

Abstract
Objectives: In this 6-month open-label extension (OLE) of NCT01491035 (a 14-day, open-label, pharmacokinetic/safety lead-in study), the long-term safety and tolerability of vortioxetine (5–20 mg/day) were investigated in children and adolescents with a DSM-IV-TR™ diagnosis of depressive or anxiety disorder in the United States or Germany. The study also was designed to provide data to inform dose selection and titration in future pediatric studies with vortioxetine.
Methods: Safety evaluations included spontaneously reported adverse events (AEs), the Columbia Suicide Severity Rating Scale (C-SSRS), and the Pediatric Adverse Events Rating Scale (PAERS; clinician administered). Clinical effectiveness was determined by Clinical Global Impressions. Comorbid attention-deficit/hyperactivity disorder was permitted, including concomitant use of stimulant medication (US sites only).
Results: Of the 47 patients who completed the lead-in period, 41 continued into the OLE. Most patients (n = 39 [95%]) continued their previous dose regimen. Twenty-one patients (51%) withdrew during the OLE; the most common primary reasons were administrative [n = 8], AEs [n = 4], and lack of efficacy [n = 3]. Thirty-five patients (85%) had ≥1 AE, 86% of which were mild or moderate in severity. Five patients (12%) reported a severe AE, none of which was considered related to study medication. The most common AEs (≥10%) were headache (27%), nausea (20%), dysmenorrhea (females; 19%), and vomiting (15%), with no relationship between AE intensity and age or dose. Five patients reported instances of suicidal ideation during the OLE, one of whom also reported this during the lead-in period. Two patients had nonsuicidal self-injurious behavior; one had a nonfatal suicide attempt. Throughout the study, there was a decrease over time in the incidence and intensity of AEs collected using the PAERS. Effectiveness assessment indicated a trend toward improvement based on numeric results.
Conclusion: This OLE confirms the findings from the lead-in study, which concluded that a dosing strategy of 5–20 mg/day is safe, well tolerated, and suitable for future clinical studies of vortioxetine in pediatric patients.
Keywords: : vortioxetine, long-term safety, pediatric patients, children, adolescents, dosing, antidepressant, depression, anxiety
Introduction
Depression in children and adolescents is a disabling condition that is associated with long-term mental and physical health problems (Keenan-Miller et al. 2007; Zisook et al. 2007; Williams et al. 2009; Merikangas et al. 2010; Rottenberg et al. 2014). The National Comorbidity Survey–Adolescent Supplement (NCS-A) demonstrated that the lifetime and 12-month prevalence of major depressive disorder (MDD) was 11.0% and 7.5%, respectively, among adolescents aged 13–18 years (Avenevoli et al. 2015). The prevalence for children appears lower (∼3%), but has not been widely studied (Costello et al. 2006; Merikangas et al. 2009, 2010; Williams et al. 2009).
Adolescents with major depression were found to have a fourfold risk of 12-month comorbidity with an anxiety disorder, behavior disorder, substance abuse disorder, or attention-deficit/hyperactivity disorder (ADHD) compared with adolescents without depression (Avenevoli et al. 2015). The NCS-A also showed a correlation between MDD and severe role impairment among adolescents (63% of the study population), with an average of 10 days “out of role” (i.e., unable to function) in the preceding 12 months (Avenevoli et al. 2015). Relative to MDD onset in adulthood, its onset in childhood and adolescence is associated with significantly more severe and recurrent forms of MDD, substantial functional impairment, and greater illness burden (Keenan-Miller et al. 2007; Zisook et al. 2007; Williams et al. 2009).
Depression is also strongly associated with suicide and suicidality (Wolitzky-Taylor et al. 2010; Hawton et al. 2013; Holma et al. 2014), and suicide is the third leading cause of death among 10–24-year-olds (Centers for Disease Control and Prevention 2015b). According to the Youth Risk Behavior Surveillance System (Kann et al. 2016) in a national survey of students in grades 9–12, 17.7% of them had seriously considered attempting suicide, 14.6% had made a plan for attempting suicide, and 8.6% had attempted suicide at least once during the 12 months preceding the survey.
Only two antidepressants (both selective serotonin reuptake inhibitors) are approved for treatment of depression in the pediatric population: fluoxetine is approved for children and adolescents (ages ≥8 years) with MDD in the United States (US) and the European Union and escitalopram is approved for adolescents (ages 12–17 years) with MDD in the US.
Vortioxetine is a multimodal-acting antidepressant that acts as a 5-HT3, 5-HT7, and 5-HT1D receptor antagonist, as well as a 5-HT1B receptor partial agonist, 5-HT1A receptor agonist, and inhibitor of the 5-HT transporter in vitro (Bang-Andersen et al. 2011; Mork et al. 2012; Westrich et al. 2012). The efficacy, safety, and tolerability of vortioxetine have been demonstrated in numerous trials of adults with MDD (Baldwin et al. 2016; Thase et al. 2016). Vortioxetine also has been investigated in a series of single- and multiple-dose pharmacokinetic studies performed to evaluate the effect of age, sex, race, renal impairment, or hepatic impairment on the pharmacokinetics of vortioxetine. The studies in patients with hepatic impairment showed no clinically meaningful differences in vortioxetine exposure or the need for dose adjustments for the intrinsic factors investigated (Chen et al. 2016).
