

Abstract
Background
In human cardiac ventricle, IK1 is mainly comprised of Kir2.1, but Kir2.2 and Kir2.3 heterotetramers occur and modulate IK1. LQT9-associated CAV3 mutations cause decreased Kir2.1 current density, but Kir2.x heterotetramers have not been studied. Here we determine the effect of LQT9-CAV3 mutation F97C on Kir2.x homo- and heterotetramers and model associated arrhythmia mechanisms.
Methods and Results
Super-resolution microscopy, Co-immunoprecipitation, cellular electrophysiology, on-cell Western blotting, simulation of Purkinje and ventricular myocyte mathematical models were used. Kir2.x isoforms have unique sub-cellular co-localization in human cardiomyocytes and co-immunoprecipitate with Cav3. F97C-Cav3 decreased peak inward Kir2.2 current density by 50% (−120mV p-value=0.019) and peak outward by 75% (−40mV p-value<0.05) but did not affect Kir2.3 current density. FRET efficiency for Kir2.2 with Cav3 is high and on-cell Western demonstrates decreased Kir2.2 membrane expression with F97C-Cav3. Cav3-F97C reduced peak inward and outward current density of Kir2.2/Kir2.1 or Kir2.2/Kir2.3 heterotetramers (p<0.05). Only Cav3 scaffolding and membrane domains co-IP with Kir2.1 and Kir2.2 and Kir2.x-N-terminal Cav3 binding motifs are required for interaction. Mathematical Purkinje, but not ventricular, myocyte model incorporating simulated current reductions, predict spontaneous delayed-after-depolarization (DAD) mediated triggered activity.
Conclusions
Kir2.x isoforms have a unique intracellular pattern of distribution in association with specific Cav3 domains, and that critically depends on interaction with N-terminal Kir2.x Cav3 binding motifs. LQT9-CAV3 mutation differentially regulates current density and cell surface expression of Kir2.x homomeric and heteromeric channels. Mathematical Purkinje cell model incorporating experimental findings suggests DAD-type triggered activity as a possible arrhythmia mechanism.
Keywords: potassium channels, model, computer-based model, ventricular tachycardia arrhythmia, arrhythmia (mechanisms), caveolar microdomains
Journal Subject Terms: Electrophysiology, Arrhythmias, Ion Channels/Membrane Transport, Translational Studies, Basic Science Research
Introduction
In the human cardiac ventricle, IK1 maintains the resting membrane potential and contributes to phase 3 repolarization1. The inward rectifier potassium channel Kir2.1 subunits, encoded by KCNJ2, assemble in tetramers and is the major molecular correlate of IK1. Gain or loss of function mutations in KCNJ2 cause genetic sudden cardiac death syndromes2–4 and loss of functional IK1 is a major contributing factor to arrhythmogenesis in failing human hearts5. Kir2.x family members Kir2.2 and Kir2.3 can create heterotetramers with Kir2.1 in the heart, whereas Kir2.x family member Kir2.4 has only been found in neural tissue associated with the heart6, 7. Kir2.1, Kir2.2 and Kir2.3 contribute to IK1 in chamber-specific ratios that change during development, with ventricle up-regulation of Kir2.1 and Kir2.2 and down-regulation of Kir2.38, 9. Homotetrameric Kir2.x channels have distinct biophysical properties including single channel conductance, pH sensitivity, strength of rectification, and response to specific agonists10–12. However, Kir2.x in heterotetramers can transfer their properties or can acquire the properties of the other channel isomers. For example, pH sensitive Kir2.3 combined with strong rectifier Kir2.1 in heterotetrameric channels create pH sensitive strongly rectifying channels11. The degree to which Kir2.x heteromerization affects arrhythmogenesis in sudden death syndromes has not been investigated.
Caveolae are lipid microdomains characterized by the presence of Cav3 that serve to localize proteins and regulatory elements for protein signaling activity13. Cav3 is encoded by CAV3 and mutations in CAV3 can cause cardiac diseases including Long QT Syndrome-9 (LQT9)14. Additionally, abnormal distribution or density of Cav3 is implicated in atrial fibrillation as well as ventricular arrhythmias in heart failure15, 16. Our lab has previously shown that CAV3 mutations associated with LQT9 increase late sodium current (INaL), decrease Kir2.1 current density, and predispose to sudden arrhythmic death17, 18.
Cav3 has 4 distinct domains: N-terminal, scaffolding, membrane and C-terminal domains13. Interestingly, most of the LQT9-associated CAV3 mutations exist in the membrane domain of Cav3 protein, raising the question as to whether this specific domain of Cav3 may be critical for its role affecting ion channels. Kir2.x isoforms contain several possible Cav3 binding motif (CBM) sites composed of a specific series of amino acids QxQxxxxQ or QxxxxQxxQ or QxQxxxxQxxQ where “Q” is an aromatic amino acid residue and a ‘”x” represents any other residue19. The site that most closely matches a CBM due to accessibility for Cav3 binding on an exposed portion of the folded Kir2.x protein20, 21 lies between amino acids 81-92 for Kir2.1. However, important details of molecular interaction between Cav3 and Kir2.x have not been elucidated22.
In our studies using induced-pluripotent stem cell-derived cardiomyocytes (iPS-CMs), LQT9 CAV3 mutation, F97C-Cav3, resulted in action potential (AP) prolongation and early-after depolarizations (EADs) due to both loss of Kir2.1 and an increased INaL23. One arrhythmia mechanism related to LQT9 may be development of EAD triggered activity, which in the whole organ can result in Torsade de Pointes and sudden arrhythmic death24. However, loss of IK1 has also been linked to delayed after-depolarization (DAD) driven ventricular arrhythmia25, 26. Purkinje cells have both a high density of caveolae27 and are known to be important drivers of arrhythmogenicity28, and we suspect that Purkinje cells may be particularly sensitive to Cav3 mutations. Thus, to understand the complex physiologic interaction between Kir2.x homo- and heterotetramers and Cav3 as well as the pathophysiologic arrhythmogenic implications of LQT9-CAV3, here we investigate: 1) immunocolocalization and immunoprecipitation of Kir2.2 and Kir2.3 with Cav3 in native human tissue and in heterologous cells respectively, 2) FRET efficiency of Kir2.x and Cav3, 3) the impact of LQT9 causing CAV3 mutation on Kir2.x current as homo- or heterotetramers, and on Kir2.x protein trafficking, 4) critical Cav3 protein domains required for interaction with Kir2.x channels and the details of the Kir2.x CBM Cav3 association, and 5) incorporate our electrophysiologic data to model the consequences of IK1 and INaL changes due to LQT9 CAV3 mutation using computational models of ventricular and Purkinje cells.
Methods
Experimental Procedures
Ethical Approval
All experiments were conducted in accordance with the University of Wisconsin-Madison and conform to National Institutes of Health guidelines. Human tissue was obtained from the UW human tissue bank and approved by the University of Wisconsin-Madison Institutional Review Board 2011-0194.
The data, analytic methods, and study materials will be made available on request to other researchers for purposes of reproducing the results or replicating the procedures.
Cav3, KCNJ4 and KCNJ12 construction and mutagenesis
The wild type (WT) human CAV3 456-bp coding sequence was cloned from human cardiac cDNA17. Site-directed mutagenesis was performed with the QuikChange Site-Directed Mutagenesis Kit (Stratagene) using the vector containing the Cav3-WT inserts as a template. Human KCNJ4 (Kir2.3) and KCNJ12 (Kir2.2) clone was previously created and reported in this lab29. Insertion of HA tag Kir2.2 extracellular residue 118 was performed using via standard cloning techniques via Geneblock (IDT) with restriction sites AfeI and ClaI. Kir2.1 and Kir2.2 NT and CT domains were created using amplicons for Kir2.1/Kir2.2 NT and CT from PCR reactions were ligated into the PCR2.1 topo vector. The NT and CT domains were excised out of the PCR2.1 vector and ligated using NT-GFP pcDNA3.1 Topo vector. PCR, bacterial transformation and cloning were performed according to manufacturer’s instructions. The Cav3 domains were constructed using primers previously described30. Briefly, the amplicons were generated and cloned into pcDNA3.1 Topo vector via restriction sites HIND III and XBA I containing a Hexa-His tag and a V5 tag at the C-terminal. A NT mCherry was added to the above Cav3 domain construct using BAMHI and HINDIII restriction site. Kir2.x dimers (Kir2.2/Kir2.3 and Kir2.2/Kir2.1) were created with a self-cleaving P2A peptide sequence ((GSG) A T N F S L L K Q A G D V E E N P G P) in the middle (Vectorbuilder.com). All constructs cloned or sub-cloned were sequenced and confirmed for correct sequence or mutagenesis. See Supplemental Table 1 for list of primers and constructs.
