

Abstract
This study identifies a novel mechanism linking IL-17A with colon tissue repair and tumor development. Abrogation of IL-17A signaling mice attenuated tissue repair of DSS-induced damage in colon epithelium and markedly reduced tumor development in AOM/DSS model of colitis-associated cancer. A novel IL-17A target gene, PLET1 (a progenitor cell marker involved in wound healing) was highly induced in DSS-treated colon tissues and tumors in an IL-17RC-dependent manner. PLET1 expression was induced in the LGR5+ colon epithelial cells after DSS treatment. LGR5+PLET1+ marks a highly proliferative cell population with enhanced expression of IL-17A target genes. PLET1 deficiency impaired tissue repair of DSS-induced damage in colon epithelium and reduced tumor formation in AOM/DSS model of colitis-associated cancer. Our results suggest that IL-17A induced PLET1 expression contributes to tissue repair and colon tumorigenesis.
Keywords: IL-17A, colon cancer, tissue repair, inflammation
Introduction
Proper maintenance of the gastrointestinal tract is essential in preventing systemic absorption of enteric toxins and bacteria. Disturbance of the intestinal epithelium homeostasis, by physical, chemical injury or microbial infections, can lead to intestinal barrier dysfunction, which is a main feature of inflammatory bowel diseases (IBD). Evidence suggests that colon cancer is also associated with microbial infection, injury, inflammation and tissue repair processes (1) . While it is well documented that chronic inflammation, is a major risk factor for the development of colon cancer, it is still an evolving research area for how inflammation and subsequent tissue repair are intrinsically linked to colon cancer.
Previous studies have shown that CD4+ IL-17A-producing, Th17 cells are abundant in the intestinal mucosa and regulated by commensal bacteria (2). Induction of Th17 cells has been shown to promote colon tumorigenesis in mouse models (3). Consistently, mice lacking IL-17A or IL17RA are protected from intestinal/colon tumor development. In Apcmin mice, which spontaneously develop intestinal adenomas, IL-17A or IL-17RA deficiency resulted in significantly fewer adenomas (4) (5) (6). Furthermore in models of colitis associated cancer (CAC), where a pre-malignant cell is induced with the carcinogen azoxymethane (AOM) and promoted by repeated cycles of dextran sulfate sodium (DSS), mice deficient in IL-17A also develop fewer colonic tumors (7) (8). However, the role of IL-17A in the intestinal tract is rather complex. Although traditionally considered only as pro-inflammatory cytokine, IL-17A exerts a protective role in the intestine as blockade of IL-17A in IBD patients exacerbates intestinal inflammation (9) (10) (11). Based on these findings, we hypothesize that additional functions of IL-17A signaling, other than its pro-inflammatory role, underline the tumor-promoting effect. In this study, we aim to further investigate the mechanism by which IL-17A signaling mediates tissue repair and tumorigenesis.
The receptor for IL-17A is a heterodimeric complex composed of IL-17RA and IL-17RC. In this study, we report that IL-17A provides a critical link between colon tissue repair and tumor development. IL-17RC deficiency compromised tissue repair with defective regenerative proliferation in the colon epithelium, and IL-17RC-deficient mice had markedly reduced tumor burdens in the AOM/DSS colon cancer model. Gene array analysis identified several novel IL-17A target genes, including PLET1 (a progenitor cell marker involved in wound healing), that were highly induced in DSS-treated colon tissues and tumors. While DSS induced PLET1 in the LGR5+ stem cells, LGR5+PLET1+ marked a group of highly proliferative cells with enhanced expression of IL-17A target genes. Importantly, PLET1 deficiency hindered the tissue repair and attenuated tumor formation in AOM/DSS model. Our results demonstrate that IL-17A-induced PLET1 expression plays a critical role in tissue repair and colon tumorigenesis.
Materials and Methods
Mice and husbandry
The IL-17RC-deficient mice, were provided by Dr. W. Ouyang (Genentech) and generated as described in (12) on C57/Bl6 background. The Act1f/f and Act1-deficient (both C57/Bl6) mice were generated as described in (13) The Lgr5EGFP-IRES-CreERT2 knock-in mice were originally generated as described (14). The Lgr5EGFP-IRES-CreERT2 mice were crossed to Act1f/f and Act1-deficient mice to generate conditional Act1f/– and Act1f/+ Lgr5cre mice. ROSA26-EGFP was purchased from The Jackson Laboratory. Plet1 knockout mice were generated with CRISPR technology in Dr. Yina Huang’s lab. For all experiments, the controls mice were littermate controls co-housed with the knockout mice. Bone marrow chimera mice were generated by irradiating C57BL/6 recipient (WT or Plet1-/-) mice twice with 600rad at a 4-h interval. Mice were reconstituted postirradiation with 1.5×107 donor bone marrow cells from WT or Plet1-/- mice by tail vein. Intraperitoneally gentamicin was administered to prevent infection. Mice were analyzed 6 weeks after the reconstitution. All animal procedures were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic Foundation.
Colitis Associated Cancer Model
8-week-old gender matched mice (IL-17RC-deficient and WT littermates) were injected with Azoxymethane (Sigma) 12.5 mg/kg. 5 days after the AOM injection, mice were treated with 2.5% DSS (36,000-50,000Da, MP Biomedicals) in sterile tap water for 5 days. Following this mice were given regular water for 16 days. This cycle was repeated for three cycles, mice were sacrificed 10 days after the end of the third DSS cycle. Tamoxifen injections during the CAC procedure in Act1;Lgr5 conditional knock-outs consisted of two weekly injections of Tamoxifen prior to AOM administration. Tamoxifen was injected on the first day of DSS treatment cycles throughout the CAC procedure. All tamoxifen injections were 5 mg/ml in corn oil. Mice were sacrificed and colons removed, they were then cut longitudinally and a person blinded to the mouse genotypes counted tumors by magnifying glass. The colon was then either placed in 10% Formalin or embedded in Optimal Cutting Temperature compound (OCT) snap frozen for tissue histology. The tumors were excised and snap frozen in liquid nitrogen for further processing for RNA or protein.