In response to regulatory requests, the first international pediatric pharmacokinetic/safety study with vortioxetine was conducted to determine if the dose range approved for adult patients (5–20 mg/day) is appropriate for pediatric efficacy and safety studies (ClinicalTrials.gov identifier: NCT01491035; EudraCT number: 2010-020170-42) (Findling et al. 2017). The objectives of the lead-in study—a prospective, multinational, multisite, 14-day, open-label, multiple-dose pharmacokinetic/safety trial of vortioxetine, 5–20 mg/day, in pediatric patients—were to assess the pharmacokinetics, safety, and tolerability of vortioxetine across the dose range, providing data for evidence-based dosing strategies for future pediatric studies.
Forty-eight patients were enrolled: 24 adolescents grouped into 4 cohorts (6 per cohort) and 24 children grouped into 4 cohorts (6 per cohort). Each cohort was assigned to receive vortioxetine, 5, 10, 15, or 20 mg, once daily for 14 days, with uptitration every 2 days for those assigned to higher dosages. Results of the 14-day lead-in period have been published and suggest that the exposure, safety, and tolerability of vortioxetine are similar to those in adults when an uptitration scheme is used to achieve higher doses (Findling et al. 2017).
In the present study, a 6-month open-label extension (OLE) of NCT01491035, the long-term safety and effectiveness of vortioxetine (5–20 mg/day) were investigated in children and adolescents with a diagnosis of a depressive or anxiety disorder. This OLE was designed to provide preliminary long-term safety data on the use of vortioxetine in children and adolescents and to provide further information regarding the dosages and uptitration scheme to be used in future pediatric studies with vortioxetine.
Methods
This study was designed and performed in accordance with the principles of the Declaration of Helsinki and conducted by seven principal investigators, each located at one study site. Six study sites were in the US and one was in Germany. Each site started patient enrollment only after approval had been granted by the appropriate ethics committee or institutional review board. Each patient provided assent to participate (written or verbal) and their parent(s) or legal representative(s) provided written informed consent before initiation in the study. Informed consent and assent were obtained separately for each study period. Patients were informed that they were free to withdraw from the study at any time, for any reason, without prejudice.
Study population
Patients who completed the lead-in study were, if judged advisable by the investigator, offered the opportunity to continue in this optional 6-month OLE with flexible-dose design.
Detailed inclusion and exclusion criteria for participation in the lead-in study were described previously (Findling et al. 2017). Briefly, pediatric outpatients aged 7–17 years (inclusive) were eligible at screening if they had a Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR™), diagnosis of a depressive or anxiety disorder that (as judged by the investigator) warranted antidepressant therapy. Patients with comorbid ADHD were allowed to participate, and concomitant stable treatment with a stimulant was permitted at sites in the US, but not in Germany.
Exclusion criteria were as follows: Axis I diagnosis of bipolar disorder, posttraumatic stress disorder, autism, pervasive developmental disorder, obsessive-compulsive disorder, schizophrenia, or schizoaffective disorder; significant risk of suicide; inability to maintain stable stimulant treatment (in the US) or taking a concomitant stimulant (in Germany); and any past or present medical condition or medication that could interfere with safety, tolerability, or effectiveness assessments or could interfere with study conduct or interpretation. Because of the limited and strictly exploratory evaluation of treatment effect in this study, there were no restrictions with respect to psychotherapy before or during the lead-in period or during the extension period.
Study design
Pharmacological interventions for depressive and anxiety disorders in children and adolescents are not expected to provide any benefit if given only for a short period (e.g., 2 weeks). Therefore, if vortioxetine treatment in the 14-day lead-in study was considered by the investigator and family to be potentially beneficial for the patient, participants who completed the lead-in period could continue treatment in the 6-month OLE.
Initially, in the OLE, patients continued on their dosage from the latter part of the lead-in period. However, during subsequent visits to the investigative site, the dosage could be uptitrated or downtitrated within the range of 5–20 mg/day based on the investigator's clinical judgment. During the lead-in study, an external expert advisory group evaluated the safety, tolerability, and preliminary pharmacokinetics before enrollment of new cohorts to allow the possibility of adjusting dose levels. Dose adjustments in the OLE were required to be within the range studied in the lead-in period (e.g., there was no titration to 20 mg until that cohort began).
Screening and baseline procedures for the OLE were performed on day 0 (visit 12), followed by a 24-week treatment period and a safety follow-up visit 14 days after the last dose of vortioxetine. After day 0, patients returned to the study site for safety and effectiveness assessments every 4 weeks until the end of the 24-week treatment period.
Safety and tolerability assessments
Safety and tolerability assessments included documenting treatment-emergent adverse events (TEAEs), conducting physical examinations, performing clinical laboratory tests, and obtaining vital signs, weight, and electrocardiograms (ECGs). Data on TEAEs, vital signs, and weight were collected at each return visit (every 4 weeks: days 28, 56, 84, 112, 140, and 168 of the OLE) until the end of the 24-week treatment period. In addition, a safety follow-up visit was conducted 2 weeks after the last dose of vortioxetine to collect information on new adverse events (AEs) and to follow-up on the outcome of AEs ongoing at the end of the treatment period. Specimens for clinical laboratory testing were collected on day 84 (visit 3) and day 168 (visit 18). ECGs and physical examination were performed on day 168.
Tolerability also was assessed using the Pediatric Adverse Event Rating Scale (PAERS) (March 2007; Shapiro et al. 2009), which was to be completed at each return visit, after the open nonleading question for TEAEs was asked and answered. The PAERS is a validated instrument for assessing the frequency and severity of solicited events frequently observed in pediatric patient populations under treatment with psychotropic medication.
Suicidality was evaluated using the Columbia Suicide Severity Rating Scale (C-SSRS) (Posner et al. 2007, 2011), a semistructured interview for systematic assessment of the presence and severity of suicidal ideation and suicidal behavior in clinical trials. Patient's history with suicidality also was assessed using the C-SSRS, which was administered following the PAERS at each return visit. Both the PAERS and C-SSRS were administered by psychiatrists or clinicians who had completed a rater training and certification program. Each individual patient was to be rated by the same certified rater throughout the OLE.