Cell culture and transfections
We chose HEK293 cells due to their lack endogenous caveolins. HEK293 cells were cultured in DMEM (Invitrogen) with 10% FBS. HEK293 cells were transiently transfected using Fugene 6 (Promega) per the manufacturer specifications. For electrophysiological experiments an equal amount of WT and/or mutant Cav3 (Pires-EGFP, Clonetech, Palo Alto, CA) along with homotetrameric Kir2.x (pcDNA3.1) were used for electrophysiology experiments. For experiments with heterotetrameric Kir2.x dimer constructs and WT and/or mutant Cav3 (Pires-EGFP, Clonetech, Palo Alto, CA) a 1:3 ratio of Kir2.x dimer: Cav3(WT/mutant) was used.
Immunostaining
Human left ventricular tissue was obtained from the UW tissue bank and prepared for immunostaining as described previously31. The rabbit anti-Kir2.2 (Abcam) and anti-Kir2.3 (Alamone) antibody has been previously shown to detect Kir2.2 and Kir2.3 in human tissue31. Other antibodies included mouse anti-Cav3 antibody (BD Transduction Laboratories), rabbit anti-HA (Applied Biological Materials Inc.) and rabbit polyclonal anti-Kir2.1 (Santa Cruz). Cells were fixed with 4% paraformaldehyde in 1X PBS and then permeabilized with Triton X-100 0.1%. Prepared slides were blocked in 5% normal goat serum for 1 hour and primary antibodies (Kir2.1, Kir2.2, Kir2.3 and Cav3) were incubated overnight. Cells were washed with PBS (0.1% Tween-20) and incubated in secondary antibodies conjugated to Alexa-568 and/or Alexa-647 (Invitrogen). Samples were then washed in PBS (0.1% Tween-20) and mounted using ProLong Gold anti-fade mounting kit (Invitrogen). A Super resolution STED Leica SP8 confocal microscope part of the UW-Madison Optical Imaging Core (UWOIC) Facility was used for image acquisition and image analysis and co-localization by ImageJ software (NIH free-ware).
Co-immunoprecipitation
HEK293 cells transfected with plasmids of interest were homogenized in ice-cold RIPA buffer (25 mM Tris-HCl (pH 7.4), 150 mM NaCl, 60 mM n-octyl D-glucoside, 1% Triton X-100, 2 mM phenylmethylsulfonyl fluoride (PMSF) and a protease inhibitor tablet/50ml). The homogenate was centrifuged at 100 × g to remove insoluble debris. A total of 4-8 mg of pre-cleared cell was used for each reaction. Lysates were incubated in primary antibody overnight at 4°C and protein-antibody complexes were recovered using recombinant protein A/G-sepharose (Pierce or Santa Cruz) beads. The isotype control IgG was used as a negative control. Cell lysates were separated on precast 10-20% SDS-PAGE gels and immunoblotted as described previously11.
Electrophysiology
Transfected HEK cells’ membrane currents were analyzed 24-48 hours after transfection using whole cell technique32 at room temperature and recorded with an Axopatch-200B amplifier. Borsilicate glass pipettes were pulled to resistances of 2–4 MΩ. Pipette solution contained (mM): KCl 30, K Aspartate 85, MgCl2 5, KH2PO4 10, K2EGTA 2, K2ATP 2, and HEPES 5(pH 7.2, KOH). Bath solution contained (mM): NaCl 140, KCl 5.4, CaCl2 1.8, MgCl2 0.5, HEPES 5, NaH2PO4 0.33, D-glucose 5.5 and (pH 7.4, NaOH). Control experiments WT-Cav3 were done contemporaneously with the F97C-Cav3 mutation. Cells were identified by GFP fluorescence. Starting at a holding potential of −50 mV, voltages were ramped from −120 to 20 mV at 25mV/sec. Data were filtered at 5 kHz and digitized using a Digidata 1400(Axon Instruments). Analysis of data was done using pClamp10 (Axon Instruments) and Origin 8 software (OriginLab Corporation).
On-cell-Western Analysis
Monolayers of HEK293 cells transiently co-transfected with HA-Kir2.2 and Cav3-WT or Cav3-F97C were fixed 22-24hrs after plating with 4% paraformaldehyde. Cells were washed with 1X-PBS and then blocked with 5% normal goat serum made in 1X-PBS for an hour and then incubated with mouse anti-HA antibody for 90 min. Cells were incubated in IRDye-680 goat anti-mouse secondary antibody for an hour. Control cells (blank HEK293) were only incubated with secondary antibodies and no primary antibody. Cells were imaged according to manufacturer specification (Li-cor Biosciences, Lincoln, NE). Integrated intensities of the 700-nm infrared signal for each well were calculated using Li-Cor Odyssey infrared imaging system software. Background intensity was subtracted from the experiment and results are reported as normalized data18, 33.
Acceptor Photobleaching FRET
FRET by acceptor photobleaching34, 35 was performed using a Leica TCS SP5 confocal microscope outfitted with a Leica inverted microscope, an argon laser, and 40×1.3 numerical aperture plan apochromatic oil immersion objective (Exton, PA). Live samples were imaged within 48 hours after transfection. The donor (GFP) was excited at 488 nm and emission collected from 492 to 505 nm, and the acceptor (mCherry) was excited at 561 nm and emission collected from 570 to 696 nm. FRET was measured by the acceptor photo- bleaching method and analyzed using Leica analysis software. FRET efficiency (E) was calculated using Leica software: E =(Dpost − Dpre)/Dpost, where Dpre and Dpost are GFP emissions before and after regional photobleaching. To calculate the distance between molecules (r) we used the following formula: FRET efficiency = (ro)6/((ro)6 + (r)6) where ro = 5.24nM35.
Mathematical modeling and simulation
The human ventricular AP was simulated with both the Grandi-Pasqualini-Bers36 (GPB, code available for download at: https://somapp.ucdmc.ucdavis.edu/Pharmacology/bers/ or http://elegrandi.wixsite.com/grandilab/downloads) and the O’Hara-Rudy (ORd, http://rudylab.wustl.edu/research/cell/code/AllCodes.html) models37. The ORd formulation of late INa (INaL) was introduced in the GPB model36. All simulations were performed in MATLAB (The MathWorks, Natick, MA) using the stiff ODE solver ode15s. The canine Purkinje cell AP was simulated using the Li-Rudy38 coded in C++ (http://rudylab.wustl.edu/research/cell/code/AllCodes.html). The rapid integration numerical technique was used to solve the system of ordinary differential equations, with minimum and maximum integration steps of 0.001 ms and 0.1 ms respectively. Simulations were run to steady-state.
Statistical Analysis
Data are expressed as mean ± SEM unless specified otherwise and analyzed using either an unpaired Student t test or ANOVA using Microsoft Excel. Values of p<0.05 were considered significant.
Results
Human ventricular tissue Kir2.x localization pattern and immunocolocalization with Cav3
Human left ventricular cryopreserved tissue sections were immunostained with Kir2.1, Kir2.2, Kir2.3 and Cav3. Images were acquired on a super resolution STED Leica SP8 confocal microscope. Shown in figure 1A, Kir2.1 (1A panel b) and Cav3 (1A panels a, d and g) stain in T-tubular striated pattern (identified by arrow), intercalated disc (identified by asterisk) and at the sarcolemma (identified by arrow head). The Pearson’s correlation coefficient, used to quantify Kir2.1 and Cav3 co-localization (1A panel c), is 0.93 (interaction ≥ 0.5 is significant). Kir2.2 (1A panel e) is found to localize at the T-tubules and Kir2.3 (1A panel h) is only present at the intercalated disk. Merged (1A panel f) image for Kir2.2 and Cav3 shows only T-tubule colocalization, with a Pearson’s correlation coefficient of 0.96. Merged (1A panel i) images of Kir2.3 and Cav3 localized to the intercalated disc, with a Pearson’s correlation coefficient of 0.86, but not T-tubule or lateral membrane. The data demonstrates a unique intracellular pattern of localization: Cav3 co-localizes with Kir2.1 at the sarcolemma, with Kir2.1 and Kir2.2 at the T-tubules, and with Kir2.1 and Kir2.3 at the intercalated disk.
Figure 1.