Gut Permeability Assay
Age and gender matched mice were treated with 3.5% DSS for 3 days. On the third day mice were gavaged with 150 μl of 80 mg/ml 4 kDa FITC-dextran (Sigma Aldrich) in PBS. Mice were sacrificed 4 hours later and blood was collected by cardiac puncture. Serum fluorescence was quantified using a VictorX3 (Perkin-Elmer Life Sciences) at excitation 485 nm, emission 530 nm for 1 second.
Histology and Immunohistochemistry
Tissues were fixed with 10% formalin and placed into paraffin tissue blocks according to routine methods by AML laboratories, or tissue was embedded in optimum cutting temperature (OCT) and sectioned. Paraffin-embedded were stained according to routine methods and antigen retrieval conducted in Citrate buffer. IHC antibodies included rat anti mouse Ki67 (Dako Cytometry), rabbit anti-mouse Ki67 (Abcam), rabbit anti CD4 and CD11b (eBioscience). Light microscopy images were obtained using an Olympus BX41 microscope (Olympus Corp.) Immunofluorescence staining was conducted with frozen tissue embedded in OCT sectioned (10μm) and fixed in acetone:methanol for 10 minutes. Secondary antibody conjugated with either, Alexa-Fluor 488 or 594 (Life Technologies) were used. Sections were mounted with VectaShield fluorescent Mounting Media (Vector Lab Inc.) containing DAPI to visualize nuclei. anti-PLET1 (clone 1D4) was as described (Depreter et al 2008). Rabbit antibody to human PLET1 (C11orf34) was from Sigma-Aldrich Prestige Antibodes by Atlas Antibodies, Visualization of GFP was conducted in fresh tissue fixed overnight in 4% paraformaldehyde, de-hydrated in 20% sucrose then placed in OCT and flash frozen and sectioned. Staining for GFP was conducted on OCT-embedded tissue, fixed for 10 minutes in 2% PFA, and incubated for 2 hours at room temperature with Rabbit anti-GFP 8334 (Santa Cruz Biotech) TUNEL assay was performed on frozen sections using TUNEL staining kit (Roche). Immunofluorescence images were generated using a Leica DM2500 or EVOS Floid (Applied Biosystems) and fluorescent images were processed using the NIH ImageJ program. Formalin-fixed, paraffin-embedded colon cancer biopsy specimens were obtained from the Department of Pathology, CWRU. Immunofluorescence images were generated as indicated above. Following immunofluorescence microscopy, the same sections were washed in PBS for 4 times and subjected to H&E staining. Quantification of images were either performed with ImageJ to calculate positive signals per view or by manually counting number of positive cells in a crypt or an area equivalent of a crypt in case no discerning structure could be found.
Gene array profiling
A total of 200 ng RNA from whole colon tissue was used for target labeling, and the target preparation was done on a Biomek FXP (Beckman Coulter, Brea, CA) using a GeneChip HT 3′ IVT Express Kit (Affymetrix, Santa Clara, CA). Labeled cRNA were hybridized on an Affymetrix GeneChip HT-MG-430PM-96 (Affymetrix). All array hybridization, washing, and scanning were performed on GeneTitan (Affymetrix), according to the manufacturer’s recommendations. Anti-log RMA data were used. P value was determined by two-tailed t-tests, p-value of <0.05 were considered significant. Volcano plots were generated by taking the –Log2 fold-ratio difference in expression value between WT and IL-17RC-deficient tissue and plotting on the x-axis and –Log10 of p-value was plotted on the y-axis and the graphs were generated in Prism 5.0 software (Graphpad). The biological replicates used for gene array profiling are as follows; Day 15, 4-WT and 4-KO; Tumor 5-WT and 6-KO.
RNA isolation and quantitative PCR
For RNA isolation of colon tissue, freshly isolated colon tissue was snap-frozen in liquid nitrogen and stored at -80°C until processing. Colon tissue was isolated using miR-Vana RNA isolation Kit (Life Technologies, Ambion) according to the provided protocol. Colon tissue was homogenized in lysis/binding buffer provided in the kit. Superscript II Reverse Transcription Kit (Invitrogen) was used to make cDNA from 0.2 – 1 μg total RNA and rt-PCR performed on a Veriti 96-well ThermoCycler (Applied Biosystems). Quantitative RT-PCR was carried out using published primers as well as those deposited in PrimerBank (Harvard), oligos were generated by Invitrogen, and SYBR Green (Applied Biosystems) on a Step-One Plus Real-time PCR machine (Applied Biosystems). All gene expression data were normalized to ms-actin or hs-gapdh as controls. Unique primers used in this study, mPlet1 forward; tcatccgtgaaaatggaaca reverse; tggctgtagtcttggctgtg. mCd177 forward; gggtgactccaaaacaatcg reverse; ctaacatccaggccgatagc. mFut2 forward; acctccagcaacgaatagtga reverse; gccgatggaattgatcgtgaa. mPigr forward; ccggcacacccggaaatac reverse; tgcctgaatactccttggaga. hPLET1 forward; agctcagccttcaagccttt reverse; gtgtgctggtgagaaaagca.
Isolation and culture conditions of colonospheres
Colonosphere culture protocol was adapted from (15) . After the isolation cells were either re-suspended in media for sorting of GFP-positive cells, DMEM-F12, 1x N2 and B27 supplements (from Invitrogen/Gibco), Pen/Strep, 10 μM HEPES, 10 μM Y27632 Rock inhibitor (Sigma Aldrich), or suspended in growth media which consisted of DMEM/F12, 1% BSA, 30 ng/ml Wnt3a (R&D systems) 500 ng/ml Rspo1(R&D systems), 50 ng/ml noggin (R&D systems), 20 ng/ml EGF (Peprotech), 50 ng/ml HGF (R&D systems), + 10 μM Y27632 (Sigma) for the first two days. The cells were further embedded in growth factor reduced Matrigel (BD biosciences) and grown on 96– or 48– well plates, the media was refreshed every two days.