Effectiveness assessments
Effectiveness was assessed with the Clinical Global Impressions–Severity of Illness (CGI-S) and Clinical Global Impressions–Improvement (CGI-I) scales. CGI-S and CGI-I scores for the OLE were collected every 4 weeks at each return visit (days 28, 56, 84, 112, 140, and 168 of the OLE) until the end of the 24-week extension treatment period. As with the safety and tolerability assessments, each patient's scoring on the CGI scales was done by a trained and certified rater who remained constant throughout the OLE.
Statistical methods
Safety and effectiveness assessments were based on the OLE data set, which included all patients who took at least one dose of vortioxetine in the lead-in period and continued into the optional 6-month extension period. Because investigators had the option to adjust the dose during the OLE, the extension-period dose was defined by the assigned cohort from the lead-in period and by modal dose (defined as the daily dose taken daily for most of both study periods). For safety and effectiveness, summary statistics are presented for continuous variables and counts and percentages are presented for categorical variables.
Safety and tolerability data are summarized by cohort and visit, using descriptive techniques. For patients who continued in the extension period, safety results are presented for the entire study (both periods), except for TEAEs and PAERS, which are provided only for the extension period.
Because the study did not include a placebo control, the effectiveness assessments (CGI-S and CGI-I) were strictly exploratory; the findings are summarized by cohort and visit for the entire study (i.e., from beginning of the lead-in period to end of OLE), using descriptive techniques. Effectiveness results are based on observed cases (OC) for the extension set, defined as all patients who received at least one dose of study medication and continued into the extension period.
Results
Of the 47 patients who completed the lead-in period, 41 (22 adolescents and 19 children) entered the OLE (Table 1). Reasons for not continuing into the OLE (one adolescent and five children) were lack of efficacy (n = 2), magnitude of commitment (n = 1), going away to college (n = 1), and missing parental consent to continue (n = 1); one patient did not specify a reason. Of the 17 patients with concurrent ADHD, 7 (2 adolescents and 5 children) were treated for ADHD during the lead-in period as well as the OLE; all 7 patients received stimulants. Medications that were discontinued before administration of the first dose of vortioxetine in the lead-in period are listed in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/cap). Comorbid psychiatric conditions are listed in Supplementary Table S2.
Table 1.
Patient Demographics at Open-Label Extension Baseline
| Child cohort | Adolescent cohort | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CC1 (n = 4) | CC2 (n = 5) | CC3 (n = 5) | CC4 (n = 5) | AC1 (n = 4) | AC2 (n = 6) | AC3 (n = 6) | AC4 (n = 6) | Total (N = 41) | |
| Assigned dose, mg/day | 5 | 10 | 15 | 20 | 5 | 10 | 15 | 20 | |
| Mean age (range), years | 10.8 (10–11) | 9.4 (8–11) | 10.4 (9–11) | 10.2 (7–11) | 15.0 (12–16) | 15.3 (13–17) | 15.2 (14–17) | 14.8 (12–17) | 12.8 (7–17) |
| Sex, n (%) | |||||||||
| Male | 2 (50) | 3 (60) | 3 (60) | 3 (60) | 3 (75) | 3 (50) | 1 (17) | 2 (33) | 20 (49) |
| Female | 2 (50) | 2 (40) | 2 (40) | 2 (40) | 1 (25) | 3 (50) | 5 (83) | 4 (67) | 21 (51) |
| Race, n (%) | |||||||||
| White | 2 (50) | 2 (40) | 3 (60) | 3 (60) | 3 (75) | 4 (67) | 4 (67) | 4 (67) | 25 (61) |
| Black/African American | 1 (25) | 3 (60) | 2 (40) | 2 (40) | 0 | 2 (33) | 1 (17) | 2 (33) | 13 (32) |
| Other | 1 (25) | 0 | 0 | 0 | 1 (25) | 0 | 1 (17) | 0 | 3 (7) |
| DSM-IV-TR diagnosis, n (%) | |||||||||
| Depressive disorder | 3 (75) | 3 (60) | 2 (40) | 1 (20) | 2 (50) | 3 (50) | 5 (83) | 5 (83) | 24 (59) |
| Anxiety disorder | 0 | 1 (20) | 2 (40) | 3 (60) | 1 (25) | 1 (17) | 0 | 0 | 8 (20) |
| Depressive and anxiety disorder | 1 (25) | 1 (20) | 1 (20) | 1 (20) | 1 (25) | 2 (33) | 1 (17) | 1 (17) | 9 (22) |
AC, adolescent cohort; CC, child cohort; DSM-IV-TR, Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision.
Although changes in vortioxetine dosing were allowed in the OLE, all patients except two adolescents continued their dosage assigned in the lead-in period for the duration of the OLE. One patient's dosage was increased from 5 to 10 mg/day due to lack of efficacy. Another patient's dosage was decreased from 20 to 10 mg/day because of fatigue (which started during the lead-in period and continued into the OLE).
Among the 41 patients who entered the OLE, 20 completed it. The primary reasons for the 21 withdrawals were AEs (n = 4), lack of efficacy (n = 3), noncompliance with treatment (n = 2), withdrawal of consent (n = 2), protocol violation (n = 1), lost to follow-up (n = 1), and administrative/other (needing concomitant ADHD stimulant [applies to Germany; n = 3], moving to another state [n = 1], miscalculating the visit window [n = 1], not returning to the site for next study visit [n = 1], poor cooperation [n = 1], and disinterest in continuing the medication [n = 1]). No trends with regard to age, sex, baseline diagnoses, comorbid ADHD, ADHD treatment, or time to withdrawal were observed for the patients who withdrew.
