

Summary
A number of mitochondrial diseases arise from Single Nucleotide Variant (SNV) accumulation in multiple mitochondria. Here we present a method for identification of variants present at the single mitochondrion level in individual mouse and human neuronal cells allowing for extremely high resolution study of mitochondrial mutation dynamics. We identified extensive heteroplasmy between individual mitochondrion, along with three high confidence variants in mouse and one in human that were present in multiple mitochondria across cells. The pattern of variation revealed by single mitochondrion data shows surprisingly pervasive levels of heteroplasmy in inbred mice. Distribution of SNV loci suggests inheritance of variants across generations resulting in Poisson jackpot lines with large SNV load. Comparison of human and mouse variants suggests that the two species might employ distinct modes of somatic segregation. Single mitochondrion resolution revealed mitochondria mutational dynamics that we hypothesize to affect risk probabilities for mutations reaching disease thresholds.
Keywords: single mitochondrion, single cell, human neuron, mouse neuron, single nucleotide variation
eTOC
Morris et al. use independent sequencing of multiple individual mitochondria from mouse and human brain cells to show high pervasiveness of mutations. The mutations are heteroplasmic within single mitochondria and within and between cells. These findings suggest mechanisms by which mutations accumulate over time, resulting in mitochondrial dysfunction and disease.
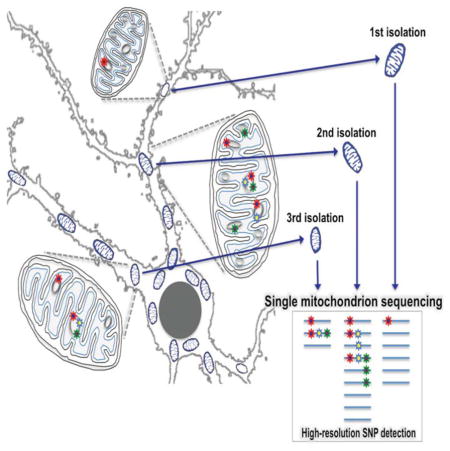
Introduction
Mitochondria are small, semi-autonomous organelles that participate in a wide range of cellular functions, including ATP production, oxidant stress, and calcium signaling (Ernster and Schatz, 1981). These organelles are unique in that they contain their own genome, often in multiple copies (multiploidy), which encodes a subset of the proteins as well as all of the tRNAs and rRNAs required for mitochondrial function (Nass and Nass, 1963; Schatz et al., 1964; Wallace et al., 1975). Maintenance and fidelity of the mitochondrial genome is imperative for proper cellular function as mutations, duplications, and other alterations of the mitochondrial genome have been implicated in a wide range of pathologies including Myoclonic Epilepsy and Ragged-Red Fiber Disease (MERFF); Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Symptoms (MELAS); Leber’s Hereditary Optic Neuropathy (LHON), as well as Type II Diabetes (Shoffner et al., 1990; Wallace et al., 1988; Pavlakis et al., 1984; van den Ouweland et al., 1992; Holt et al., 1990). For many of these mutations, there is not a straightforward association between mutational burden and when a patient will present with symptoms, though a clear causal relationship between the specific mutations and disease can be shown (Wallace et al., 2013).
Mitochondrial mutations result in a genomic effect referred to as heteroplasmy, in which the organism contains a mixture of wild type and mutated genomes. Dynamics of somatic and germline inheritance modulate levels of heteroplasmy (Schon et al., 2012). Rapid loss or fixation of new mutations resulting in reversion to homoplasy is promoted by uniparental inheritance (maternal) and bottleneck during oogenesis as well as stochastic drift during vegetative segregation (Bergstrom et al., 1998; Birky Jr, 2001). However, dynamics of intracellular selection and alternative mechanisms of vegetative segregation may maintain heteroplasmic states (Birky Jr, 2001; Stewart et al., 2015; Lehtinen et al., 2000). A high load of cells with heteroplasmic mitochondrial mutations at low to intermediate frequencies might increase the risk to an individual for mitochondrial disease where sub-threshold levels of variants might increase through vegetative segregation. Thus, measuring heteroplasmy early on, when the mutant genome is a small portion of the whole, and tracking it over time, may be informative in monitoring disease progression as well as treatment responsiveness.
While others have measured genetic variation of mtDNA isolated from single mitochondrion, these studies were limited to specific regions of the mtDNA, focused specifically on deletions, or measured a single mitochondrion isolated from a complex mixture of cell types (Cavelier et al., 2000; Reiner et al., 2010; Poe III et al., 2010)). This precludes assessment of total mutational burden across the entire genome for all genomes present. To this end, we have developed a method for isolation and analysis of the genomic DNA from a single mitochondrion, without loss of its spatial origin within a cell, in order to identify the presence of variants at single mitochondrion resolution. Using this approach, we investigated the nature of mitochondrial genome variation in human and mouse brain cells at multiple scales —from different cells in a single individual to different subcellular locations within a single cell. Our data suggests that even in inbred strains of mice, there is a broad segregating mitochondrial variation, within and across individuals, resulting in a large variation in individual heteroplasmy load. Although the data is more limited, we also find that human samples show unusual levels of heteroplasmy arising from within single mitochondrion polymorphism.