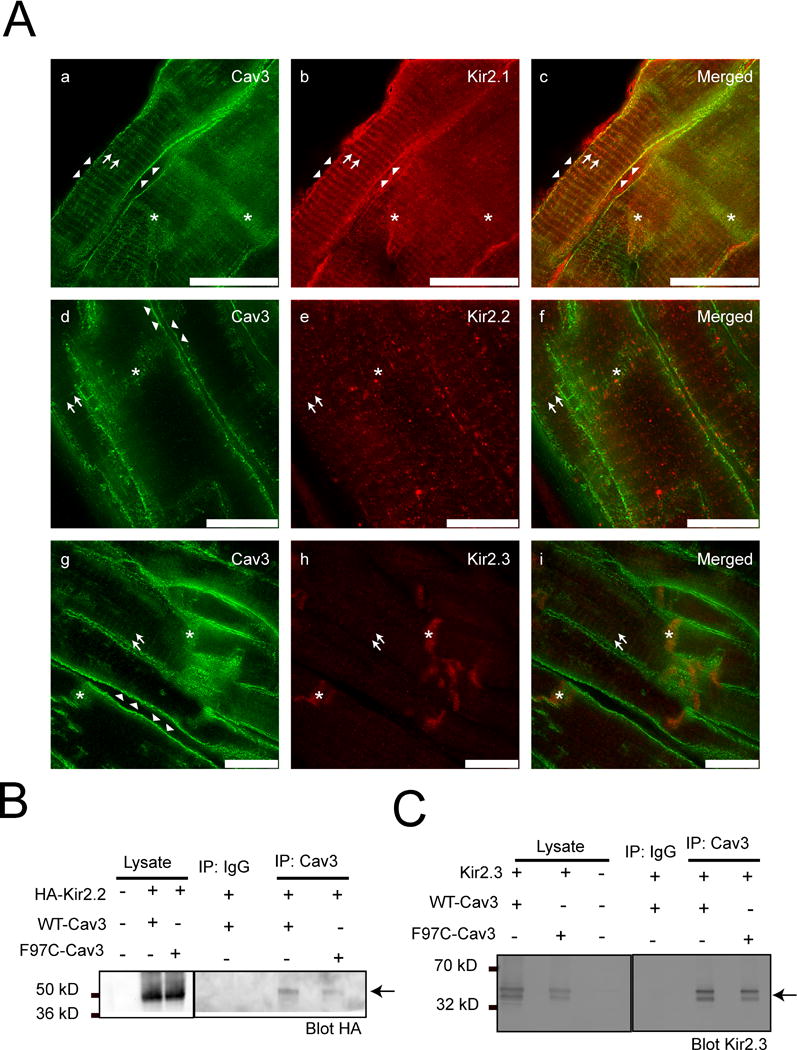
A) Kir2.1, Kir2.2 and Kir2.3 coimmunolocalize with Cav3 at specific subcellular domains in human ventricle. STED images of human left ventricle tissue stained for Kir2.1 (b, red), Kir2.2 (e, red), Kir2.3 (h, red) and Cav3 (a, d and g in green). The merged panels are c, f and i. Arrows represent T-tubular pattern of staining. Arrow heads identify the lateral membrane. Asterix identify intercalated disk. Scale bar = 10 μm. B) and C) Kir2.2 and Kir2.3 co-imunoprecipitate with Cav3. HA-Kir2.2 (B) and Kir2.3 (C) co-immunoprecipate with WT-Cav3 and F97C-Cav3 in HEK293 cells as shown by arrows.
Kir2.x channels co-immunoprecipitate with WT and LQT9 Cav3 mutation
Co-immunoprecipitation (Co-IP) and Western blot analysis was performed for HEK293 cells transfected with either extracellular hemagglutinin-Kir2.2 (HA-Kir2.2) with WT-Cav3 or F97C-Cav3. Cell lysates were incubated with anti-Cav3 antibody or rabbit IgG (negative control). The Co-IP experiment in figure 1B demonstrates that Kir2.2 associates with WT-Cav3 and F97C-Cav3 (right panel figure 1B), indicated by arrows, n=4. In a separate experiment, HEK293 cells were transfected with Kir2.3 with either WT-Cav3 or F97C-Cav3. A representative Western blot of cell lysates incubated with anti-Cav3 or mouse IgG is shown in the left panel of figure 1C. The Co-IP experiment demonstrates Kir2.3 associates with WT-Cav3 and F97C-Cav3 (right panel figure 1C), indicated by arrows, n=5.
Kir2.x homomeric channel currents are differentially modulated by LQT9 Cav3 mutation
In a previous study we found that the LQT9-associated CAV3 mutation F97C-Cav3 decreased Kir2.1 current density, but WT-Cav3 had no effect on Kir2.1 current compared to control without Cav334. Because human ventricular IK1 also contains Kir2.2 and to a lesser degree Kir2.3, we studied the effect of F97C-Cav3 or WT-Cav3 on Kir2.2 or Kir2.3 when co-expressed in HEK293 cells. A sample trace of Kir2.2 current with WT-Cav3 or F97C-Cav3 using ramp protocol from −120 to +20 mV at 25mV/sec is displayed in figure 2A. Summary data of the Kir2.2 and Kir2.3 relationships with WT-Cav3 or F97C-Cav3 is illustrated in figure 2B and 2C, respectively, and quantitatively in Table 1. Average peak outward (−40mV) and inward (−120mV) Kir2.2 current density was decreased when expressed with F97C-Cav3 compared to WT-Cav3, p<0.01. In contrast, neither peak outward (−40mV) nor inward (−120mV) Kir2.3 current density was different between cells with either WT-Cav3 or F97C-Cav3, p>0.05.
Figure 2. F97C Cav3 reduces both inward and outward current density of Kir2.2 but not Kir2.3 channels in HEK293 Cells.
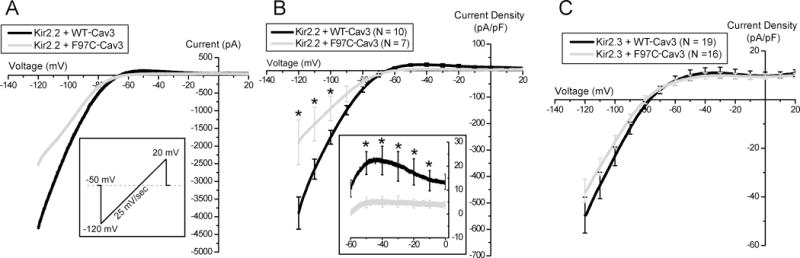
A) Sample traces of ramp protocol of Kir2.2 with WT-Cav3 (black) vs. F97C-Cav3 (grey). B) Current- voltage relationship of Kir2.2 channels co-expressed with WT-Cav3 (black) or F97C-Cav3 (grey). Inset shows the zoomed in of the I-V trace between −60 and 0 mV. C) Current- voltage relationship of Kir2.3 channels co-expressed with WT-Cav3 (black) or F97C-Cav3 (grey). * indicates p<0.01.
Table 1.
Summary Electrophysiology Data from Kir2.x Monomers
| Construct | WT-Cav3 | F97C-Cav3 | ||||
|---|---|---|---|---|---|---|
| max inward (pA/PF) | max outward (pA/PF) | n | max inward (pA/PF) | max outward (pA/PF) | n | |
| Kir2.2 | −538.45 ± 60.72 | 22.32 ± 6.01 | n=10 | −271.21 ± 84.77 | 5.04 ± 2.21 | n=7 |
| Kir2.3 | −65.49 ± 8.92 | 14.71 ± 3.51 | n=19 | −42.33 ± 6.07 | 10.11 ± 1.48 | n=16 |
Cav3 and Kir2.2 Association by FRET
We studied the association of Kir2.2 and Cav3 by FRET using the acceptor photobleaching method. We designed Kir2.2 as the donor with N-terminal GFP and Cav3 as acceptor with N-terminal mCherry. HEK293 cells were transfected with Kir2.2-GFP and Cav3-mCherry vs. vector-mCherry (as the negative control). Pre-bleached, live cells expressing both fluorophores were chosen for study and regions of interest were selected at the cell membrane, representative images shown in Supplemental Figure 1. Kir2.2 and Cav3 FRET efficiency is 25.0+/−1.76% vs. 6.86+/−1.05% for Kir2.2 and vector alone (p<0.0001). Summary results are shown by the bar graph in figure 3A. The calculated distance between proteins based on FRET efficiency is 6.61nM.
Figure 3. F97C-Cav3 significantly reduces Kir2.2 cell surface expression in HEK293 Cells.