Overall, the mean (±standard deviation) exposure to vortioxetine was 133 (±61) days. Nearly half of the patients (n = 19; 46%) took vortioxetine for between 183 and 196 days. The majority of patients received all scheduled doses of vortioxetine based on patient reports and the count of returned tablets at each visit. The mean overall compliance rate was 97% (range, 83%–100%).
Safety and tolerability
Spontaneous AEs
An overview of TEAEs during the extension period is provided in Table 2. Thirty-five patients (85%) experienced a TEAE during the OLE, and AE rates were similar among cohorts. There were 57 TEAEs among the adolescent cohorts and 32 among the child cohorts. Overall, the TEAE profile in children and adolescents appeared similar to that of adults (Baldwin et al. 2016). The most common TEAEs (incidence ≥5% in the overall population) in the extension period were headache, nausea, dysmenorrhea (females), vomiting, toothache, upper respiratory tract infection, and increased weight. Most TEAEs (86%) were mild or moderate in severity, and none of the severe TEAEs was considered related to vortioxetine by the investigator. There was no relationship between the intensity of TEAEs and age or dose.
Table 2.
Summary of Treatment-Emergent Adverse Events
| Total (N = 41) | |
|---|---|
| Any TEAE | 35 (85) |
| Serious TEAE | 3 (7) |
| TEAE leading to withdrawal | 4 (10) |
| TEAEs with overall incidence ≥5% | |
| Headache | 11 (27) |
| Nausea | 8 (20) |
| Dysmenorrheaa | 4 (19) |
| Vomiting | 6 (15) |
| Toothache | 3 (7) |
| Upper respiratory tract infection | 3 (7) |
| Weight increase | 3 (7) |
Values are number of patients (percentage).
Sex-specific event; data represent percentage of female patients.
TEAE, treatment-emergent adverse event.
Approximately half of the patients (n = 21; 51%) experienced TEAEs that were considered related to vortioxetine; the most common of these (occurring in ≥5 patients overall) were headache (n = 8) and nausea (n = 7). All treatment-related AEs were mild or moderate in severity, and there was no relationship between the incidence of these TEAEs and age or dose. Three patients (all adolescents) experienced a serious AE (suicidal ideation [n = 1], intentional overdose and nonfatal suicide attempt [n = 1], and appendicitis [n = 1]), all of which were considered unrelated to the study drug. All patients recovered from these events. Four patients (all adolescents) withdrew due to an AE (irritability [n = 1], headache [n = 1], nausea [n = 1], and nonfatal suicide attempt [n = 1]). The patient who attempted suicide was found to have stopped taking the study medication and was withdrawn from the study for that reason; the suicide attempt was considered unrelated to vortioxetine by the investigator. There were no deaths in this study.
The mean changes in body mass index (BMI) from lead-in baseline to the last day of dosing in the OLE ranged from −0.35 to 0.86 kg/m2 for adolescents and from 0.42 to 2.43 kg/m2 for children. The mean changes in weight for the same period ranged from −0.91 to 2.33 kg for adolescents and from 0.83 to 5.06 kg for children. Fourteen patients (five adolescents and nine children) had potentially clinically significant (PCS) changes in weight (increase or decrease of ≥7%), which were reported only during the extension period; 12 patients had an increase and 2 had a decrease. The two patients with PCS weight decrease were obese [BMI for age, ≥95th percentile (Centers for Disease Control and Prevention 2015a)] at baseline, and the reasons for weight loss were attributed to dieting and concomitant treatment with ADHD medication.
Of the 12 patients with PCS weight increase, 3 children were overweight [BMI range for age, 85th–94th percentile (Centers for Disease Control and Prevention 2015a)], 1 child was obese, and 3 adolescents were obese. The main reasons for weight increase in these patients were height increase, growth spurt, and dietary changes; 1 event was considered possibly related to the study drug, occurring in an 11-year-old girl who was obese at baseline (BMI, 31.8 kg/m2) and whose BMI increased by 5.7 kg/m2. In 4 of the 12 patients who had PCS weight increase (2 adolescents and 2 children), the increase was observed only on the last visit of the extension period.
There were no clinically relevant changes in mean clinical laboratory values, vital signs, or ECG parameter values, nor were there any meaningful differences in these parameters between age groups or dosage cohorts. The majority of mean clinical safety laboratory values were within reference ranges, and no PCS laboratory values were reported.
Mean vital sign values outside the reference range were generally isolated, and postbaseline PCS vital sign values were reported for 21 patients (13 adolescents and 8 children), including PCS low standing pulse rate (n = 6), PCS high standing pulse rate (n = 9), PCS low supine pulse rate (n = 12), PCS high supine pulse rate (n = 1), PCS low standing systolic blood pressure (n = 3), PCS low supine systolic blood pressure (n = 1), and PCS low supine diastolic blood pressure (n = 1). For nine patients, at least one of the postbaseline PCS vital sign values also had been PCS at the lead-in screening or baseline visit. Among the patients with PCS vital signs, one adolescent (vortioxetine 10 mg/day) had a corresponding TEAE (orthostatic heart rate response increased), which was considered unrelated to the study medication. On day 140 of the OLE, this patient's standing and supine pulse rates were 146 and 128 beats/min, respectively, and both returned to normal by day 168 (118 and 94 beats/min, respectively).