Results
PCR amplification of single mitochondrial DNA
Primary mouse neuronal or astrocyte cultures were loaded with Mitotracker Red to identify single mitochondrion for collection by micropipette (Fig. 1AB, 1DE). Following collection (Fig. 1C, 1F), a single mitochondrion was lysed to release the mitochondrial DNA, which was then PCR amplified through two rounds of PCR. Round 1 PCR produced three fragments averaging 5.5 Kb in size, which covered 99% of the mitochondrial genome. Round 1 PCR products were then input into thirty individual second round semi-nested PCR reactions, which were offset from each other to achieve full coverage of the Round 1 fragments (Fig. 1G). Round 2 PCR reactions were pooled, sheared, and input into library construction for Illumina NGS deep sequencing. Two different enzymes: either Platinum TAQ or NEB Q5, were used for the first round of PCR to avoid potential systematic biases introduced by any one enzyme. This design also allowed for production of three types of control replicates: at the round 1 PCR step, at the round 2 PCR step, and at the library construction step-each of which allowed for error rate calibration.
Figure 1. Overview of sample collection and processing.

See also Table S1 and Table S2. Primary cultures of astrocytes (A–C) or neurons (D–F) loaded with Mitotracker Red with pre- and post- isolation images of target mitochondrion (red arrows) are shown (B vs. C) and (E vs. F). Scale bars in full size images: 20 microns (A,D), in zoomed images: 5 microns (B,C,E,F). Pixels in the zoomed images were resampled for clarity. (G) Schematic of mtDNA coverage by Round 1 and 2 PCR amplicons. Round 2 PCR reactions were pooled (G, Pooled PCR) prior to acoustic shearing (G, Sheared PCR), and standard Illumina library construction (G, DNA libraries).
A total of 165 biologically unique single mitochondrion samples isolated either from mouse cortical astrocyte soma or neuronal dendrites were sequenced along with 21 samples derived from populations of mitochondria isolated from tissue (Fig. S1). This dataset was curated to a final number of 118 after filtering out samples that did not meet quality criteria. These 118 samples included single mitochondrion isolated from 103 unique cells-60 astrocytes and 58 neurons. In addition, multiple mitochondria were isolated from each of eight neurons as well as each of eight astrocytes.
Mouse single mitochondria carry surprisingly high levels of SNV loci
Despite the fact that all samples were derived from the inbred C57BL/6N strain, we saw a broad distribution of SNV loci at multiple levels of sampling: across different animals, different cells from the same animal, and different mitochondrion from the same cell. Figure S2 shows the sample lineage tree scaled by SNV events mapped onto the lineage branch lengths. The figure shows a broad distribution of SNV differences between samples (indicated by non-zero branch lengths) at every level of sampling. Several samples stand out with a high level of variants, at least 2-fold more than expected from the technical false positive rate (Supplemental Materials): M1C1D1Nh (17 variants), M11C45D0Ac (18 variants), M19C200aD1Nc (22 variants), M4C6D0Ac (41 variants) (Fig. S2, black arrows). The relationships between these samples and others derived from the same mothers suggest that these are true biological SNVs and not due to technical error. For example, one type of technical error that can be discounted is the reported oxidative adducts that may occur at specific positions within the mtDNA that would be read as mutations in the early stages of the PCR scheme as our analyses are based on variants that are found in multiple samples from multiple pups. In addition, the high confidence positions were also found in population samples prepared from tissue isolated from freshly sacrificed animals. For instance, M19C200aD1Nc is one of three single mitochondria sequenced from mother 19 and all of these single mitochondrion samples have elevated numbers of variants (11, 15, or 22) (Fig. 2A). Of note, two of these mitochondrion, M19C200aD1Nc and M19C200bD1Nc were isolated from the same cell and share a variant unique to the pair at site 9461, separate from the third mitochondrion M19C199D1Nc, which was isolated from a distinct cell.
Figure 2. Allele frequencies inherited from the mother between mouse single mitochondrion samples at high confidence positions.

See also Fig. S1, Fig. S2, and Fig. S3. (A) Sample lineage tree for single mitochondrion samples of pups from mother IDs 4,11, and 19 scaled to the number of SNVs per sample indicated by the key to the right. (B) Histogram of read frequencies (10%–100%) of the most abundant non-reference alleles across all single mitochondrion (n=118). The inset plots the log2 transformed frequencies against the minor allele frequencies. (C) Dot plot of the most frequent non-reference allele frequency for all 118 single mouse mitochondrion for position 3816, 9027, or 9461. Each dot represents up to 4 observations, each of which is color coded based on the mother ID.