Flow cytometry analysis of intratumoral T cells
Tumors excised from mouse colon were cleaned in HBSS, cut into small pieces, and resuspended in HBSS supplemented with 0.5 μM EDTA and 15 μg ml−1 dithiothreitol, followed by orbital shaking for 15 min twice at room temperature. Tumor tissues were then resuspended in complete RPMI supplemented with 400 μg ml−1 DNase and 1 mg ml−1 collagenase and shaken at 37 °C for additional 90 min. Supernatant and tumor pieces were passed through a 70-μM cell strainer and washed. Cells were resuspended in a 33% Percoll gradient (GE healthcare, Piscataway, NJ) and centrifuged at room temperature for 20 min. Pellet was collected, washed, and used for PMA and ionomycin stimulation for 5 hours. Stimulated cells are treated with Golgi-Stop (BD biosciences) 2 hours prior to harvest. Harvested cells were washed and stained for CD4 (eBioscience), IL-17 (eBioscience) and IL-22 (eBioscience). Analysis of flow cytometry data was performed using FlowJo by gating on the CD4+ population.
Statistical Analysis
Raw datasets were first tested for normality by the Kolmogorov-Smirnov test. Data were then analyzed using unpaired t test or the Mann-Whitney rank-sum test where appropriate. Two-way analysis of variance (ANOVA) was conducted for grouped analyses. P-values less than or equal to 0.05 were considered statistically significant. All statistics and graphical analyses were conducted with Mac Prism 5.0 software (GraphPad).
Results
IL-17RC-deficiency attenuates tumor development
While IL-17RA is viewed as a common receptor subunit for IL-17A family members, IL-17A, as a homodimer or a heterodimer with IL-17F, specifically binds to IL-17RA/IL-17RC heterodimeric receptor. To investigate the role of IL-17A in colon tumorigenesis, we tested how deletion of IL-17RC affected colon cancer development and progression in CAC. In IL-17RC–/– mice, AOM/DSS-induced tumor numbers and sizes were significantly reduced (Fig. 1a, b). Interestingly, the immune cell infiltration into the tumor, including CD4+ T cells and CD11b+ myeloid cells, were similar between WT and IL-17RC–/– mice (Fig. 1c). Flow cytometry analysis indicated there were comparable numbers of IL-17A- and IL-22-producing CD4+ T cells in the tumors from WT and IL-17RC–/– mice (Supple. Fig. 1a). Notably, compared to the tumors in the controls, IL-17RC–/– tumors exhibited reduced staining for the proliferative marker Ki67, although no differences in nuclear localization of β-catenin were detected (Fig. 1c). Consistent with reduced proliferation in the tumors, CYCLIN-D levels were markedly reduced in the IL-17RC–/– tumors (Fig. 1d). Activation of transcription factors NFkB and STAT3, were however comparable between the WT and IL-17RC–/– tumors (Fig. 1d). Together these results suggest that the reduced tumorigenesis is unlikely due to a decrease in inflammation in the IL-17RC-/- mice.
Figure 1. IL-17RC mediates colon tumorigenesis in CAC model.
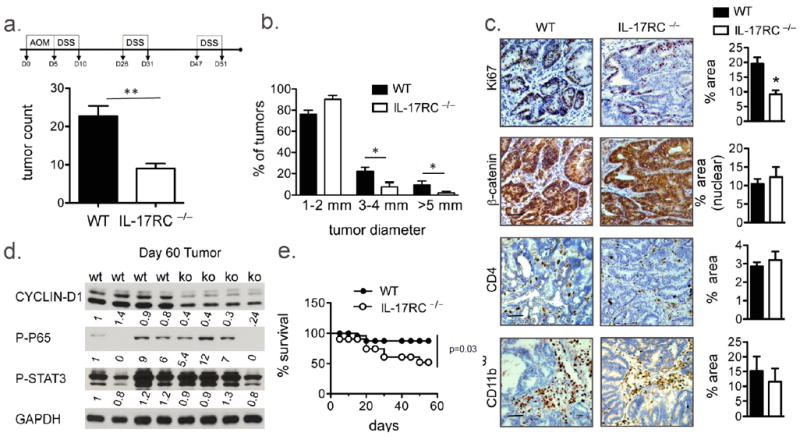
(a, top) Schematic of CAC model (a) Tumor number (b) Tumor size distribution (a and b n=12 per group), (c) IHC images from WT and IL-17RC-deficient tumor tissue stained for the indicated markers. (d) Immunoblot analysis of colon tumor tissue from the indicated mice, each lane represents one mouse. (e) Percent survival of the indicated mice throughout the CAC model. Representative images and data are shown in (c). Quantification was performed on staining of 5 tumors from each group and in 3 views per sample. Error bars represent mean ± SEM scale bars =100μm, p values shown are * <0.05.
IL-17RC-deficiency impairs regenerative response following DSS-induced injury
Intriguingly, the IL-17RC–/– mice exhibited reduced survival compared to WT mice in spite of a decreased tumor burden (Fig. 1e), which led us to hypothesize that IL-17A plays a protective role in gut injury/inflammation. Indeed, the IL-17RC–/– mice lost significantly more weight after DSS treatment compared to WT (Fig. 2a). DSS treatment induces colonic inflammation that leads to tissue damage and upon its withdrawal tissue repair process predominates. Thus, IL-17RC–/– and control mice were treated with DSS for 5-days followed by 5 days of regular water to allow for the epithelium to recover. Notably, the IL-17RC–/– and control mice showed similar tissue injury at day 5 of DSS treatment (Fig. 2b). On the other hand, whereas WT mice had replenished the DSS-injured epithelium with highly proliferative nascent crypts 5 days after DSS removal (termed d10), this tissue repair was markedly impaired in the IL-17RC–/–mice (Fig. 2b). In support of this, we detected less Ki67 staining in the colon of IL-17RC–/– mice on day 10 (5 days after DSS removal) compared to that of wild-type mice (Fig. 2b and c). We then checked the gut permeability in these mice on day 10 (5 days after DSS removal) using FITC-dextran assay. Consistently, we detected more FITC in the serum of IL-17RC–/– mice than that in littermate controls (Fig. 2d), suggesting that DSS-treated IL-17RC–/– mice have a defective in tissue repair.
