

Abstract
RNA-binding proteins are altered in large-scale, genome-wide cancer studies, although it is unclear how these proteins control tumor progression. Using unbiased gene expression and immunohistochemical analysis of 802 stage I lung adenocarcinoma (LUAD) patients, we show that increased adenosine deaminase acting on double-stranded RNA (ADAR) expression correlates with tumor recurrence. Knockdown of ADAR in LUAD cells reduces mesenchymal properties, cellular migration, and invasion. Analysis of gene expression patterns following the loss of ADAR identifies enrichment in cell migration pathways, most notably focal adhesion kinase (FAK). We show that ADAR posttranscriptionally increases the FAK oncogene through physical interaction with its RNA binding domain and editing a specific intronic site, resulting in stabilization and increase of FAK transcript. Moreover, pharmacological inhibition of FAK blocks ADAR-induced increase of cell invasion in LUAD cells, suggesting a potential therapeutic application for ADAR high-expressing LUAD. Collectively, we identify ADAR as an important regulator of LUAD progression through its ability to stabilize FAK.
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer-related death in the United States, with a dismal 5-year survival of 15%. The primary cause of death is the development of metastatic disease (1). Lung adenocarcinoma (LUAD), the most common NSCLC histologic subtype, is characterized by specific oncogenes and driver mutations or translocations that initiate and maintain tumorigenesis (2). One of the biggest challenges in LUAD is the identification and characterization of novel aberrant oncogenic drivers and mutations that are responsible for disease progression. This is a crucial step for the continued development of targeted therapies.
Cancer cells create tumor-specific transcripts through dysregulation of posttranscriptional processes such as alternative splicing, 3′ processing, and RNA editing (3). Pan-cancer studies have demonstrated the presence of widespread dysregulated RNA-editing patterns in cancer, of which adenosine-to-inosine (A-to-I) RNA editing plays a major role (4–6). Adenosine deaminase acting on double-stranded RNA (ADAR) catalyzes the deamination of A to I (7, 8), which results in the translational machinery reading inosine as guanosine, therefore effectively creating A-to-G changes in the RNA. There are three ADAR gene family members: ADAR1, 2, and 3. ADAR1 (ADAR) and ADAR2 (ADARB1) are ubiquitously expressed, whereas ADAR3 (ADARB2) is expressed only in the brain (9). The editing activity of ADAR affects gene expression and function by (a) changing codons and, thus, amino acid sequences of proteins; (b) altering RNA sequences, which can lead to pre-mRNA splice site changes; (c) altering the seed sequences of miRNAs targets; and (d) affecting the stability of the RNA (10, 11). A recent study suggested that amplification of ADAR is associated with poor outcomes in patients with NSCLC (12). However, the mechanism(s) of increased ADAR expression and their downstream effectors in the progression of lung cancer remain unclear.
Focal adhesion kinase (FAK) is overexpressed in solid tumors (13) and correlates with tumor progression (14). FAK is a cytosolic tyrosine kinase that is a crucial regulator of cell migration (15), invasion (16, 17), adhesion (18) and tumor metastasis (13, 14). Given the importance of FAK in tumor progression, pharmacological inhibitors of FAK are currently in phase I/II clinical trials (clinicaltrials.org).
In this study, we confirm that ADAR is amplified and overexpressed in LUAD. Using a large cohort of patients with stage I LUAD (N = 802), we show that high ADAR expression is an independent predictor of tumor recurrence. Knockdown of ADAR in LUAD cells with amplified ADAR leads to decreased migration and invasion. Mechanistically, we identify FAK as a novel target of ADAR in LUAD. ADAR increases FAK expression through stabilization of FAK mRNA in an RNA editing–dependent manner. Finally, by manipulating FAK activity through either ectopic expression of FAK or treatment with specific FAK small molecule inhibitors, we show that FAK plays a key role in ADAR-induced increases in migration and invasion of LUAD cells. These findings suggest that small molecule inhibition of FAK activity may be a potential therapeutic strategy for the treatment of LUAD with high ADAR expression.