For those samples with at least three mitochondria on from each mother, we carried out ANOVA analysis of the effect of mother, cell type (astrocyte versus neuron), and cell location (cortical versus hippocampus) on SNV numbers. Cell type and cell location had no effect on number of SNVs but the mother identity was highly significant (p < 10−14). Because the variances were nearly heteroscedastic, we carried out a Kruskal-Wallis test for the effect of the mother, which was also highly significant (p < 10−4). Posthoc Tukey’s test suggests significantly elevated levels of SNV for mothers 4, 11, and 19 (Fig. 2A). There is an average of 5.4 SNV sites per mother with coefficient of variation of 0.99, consistent with a Poisson count for the number of SNV positions per mother. This suggests that SNV positions arise in random positions and then are randomly accumulated in different individual lineages. Accumulation in lineages would require inheritance of mitochondrion variants, perhaps through polymorphic multiploid genomes within individual mitochondrion. This pattern of variant inheritance would also predict rare jackpot animals from Poisson sampling with an unusually high load of SNV sites that might predispose those animals towards higher probability of reaching disease phenotype thresholds.
Individual mitochondrion also varied considerably in the number of SNV sites and the variant allelic frequency at these sites as measured by read frequency. Among the 118 single mitochondrion samples from mice, there were on average 3.9 SNV sites per mitochondrion with a standard deviation of 5.71, indicating over-dispersion of SNV sites with some mitochondrion containing an unexpected high or low mutational load. Such over-dispersion might arise from preferential birth-death of certain multi-loci variant combinations leading to fitness effects (cf., Jokinen et al, 2015.; Jenuth et al., 1997). The majority of the single mitochondria had some SNV sites with high frequency non-reference alleles. On average, each mitochondrion contained 1.2 SNV sites (S.D. = 0.825) with a non-reference allele frequency of at least 0.5, with 82% of the mitochondrion containing at least one such site. Figure 2B shows a histogram of the allele frequencies of the most frequent non-reference allele at the SNV sites in our data that pass the 10% threshold (Supplemental Materials). The inset in the figure shows log2 transformed counts, which clearly deviates from the straight line that would be expected if the non-reference alleles were due to PCR errors (Supplemental Materials). If we assume each site and each mitochondrion were independent units of drift and the variant alleles were fitness neutral, we would expect a symmetric U-shaped distribution (Birky Jr et al., 1983). The multi-level units of segregation for mitochondrion makes the expectation difficult to derive, but the slight U-shape with a deficiency of frequencies near 100% suggests a mixture of neutral alleles and deleterious alleles.
SNVs with high frequency variants suggest independent segregation of haplotypes with different levels of purifying and balancing selection
For a more stringent and detailed analysis, we filtered for SNV sites independently found in four or more samples, which is expected to occur with probability less than 0.01 after Bonferroni correction for the size of the genome (Supplemental Materials). We found SNVs in more than four samples at four different sites: 3816 (variant: C; Mt-Tq); 9027 (variant: A; Mt-Co3); 9461 (variant: C; Mt-Nd3); 16265 (variant: T; D-loop) supported by 5, 59, 70, or 17 samples respectively (Table 1). The variant allele was the major frequency allele (>90%) for 39 samples at position 9027 and 22 samples at position 9461. Support for the SNV identified at positions 3816, 9027, and 9461 was provided in mitochondrial population samples derived from the mothers of the pups from which the cultures were made as well as population mitochondria from the pups themselves (Fig. S3A). Further support was sought by assaying for variants in mitochondrial transcribed RNA using single cell RNASeq analysis of neurons and astrocytes from the same strain of mouse (Van Gelder et al., 1990; Eberwine et al., 1992). The variant at positions 9027 and 3816 were supported by the RNASeq data (Table 1). We note that some of the RNAseq data had low coverage, which made these expressed RNA validations weaker (last column, Table 1). In fact, position 9461 and 16265 did not have sufficient read coverage for statistical validation by RNASeq data but we did identify some samples with a small number of reads supporting the alternate allele.
Table 1. Alternate major and minor alleles found in mouse or human supported by at least two samples.
For each mitochondrial DNA position, the number of samples supporting the variant allele either as a major alternate allele (frequency >90%) or a minor alternate allele (frequency ≤ 10%) is shown for positions where at least 2 samples support a variant. Also shown is the consequence of the mutation (mutation type/impact) and the number of single cell RNASeq samples containing reads that support the variant out of all single cells with reads covering the indicated position.