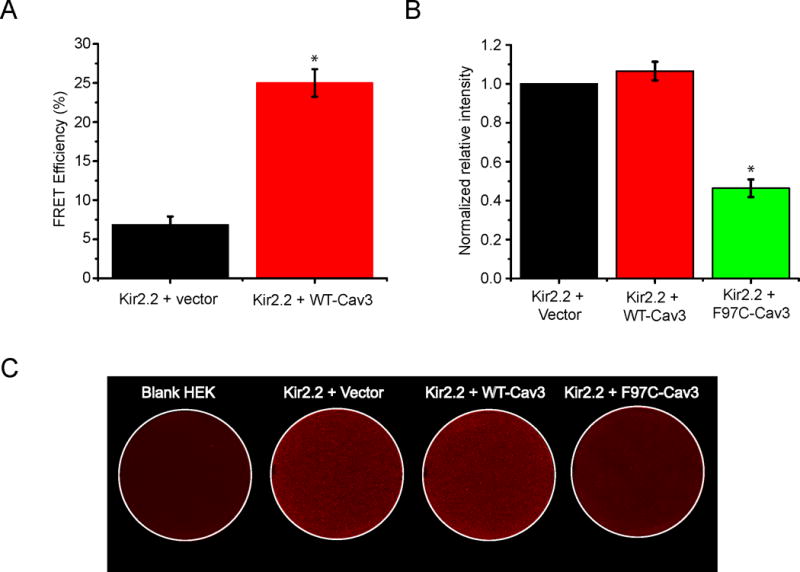
A) The bar graph describes the summary data from multiple FRET experiments between Kir2.2 (GFP tagged) and Cav3 (mCherry tagged), showing that the average FRET efficiency is 25+/−1.76% (n=18) as compared with 6.86+/−1.05% (n=19) in control (Kir2.2 (GFP tagged) + vector (mCherry)) p<0.001. (* indicates p<0.001) B) The bar graph displays quantitative results of eight independent experiments p<0.001. (* indicates p<0.001) C) Representative on-cell Western blot analysis illustrating intensity levels of extracellular HA-tagged Kir2.2 surface expression in wells co-transfected with HA-Kir2.2. Panel (a) shows vector only, Kir2.2 alone panel (b), Kir2.2 with WT-Cav3 (c), and F97C-Cav3 (d). a shows untreated cells stained with both primary and secondary antibody.
CAV3 Mutation decreases cell surface expression of Kir2.2 channels
We previously found that Kir2.1 surface membrane expression decreases in the presence of F97C-Cav318. As shown in figure 3, Kir2.2 but not Kir2.3 current density was affected by F97C-Cav3. Therefore, we studied Kir2.2 surface membrane expression by on-cell Western blotting technique using transiently transfected HEK293 cells with Kir2.2 with an extracellular HA-tag and WT-Cav3 or F97C-Cav3. The density of surface Kir2.2 expression is determined from 4 analyzed plates, blank HEK cells with empty vector (negative control), Kir2.2 alone (positive control), Kir2.2 with WT-Cav3, and Kir2.2 with F97C-Cav3. As shown in summary data for figure 3B, F97C-Cav3 decreased the intensity level of Kir2.2 by 53.7+/−4.8% but WT-Cav3 showed no difference (107+/− 4.8%) from control experiment (n=8, p<0.001). A representative example of on-cell Western blotting technique is shown in figure 3C. In a separate set of experiments, we investigated the co-localization of Kir2.2 with either WT-Cav3 or F97C-Cav3 in association with the Golgi by immunostaining. In HEK cells, transfected Kir2.2 with F97C-Cav3 but not with WT-Cav3 co-localized in the Golgi, as evidenced by Golgin-97 staining (images are shown in Supplemental Figure 2).
Heterotetrameric channels Kir2.1-Kir2.2 and Kir2.3-Kir2.2 remain susceptible to F97C-Cav3
Heterotetrameric channels containing combinations of Kir2.1, Kir2.2 or Kir2.3 exist in the heart. To understand the physiologic consequences of Kir2.x combinations in the presence of LQT9-associated CAV3 mutation F97C, particularly given the differential effect on homomeric channels (figure 2), we studied Kir2.1-Kir2.2 or Kir2.2-Kir2.3 dimer constructs transfected with WT-Cav3 vs. F97C-Cav3. Example barium sensitive traces recorded using the ramp protocol are shown in figure 4A for Kir2.1-Kir2.2 and 4C for Kir2.2-Kir2.3. Ramp protocol is depicted in the inset in the same panels. Summary data of the Kir2.1-Kir2.2 relationship with WT-Cav3 or F97C-Cav3 is illustrated in figure 4B and in Table 2. Average peak outward (−50 mV, panel B inset) and inward (−120mV) Kir2.1-Kir2.2 dimer current with F97C-Cav3 was decreased compared to WT-Cav3, p<0.05. Despite the lack of F97C-Cav3 on Kir2.3 monomers, both peak outward (−60mV, panel D inset) and inward (−120mV) Kir2.2-Kir2.3 dimer current decreased in the presence of F97C-Cav3 compared to WT-Cav3, shown in figure 4D and summarized in Table 2, p<0.05.
Figure 4. F97C-Cav3 reduces both inward and outward current density of Kir2.1-Kir2.2 and Kir2.3-Kir2.2 heterotetramers in HEK293 Cells.
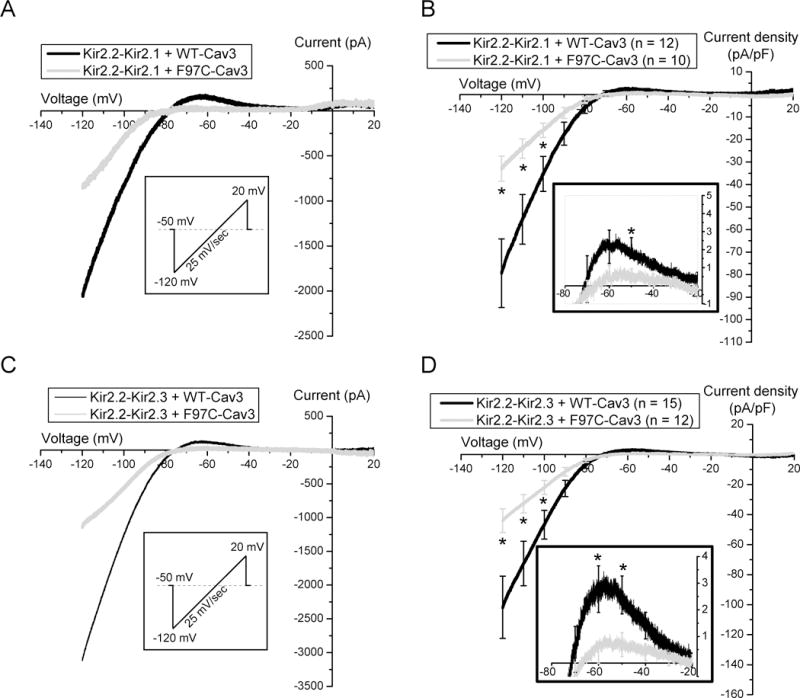
A) Sample traces of ramp protocol of Kir2.1-Kir2.2 with WT-Cav3 (black) vs. F97C-Cav3 (grey). B) Current- voltage relationship of Kir2.1-Kir2.2 heteromeric channels co-expressed with WT-Cav3 (black), F97C-Cav3 (grey). Inset shows the zoomed in of the I-V trace between −80 and −20 mV. C) Sample traces of ramp protocol of Kir2.2-Kir2.3 with WT-Cav3 (grey) vs. F97C-Cav3 (grey). D) Current- voltage relationship of Kir2.3-Kir2.2 heteromeric channels co-expressed with WT-Cav3 (black) or F97C-Cav3 (grey). Inset shows the zoomed in of the I-V trace between −80 and −20 mV. * denotes p≤0.05.
Table 2.
Summary Electrophysiology Data from Kir2.x Dimers
| Construct | WT-Cav3 | F97C-Cav3 | ||||
|---|---|---|---|---|---|---|
| max inward (pA/PF) | max outward (pA/PF) | n | max inward (pA/PF) | max outward (pA/PF) | n | |
| Kir2.1-Kir2.2 | −79.39 ± 15.25 | 2.09 ± 0.58 | n=12 | −32.98 ± 5.62 | 0.61 ± 0.34 | n=10 |
| Kir2.2-Kir2.3 | − 101.76 ± 20.74 | 2.78 ± 0.87 | n=15 | −44 ± 7.79 | 0.38 ± 0.39 | n=12 |
N-terminal Kir2.1 and Kir2.2 binding motifs Co-immunoprecipitate with Cav3 while the scaffolding and membrane domain of Cav3 co-imunoprecpitates with Kir2.1/Kir2.2
Kir2.1 and Kir2.2 have several possible CBMs composed of a specific sequence of aromatic amino acids including QxQxxxxQ or QxxxxQxxQ or QxQxxxxQxxQ where “Q” is an aromatic amino acid residue and a ‘”x” represents any other residue19. This potential CBM is conserved across isoforms, as depicted in figure 5E39. In previous studies, the Kir2.1 Golgi export signal requires association of critical N-terminus (NT) and C-terminus (CT) residues40. Additionally, we previously found that F97C-Cav3 caused accumulation of Kir2.1 in the Golgi34 and here we find decreased Kir2.2 trafficking in the presence of F97C-Cav3. Thus, there may be disruption of the Golgi export signal in the presence of F97C-Cav3. To determine the critical region for Cav3 and Kir2.x interaction, we created HA-tagged constructs containing the intracellular CBM for Kir2.1 and Kir2.2 (see Supplemental Table 1 for construct primer details). HEK293 cells were co-transfected with Cav3 and one of the four constructs: HA-Kir2.1-NT, HA-Kir2.1-CT, HA-Kir2.2-NT, or HA-Kir2.2-CT. Cell lysates were analyzed by Western blot as shown in figure 5A and 5C, left panels. The co-IP demonstrates that HA-Kir2.2-NT associates with Cav3 but not HA-Kir2.2-CT or IgG (negative control) (figure 5A, right panel), indicated by arrow heads. Similarly, HA-Kir2.1-NT, but not HA-Kir2.1-CT or IgG, co-IPs with Cav3 (figure 5C, right panel), indicated by arrow heads. This suggests that for both Kir2.2 and Kir2.1 the N-terminal CBM is necessary for interaction with Cav3.