Figure 2. IL-17RC-deficiency impacts colon epithelial integrity and wound healing response.
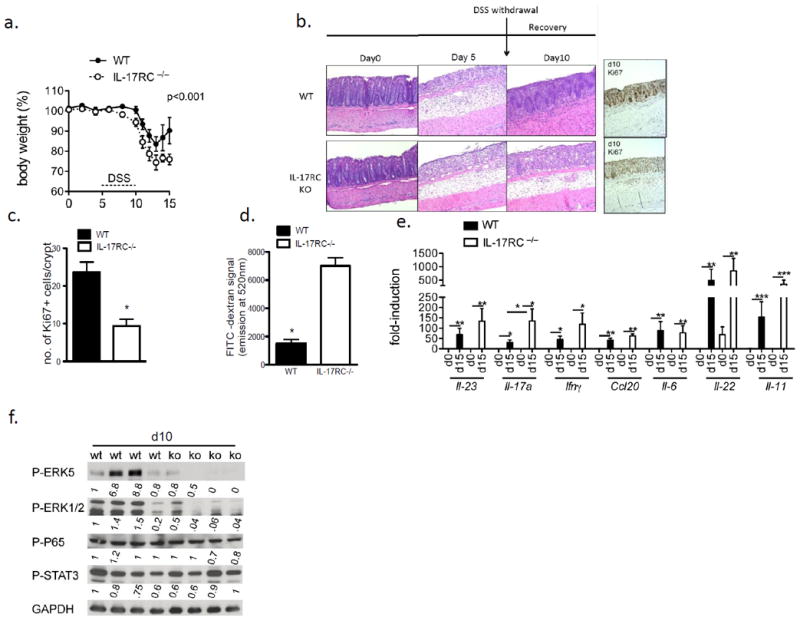
(a) Weight loss during first 15 days of CAC (b) H&E or IHC staining for Ki67 in colon tissue from DSS-treated WT and IL-17RC-deficient mice. Mice were treated with DSS for 5-days followed by 5 days of regular water to allow for the epithelium to recover. (c) Quantification of Ki67 staining. (d) Serum FITC signal from mice treated with 3.5% DSS for 5 days followed by DSS withdrawal for 5 days, and then gavaged with FITC-dextran. Data shown is from a representative experiment with n=4 mice per group. (e) RT-qPCR from colon tissue of mice from day 10 of the DSS treatment, n=4 mice per group. All gene expression values were normalized to β-actin. (f) Immunoblot analysis of DSS treated colon tissue from WT or IL-17RC-deficient (KO) mice. Each lane represents a single mouse. Quantification was performed on staining of 3 colon sections from each group and in 3 views per sample. Experiments were reproduced 3 times, error bars represent mean ± SEM scale bars =100μm, p values shown are * <0.05, ** <0.01, *** <0.001.
Several studies report that STAT3-activating cytokines, IL-6, IL-11 and IL-22 are critical mediators of the colon tissue repair response and colon tumorigenesis (16) (17) (18) (19). Nevertheless, expression of these cytokines in the colonic tissue of IL-17RC-deficient mice was comparable to that in WT controls (Fig. 2e), while IL-22 was even slightly increased at the protein level in the colonic tissue of IL-17RC-deficient mice (Supple. Fig. 1b). Furthermore, STAT3 and NFkB activation were similarly active within the IL-17RC–/– colon tissue compared to littermate controls (Fig. 2f). Thus, the impact of IL-17A on colonic tissue repair and barrier integrity may not be due to defective production of STAT3 activating cytokines. On the other hand, it is intriguing that ERK1/2 and ERK5 activation were dramatically reduced in the IL-17RC-deficient colon tissue compared to littermate controls (Fig. 2f), suggesting a critical role of ERKs in IL-17A-dependent tissue repair in response to DSS-induced injury.
IL-17A novel target genes are highly induced in regenerating and tumor tissues
These data thus far suggest that the impact of IL-17A in colon tumorigenesis might be linked to its role in the tissue regeneration/repair process. Previous studies have suggested that genes involved in tissue repair may also contribute to the survival of pre-malignant cells during tumor growth (20). Thus, we sought to determine IL-17A-responsive genes that might link tissue repair and tumor development. We performed gene array analysis on colonic tissue during recovery-phase after DSS withdrawal (day 10) and tumor tissue (tumor) from IL-17RC–/– and WT mice. The impact of IL-17RC deficiency on the global gene expression profiles of the day 10 colon tissues and tumor tissues were determined (Fig. 3a, b). Interestingly, several genes with possible functions at barrier surfaces were significantly reduced in the IL-17Rc–/– colon tissues and/or tumors, including Pigr, Fut2, Cd177 and Plet1 (Fig. 3a,b,c). These genes were further confirmed by quantitative RT-PCR (Fig. 3d,e).
Figure 3. IL-17RC-dependent gene analysis during regeneration and in established tumors.