| Mouse | |||||||
|---|---|---|---|---|---|---|---|
| MAJOR ALLELE | |||||||
| Position | Gene | Reference | Variant | #Samples | Mutation Type | Impact | RNASeq |
| 6543 | Mt-Co1 | G | A | 3 | missense | moderate | 0/297 (0%) |
| 9027 | Mt-Co3 | G | A | 39 | missense | moderate | 178/293 (61%) |
| 9461 | Mt-ND3 | T | C | 22 | Alternate START | high | No read coverage |
| MINOR ALLELE | |||||||
| Position | Gene | Reference | Variant | #Samples | Mutation Type | Impact | RNASeq |
| 1317 | Mt-rnr1 | T | A | 1 | Non-coding | Not annotated | 31/288(11%) |
| C | 1 | 21/288(7%) | |||||
| 2651 | Mt-rnr2 | T | C | 2 | Non-coding | Modifying | 6/31(19%) |
| 3079 | Mt-ND1 | G | A | 1 | Missense | Moderate | 12/221(5%) |
| T | 1 | 10/221(5%) | |||||
| 3739 | Mt-Ti | G | A | 2 | Non-coding | Modifying | 1/22(5%) |
| 3816 | Mt-Tq | T | C | 5 | Non-coding | Modifying | 2/7(29%) |
| 5613 | Mt-Co1 | C | T | 2 | Missense | Moderate | 2/149(14%) |
| 7612 | Mt-Co2 | T | C | 2 | Synonymouse | Low | 41/309(13%) |
| 9027 | Mt-Co3 | G | A | 20 | Missense | Moderate | 8/293(3%) |
| 9221 | Mt-Co3 | A | G | 2 | Synonymous | Low | 76/297(26%) |
| 9461 | Mt-Nd3 | C | T | 48 | Alternate START | High | No read coverage |
| 12913 | Mt-Nd5 | C | A | 2 | STOP gained | High | 3/108(3%) |
| 13776 | Mt-Nd6 | C | T | 2 | Missense | Moderate | 10/224(5%) |
| 15191 | Mt-Cyb | C | T | 2 | Synonymous | Low | 16/303(5%) |
| 15475 | D-loop | A | G | 2 | Non-coding | Not annotated | 1/34(3%) |
| 15706 | D-loop | C | A | 2 | Non-coding | Not annotated | 0/37 (0%) |
| 16029 | D-loop | A | G | 2 | Non-coding | Not annotated | 4/59(7%) |
| 16265 | D-loop | C | T | 17 | Non-coding | Not annotated | No read coverage |
| HUMAN | |||||||
| MAJOR ALLELE | |||||||
| 309 | D-loop | C | T | 5 | Non-coding | Unknown | 0/62(0%) |
| 13928 | Mt-Nd5 | C | G | 3 | Non-coding | Unknown | 5/187(3%) |
| MINOR ALLELE | |||||||
| 309 | D-loop | C | T | 15 | Non-coding | Unknown | 45/62(73%) |
| 310 | D-loop | C | T | 3 | Non-coding | Unknown | 53/54(98%) |
| 961 | Mt-rnr1 | T | C | 2 | Non-coding | Unknown | 66/224(29%) |
| 13928 | Mt-Nd5 | C | G | 3 | Missense | Moderate | 56/187(30%) |
High Confidence SNVs in Single Mitochondria
The variants detected at two of the three high confidence positions, 3816 and 9027, result in mutations annotated as moderate or high impact. Position 3816 is located within the gene encoding the mitochondrial tRNA for glutamine and results in an A->G substitution in the second position of the anticodon stem (Jühling et al., 2009). Note that the alleles are complementary to those mentioned above (T->C) since this tRNA is transcribed from the opposite strand from which variants were called. Position 9027 is located within the gene coding for cytochrome c oxidase subunit III. The variant found here (G->A) results in a missense mutation in the gene, replacing a glycine residue with serine at amino acid 141. This position is predicted to occur within an alpha-helical membrane spanning region of the protein where the polar alcohol side group of serine could impact the protein side chain interactions within the alpha-helix and thus secondary and tertiary structure of the protein based on sequence homology with other highly conserved Eukaryotes for which the structure is experimentally known (Schwede et al., 2009).
Position 9461 is the third position of the start codon of the gene encoding NADH Dehydrogenase, subunit III (MT-ND3). In the C57Bl/6J reference strain, this codon is the mammalian mitochondrial alternative start codon ATT (Sachadyn et al,, 2008). The observed SNV allele “C” in the C57Bl/6N strain used here changes this start codon to another alternative start codon, ATC. Of the 50 Mus Musculus mtDNA variant sequences in Genbank, 46 strains use the codon ATC, while only four strains (C57BL/6J, MOLF/EiJ, SOD1/EiJ, and PWD/PhJ) use the codon ATT. In the 14 sequenced species of the outgroup genus Rattus spp, nine species use ATT, four species use ATC, and one species uses ATA (same as humans). The SNV site within our single mitochondrion data alternates between ATT and ATC, suggesting the possibility that this position might be ancestrally polymorphic with some lineages fixed for one or the other variant while some lineages maintain ancestral heteroplasmy. These data suggest unexpected macro-evolutionary time scale maintenance of SNV variants, which might indicate balancing polymorphisms.