Figure 5. Co-Immunoprecipitation of Kir2.1 and Kir2.2 N-terminal domain and Cav3 scaffolding and membrane domains.
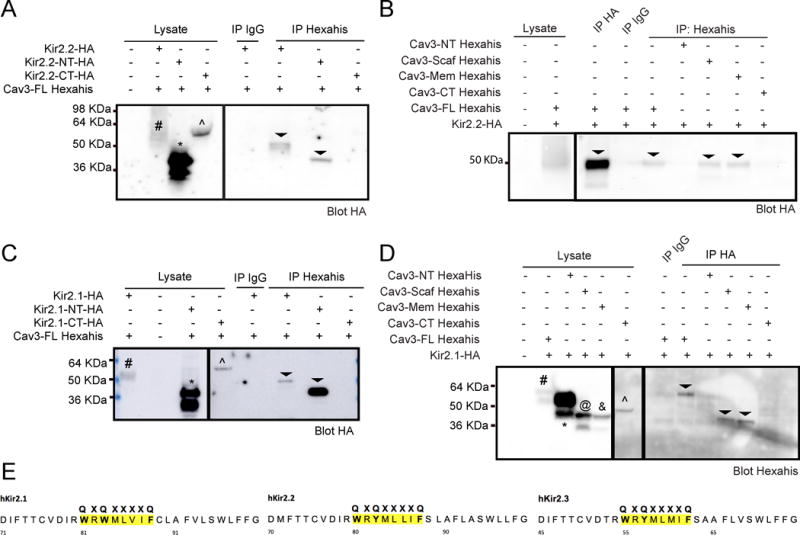
A) Kir2.2-HA (#) and Kir2.2-NT domain (*) associates with Cav3 but not Kir2.2-CT domain(^) or IgG (negative control), indicated by arrow heads in right panels, n=4. C) Co-IP demonstrates that Kir2.1-HA (#) and Kir2.1-NT domain (*) associates with Cav3 but not Kir2.1-CT domain(^) or IgG, indicated by arrow heads in right panels, n=4. B) and D) Co-IP of Cav3-full length(#), Cav3-scaffolding (@) and Cav3-membrane (&) domains associate with B) Kir2.2-HA (n=5) and D) Kir2.1-HA (n=4), indicated by arrow heads in the right panels. Cav3-NT (*) and Cav3-CT (^) do not associate with Kir2.2-HA and Kir2.1-HA. IgG is run as a negative control. E) Amio acid sequence of CBM conserved across Kir2.1, Kir2.2 and Kir2.3.
Cav3 has 4 protein domains: N-terminal (NT; aa 1-54), scaffolding (aa 55-73), membrane (aa 74-106), and C-terminal (CT; aa 107-151)14. LQT9-associated CAV3 mutations are most frequently found in the membrane and scaffolding domains of Cav3. To investigate which domain(s) of Cav3 associate with Kir2.1 or Kir2.2, we created constructs of the 4 Cav3 domains with Hexa-His tag. HEK293 cells were transfected with either Kir2.1 or Kir2.2 and either Cav3-full length or Cav3-NT, Cav3-scaffolding, Cav3-membrane, or Cav3-CT domain. Cell lysates were analyzed by Western blot as shown in figure 5B, left panel, for Kir2.2 with Cav3 or its domains and 5D, left panel for Kir2.1 with Cav3 or its domains. Co-IP analysis shown in figure 5B, right panel, for Kir2.2 (n=5) and figure 5D, right panel, for Kir2.1 (n=4), demonstrates the Cav3-scaffolding and Cav3-membrane domains associate with Kir2.2 and Kir2.1, while the NT and CT Cav3 domains do not associate with either Kir2.2 or Kir2.1.
Implications for Arrhythmogenesis
We sought to determine the consequences of Cav3-mediated Kir2.x downregulation and INaL enhancement for arrhythmia triggers by simulating human ventricular and canine Purkinje cell computational models36–38. To elicit EADs, we reduced the repolarization reserve by pacing the virtual cells at slow rates (4000 and 2000 ms BCL). DAD occurrence was assessed following rapid pacing (300 ms BCL) and subsequent pause after pacing cessation.
With both the GPB36 and ORd37 human ventricular cell models, increase of INaL and IK1 reduction result in AP prolongation, especially at slow rates, but EADs are only seen when the maximal conductance of INaL is increased by several fold. As shown in figure 6A, a 12-fold increase in INaL causes EAD in the GPB model paced at 4000 ms. In this setting, IK1 reduction depolarizes the resting membrane potential and reduces INa, thus relatively decreasing INaL availability, and counteracts the formation of EADs. We obtained the same results when simulating a condition in which IK1 is carried by the Kir2.1 isoform only (i.e., not by a mix of isoforms, see figure 6C). However, with the single isoform, a greater reduction of IK1 is required to prevent EADs (figure 6B). Similar results were predicted at faster pacing rates (BCL=2000 and 1000 ms). In the ORd model, a much larger increase in INaL is needed to elicit EADs, and IK1 downregulation has little effect on EAD formation, but prolongs final repolarization (data not shown). No DADs are elicited in the human ventricular cell models with the described changes in IK1 and INaL densities (data not shown).
Figure 6.
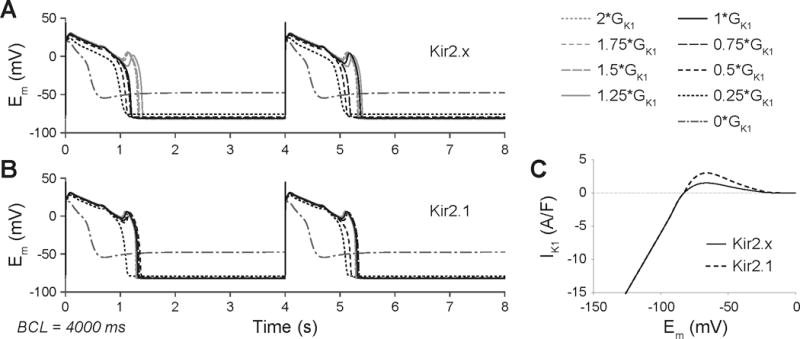
APs elicited by INaL enhancement (12×) in the GPB human ventricular myocyte model during constant pacing at 0.25 Hz (BCL = 4000 ms) at varying IK1 maximal conductance (GK1). Traces in A) are obtained with IK1 carried by Kir2.x mix), while traces in B) are obtained with IK1 carried by Kir2.1 only. C) IK1-Em relationships for Kir2.x and Kir2.1 models.
In contrast, using the Li-Rudy canine Purkinje cell model38, triggered activity is readily revealed when simulating concomitant IK1 downregulation (−50%) and INaL upregulation (2-fold increase). In figure 7 we demonstrate the Li-Rudy Purkinje model showing simulated AP (top panels), calcium transient (middle panels), and sodium-calcium exchange (NCX) activity (lower panels). At slow pacing rate (both 4000 and 2000 ms BCL), spontaneous APs are generated between paced APs (figure 7A and D), via a mechanism involving INaL-induced calcium loading, calcium release, and consequent NCX enhancement leading to resting membrane potential depolarization. This dual effect of calcium loading and unstable resting membrane potential increases the likelihood of calcium overload mediated diastolic calcium release and decreases the threshold for DADs as shown in figure 7. Notably, blocking If does not prevent the occurrence of unstimulated beats. At faster pacing rates, under control conditions or with 50% IK1 reduction alone, no triggered activity is observed and with 50% IK1 reduction there is slight resting membrane potential depolarization (figure 8A and B). When simulating INaL upregulation (+100%) alone, DAD-mediated triggered activity is predicted causing a few unstimulated beats before returning to a stable resting membrane potential (figure 8C). Notably, when concomitant IK1 downregulation and INaL enhancement are combined, as is the hypothesized effect of F97C-Cav3 mutation, the synergistic interaction of increasing calcium load and reducing DAD threshold lead to AP oscillations, which are not prevented by If inhibition (figure 8D and E).
Figure 7.
