. doi: 10.1038/nature16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonoda Y, et al. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001;61:4956–4960. [PubMed] [Google Scholar]
- 52.Rutka JT, et al. Establishment and characterization of five cell lines derived from human malignant gliomas. Acta Neuropathol. 1987;75:92–103. doi: 10.1007/BF00686798. [DOI] [PubMed] [Google Scholar]
- 53.Malina A, et al. Adapting CRISPR/Cas9 for functional genomics screens. Methods Enzymol. 2014;546:193–213. doi: 10.1016/B978-0-12-801185-0.00010-6. [DOI] [PubMed] [Google Scholar]
- 54.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao X, et al. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol. 2008;10:643–653. doi: 10.1038/ncb1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 57.Ajaz S, Czajka A, Malik A. Accurate measurement of circulating mitochondrial DNA content from human blood samples using real-time quantitative PCR. Methods Mol Biol. 2015;1264:117–131. doi: 10.1007/978-1-4939-2257-4_12. [DOI] [PubMed] [Google Scholar]
- 58.Vazquez F, et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23:287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lasorella A, Rothschild G, Yokota Y, Russell RG, Iavarone A. Id2 mediates tumor initiation, proliferation, and angiogenesis in Rb mutant mice. Mol Cell Biol. 2005;25:3563–3574. doi: 10.1128/MCB.25.9.3563-3574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu S, et al. The bHLH repressor Deadpan regulates the self-renewal and specification of Drosophila larval neural stem cells independently of Notch. PLoS One. 2012;7 doi: 10.1371/journal.pone.0046724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.