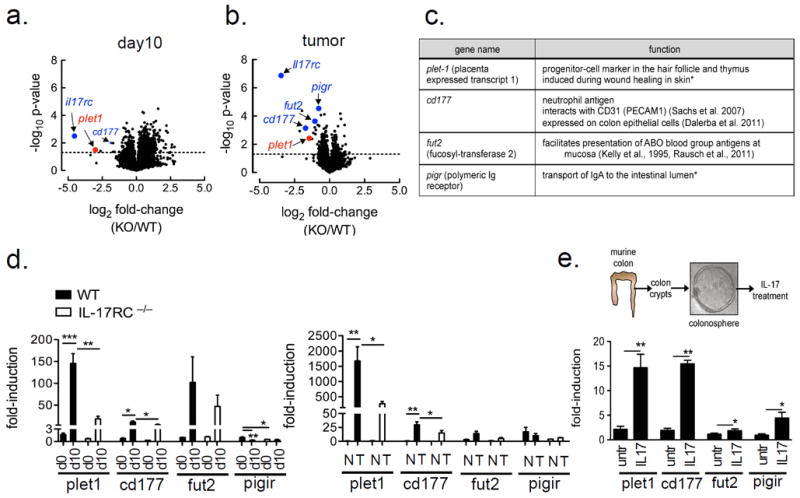
Volcano plots from Affymetrix gene-array comparing (a) day 10 (3.5% DSS for 5 days followed by DSS withdrawal for 5 days) colon tissue (n=4 per group) and (b) tumor tissue (n=5-6 per group) mRNA between WT and IL-17RC-deficient mice. Expression data were averaged and p-values derived by fold-change in expression between IL-17RC-deficient and WT, dotted line represents a p-value of 0.05 as determined by unpaired two-tailed t-test. (c) Table of genes significantly reduced in the tumors of IL-17RC-deficient mice, and their known functions, * references as cited in the text. (d) Gene expression by RT-qPCR from untreated and DSS Day 10 treated colon tissue, n=5 mice per group or (e) RT-qPCR results from normal-adjacent (N) and tumor tissue (T), n=5-6 mice per group. (f, top) Schematic of colonosphere isolation and IL-17A treatment procedure. (f, bottom) Gene expression by RT-qPCR from colonospheres generated from WT mice. Spheres were grown for one week and then treated with IL-17A (50 ng/ml) for 24 hours, shown are representative data from 3 independent experiments, RT-qPCR data in d-f were normalized to β-actin. Data presented as mean ± SEM, p values shown are * <0.05, ** <0.01, ***<0.001.
Next, we sought to determine whether these genes (Plet1, Cd177, Fut2 and Pigr) are directly regulated by IL-17A in colon epithelial cells. Thus we isolated primary murine colon crypt cells, which is comprised of LGR5+ stem cells and differentiated cells, and embedded these cells into a 3-D matrix (matrigel) for colonosphere formation in the presence and absence of exogenous IL-17A (Fig. 3e). While Pigr is a known IL-17A target gene (21) (22), IL-17A also highly induced the expression of Plet1 and Cd177 in colonosphere culture (Fig. 3f). Although IL-17A-induced Fut2 expression was modest in the colonospheres, its high expression in the DSS-treated and tumor tissues was partially dependent on IL-17RC, suggesting that IL-17A may cooperate with other extrinsic factors to promote its expression in vivo. Notably, Plet1 was the most highly induced gene in vivo both in colonic tissue during recovery-phase after DSS withdrawal (day 10) and tumor tissue (tumor).
PLET1+LGR5+ mark a highly proliferative cell population expressing IL-17A-target genes
Previous studies have shown that intestinal stem cells (including the LGR5+ cells) are essential for tissue repair as well as tumorigenesis. We then sought to determine the cellular origin of PLET1-expression in relation to LGR5+ cells in response to DSS. We isolated colonic epithelial cells from naïve or DSS-treated Lgr5-eGFP-creERt2 mice, followed by FACS for PLET1 and LGR5 expression (Fig. 4a). DSS-induced PLET expression was primarily restricted to the LGR5+ cells, giving rise to two distinct cell populations, LGR5+ (G) and PLET1+/LGR5+ (G+P) (Fig. 4a). Interestingly, the PLET1–expressing cells exhibited higher expression levels of late S-phase cyclins, Cyclin A1 and Cyclin B (Fig. 4b). In addition, Plet1 expression co-localized with Ki67+ cells in DSS-treated colon tissue and tumor (Fig. 4c). These results suggest that PLET1+/LGR5+ (G+P) marked highly proliferative cells. Furthermore, other IL-17A-regulated genes, Fut2, Pigr and Cd177 were also increased in the G+P cells (Fig. 4b), implicating that PLET1+/LGR5+ (G+P) might be an IL-17A-responsive cell population. Indeed LGR5-specific Act1-deficiency (abrogating IL-17A pathway) resulted in a substantial reduction of PLET1+/LGR5+ (G+P) cells compared to that from control mice in response to DSS (Fig. 4e), confirming that this DSS-induced cell population was IL-17A-dependent. Consistently, the expression levels of the other IL-17A target genes were also ablated from the Act1F/–Lgr5cre cells (Fig. 4f). Moreover, the reduction of PLET1+/LGR5+ (G+P) cell population in Act1F/–Lgr5cre mice was accompanied by impaired tissue repair (Supple. Fig 2a and 2b) and attenuated AOM/DSS colon tumorigenesis (Supple. Fig 2c), implicating the functional importance of the PLET1+/LGR5+ cell population.
Figure 4. PLET1 is induced from colonic crypts during DSS treatment.