Results
High ADAR expression is associated with tumor recurrence in LUAD patients
We analyzed The Cancer Genome Atlas (TCGA) LUAD and squamous carcinoma (SQ) patient cohorts, using the cBioPortal for Cancer Genomics (19). This revealed that ADAR is significantly amplified and overexpressed in LUAD, compared with SQ (ADAR DNA copy number amplification: LUAD 14.3% vs. SQ 1.7%; ADAR mRNA overexpression: LUAD 23% vs SQ 8.4%) (Figure S1). We next examined ADAR copy number and mRNA expression in LUAD cells and normal human bronchial epithelial cells (HBECs) by Droplet Digital PCR and quantitative reverse-transcription PCR (qRT-PCR), respectively. Consistent with observations from the TCGA cohort, ADAR was amplified and overexpressed in most tested LUAD cells, compared with HBECs (Figures 1A and 1B). Moreover, ADAR protein were also substantially higher in all tested LUAD cells compared to HBEC (Figure 1C).
Figure 1. ADAR is overexpressed in lung adenocarcinoma (LUAD) and correlates with tumor recurrence.
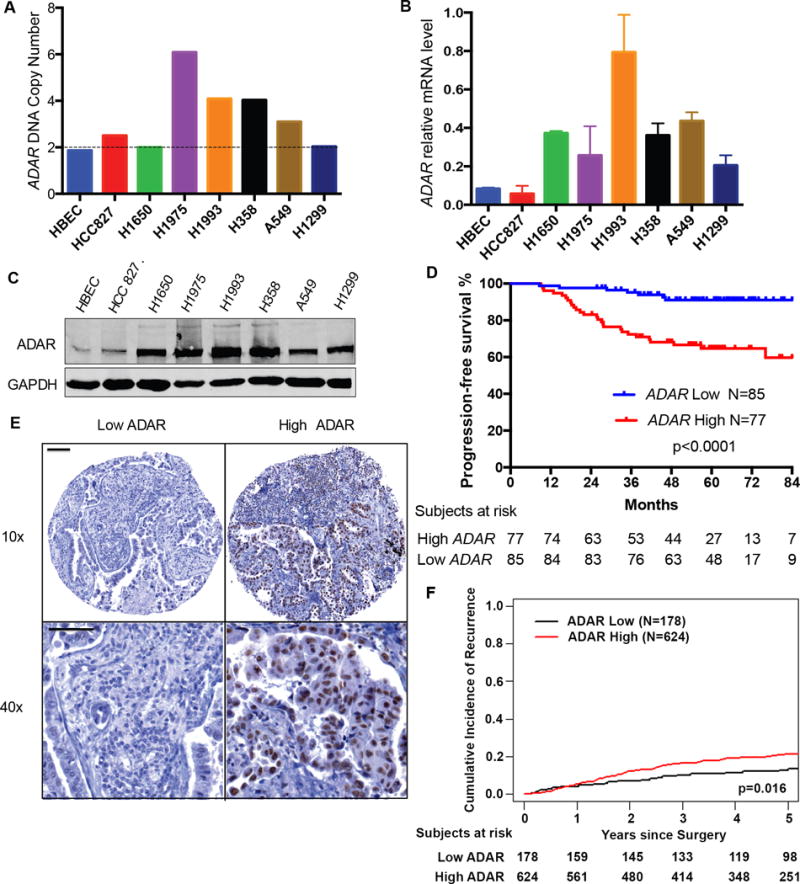
(A) ADAR DNA copy numbers were determined by droplet digital PCR in human bronchial epithelial cells (HBECs) and the indicated LUAD cells. Data are in triplicate from three experiments. (B) ADAR mRNA expression in HBEC and the indicated LUAD cells were assessed by qRT-PCR. HPRT was amplified as a reference. Data are means ± SEM and in triplicate from three experiments. (C) Western blot of ADAR protein expression in HBEC and LUAD cells. N = 3 experiments. (D) Kaplan-Meier curve of progression-free survival based on ADAR mRNA expression in 162 stage I LUAD patients in the NCCRI cohort (log-rank test: p<0.0001). (E) Immunohistochemical analysis showing low and high ADAR expression in two representative stage I LUAD tumors. Scale bars: 100μm (Upper), 50μm (Lower) (F) Cumulative incidence of recurrence based on ADAR protein expression in 802 patients with stage I LUAD (Gray’s test: p=0.016).