Figure 2C shows a dot plot of the most frequent non-reference allele frequency for the three sites: 3816, 9027, and 9461. The variant allele frequency for site 3816 tends to be mostly low while 9027 shows moderate levels of higher frequency alleles. These SNV sites might be subject to higher levels of purifying selection. The variant frequency of 9461 shows the greatest variation with significant numbers of mitochondrion with greater than 50% alternative allele “C”, suggesting neutrally drifting variation or perhaps balancing selection at the level of the whole cell. The plot shows that the variant alleles span different mothers as well as mitochondrion from the same animal with widely varying frequencies of variant alleles. We computed whether the allele frequencies of the three SNV positions show evidence of similar frequencies in individual mitochondrion. Every sample rejected the null hypothesis of similar proportionality (chi-square test), suggesting that the variants within a mitochondrion are segregating independently. This result is consistent with the idea the nucleoids segregate the mutant alleles independently (Carling et al., 2011).
Human mitochondrion SNV show lower levels of allele frequency variation compared to mouse
To determine whether we could use this same approach to evaluate heteroplasmy of single human mitochondrion, we analyzed mitochondria isolated from primary human brain cell cultures (Spaethling et al., 2017). These cultures were prepared from residual surgically removed brain tissue upon glioblastoma resection from the left frontal cortex of a 63 yr. old female subject (Patient Number 50). Cultures were approximately 3 weeks old at the time of mitochondrial isolation. A total of 21 mitochondrion were patch pipette isolated and sequenced from multiple live human cells. In parallel, population mitochondrial DNA derived from tissue from a different patient diagnosed with Normal Pressure Hydrocephalus (Patient Number 8) was also sequenced. Five (S1–S5), two (S7,S8), and nine single mitochondria (S9–S17) were isolated from each of three different cells with neuronal morphology (Fig. S3B, “N”). The five remaining mitochondria were isolated from five different cells with non-neuronal morphology (Fig. S3B, “X”). The neuron from which nine mitochondria were isolated is shown in Fig. 3A along with images of the mitochondrion isolated from this cell. The processes on this human neuron were hundreds of microns long, which provided the opportunity for multiple isolations from this one cell.
Figure 3. Relationship between human single mitochondrion and comparison to mouse. See also Fig. S3.

(A) Image of human cell number 5. Scale bar = 100 microns. Pre- and post- isolation images and enlarged mitochondrion images shown adjacent. Scale bar = 10 microns. (B) Dot plot of arcsin transformed allele frequency deviations from means for human or mouse single mitochondrion. The top two panels show between cell deviations (human: n= 3 cells from 1 individual; mouse: n=19 individuals each with 3,2,2,9,6,3,7,3,3,3,2,4,7,2,12,8,3,4, or 2 cells), while the bottom panel shows within cell deviations (human: n=3 cells each with 2,5, or 9 mitochondrion (DF = 13), mouse: n=16 cells with 2,2,5,2,2,3,2,2,3,2,3,2,3,2,3, or 2 mitochondrion (DF = 24).
Isolated single mitochondria were processed for sequencing using the same approach used for the mouse samples, however, with a unique set of primers due to the sequence divergence between mouse and human mitochondrial DNA sequences (Table S2). Table 1 lists the positions for which a minor allele was detected with at least 10% read frequency in two or more single mitochondrion samples. There was one Bonferroni corrected significant SNV position at 309 (D-loop) that was detected in 20 of the 21 single mitochondrion samples as well as the population sample (Supplemental Materials). Five of these samples displayed a major allele switch from the reference sequence (showing T rather than C), while the remainder of the samples contained the minor “T” allele in addition to the major reference allele “C”. Using the same library quality filter criterion imposed upon the mouse samples, however, only 4 of the 21 mitochondria pass (S2, S12, S14, and S18) and support position 309 (S12 and S14 are derived from the cell pictured in Fig. 3A). Nevertheless, the variant alleles at this position were high frequency with an average of 45.79% and a standard deviation of 6.274%, resulting in most samples containing similar proportions of both C and T alleles.
We examined the pattern of allele frequencies for human position 309 against that of the most variable mouse position 9461 (Fig. 3B). While the human sample sizes were small, we found significantly greater non-reference allele frequency variation of mitochondrion in the same cell for mouse compared to humans (arcsin transformed F-test, F= 7.00, df = 24, 13; p < 0.00036). We also found significantly greater non-reference allele frequency variation across cells of the same individual for mouse compared to humans (arcsin transformed F-test, F= 82.76, df = 66, 2; p < 0.012). With the caveat that the results may be influenced by the difference in human and mouse sample sizes, the variant allele frequencies of different mitochondria in the same cell and average allele frequencies of mitochondria from each cell tended to be similar in humans but different in mouse, suggesting a difference in the mode of vegetative segregation. Neupane et al. (2015) studied levels of heteroplasmy in mouse embryonic stem cells and found greater level of allele frequency variation between single cells of the same cell colony compared to that between different cell colonies. This suggests that sampling variation during mitochondrial replication within a cell (either by differential birth-death of different homozygous organelles or by different replication of nucleoids within an organelle), might play a greater role in determining levels of heteroplasmy in mouse. Our results shown in Fig 3B are consistent with this observation.