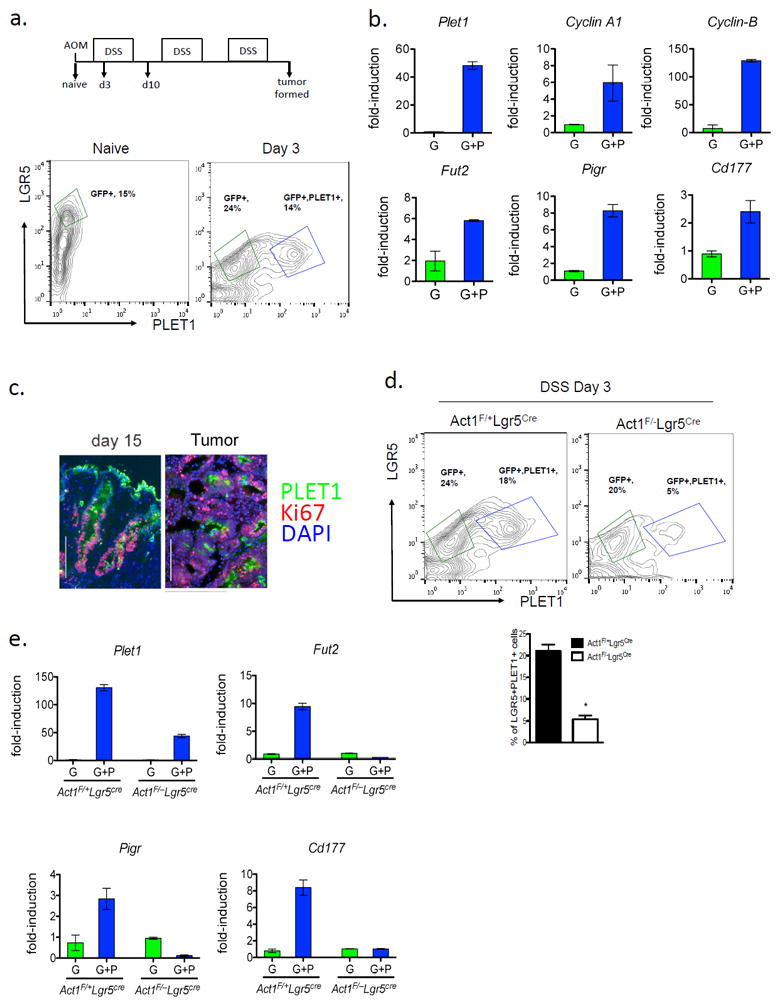
(a) FACS analysis of colonic epithelial cells derived from DSS treated Lgr5EGFP-IRES-CreERT2 mice, n=3 mice combined for each treatment group. (b) Gene expression from FACS-sorted cell populations from day 3 of DSS treatment; G, LGR5-eGFP-positive, GP, LGR5-eGFP/PLET1-positive, all RT-qPCR results are relative to Gapdh. (c) Immunofluorescence staining of PLET1 and Ki67 in colon tissue and tumors from AOM-DSS treated mice. (d) Representative flow cytometry analysis of isolated colonic epithelial cells and quantification of FACS data from the indicated mice, n=3 mice combined for each group. (e) RT-qPCR from the indicated FACS sorted cell populations relative to Gapdh. Data are representative of 2 independent experiments are presented as mean ± SEM.
PLET1 promotes ERK1/2 activation, tissue repair and tumorigenesis
We then studied how IL-17A signaling leads to up-regulation of Plet1 expression. It is noteworthy that the activation of ERKs was dramatically reduced in the DSS-treated IL-17RC-deficient colon tissue compared to littermate controls (Fig. 2e). We recently reported that IL-17A induced ERK5 activation controls the keratinocyte proliferation in the inflammation associated skin cancer model. We hypothesized that the same pathway also operates in the colonic epithelial cells. ERK5 activation was indeed markedly increased in colon crypt cells following IL-17A stimulation (Fig. 5a). Furthermore, inhibitor of MEK5 (BIX01289, the upstream MAP2K for ERK5), reduced the expression of PLET1 in DSS-treated colon crypts (Fig. 5c), and increased the gut permeability (Fig. 5b), indicating that IL-17A-induced plet1 expression is ERK5-dependent during tissue repair process in the colon.
Figure 5. PLET1 is an ERK5-target and promotes cell transformation and P-ERK1/2 signaling.

(a) Lysates from colon crypts of WT mice untreated or stimulated with IL-17A (50 ng/ml) were analyzed by western blotting with the indicated antibodies. (b) Serum FITC-dextran from DSS treated mice injected with BIX02189 or vehicle control, shown are representative results from two independently performed experiments. (c) Western analysis of lysates from colon epithelial cells of mice treated with BIX01289 and DSS for 5 days followed by 5 days of DSS removal. Each lane is from one mouse. Band intensity normalized to β-ACTIN is indicated below the blots. (d-f) Plet1-/- (n=5) and Plet1+/- (n=6) littermate control mice were subjected to 3.5% DSS treatment for 5 days followed by 5 days of DSS withdrawal. On day 10 mice were subjected to gut permeability assay using FITC dextran (e). The mice were sacrificed and the colons were taken for H&E and Ki67 staining (d) or western blot analysis (f). (g) Plet1+/- (n=15) and Plet1-/- (n=10) mice were subjected to AOM-DSS regiment. Number of tumors per mouse and tumor size distribution were plotted. (h) Representative image of H&E and Ki67 staining of tumors from mice of indicated genotypes. Quantification is shown as bar graph to the right. Quantification of histology was performed on staining of 3 colon sections from each group and in 3 views per sample. All data presented are means ± SEM, p values shown are * <0.05.
To investigate the functional importance of PLET1, we generated Plet1 knockout mice using CRISPR-Cas9 technology. We introduced a frameshift in the coding region of PLET1 to abolish the protein expression (Supple. Fig 3). We subjected the Plet1-/- and Plet1+/- littermate control mice to the same DSS treatment as described for figure2: mice were treated with DSS for 5-days followed by 5 days of regular water to allow for the epithelium to recover. Histologically, the Plet1-/- and Plet1+/- littermate control mice exhibited similar epithelial erosion, cell infiltration and edema on day 5 (d5) of DSS treatment (Fig. 5d). On the other hand, whereas Plet+/- mice had replenished the DSS-injured epithelium with highly proliferative nascent crypts 5 days after DSS removal (termed d10), this tissue repair was markedly impaired in the Plet1-/- mice (Fig. 5d). In addition, Plet1 deficiency also reduced the number of proliferating cells in the recovering epithelium 5 days after withdrawal of DSS (d10, Fig. 5d). Consistently, 5 days after withdrawal of DSS, Plet1-/- mice exhibited significantly increased gut permeability compared to the Plet1+/- littermate control (Fig. 5e). Bone marrow transfer experiment indicated that the protective role of Plet1 was primarily contributed by the non-hematopoietic compartment (Supple. Fig. 4). Interestingly, while ERK5 activation was comparable between Plet1+/- and Plet1-/- colon tissue, p-ERK1/2 levels were reduced in the Plet1-/- colon tissue from day 10 (Fig. 5f), suggesting that Plet1-dependent ERK1/2 activation might play an important role during the tissue repair process. Plet1-/- and Plet1+/- littermate control mice were subjected to the AOM/DSS colon cancer model. PLET1 deficiency substantially reduced the tumor numbers (Fig 5g). The tumors from PLET1-deficient mice showed reduced Ki67 staining, suggestive of an attenuated tumor growth (Fig 5h).