To assess the clinical relevance of increased ADAR mRNA expression in LUAD specimens, we performed an unbiased analysis using a publicly available gene expression microarray data set including 162 patients with stage I LUAD (NCCRI cohort http://www.abren.net/PrognoScan) (20). Patients with high ADAR mRNA expression had decreased progression-free survival (Figure 1D). To confirm that ADAR overexpression correlates with the progression of LUAD in a larger cohort of patients with stage I LUAD, we examined ADAR expression in Memorial Sloan Kettering Cancer Center (MSKCC) LUAD tissue microarray of stage I LUAD specimens. Immunostaining showed that ADAR was primarily located in the nucleus (Figure 1E). As expected, high intratumoral ADAR expression was associated with higher cumulative incidence of tumor recurrence (Figure 1F). Collectively, our data show that high ADAR expression correlates with tumor recurrence and poor prognosis in patients with early-stage LUAD.
ADAR is involved in LUAD cell migration and invasion
To examine the functional mechanisms of ADAR in LUAD cells, we stably knocked down ADAR, using two shRNAs targeted to different regions in ADAR coding sequences (ADAR A, ADAR B) in endogenously ADAR-amplified H1975 and H358 cells. The efficiency of ADAR knockdown (KD) was confirmed by qRT-PCR and Western blot (Figures 2A and 2B). To validate the successful knockdown of ADAR, we also detected the expression of two known ADAR downstream targets: F11R and CCND1. There was decreased abundance of F11R and CCND1 mRNA in ADAR KD cells, compared to scramble control cells (Figure S2A and S2B). To confirm that deaminase activity was also reduced in ADAR KD cells, we performed RESSq-PCR (21) to quantify ADAR-edited AZIN1 transcripts (5). A decrease in edited AZIN1 transcripts was seen in ADAR KD cells, compared to scramble control cells (Figure S2C).
Figure 2. ADAR KD inhibits cell migration and invasion.
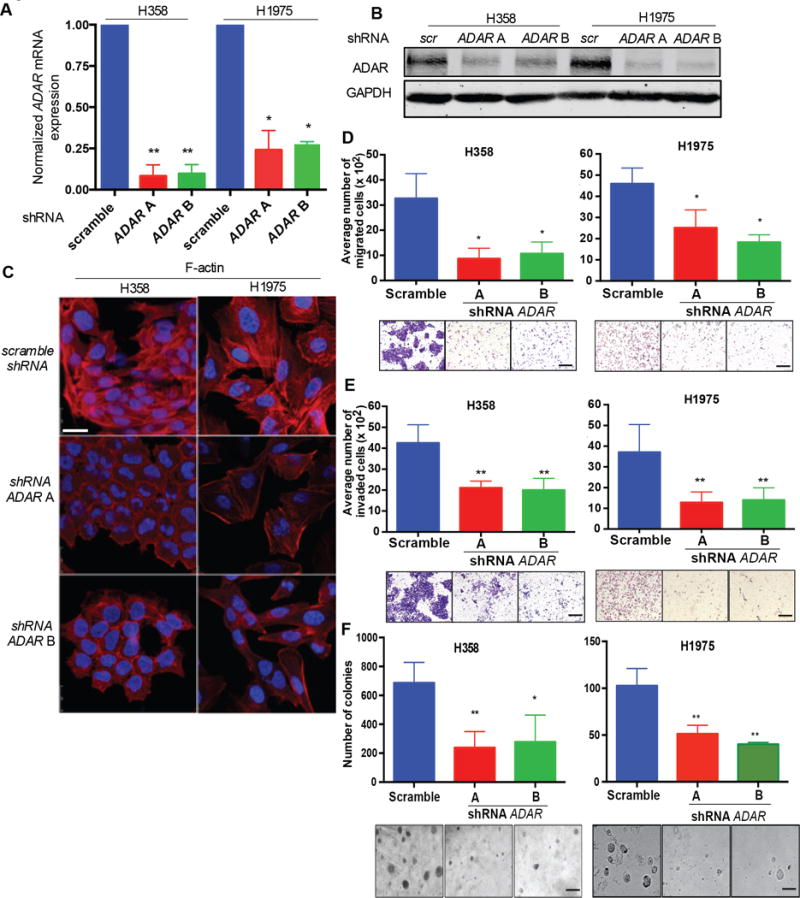
(A) qRT-PCR for ADAR mRNA relative to 18s in H358 and H1975 scramble and ADAR KD, cells. *p<0.05 and **p<0.01, compared with scramble (Mann-Whitney test). Data are means ± SEM from three independent experiments. (B) Western blot of ADAR expression in H358 and H1975 scramble and ADAR KD cells. N = 3 independent experiments. (C) Immunofluorescent staining with phalloidin (Red: F-actin) in H358 and H1975 scramble and ADAR KD cells. Scale bar: 25μm. N = 3 independent experiments. (D) Cell migration of H358 and H1975 scramble control and ADAR KD cells. cells. *p<0.05, compared with scramble (Mann-Whitney test). Data are means ± SEM from three independent experiments. Scale bar: 100μm. (E) Cell invasion of H358 and H1975 scramble and ADAR KD cells. cells. **p<0.01, compared with scramble (Mann-Whitney test). Data are means ± SEM from three experiments. Scale bar: 100μm. (F) Soft agar colony formation of H358 and H1975 scramble and ADAR KD cells. *p<0.05 and **p<0.01, compared with scramble (Mann-Whitney test). Data are means ± SEM from three independent experiments. Scale bar: 200μm.