Discussion
Generally, in the case of the mouse samples, when the mother and pup population mitochondrial DNA sequence was available, the single mitochondrion sequence agreed at the high confidence variable positions. This suggests that at least one copy of mitochondrial genome per mitochondrion may contain a variant allele observed at the population level. Other genomes within the same mitochondrion may complement possibly deleterious mutant variants until a threshold frequency is reached within each mitochondrion (Yoneda et al., 1994; Schon et al., 2010). The pathogenicity of a particular mutation will be a function of its frequency in the cell, its impact on mitochondrial function, and the dependence of a particular tissue on mitochondrial function (Wallace, 1992). The probability of reaching phenotypically significant frequencies of mutation will depend on the initial distribution of inherited sub-threshold variants (heteroplasmy load), new mutations, somatic drift, and selection. In our study, we leveraged single mitochondrion sequencing and variation analysis to infer patterns of sub-threshold heteroplasmy and somatic patterns of segregating variation.
We note several complicating factors may influence our results including unusual cellular environment within in vitro cell cultures leading to de novo mutations as well as possible effects of somatic selection which may depend on variation at other genomic loci or tissue specific selection (e.g., Jokinen et al., 2015; Jenuth et al., 1997). Unusual environment leading to mutations can influence the total number of SNV loci but we believe it unlikely to lead to SNVs shared in multiple samples as those described in Table 1. Somatic selection may be an important determinant of mitochondrial variation patterns, but most of the selection is likely to be purifying selection leading to homogenization. In addition, for these sample sizes we did not observe significant effects of cell type or anatomical location in determining SNV variants. We also add the caveat that our study involves measurement of standing variation and we do not directly measure inheritance or somatic segregation patterns. We infer dynamical processes from these static patterns of variation that may be limited by a lack of detailed mathematical models at the level of individual mitochondrial genomes.
The data presented here suggests a pervasive level of low to moderate frequency heteroplasmy at multiple loci, that in mice is inherited and accumulated in individual lineages resulting in random rare jackpot animals with large number of SNVs from Poisson sampling. Given that most of the SNV variation we observed was from within mitochondrion polymorphic polyploidy, single mitochondrion heteroplasmy may be the major mode of inheritance of low frequency heteroplasmy. For the human samples, we observed lower numbers of SNV sites (total of 65 including singletons) compared to the mouse samples (total of 289 including singletons). The difference in sample sizes would lead to approximately 2.4-fold (square root of sample size ratios) lower power in humans. Thus, we see ~50% of expected levels of SNV in humans compared to mouse. This is most likely due to sampling from a single individual (recall large mother effect in mouse), but it may also be due to differences in somatic segregation mechanisms.
As discussed, our results suggest mouse and human mitochondria may have differing mechanisms of somatic segregation that might affect probability of mutant frequencies exceeding critical thresholds. Segregation differences may arise as a result of mutations, but specific effects due to specific mutations are not possible to infer from our data. Here, if there are differences in segregation mechanisms, we suggest a species-specific effect rather than mutation-specific effect. These results emphasize the need for human based model systems for the study of mitochondrial genomics and disease.
Experimental Procedures
Single mitochondrion mtDNA isolation, amplification
Isolation of single mitochondrion
Micropipettes were purchased from Eppendorf (Femtotips II, 930000043) and affixed to a motorized micropipette holder controlled by an Eppendorf Transferman NK2. A single mitochondrion was identified in a live cell based on continuous matrix staining with Mitotracker Red and chosen for isolation if they could be resolved visually from neighboring mitochondria (see supplemental materials). While exerting positive pressure, the tip of the micropipette was positioned at the lateral most end of the mitochondria before the rat saline perfusion was turned off. Positive pressure was removed and gentle suction permitted collection of the single mitochondrion into the tip of the micropipette. The tip was removed and broken into a collection tube containing 4 μL of lysis buffer (100 mM Tris-HCl pH 8, 150 mM NaCl, 20 mM EDTA, 0.2% SDS)) and spun down briefly before storing on ice until ready for processing.
mtDNA isolation and amplification
A single mitochondrion was lysed by adding 1 μL of 0.05 mg/mL Proteinase K and incubating at 55°C for 30 minutes. Lysates were diluted with nuclease free water prior to Proteinase K inactivation at 95°C for 10 minutes. The full volume of the lysate was then input into a 100 μL multiplexed long PCR reaction using either the Platinum Taq (version 1 long PCR primers) or Q5 systems (version 2 long PCR primers) and cycled 10 ten times through the appropriate program. For each sample, the long PCR amplicons were then input into 30 individual semi-nested or nested short PCR reactions using the Platinum Taq Supermix for 40 cycles to achieve further amplification using 1 cycle of 94°C for 2 minutes; 40 cycles of 94°C for 30 seconds, 60°C for 30 seconds, 72°C for 2 minutes 30 seconds; 1 cycle of 72°C for 5 minutes. For mouse samples, the following first round PCR program was run using the Q5 system with version 2 Long PCR primers: 1 cycle of 98°C for 30 seconds; 10 cycles of 98°C for 5 seconds, 60°C for 30 seconds, 72°C for 2 minutes 45 seconds; 1 cycle of 72°C for 2 minutes. For the TAQ system with Version 1 Long PCR primers: 1 cycle of 94°C for 2 minutes; 10 cycles of 94°C for 30 seconds, 65°C for 30 seconds, 72°C for 5 minutes 30 seconds; 1 cycle of 72°C for 5 minutes. For the human samples, the following program was used for the first round long PCR with the Q5 system: 1 cycle of 98°C for 30 seconds; 10 cycles of 98°C for 5 seconds, 65°C for 30 seconds, 72°C for 2 minutes 45 seconds; 1 cycle of 72°C for 2 minutes. Mouse primer sequences are listed in Table S1, human primer sequences in Table S2. See Supplemental Materials.