Discussion
In this study, we demonstrate that IL-17A signaling links colon tissue repair and tumor development. We found that PLET1, a progenitor cell marker involved in cell proliferation is highly induced in the Lgr5+ cells in response to IL-17A stimulation. PLET1 was highly enriched in regenerating and tumor tissue in an IL-17RC–dependent manner, and marks a population of highly proliferative cells enriched for IL-17A target genes. While ERK5-dependent emergence of PLET1 correlated with p-ERK1/2 signaling and colonic barrier function, PLET1 deficiency impaired tissue repair and tumor development, respectively, in DSS-induced colitis and AOM/DSS colitis-associated cancer models. Our results suggest that a unique stem-cell IL-17A response, denoted in part by PLET1 expression, contributes to tissue repair and colon tumorigenesis.
Through gene-array analysis of the IL-17RC-deficient colon tissues and tumors, we identified several novel IL-17A-regulated genes including Pigr, Fut2, Cd177 and Plet1. We further confirmed these genes as being regulated by IL-17A in the colonosphere culture. Functionally, these genes have been demonstrated to be important for intestinal/colonic homeostasis. Pigr, a known IL-17A target gene, functions to facilitate transport of mucosal IgA via intracellular vesicles to the gastrointestinal lumen where it can be processed into soluble IgA. In humans Fut2 is critical for processing the H-blood group antigen, the precursor to ABO antigens, at mucosal sites. What is noteworthy is that a reported 20% of Caucasians, homozygous for Fut2 null alleles, do not express ABO antigens in saliva or mucus (23). Interestingly lack of ABO blood group antigens into body fluids has been associated with Crohn’s disease, development of oral candidiasis, recurrent urinary tract infection and infection with meningococcus to name a few (24). Additionally, Cd177 is a documented neutrophil antigen as well as a marker of human colonic epithelial cells and prognostic indicator in colon cancer (25). Moreover, Cd177 has been reported to interact with CD31 (PECAM) expressed on endothelial cells (26). Thus it is conceivable that IL-17A-dependent regulation of these genes promotes colonic homeostasis all of which may contribute to processes of tissue regeneration and colonic tumorigenesis.
Plet1 showed the highest induction in the wounded tissue and the tumor. It has been shown that Plet1 is up-regulated during wound healing in the skin and can directly influence migration of keratinocytes (27, 28). During homeostasis, PLET1 expression is restricted to a region within the hair follicle near the upper isthmus and sebaceous gland (29) . While this region contains other distinct progenitor cell populations, upon injury, only the PLET1 expressing cells are drawn out of this niche. Furthermore, PLET1-emergence observed with epidermal Snail-overexpression in skin tumor progression, where PLET1+ cells exhibited elevated expression of cyclins, Foxm1 and other molecules implicated in G2/M cell cycle progression and cytokinesis (30) . Consistent with this report we found that PLET1 was associated with the Ki67 proliferation marker in DSS-treated colon tissue and tumors. Interestingly, cell surface staining of PLET1 revealed a unique cell population that is LGR5+/PLET1+–double positive. Furthermore, the double-positive cells expressed high levels of the identified IL-17A-dependent genes and cyclin mRNAs. Together these data are supportive of a plausible LGR5+ stem-cell intrinsic response to IL-17A signaling.
We analyzed the colon tissue for IL-17A-dependent signaling pathways that are activated during the tissue repair following DSS-induced injury. Although STAT3 and NFkB have clearly been implicated in mucosal wound healing and tumorigenesis, we did not observe a substantial defect in the activation of these pathways in IL-17Rc–/– tissue. Notably, there was a marked impairment in the activation of ERK5 and ERK1/2. Inhibiting the activity of MEK5 (BIX02189), the upstream MAP2K for ERK5, resulted in increased FITC-signal in the serum as well as a significant reduction in PLET1 levels. Importantly, PLET1 deficiency indeed reduced DSS-induced ERK1/2 phosphorylation, compromised tissue repair and attenuated tumor development in AOM/DSS cancer model.
Like IL-17A, IL-22 has been demonstrated to promote wound healing in the colon epithelium by activating the STAT3 signaling. In support of this, IL-22-deficient mice are highly susceptible to DSS challenge. We found the protein level of IL-22 was slightly elevated in the colon tissue of DSS-treated IL-17RC-/- mice compared to that in WT mice (Supple. Figure.1b). Considering the protective role of IL-22 in colon tissue repair, the attenuated tissue repair in DSS-treated IL-17RC-/- mice is unlikely due to the expression of IL-22 in these mice. Notably, the IL-17A level in the IL-17RC-/- mice was significantly elevated in response to DSS treatment compared to the littermate control (Figure 2e). Together with the elevated IL-22 expression, the data collectively suggest a dysregulation of Th17/Th22 balance in the IL-17RC-/- mice. Given the critical role of Th17/Th22 in the colon, one might speculate that such desregulation of Th17/Th22 may affect the homeostasis of colon epithelium via mechanisms beyond IL-17A/IL-22. Future studies are required to investigate such possibility.
Supplementary Material
Acknowledgments
The authors would like to thank the Imaging Core and the Genomics Core at Cleveland Clinic Lerner Research Institute for their technical support; and the Li lab members for constructive discussion.
This work was supported by NIH grants P01-CA062220 (NCI) to X.L., R01-NS071996 (NINDS) to X.L. and P01-HL103453 (NHLBI) to X.L. and J.Z. is a Ph.D. student in the Molecular Medicine Ph.D. Program of Cleveland Clinic and Case Western Reserve University, funded in part, by the Med into Grad initiative of the Howard Hughes Medical Institute and T32GM088088 from NIH.
Footnotes
Conflict of Interests
The authors declare no conflict of interest.