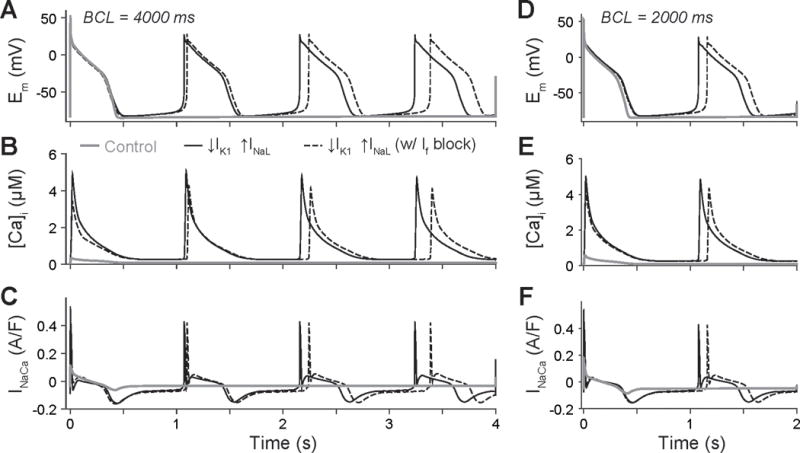
(A, D) APs, (B, E) Ca transients, and (C, F) NCX currents elicited by INaL enhancement (2×) and IK1 downregulation (−50%) in the Li-Rudy canine Purkinje cell model during constant pacing at 2000 ms (left) and 4000 ms (right) BCL.
Figure 8.
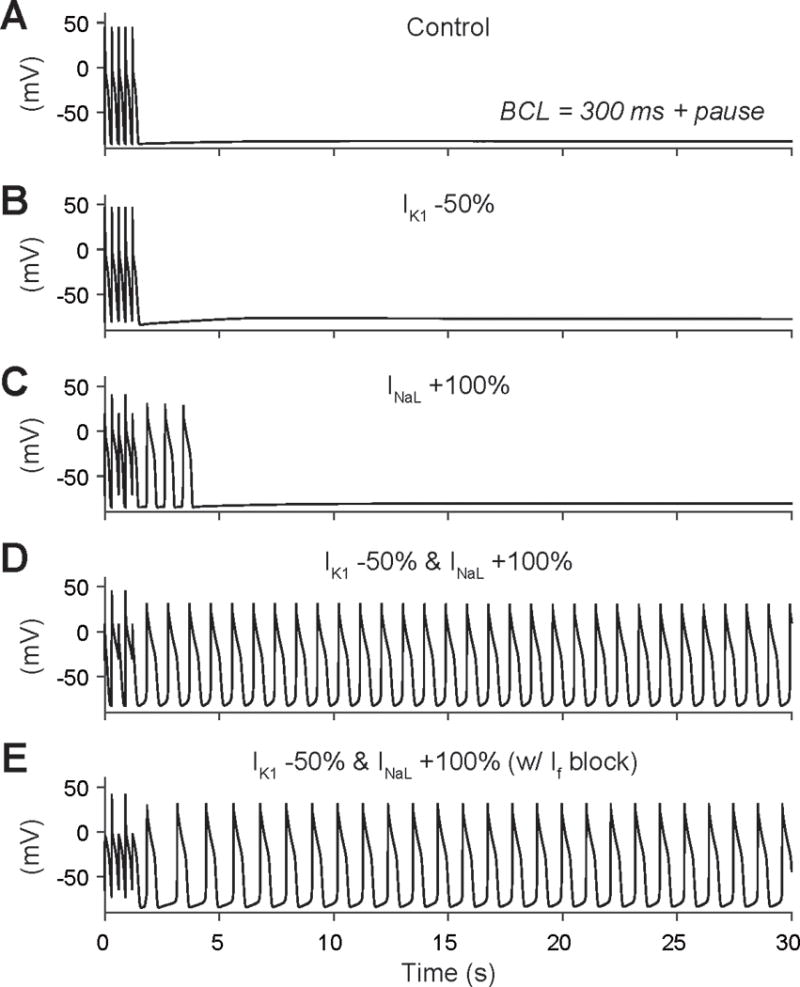
Membrane potential during 300 ms BCL stimulations followed by a pause is shown in (A) control conditions, (B) with IK1 downregulation (−50%), (C) INaL enhancement (2-fold), or (D) with concomitant IK1 reduction and INaL increase (fourth row). (E) Triggered activity in the latter condition cannot be prevented by If inhibition.
Discussion
We demonstrated the differential and protein domain specific distribution of Kir2.x channels in association with Cav3 and explore the impact on Kir2.x homomeric and heteromeric channel current densities when expressed with LQT9-associated CAV3 mutation. We have then combined these findings and our previously established current changes with LQT9 CAV3 mutation and incorporated these into a mathematical Purkinje and ventricular myocyte models. Co-localization of Kir2.1 at the sarcolemma, intercalated disc, and T-tubules with Cav3 and Kir2.2 at T-tubules by STED correlates with the vulnerability of Kir2.1 and Kir2.2 when expressed with a LQT9 CAV3 mutation. Our mathematical modeling suggests an unexpected arrhythmia mechanism for a LQT mutation, but one that is consistent with loss of IK1.
Differential Human Ventricular Intracellular Distribution of Kir2.x
Kir2.x isoforms have differential distribution in cardiac tissue8 and we show here their intracellular relationship in human ventricular tissue in association with Cav3. Kir2.1 is the dominant isoform and co-localizes with Cav3 by STED super-resolution microscopy to the sarcolemma, T-tubule and intercalated disc. There appears to be overlapping distribution of Cav3 and Kir2.1 with Kir2.2 in the T-tubule, and with Kir2.3 at the intercalated disc. While similar Kir2.x distribution patterns have been noted in the canine ventricle41, to our knowledge this is the first detailed description in human ventricular tissue. Interestingly, we did not see an overlap of Kir2.2 and Kir2.3, despite previous work demonstrating function heterotetramers of these isoforms11. Note that we did not analyze human atrial tissue, where there is relatively more Kir2.38. The lack of effect of F97C-Cav3 on Kir2.3 may be one explanation for the absence of atrial arrhythmias in these patients with CAV3 mutations. Given the intracellular distribution of Kir2.1 and Kir2.2, and our finding of loss of function of Kir2.118 and Kir2.2 in the presence of CAV3 mutations, we propose that ventricular myocytes have disrupted caveolar membrane domain presence of inward rectifier current and subsequent arrhythmia vulnerability.
Cav3 creates cellular microdomains to efficiently compartmentalize ion channels, G-protein coupled receptors and regulatory proteins. Disruption of those microdomains can result in ion channel dysfunction and arrhythmogenesis. The disruption of Kir2.1 and Kir2.2 in caveolar membranes may also be pro-arrhythmic for other clinical entities including dilated cardiomyopathy. Kir2.x isoforms change expression patterns in dilated cardiomyopathy42 and Cav3 has been shown to be down-regulated13. Namely, Kir2.1 and Kir2.3 protein and transcripts increase, while Kir2.2 is down-regulated42, despite overall loss of IK1.The loss of caveolar membrane in heart failure results in dysregulation of the L-type calcium channel43, 44. Overall, we suspect that one of the mechanisms involved in lK1 loss in heart failure is related to the loss of caveolar membranes causing abnormal distribution or failure to traffic of Kir2.1 and Kir2.2, leading to loss of repolarization reserve and arrhythmogenesis. This is an active area of research in our lab and the current study supports this hypothesis.
Cav3 Domain Specificity of Kir2.x Interaction
Mutations in CAV3 cause a range of degenerative muscular disorders in skeletal muscle including: limb-girdle muscular dystrophy autosomal dominant type C, isolated hyperCKemia, rippling muscle disease, and distal myopathy45. In the heart, CAV3 mutations are associated with hypertrophic cardiomyopathy and LQTS, so called LQT917, 45. Of the known mutations related to caveolinopathies, the majority are in the highly conserved scaffolding domains46 while in LQT9 the majority are in the scaffolding and membrane domains. Here we find that the scaffolding and membrane Cav-3 domains interact (indirectly or directly) with Kir2.1 and Kir2.2. This is intriguing from a pathophysiology perspective given that LQT9 mutations most often occur in these Cav3 domains. This raises the question as to whether this Cav3 specific domain may be critical for its role affecting ion channels vs. degenerative muscular disorders, however will require additional sequence analyses for LQT and other genetic cardiac disorders.