Analysis
Variant calling from whole genome sequencing
Prior to calling variants, samples in which less than 50ng of DNA was input into library construction were omitted since there was a significant effect of average library input amount on the false positive rate (linear regression, p = 0.011) calculated from the technical replicate samples. For samples that passed this library filter, positions covered by a PCR primer used for PCR as well as 10 positions upstream and 2 positions downstream of the primer binding site were removed to avoid contribution of mutant primers to variable positions. A position was then identified as a variable position (ie the presence of a minor allele) if the following conditions were met: (A) read depth coverage >1000, (B) baseQuality > 30, (C) mappingQuality > 60, (D) read frequency of allele ≥ 10%. We obtained an average of ~17,500-fold coverage of each base across all of the mitochondrial genomes derived from a single mitochondrion. A single mitochondrion may contain multiple genomes. If a mitochondrion has k genomes, then at a given genomic position a variant will manifest as Single Nucleotide Variant (SNV) with a frequency of 0, 1/k, 2/k…k/k and corresponding expected read frequencies. Errors arising from sequencing and any of the PCR steps will generate variant artifacts at a frequency that depends on when the error arose during amplification with high frequency errors being exponentially unlikely. Therefore, to guard against false positive calls of an SNV site, we set a read frequency threshold parameter, θ. Using replicate control data, we examined setting θ at 10%, 5%, and 1% of the reads per single mitochondrion. We settled on using the most conservative 10% threshold that resulted in observed false positive rate of SNV calls of 0.57 (SE=0.300) positions per genome for the Round 1 PCR replicates, 5.73 (SE=1.456) positions per genome for the Round 2 PCR and 1.91(SE=0.756) for library construction. Assuming independent errors at each of the steps, using the 10% frequency threshold, we expect 8.21 false positive positions per genome, or approximately 5.13x10−4 per position, which is within the bounds of our model-based estimates (see below) of Taq-PCR error rates between 7x10−4 and 7x10−5. For estimating the mutation events along the branches of the cell lineage tree, we used the program Mesquite 3.2 (Maddison, W.P. and Maddison, D.R., http://mesquiteproject.org) with the default Maximum Parsimony character reconstruction method.
Variant calling from RNASeq
Effect of variants on tRNA and protein structure
Supplementary Material
Highlights.
Single mitochondrion sequencing shows single mitochondrion heteroplasmy
The distribution of SNV loci suggests inheritance of variants across generations
Analysis of SNVs in human and mouse suggests distinct modes of somatic segregation
Acknowledgments
This study was supported by the NIH Single Cell Analysis Program U01 MH098953, NIMH R33 MH106637, NARSAD Young Investigator Grant (JM) and the Brain Research Foundation (JE). Research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number TL1TR001880 (JM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Kevin Miyashiro for preparing primary mouse cell cultures, and our clinical research coordinators: Tim Prior, Kelsey Nawalinski, Katherine Murphy, and Eileen Maloney.