The only postbaseline PCS ECG parameter reported in the entire study (both periods) was PCS low heart rate that occurred in six patients (five adolescents and one child), which was reported in the lead-in period. Four patients had isolated postbaseline PCS low heart rate. In two of them, the low heart rate also had been PCS at the screening and baseline visit. None of the PCS ECG values was reported as an AE.
Pediatric Adverse Events Rating Scale
Similar to spontaneously reported TEAEs, the majority of PAERS symptoms were mild or moderate in intensity. The PAERS symptoms of extreme intensity were single events reported for individual patients. Figure 1 shows the distribution of patients according to their highest intensity of any PAERS item.
FIG. 1.

Distribution of patients according to highest intensity of any PAERS symptom by visit (OC). Patients affected by ≥1 PAERS symptom: baseline, n = 38; week 2 (end of lead-in treatment period), n = 37; and week 26 (end of OLE treatment period), n = 15. PAERS, Pediatric Adverse Events Rating Scale; OC, observed cases; OLE, open-label extension.
Columbia Suicide Severity Rating Scale
Among patients who participated in the OLE, the lifetime assessment performed at the lead-in screening showed that two adolescents had experienced a nonfatal suicide attempt beforehand and that another two adolescents had previously demonstrated preparatory acts or behavior. Fifteen patients (37%), including children and adolescents, had a history of suicidal ideation before the lead-in period. No suicidal ideation or suicidal behavior was reported at OLE baseline. During the OLE, two adolescents had nonsuicidal self-injurious behavior, one of whom also reported nonsuicidal self-injurious behavior in the lead-in period. During the OLE, five patients reported suicidal ideation only. Another patient reported suicidal ideation as well as suicidal behavior (via the C-SSRS) and experienced related serious AEs (intentional overdose and suicide attempt). All patients who reported suicidal ideation or suicidal behavior during the OLE had experienced suicidal ideation before any treatment with vortioxetine.
Effectiveness
In general, children and adolescents experienced improvement in illness severity as measured by CGI-S (Fig. 2). The mean change in CGI-S score from pretreatment lead-in baseline for individual cohorts ranged from −0.8 to −1.6 at the end of the lead-in period and from −1.0 to −3.0 at the end of the extension period. At lead-in baseline, the mean CGI-S scores for children and adolescents continuing into the OLE were 4.2 and 4.1, respectively. At the end of the OLE, the mean CGI-S scores were 2.8 and 2.0, respectively. Similarly, CGI-I results showed overall improvement in illness; the improvements in mean CGI-I scores evident during the lead-in period were maintained throughout the OLE among children and adolescents (Fig. 3). On day 2 of treatment in the lead-in period, the mean CGI-I score was 3.8 (no change from treatment initiation) for children and adolescents who continued into the OLE. At the end of the extension period, the mean CGI-I score was 2.3 (much improved) for children and 1.9 (much improved) for adolescents. The mean CGI-I scores for the individual child and adolescent dose cohorts ranged from 2.2 (much improved) to 3.0 points (minimally improved) at the end of the lead-in period and from 1.8 (much improved) to 3.0 points (minimally improved) at the end of the OLE.
FIG. 2.
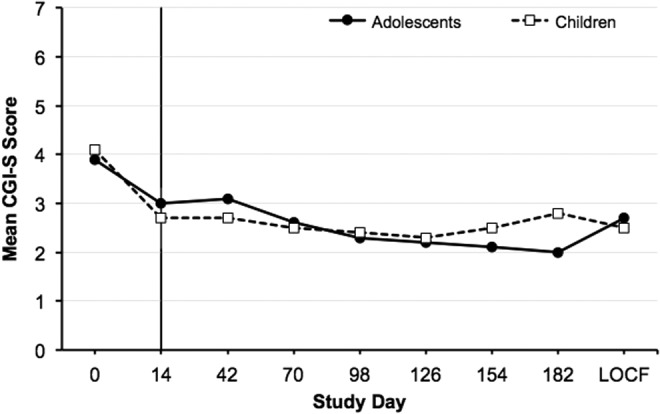
Mean CGI-S score during the lead-in period (days 1–14) and extension period (days 14–182) in children and adolescents (OC). LOCF reported for day 182. CGI-S, Clinical Global Impressions–Severity of Illness; LOCF, last observation carried forward; OC, observed cases.
FIG. 3.
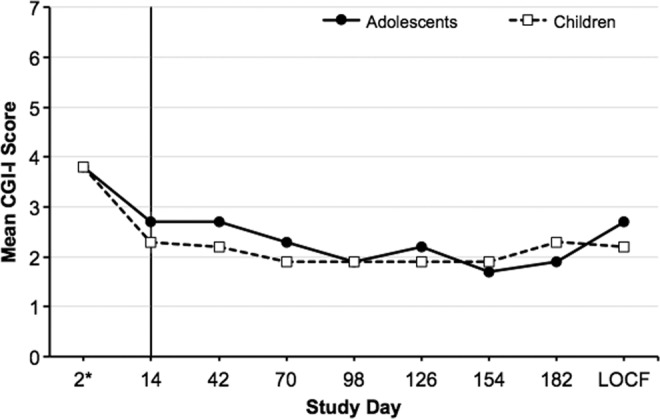
Mean CGI-I score during the lead-in period (days 1–14) and extension period (days 14–182) in children and adolescents (OC). LOCF reported for day 182. *CGI-I is scored based on change; therefore, no score can be reported for day 1. CGI-I, Clinical Global Impressions–Improvement; LOCF, last observation carried forward; OC, observed cases.