Kir2.1 and Kir2.2 Cav3 Binding Sequence Homology and Protein Trafficking
Despite their unique properties and regulation, both Kir2.1 and Kir2.2 have sequence homology with Cav3 binding motifs. We determined that the N-terminal Cav3 binding motif is required for interaction of both Kir2.1 and Kir2.2. In our previous work, we found that the F97C mutation caused disruption of Kir2.1 surface membrane expression with retention of protein in the Golgi18. For Kir2.1, the Golgi export signal involves conserved basic residues from the N-terminus and hydrophobic C-terminal residues which interact to form a salt-bridge patch of residues to form the AP-1 binding signal for sarcolemma distribution40, 47. We suspect that F97C-Cav3 disrupts the salt bridge formation in normal N-terminal residues association of Kir2.1 or Kir2.2. This interferes with AP-1 binding signal formation and thus loss of protein at the surface membrane. Alternatively, the N-terminus Cav3 binding motif overlaps with the cholesterol sensitivity belt48, 49 and F97C-Cav3 may disrupt the membrane channel insertion due to lack of capability to bind cholesterol. It is unclear if Cav3 and cholesterol independently regulate Kir2.x membrane localization or if these mechanisms combine in cardiomyocytes to regulate Kir2.x channel distribution and function.
Loss of IK1 and increase in INaL in LQT9: Vulnerable “sink” with arrhythmogenic “source”
Loss of Kir2.1 and Kir2.2, whether as homo- or heterotetramers, can lead to decreased membrane stability and AP prolongation in the ventricle. In our previous studies we have evaluated the impact of a LQT9 CAV3 mutation on Kir2.1 current18 and here we demonstrate not only the impact of the mutation on Kir2.2 current, the Kir2.x isoform also commonly found in human ventricular myocytes, but also on Kir2.3. Interestingly, Kir2.3 current was not affected by LQT9 CAV3 mutation, but the heterotetramer Kir2.2-Kir2.3 demonstrated loss of function. This highlights the importance of the Kir2.1/Kir2.2-Cav3 relationship in the ventricle and the potential for arrhythmogenesis.
The signature arrhythmia in LQTS is Torsade de Pointes, which occurs in the setting of decreased repolarization reserve and susceptibility for EADs50. Unlike genetic arrhythmia syndromes related to an ion channel mutation, LQT9 and other channel-associated proteins can affect more than one channel or regulatory pathway. As such, another arrhythmogenic factor for LQT9 CAV3 mutations is an increase in INaL17 by channel nitrosylation51. Taken together, loss of IK1 and increase in INaL can decrease repolarization reserve and cause AP prolongation and EADs in iPS-CMs23. When combining the effects of F97C-Cav3 on both IK1 and INa, we hypothesized a mechanism of EAD-induced triggered activity for arrhythmogenesis. The mathematical myocyte modeling revealed INaL-associated EADs in myocytes (opposed by IK1 downregulation) but no sustained arrhythmic activity, similar to our iPS-CM experiments23. Interestingly, the Purkinje model revealed a DAD-driven triggered activity and sustained arrhythmia. Taken in context, the ventricular arrhythmia mechanism related to Cav3 mutations may be a combined mechanism: Purkinje cell-generated DADs are able to overcome an anticipated “source-sink” mismatch because the myocytes (“sink”) are EAD susceptible, decreasing the arrhythmia threshold and number of required Purkinje cells (“source”) to initiate a sustained arrhythmia. This proposed model, while only speculative and warranting further theoretical and experimental testing52, 53, is akin to Purkinje-dependent DAD perpetuation in EAD-susceptible myocytes in heart failure. Development of an in vivo model to help address and answer these questions of arrhythmogenesis is an area of active investigation in our lab.
Conclusions
Cav3 is an important regulator of ion channel function and here we provide detailed analysis of the interaction with Kir2.x isoforms, domains and CBM for interaction, as well as integration of these findings into myocyte and Purkinje modeling system to identify a putative arrhythmia mechanism, which warrants future validation in an in vivo model. Unique from other LQTS, patients with LQT9 might be vulnerable to ventricular arrhythmias due to Purkinje-dependent DAD triggered activity, which may perpetuate due to altered repolarization reserve in ventricular myocytes.
Supplementary Material
What is Known?
Kir2.1, Kir2.2 and Kir2.3 are the molecular correlates of IK1 in the heart.
LQT9 is caused by CAV3 mutations
F97C-Cav3 mutation increase late sodium current (INaL) and decreases Kir2.1 current density by distinctive mechanisms.
What the study adds?
Kir2.x isoforms have a unique intracellular pattern of distribution in association with Cav3.
LQT9 causing CAV3 mutation differentially regulates current density and cell surface expression of Kir2.x homomeric and heteromeric channels.
Mathematical Purkinje cell and myocyte model suggests that both loss of IK1 and increased INaL are required for arrhythmia generation in LQT9
Acknowledgments
We thank Dr. Louise Reilly for molecular biological assistance, Sara Abozeid for assistance with cell culture, and Drs. Erick Schumuck and Amish Raval for human tissue sharing. We thank Dr. Pan Li for help with troubleshooting the Purkinje cell model code.
Sources of Funding: This project was supported by National Institutes of Health (NIH) Grants R01-HL-128598 (L. L. Eckhardt) and R01-HL-131517 (E. Grandi), the American Heart Association Scientist Development Grant 15SDG24910015 (E. Grandi), and the Heart Rhythm Society post-doctoral fellowship 16OA9HRS (S. Morotti).
Footnotes
Disclosures: none
References
- 1.Lopatin AN, Nichols CG. Inward rectifiers in the heart: An update on i(k1) Journal of molecular and cellular cardiology. 2001;33:625–638. doi: 10.1006/jmcc.2001.1344. [DOI] [PubMed] [Google Scholar]
- 2.Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R. Functional and clinical characterization of kcnj2 mutations associated with lqt7 (andersen syndrome) J Clin Invest. 2002;110:381–388. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J. A novel form of short qt syndrome (sqt3) is caused by a mutation in the kcnj2 gene. Circulation research. 2005;96:800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 4.Tester D, Arya P, Will M, Haglund C, Farley A, Makielski J, Ackerman M. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm. 2006;3:800–805. doi: 10.1016/j.hrthm.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 5.Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90:2534–2539. doi: 10.1161/01.cir.90.5.2534. [DOI] [PubMed] [Google Scholar]
- 6.Nichols CG, Makhina EN, Pearson WL, Sha Q, Lopatin AN. Inward rectification and implications for cardiac excitability. Circulation research. 1996;78:1–7. doi: 10.1161/01.res.78.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Liu GX, Derst C, Schlichthorl G, Heinen S, Seebohm G, Bruggemann A, Kummer W, Veh RW, Daut J, Preisig-Muller R. Comparison of cloned kir2 channels with native inward rectifier k+ channels from guinea-pig cardiomyocytes. J Physiol. 2001;532:115–126. doi: 10.1111/j.1469-7793.2001.0115g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Yue L, White M, Pelletier G, Nattel S. Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation. 1998;98:2422–2428. doi: 10.1161/01.cir.98.22.2422. [DOI] [PubMed] [Google Scholar]
- 9.Liu A, Tang M, Xi J, Gao L, Zheng Y, Luo H, Hu X, Zhao F, Reppel M, Hescheler J, Liang H. Functional characterization of inward rectifier potassium ion channel in murine fetal ventricular cardiomyocytes. Cell Physiol Biochem. 2010;26:413–420. doi: 10.1159/000320565. [DOI] [PubMed] [Google Scholar]
- 10.Leonoudakis D, Mailliard W, Wingerd K, Clegg D, Vandenberg C. Inward rectifier potassium channel kir2.2 is associated with synapse-associated protein sap97. J Cell Sci. 2001;114:987–998. doi: 10.1242/jcs.114.5.987. [DOI] [PubMed] [Google Scholar]
- 11.Vaidyanathan R, Taffet SM, Vikstrom KL, Anumonwo JM. Regulation of cardiac inward rectifier potassium current (i(k1)) by synapse-associated protein-97. J Biol Chem. 2010;285:28000–28009. doi: 10.1074/jbc.M110.110858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen NA, Sha Q, Makhina EN, Lopatin AN, Linder ME, Snyder SH, Nichols CG. Inhibition of an inward rectifier potassium channel (kir2.3) by g-protein betagamma subunits. J Biol Chem. 1996;271:32301–32305. doi: 10.1074/jbc.271.50.32301. [DOI] [PubMed] [Google Scholar]
- 13.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 14.Balijepalli RC, Kamp TJ. Caveolae, ion channels and cardiac arrhythmias. Prog Biophys Mol Biol. 2008;98:149–160. doi: 10.1016/j.pbiomolbio.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright PT, Nikolaev VO, O’Hara T, Diakonov I, Bhargava A, Tokar S, Schobesberger S, Shevchuk AI, Sikkel MB, Wilkinson R, Trayanova NA, Lyon AR, Harding SE, Gorelik J. Caveolin-3 regulates compartmentation of cardiomyocyte beta2-adrenergic receptor-mediated camp signaling. Journal of molecular and cellular cardiology. 2014;67:38–48. doi: 10.1016/j.yjmcc.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volonte D, McTiernan CF, Drab M, Kasper M, Galbiati F. Caveolin-1 and caveolin-3 form heterooligomeric complexes in atrial cardiac myocytes that are required for doxorubicin-induced apoptosis. Am J Physiol Heart Circ Physiol. 2008;294:H392–401. doi: 10.1152/ajpheart.01039.2007. [DOI] [PubMed] [Google Scholar]