Footnotes
Author Contributions: JK and JE designed experiments and wrote the manuscript. JHL, JM developed, optimized and implemented mitochondrial isolations in consultation with JYS. JHL maintained human cell cultures using the procedure developed by JYS. HG designed the PCR primers. JM and HZ performed experiments. YJN and JK designed and performed data analysis. GHB, SB, HIC, DKK, THL, DMO performed surgeries. MSG performed surgeries and recruited patients. AVU and JAW developed SOPs for enrollment/consent of patients/live tissue transport, and maintained de-identified patient database. All authors reviewed the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bergstrom CT, Pritchard J. Germline bottlenecks and the evolutionary maintenance of mitochondrial genomes. Genetics. 1998;149:2135–2146. doi: 10.1093/genetics/149.4.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birky CW., Jr The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu Rev Genet. 2001;35:125–148. doi: 10.1146/annurev.genet.35.102401.090231. [DOI] [PubMed] [Google Scholar]
- Birky CW, Jr, Maruyama T, Fuerst P. An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics. 1983;103:513–527. doi: 10.1093/genetics/103.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling PJ, Cree LM, Chinnery PF. The implications of mitochondrial DNA copy number regulation during embryogenesis. Mitochondrion. 2011;11:686–692. doi: 10.1016/j.mito.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Cavelier L, Johannisson A, Gyllensten U. Analysis of mtDNA copy number and composition of single mitochondrial particles using flow cytometry and PCR. Exp Cell Res. 2000;259:79–85. doi: 10.1006/excr.2000.4949. [DOI] [PubMed] [Google Scholar]
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P. Analysis of gene expression in single live neurons. Proc Natl Acad Sci USA. 1992;89:3010–3014. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernster L, Schatz G. Mitochondria: A historical review. J Cell Biol. 1981;91:227s–255s. doi: 10.1083/jcb.91.3.227s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–433. [PMC free article] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Shoubridge EA. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- Jokinen R, Lahtinen T, Marttinen P, Myöhänen M, Ruotsalainen P, Yeung N, Shvetsova A, Kastaniotis AJ, Hiltunen JK, Öhman T, et al. Quantitative changes in Gimap3 and Gimap5 expression modify mitochondrial DNA segregation in mice. Genetics. 2015;200:221–235. doi: 10.1534/genetics.115.175596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jühling F, Mörl M, Hartmann RK, Sprinzl M, Stadler PF, Pütz J. tRNAdb 2009: compilation of tRNa sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Lehtinen SK, Hance N, El Meziane A, Juhola MK, Juhola KM, Karhu R, Spelbrink JN, Holt IJ, Jacobs HT. Genotypic stability, segregation and selection in heteroplasmic human cell lines containing np 3243 mutant mtDNA. Genetics. 2000;154:363–380. doi: 10.1093/genetics/154.1.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The sequence/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass MMK, Nass S. Intramitochondrial fibers with DNA characteristics. J Cell Biol. 1963;19:593–611. doi: 10.1083/jcb.19.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane J, Ghimire S, Vandewoestyne M, Lu Y, Gerris J, Van Coster R, Deroo T, Deforce D, Vansteelandt S, De Sutter P, et al. Cellular heterogeneity in the level of mtDNA heteroplasmy in mouse embryonic stem cells. Cell Reports. 2015;13:1304–1309. doi: 10.1016/j.celrep.2015.10.019. [DOI] [PubMed] [Google Scholar]
- Pavlakis SG, Phillips PC, DiMauro S, De Vivo DE, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann Neurol. 1984;16:481–488. doi: 10.1002/ana.410160409. [DOI] [PubMed] [Google Scholar]
- Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-Seq data. Am J Hum Genet. 2013;93:641–651. doi: 10.1016/j.ajhg.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poe BG, III, Duffy CF, Greminger MA, Nelson BJ, Arriaga EA. Detection of heteroplasmy in individual mitochondrial particles. Anal Bioanal Chem. 2010;387:3397–3407. doi: 10.1007/s00216-010-3751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner JE, Kishore RB, Levin BC, Albanetti T, Boire N, Knipe A, Helmerson K, Deckman KH. Detection of heteroplasmic mitochondrial DNA in single mitochondria. PLoS One. 2010;5:e14359. doi: 10.1371/journal.pone.0014359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachadyn P, Zhang XM, Clark LD, Naviaux RK, Heber-Katz E. Naturally-occurring mitochondrial DNA heteroplasmy in the MRL mouse. Mitochondrion. 2008;8:358–366. doi: 10.1016/j.mito.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz G, Haslbrunner E, Tuppy H. Deoxyribonucleic acid associated with yeast mitochondria. Biochem Biophys Res Commun. 1964;15:127–132. doi: 10.1016/0006-291x(64)90311-0. [DOI] [PubMed] [Google Scholar]
- Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Gilkerson RW. Functional complementation of mitochondrial DNAs: mobilizing mitochondrial genetics against dysfunction. Biochim Biophys Acta. 2010;1800:245–249. doi: 10.1016/j.bbagen.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Schwede T, Sali A, Honig B, Levitt M, Berman HM, Jones D, Brenner SE, Burley SK, Das R, Dokholyan NV, et al. Outcome of a Workshop on Applications of Protein Models in Biomedical Research. Structure. 2009;17:151–159. doi: 10.1016/j.str.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- Spaethling JM, Na YJ, Lee J, Ulyanova AV, Baltuch GH, Bell TJ, Brem S, Chen HI, Dueck H, Fisher SA, et al. Primary cell culture of live neurosurgically resected aged adult human brain cells and single cell transcriptomics. Cell Reports. 2017;18:791–803. doi: 10.1016/j.celrep.2016.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–542. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, Struyvenberg PA, de Kamp JJ, Massen JA. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet. 1992;1:368–371. doi: 10.1038/ng0892-368. [DOI] [PubMed] [Google Scholar]
- Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Diseases of the mitochondrial DNA. Annu Rev Biochem. 1992;61:1175–1212. doi: 10.1146/annurev.bi.61.070192.005523. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Bunn CL, Eisenstadt JM. Cytoplasmic transfer of chloramphenicol resistance in human tissue culture cells. J Cell Biol. 1975;67:174–188. doi: 10.1083/jcb.67.1.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, 2nd, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- Yoneda M, Miyatake T, Attardi G. Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol Cell Biol. 1994;14:2699–2712. doi: 10.1128/mcb.14.4.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.