The data sets presented in this article have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus database under accession number GSE104122 via ULR https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104122
References
- 1.Kluwe J, Mencin A, Schwabe RF. Toll-like receptors, wound healing, and carcinogenesis. Journal of molecular medicine. 2009;87:125–138. doi: 10.1007/s00109-008-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Punkenburg E, Vogler T, Buttner M, Amann K, Waldner M, Atreya R, Abendroth B, Mudter J, Merkel S, Gallmeier E, Rose-John S, Neurath MF, Hildner K. Batf-dependent Th17 cells critically regulate IL-23 driven colitis-associated colon cancer. Gut. 2016;65:1139–1150. doi: 10.1136/gutjnl-2014-308227. [DOI] [PubMed] [Google Scholar]
- 4.Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5540–5544. doi: 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G, Karin M. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17A-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, Zhang W, Zhong Z, Sanchez-Lopez E, Wu LW, Taniguchi K, Feng Y, Fearon E, Grivennikov SI, Karin M. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity. 2014;41:1052–1063. doi: 10.1016/j.immuni.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong Z, Yang XO, Yan H, Liu W, Niu X, Shi Y, Fang W, Xiong B, Wan Y, Dong C. A protective role by interleukin-17F in colon tumorigenesis. PloS one. 2012;7:e34959. doi: 10.1371/journal.pone.0034959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyun YS, Han DS, Lee AR, Eun CS, Youn J, Kim HY. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis. 2012;33:931–936. doi: 10.1093/carcin/bgs106. [DOI] [PubMed] [Google Scholar]
- 9.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17AF. The Journal of experimental medicine. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Connor W, Jr, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nature immunology. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, Karczewski J, Pezous N, Bek S, Bruin G, Mellgard B, Berger C, Londei M, Bertolino AP, Tougas G, Travis SP G. Secukinumab in Crohn’s Disease Study. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature medicine. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 13.Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nature immunology. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 14.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 15.Yui S, Nakamura T, Sato T, Nemoto Y, Mizutani T, Zheng X, Ichinose S, Nagaishi T, Okamoto R, Tsuchiya K, Clevers H, Watanabe M. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nature medicine. 2012;18:618–623. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 16.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, Ouyang W, Neurath MF, Becker C. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. The Journal of experimental medicine. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. The Journal of experimental medicine. 2013;210:917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, Ziegler PK, Nguyen PM, Preaudet A, Farid R, Edwards KM, Boglev Y, Luwor RB, Jarnicki A, Horst D, Boussioutas A, Heath JK, Sieber OM, Pleines I, Kile BT, Nash A, Greten FR, McKenzie BS, Ernst M. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer cell. 2013;24:257–271. doi: 10.1016/j.ccr.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011;35:467–477. doi: 10.1016/j.immuni.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao AT, Yao S, Gong B, Elson CO, Cong Y. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. Journal of immunology. 2012;189:4666–4673. doi: 10.4049/jimmunol.1200955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon C, VanDussen KL, Miyoshi H, Stappenbeck TS. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal immunology. 2014;7:818–828. doi: 10.1038/mi.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly RJ, Rouquier S, Giorgi D, Lennon GG, Lowe JB. Sequence and expression of a candidate for the human Secretor blood group alpha(1,2)fucosyltransferase gene (FUT2). Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype. The Journal of biological chemistry. 1995;270:4640–4649. doi: 10.1074/jbc.270.9.4640. [DOI] [PubMed] [Google Scholar]
- 24.Rausch P, Rehman A, Kunzel S, Hasler R, Ott SJ, Schreiber S, Rosenstiel P, Franke A, Baines JF. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19030–19035. doi: 10.1073/pnas.1106408108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, Sim S, Okamoto J, Johnston DM, Qian D, Zabala M, Bueno J, Neff NF, Wang J, Shelton AA, Visser B, Hisamori S, Shimono Y, van de Wetering M, Clevers H, Clarke MF, Quake SR. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nature biotechnology. 2011;29:1120–1127. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sachs UJ, Andrei-Selmer CL, Maniar A, Weiss T, Paddock C, Orlova VV, Choi EY, Newman PJ, Preissner KT, Chavakis T, Santoso S. The neutrophil-specific antigen CD177 is a counter-receptor for platelet endothelial cell adhesion molecule-1 (CD31) The Journal of biological chemistry. 2007;282:23603–23612. doi: 10.1074/jbc.M701120200. [DOI] [PubMed] [Google Scholar]
- 27.Raymond K, Richter A, Kreft M, Frijns E, Janssen H, Slijper M, Praetzel-Wunder S, Langbein L, Sonnenberg A. Expression of the orphan protein Plet-1 during trichilemmal differentiation of anagen hair follicles. The Journal of investigative dermatology. 2010;130:1500–1513. doi: 10.1038/jid.2010.4. [DOI] [PubMed] [Google Scholar]
- 28.Tatefuji T, Arai C, Mori T, Okuda Y, Kayano T, Mizote A, Okura T, Takeuchi M, Ohta T, Kurimoto M. The effect of AgK114 on wound healing. Biological & pharmaceutical bulletin. 2006;29:896–902. doi: 10.1248/bpb.29.896. [DOI] [PubMed] [Google Scholar]
- 29.Nijhof JG, Braun KM, Giangreco A, van Pelt C, Kawamoto H, Boyd RL, Willemze R, Mullenders LH, Watt FM, de Gruijl FR, van Ewijk W. The cell-surface marker MTS24 identifies a novel population of follicular keratinocytes with characteristics of progenitor cells. Development. 2006;133:3027–3037. doi: 10.1242/dev.02443. [DOI] [PubMed] [Google Scholar]
- 30.De Craene B, Denecker G, Vermassen P, Taminau J, Mauch C, Derore A, Jonkers J, Fuchs E, Berx G. Epidermal Snail expression drives skin cancer initiation and progression through enhanced cytoprotection, epidermal stem/progenitor cell expansion and enhanced metastatic potential. Cell death and differentiation. 2014;21:310–320. doi: 10.1038/cdd.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.