Discussion
This OLE study was conducted in a binational sample of pediatric patients and allowed for continuation of vortioxetine treatment for up to 6 months, with a goal of determining whether the therapeutic doses of vortioxetine approved for adults (5–20 mg/day) are appropriate for future clinical studies of vortioxetine in pediatric patients. Most patients from the lead-in period continued into the OLE, and the results provide further evidence that vortioxetine 5–20 mg/day is generally safe and well tolerated and is associated with continued effectiveness in children (aged 7–11 years) and adolescents (aged 12–17 years) with a depressive and/or anxiety disorder. The findings of this extension study are consistent with those of the lead-in period, which concluded that an uptitrated dosing strategy of vortioxetine 5–20 mg/day is suitable for future clinical studies of vortioxetine in pediatric patients (Findling et al. 2017).
Overall, the TEAE profile in children and adolescents appeared similar to that for adults; headache and nausea also were the most common TEAEs in studies with adult patients (Baldwin et al. 2016). Of the 13 headache events in the present OLE, most (n = 10) lasted between 2 hours and 6 days. Of the nine nausea events in the OLE, most (n = 6) lasted between 1 and 6 hours. As for the 14 patients with PCS weight change, the mean (±standard deviation) BMI increased by 1.80 (±1.99) kg/m2 during the entire study. Of the 17 patients with concurrent ADHD, 7 were treated with stimulants in both study periods. No safety issues related to concomitant treatment with stimulants were identified. This is important given that pediatric patients with ADHD often have a comorbid depressive or anxiety disorder, and there is a frequent use of stimulants in these patients (Daviss 2008; Merikangas et al. 2010; Larson et al. 2011; Cuffe et al. 2015).
Based on the PAERS, the signs and symptoms reported during the extension period and during the entire study (combined periods) reflected symptoms of the patients' illness and the TEAEs reported spontaneously. Moreover, there was generally a decrease over time in the incidence and intensity of the signs and symptoms collected using the PAERS. At OLE conclusion, the majority of symptoms were mild. C-SSRS findings showed that six patients experienced suicidal ideation or behavior during the OLE, all of whom were adolescents and had reported these symptoms at lead-in baseline. None of these events was considered related to vortioxetine or resulted in suicide completion, which is consistent with the currently understood relationship between antidepressants and suicidality risk (Birmaher and Brent 2005; Dubicka et al. 2006; Bridge et al. 2007; Gibbons et al. 2012; Ho 2012).
Although assessment of effectiveness was not a main objective of the OLE, CGI-S scores were found to improve during vortioxetine treatment. Most of the improvements occurred in the lead-in period and were maintained throughout the 6-month OLE. Similarly, CGI-I results indicated substantial improvement in illness among children as well as adolescents.
Limitations
This was an open-label study in a relatively small sample of pediatric patients. Because it was a long-term study in pediatric patients, a withdrawal rate of ∼50% would not be unexpected (Atkinson et al. 2014). Although self-reported adherence was very high, this could not be definitively confirmed because patients were not required to remain at the study site throughout the OLE. Due to the small sample size and lack of placebo control, effectiveness results are strictly exploratory and thus should be interpreted with caution. Throughout the study (both periods), there were no restrictions with respect to concomitant psychotherapy; therefore, the effect that concomitant psychotherapy may have had on the safety and effectiveness outcomes is not known.
Conclusions
This OLE provides further evidence that vortioxetine, 5–20 mg/day, is often well tolerated and is associated with effectiveness for up to 6 months in children and adolescents with a depressive or anxiety disorder. Results from the current study suggest that vortioxetine at dosages of 5–20 mg/day and the employed uptitration scheme are appropriate for use in clinical trials that will more definitively evaluate vortioxetine in pediatric patients.
Clinical Significance
There is a lack of currently approved antidepressant medications for pediatric patients. The two approved medications for depression in youth are selective serotonin reuptake inhibitors, and no alternatives exist for patients who do not respond to or cannot tolerate either drug. Results from this OLE study suggest that vortioxetine, at dosages of 5–20 mg/day, is generally well tolerated and could be efficacious in pediatric patients. Future clinical trials evaluating the efficacy and safety of vortioxetine in larger pediatric populations are warranted.