- 17.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-qt syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 18.Vaidyanathan R, Vega AL, Song C, Zhou Q, Tan B, Berger S, Makielski JC, Eckhardt LL. The interaction of caveolin 3 protein with the potassium inward rectifier channel kir2.1: Physiology and pathology related to long qt syndrome 9 (lqt9) J Biol Chem. 2013;288:17472–17480. doi: 10.1074/jbc.M112.435370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 20.Byrne DP, Dart C, Rigden DJ. Evaluating caveolin interactions: Do proteins interact with the caveolin scaffolding domain through a widespread aromatic residue-rich motif? PLoS One. 2012;7:e44879. doi: 10.1371/journal.pone.0044879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins BM, Davis MJ, Hancock JF, Parton RG. Structure-based reassessment of the caveolin signaling model: Do caveolae regulate signaling through caveolin-protein interactions? Dev Cell. 2012;23:11–20. doi: 10.1016/j.devcel.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willis BC, Ponce-Balbuena D, Jalife J. Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am J Physiol Heart Circ Physiol. 2015;308:H1463–1473. doi: 10.1152/ajpheart.00176.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaidyanathan R, Markandeya YS, Kamp TJ, Makielski JC, Janaury CT, Eckhardt LL. Ik1-enhanced human induced pluripotent stem cell-derived cardiomyocytes: An improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am J Physiol Heart Circ Physiol. 2016 doi: 10.1152/ajpheart.00481.2015. ajpheart 00481 02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.January CT, Riddle JM, Salata JJ. A model for early afterdepolarizations: Induction with the ca2+ channel agonist bay k 8644. Circulation research. 1988;62:563–571. doi: 10.1161/01.res.62.3.563. [DOI] [PubMed] [Google Scholar]
- 25.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circulation research. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 26.Myles RC, Wang L, Bers DM, Ripplinger CM. Decreased inward rectifying k+ current and increased ryanodine receptor sensitivity synergistically contribute to sustained focal arrhythmia in the intact rabbit heart. J Physiol. 2015;593:1479–1493. doi: 10.1113/jphysiol.2014.279638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mobley BA, Page E. The surface area of sheep cardiac purkinje fibres. J Physiol. 1972;220:547–563. doi: 10.1113/jphysiol.1972.sp009722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyden PA, Dun W, Robinson RB. Cardiac purkinje fibers and arrhythmias; the gk moe award lecture 2015. Heart Rhythm. 2016;13:1172–1181. doi: 10.1016/j.hrthm.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eckhardt LL, Farley AL, Rodriguez E, Ruwaldt K, Hammill D, Tester DJ, Ackerman MJ, Makielski JC. Kcnj2 mutations in arrhythmia patients referred for lqt testing: A mutation t305a with novel effect on rectification properties. Heart Rhythm. 2007;4:323–329. doi: 10.1016/j.hrthm.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markandeya YS, Fahey JM, Pluteanu F, Cribbs LL, Balijepalli RC. Caveolin-3 regulates protein kinase a modulation of the ca(v)3.2 (alpha1h) t-type ca2+ channels. J Biol Chem. 2011;286:2433–2444. doi: 10.1074/jbc.M110.182550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaidyanathan R, Taffet SM, Vikstrom KL, Anumonwo JM. Regulation of cardiac inward rectifier potassium current (ik1) by synapse associated protein-97. J Biol Chem. 2010 doi: 10.1074/jbc.M110.110858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 33.Delisle BP, Anderson CL, Balijepalli RC, Anson BD, Kamp TJ, January CT. Thapsigargin selectively rescues the trafficking defective lqt2 channels g601s and f805c. J Biol Chem. 2003;278:35749–35754. doi: 10.1074/jbc.M305787200. [DOI] [PubMed] [Google Scholar]
- 34.Vaidyanathan R, O’Connell RP, Deo M, Milstein ML, Furspan P, Herron TJ, Pandit SV, Musa H, Berenfeld O, Jalife J, Anumonwo JM. The ionic bases of the action potential in isolated mouse cardiac purkinje cell. Heart Rhythm. 2013;10:80–87. doi: 10.1016/j.hrthm.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akrap N, Seidel T, Barisas BG. Forster distances for fluorescence resonant energy transfer between mcherry and other visible fluorescent proteins. Anal Biochem. 2010;402:105–106. doi: 10.1016/j.ab.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and ca transient. Journal of molecular and cellular cardiology. 2010;48:112–121. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Hara T, Virag L, Varro A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput Biol. 2011;7:e1002061. doi: 10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li P, Rudy Y. A model of canine purkinje cell electrophysiology and ca(2+) cycling: Rate dependence, triggered activity, and comparison to ventricular myocytes. Circulation research. 2011;109:71–79. doi: 10.1161/CIRCRESAHA.111.246512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han H, Rosenhouse-Dantsker A, Gnanasambandam R, Epshtein Y, Chen Z, Sachs F, Minshall RD, Levitan I. Silencing of kir2 channels by caveolin-1: Cross-talk with cholesterol. J Physiol. 2014;592:4025–4038. doi: 10.1113/jphysiol.2014.273177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma D, Taneja TK, Hagen BM, Kim BY, Ortega B, Lederer WJ, Welling PA. Golgi export of the kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell. 2011;145:1102–1115. doi: 10.1016/j.cell.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melnyk P, Zhang L, Shrier A, Nattel S. Differential distribution of kir2.1 and kir2.3 subunits in canine atrium and ventricle. Am J Physiol Heart Circ Physiol. 2002;283:H1123–1133. doi: 10.1152/ajpheart.00934.2001. [DOI] [PubMed] [Google Scholar]
- 42.Szuts V, Menesi D, Varga-Orvos Z, Zvara A, Houshmand N, Bitay M, Bogats G, Virag L, Baczko I, Szalontai B, Geramipoor A, Cotella D, Wettwer E, Ravens U, Deak F, Puskas LG, Papp JG, Kiss I, Varro A, Jost N. Altered expression of genes for kir ion channels in dilated cardiomyopathy. Can J Physiol Pharmacol. 2013;91:648–656. doi: 10.1139/cjpp-2012-0413. [DOI] [PubMed] [Google Scholar]
- 43.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of t-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6854–6859. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, Streich JH, Korff B, Tuan HT, Hagen B, Luther S, Hasenfuss G, Parlitz U, Jafri MS, Hell SW, Lederer WJ, Lehnart SE. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of t-tubule membrane structures after myocardial infarction. Circulation research. 2012;111:402–414. doi: 10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aboumousa A, Hoogendijk J, Charlton R, Barresi R, Herrmann R, Voit T, Hudson J, Roberts M, Hilton-Jones D, Eagle M, Bushby K, Straub V. Caveolinopathy–new mutations and additional symptoms. Neuromuscul Disord. 2008;18:572–578. doi: 10.1016/j.nmd.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 46.Stavusis J, Inashkina I, Jankevics E, Radovica I, Micule I, Strautmanis J, Naudina MS, Utkus A, Burnyte B, Lace B. Cav3 gene sequence variations: National genome database and clinics. Acta Neurol Scand. 2015;132:185–190. doi: 10.1111/ane.12369. [DOI] [PubMed] [Google Scholar]
- 47.Li X, Ortega B, Kim B, Welling PA. A common signal patch drives ap-1 protein-dependent golgi export of inwardly rectifying potassium channels. J Biol Chem. 2016;291:14963–14972. doi: 10.1074/jbc.M116.729822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenhouse-Dantsker A, Logothetis DE, Levitan I. Cholesterol sensitivity of kir2.1 is controlled by a belt of residues around the cytosolic pore. Biophys J. 2011;100:381–389. doi: 10.1016/j.bpj.2010.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furst O, Nichols CG, Lamoureux G, D’Avanzo N. Identification of a cholesterol-binding pocket in inward rectifier k(+) (kir) channels. Biophys J. 2014;107:2786–2796. doi: 10.1016/j.bpj.2014.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185–1192. doi: 10.1172/JCI112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng J, Valdivia CR, Vaidyanathan R, Balijepalli RC, Ackerman MJ, Makielski JC. Caveolin-3 suppresses late sodium current by inhibiting nnos-dependent s-nitrosylation of scn5a. Journal of molecular and cellular cardiology. 2013 doi: 10.1016/j.yjmcc.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: Requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local beta-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circulation research. 2012;110:1454–1464. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.