Supplementary Material
Disclosures
R.L.F. receives or has received research support from, acted as a consultant for, and/or served on a speaker's bureau for Actavis, Akili, Alcobra, American Academy of Child and Adolescent Psychiatry, American Psychiatric Press, Bracket, CogCubed, Cognition Group, Coronado Biosciences, Elsevier, Epharma Solutions, Forest, Genentech, GlaxoSmithKline, Guilford Press, Ironshore, Johns Hopkins University Press, KemPharm, Lundbeck, Medgenics, Merck, NIH, Neurim, Novartis, Otsuka, PCORI, Pfizer, Physicians Postgraduate Press, Purdue, Rhodes Pharmaceuticals, Roche, Sage, Shire, Sunovion, Supernus Pharmaceuticals, Syneurx, Takeda, Teva, TouchPoint, Tris, Validus, and WebMD. A.S.R. receives or has received research support from, acted as a consultant for, received travel support from, and/or served on a data safety monitoring board for Aevi Genomics, Allergan/Actavis/Forest, American Academy of Child and Adolescent Psychiatry, American Academy of Pediatrics, Bracket, Case Western Reserve University, College of Psychiatric and Neurologic Pharmacists, Eli Lilly, GlaxoSmithKline, Guilford Press, Ironshore, Johnson & Johnson/Janssen, Lundbeck, Neuronetics, Neuroscience Education Institute, Nevada Psychiatric Association, National Institutes of Health, North American Center for Continuing Medical Education, Otsuka, Pfizer, Rhodes Pharmaceuticals, Sunovion, Supernus, SyneuRx, Takeda, and University of Cambridge. M.P.D.-B. receives or has received research support from, acted as a consultant for, received travel support from, and/or served on a speaker's bureau for Actavis, American Academy of Child and Adolescent Psychiatry, Bracket, Eli Lilly, GlaxoSmithKline, Guilford Press, Johnson & Johnson, Lundbeck, Pfizer, Purdue, Shire, Sunovion, Supernus, and Takeda. M.H. receives or has received research support from, acted as a consultant for, received travel support from, and/or served on a speaker's bureau for Actelion, Eli Lilly, Engelhardt, Janssen-Cilaq, Lundbeck, Medice, Novartis, Shire, and Steiner-Arzneimittel and has an international patent on Doppler radar to assess ADHD (Government Research Grants: BMBF, BMFT, and BfArM). N.K.M. receives or has received research support from Forest Pharmaceuticals, Lundbeck, Otsuka, National Institute of Mental Health, Roche, Sunovion, Shire, and Tonix and received an honorarium for participation and travel support for a meeting sponsored by Sunovion. E.H.S. receives or has received research support from Alcobra, Alkermes, Allergan, Assurex, Auspexm, Daiichi-Sankyo, Eli Lilly, Forest Pharmaceuticals, Lundbeck, Indivior, Ironshore, Medgenics, Otsuka, Palatin, Pfizer, Polus, Sage, Shire, Sunovion, Supernus, Takeda, Tal Medical, TEVA, and Tonix. R.E.S. has no outside conflicts of interest and has not received a stipend or any other payment. At the time of this study, L.H.P. was an employee of Lundbeck. G.C. is an employee of Takeda Pharmaceuticals Company, Ltd. O.M.L. is an employee of Lundbeck. At the time of this study, P.A. was an employee of Lundbeck.
References
- Atkinson SD, Prakash A, Zhang Q, Pangallo BA, Bangs ME, Emslie GJ, March JS: A double-blind efficacy and safety study of duloxetine flexible dosing in children and adolescents with major depressive disorder. J Child Adolesc Psychopharmacol 24:180–189, 2014 [DOI] [PubMed] [Google Scholar]
- Avenevoli S, Swendsen J, He JP, Burstein M, Merikangas KR: Major depression in the national comorbidity survey-adolescent supplement: Prevalence, correlates, and treatment. J Am Acad Child Adolesc Psychiatry 54:37–44.e2, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin DS, Chrones L, Florea I, Nielsen R, Nomikos GG, Palo W, Reines E: The safety and tolerability of vortioxetine: Analysis of data from randomized placebo-controlled trials and open-label extension studies. J Psychopharmacol 30:242–252, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang-Andersen B, Ruhland T, Jorgensen M, Smith G, Frederiksen K, Jensen KG, Zhong H, Nielsen SM, Hogg S, Mork A, Stensbol TB: Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): A novel multimodal compound for the treatment of major depressive disorder. J Med Chem 54:3206–3221, 2011 [DOI] [PubMed] [Google Scholar]
- Birmaher B, Brent D: Should we use antidepressants for the treatment of major depressive disorder in children and adolescents? Rev Bras Psiquiatr 27:89–90, 2005 [DOI] [PubMed] [Google Scholar]
- Bridge JA, Iyengar S, Salary CB, Barbe RP, Birmaher B, Pincus HA, Ren L, Brent DA: Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: A meta-analysis of randomized controlled trials. JAMA 297:1683–1696, 2007 [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention: About Child & Teen BMI. 2015a. Available at: https://www.cdc.gov/healthyweight/assessing/bmi/childrens_bmi/about_childrens_bmi.html Accessed December15, 2016
- Centers for Disease Control and Prevention: Suicide Prevention, Youth Suicide. 2015b. Available at: https://www.cdc.gov/violenceprevention/suicide/youth_suicide.html Accessed December, 15, 2016
- Chen G, Nomikos G, Affinito J, Jacobson W, Zhao Z, Wang S, Xie J: Single-dose pharmacokinetics of vortioxetine in subjects with mild, moderate, or severe hepatic impairment [abstract LB-005]. In: American Society for Clinical Pharmacology and Therapeutics (ASCPT). San Diego, 2016. Available at: www.ascpt.org/Portals/8/docs/Meetings/2016%20Annual%20Meeting/Final%20Program.pdf Accessed 3/21/2017 [Google Scholar]
- Costello EJ, Erkanli A, Angold A: Is there an epidemic of child or adolescent depression? J Child Psychol Psychiatry 47:1263–1271, 2006 [DOI] [PubMed] [Google Scholar]
- Cuffe SP, Visser SN, Holbrook JR, Danielson ML, Geryk LL, Wolraich ML, McKeown RE: ADHD and psychiatric comorbidity: Functional outcomes in a school-based sample of children. J Atten Disord 2015. [Epub ahead of print]; DOI: 10.1177/1087054715613437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daviss WB: A review of co-morbid depression in pediatric ADHD: Etiology, phenomenology, and treatment. J Child Adolesc Psychopharmacol 18:565–571, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubicka B, Hadley S, Roberts C: Suicidal behaviour in youths with depression treated with new-generation antidepressants: Meta-analysis. Br J Psychiatry 189:393–398, 2006 [DOI] [PubMed] [Google Scholar]
- Findling RL, Robb AS, DelBello M, Huss M, McNamara N, Sarkis E, Scheffer R, Poulsen LH, Chen G, Lemming OM, Areberg J, Auby P: Pharmacokinetics and safety of vortioxetine in pediatric patients. J Child Adolesc Psychopharmacol. DOI: 10.1089/cap.2016.0155, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RD, Brown CH, Hur K, Davis J, Mann JJ: Suicidal thoughts and behavior with antidepressant treatment: Reanalysis of the randomized placebo-controlled studies of fluoxetine and venlafaxine. Arch Gen Psychiatry 69:580–587, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawton K, Casanas ICC, Haw C, Saunders K: Risk factors for suicide in individuals with depression: A systematic review. J Affect Disord 147:17–28, 2013 [DOI] [PubMed] [Google Scholar]
- Ho D: Antidepressants and the FDA's black-box warning: Determining a rational public policy in the absence of sufficient evidence. Virtual Mentor 14:483–488, 2012 [DOI] [PubMed] [Google Scholar]
- Holma KM, Haukka J, Suominen K, Valtonen HM, Mantere O, Melartin TK, Sokero TP, Oquendo MA, Isometsa ET: Differences in incidence of suicide attempts between bipolar I and II disorders and major depressive disorder. Bipolar Disord 16:652–661, 2014 [DOI] [PubMed] [Google Scholar]
- Kann L, McManus T, Harris WA, Shanklin SL, Flint KH, Hawkins J, Queen B, Lowry R, Olsen EO, Chyen D, Whittle L, Thornton J, Lim C, Yamakawa Y, Brener N, Zaza S: Youth risk behavior surveillance—United States, 2015. MMWR Surveill Summ 65:1–174, 2016 [DOI] [PubMed] [Google Scholar]
- Keenan-Miller D, Hammen CL, Brennan PA: Health outcomes related to early adolescent depression. J Adolesc Health 41:256–262, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson K, Russ SA, Kahn RS, Halfon N: Patterns of comorbidity, functioning, and service use for US children with ADHD, 2007. Pediatrics 127:462–470, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- March J, Karayal O, Chrisman A: CAPTN: the Pediatric Adverse Event Rating Scale. In: Novins DK, DeYong A, eds. Scientific Proceedings of the Annual Meeting of the American Academy of Child and Adolescent Psychiatry Boston, Massachusetts, 2007, p. 241 (abstract) [Google Scholar]
- Merikangas KR, He JP, Brody D, Fisher PW, Bourdon K, Koretz DS: Prevalence and treatment of mental disorders among US children in the 2001–2004 NHANES. Pediatrics 125:75–81, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merikangas KR, Nakamura EF, Kessler RC: Epidemiology of mental disorders in children and adolescents. Dialog Clin Neurosci 11:7–20, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mork A, Pehrson A, Brennum LT, Nielsen SM, Zhong H, Lassen AB, Miller S, Westrich L, Boyle NJ, Sanchez C, Fischer CW, Liebenberg N, Wegener G, Bundgaard C, Hogg S, Bang-Andersen B, Stensbol TB: Pharmacological effects of Lu AA21004: A novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther 340:666–675, 2012 [DOI] [PubMed] [Google Scholar]
- Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, Currier GW, Melvin GA, Greenhill L, Shen S, Mann JJ: The Columbia-Suicide Severity Rating Scale: Initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168:1266–1277, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner K, Oquendo MA, Gould M, Stanley B, Davies M: Columbia Classification Algorithm of Suicide Assessment (C-CASA): Classification of suicidal events in the FDA's pediatric suicidal risk analysis of antidepressants. Am J Psychiatry 164:1035–1043, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg J, Yaroslavsky I, Carney RM, Freedland KE, George CJ, Baji I, Dochnal R, Gadoros J, Halas K, Kapornai K, Kiss E, Osvath V, Varga H, Vetro A, Kovacs M: The association between major depressive disorder in childhood and risk factors for cardiovascular disease in adolescence. Psychosom Med 76:122–127, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro M, Silva SG, Compton S, Chrisman A, DeVeaugh-Geiss J, Breland-Noble A, Kondo D, Kirchner J, March JS: The child and adolescent psychiatry trials network (CAPTN): Infrastructure development and lessons learned. Child Adolesc Psychiatry Ment Health 3:12, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thase ME, Mahableshwarkar AR, Dragheim M, Loft H, Vieta E: A meta-analysis of randomized, placebo-controlled trials of vortioxetine for the treatment of major depressive disorder in adults. Eur Neuropsychopharmacol 26:979–993, 2016 [DOI] [PubMed] [Google Scholar]
- Westrich L, Pehrson A, Zhong H, Nielsen SM, Frederiksen K, Stensbol TB, Boyle NJ, Hentzer M, Sanchez C: In vitro and in vivo effects for the multimodal antidepressant vortioxetine (Lu AA21004) at human and rat targets. Int J Psychiatry Clin Pract 16:47, 2012 [Google Scholar]
- Williams SB, O'Connor E, Eder M, Whitlock E: U.S. Preventive Services Task Force Evidence Syntheses, formerly Systematic Evidence Reviews. In: Screening for Child and Adolescent Depression in Primary Care Settings: A Systematic Evidence Review for the U.S. Preventive Services Task Force. Rockville (MD), Agency for Healthcare Research and Quality (US), 2009 [PubMed] [Google Scholar]
- Wolitzky-Taylor KB, Ruggiero KJ, McCart MR, Smith DW, Hanson RF, Resnick HS, de Arellano MA, Saunders BE, Kilpatrick DG: Has adolescent suicidality decreased in the United States? Data from two national samples of adolescents interviewed in 1995 and 2005. J Clin Child Adolesc Psychol 39:64–76, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisook S, Lesser I, Stewart JW, Wisniewski SR, Balasubramani GK, Fava M, Gilmer WS, Dresselhaus TR, Thase ME, Nierenberg AA, Trivedi MH, Rush AJ: Effect of age at onset on the course of major depressive disorder. Am J Psychiatry 164:1539–1546, 2007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.