Following knockdown of ADAR, we observed a change in LUAD cell morphology, compared with scramble control cells. ADAR KD cells exhibited a cobblestone, epithelioid morphological appearance, whereas scramble control cells had an elongated, mesenchymal shape (Figure 2C). Immunofluorescence with F-actin staining revealed reduced stress fiber formation and a more boundary-like staining pattern in ADAR KD cells compared to scramble control cells (Figure 2C). Furthermore, knockdown of ADAR significantly inhibited cell migration (Figure 2D) and invasion (Figure 2E) in LUAD cells compared to scramble control cells. To confirm that ADAR KD-mediated inhibition of cell migration and invasion were not secondary to decreased cell viability, we performed cell proliferation assays. Consistent with prior studies(12), loss of ADAR does not affect cell growth within post-plating 3 days (Figure S2D). However, scramble control cells exhibited increased growth rates post-plating 5 and 7 days, compared to ADAR KD cells (Figure S2D). Cell doubling time assays revealed that, at the low cell density, there is no difference of growth rate between scramble and ADAR KD cells (Figure S2E). In contrast, increasing cell density robustly promotes the growth of scramble cells, compared to ADAR KD cells (Figure S2E). Collectively, these data suggest that cell-cell contact affects the growth of cells with ADAR wild-type, but not with ADAR KD.
The ability to grow in an anchorage-independent manner is a hallmark of cancer. Knockdown of ADAR in LUAD cells leads to a reduction in growth and smaller colonies in soft agar assays compared to scramble cells (Figure 2F). Collectively, our data shows that the presence of ADAR correlates with an altered cellular epithelial phenotype, which is associated with enhanced LUAD cell migration and invasion.
ADAR affects the FAK signaling pathway
To investigate the putative pathway(s) through which ADAR promotes cell migration and invasion in LUAD, we performed Illumina microarray analysis using H358 ADAR KD and scramble cells. We identified 2207 genes that were differentially expressed between ADAR KD and scramble cells, with a fold change of −32 to +243 (adjusted p value, p < 0.01). To functionally annotate our microarray data and investigate relevant pathways, we analyzed the top 20% most significant genes, using Ingenuity software (Redwood City, CA). Cellular movement emerged as one of the most affected pathways following the loss of ADAR. Differential expression of genes between ADAR KD cells and scramble cells reveal FAK to be the most significantly differentially expressed (Figure 3A). FAK promotes the reorganization of cytoskeletal components and cell motility (22). Hence, we hypothesized that FAK plays a role in ADAR-mediated cell migration and invasion.
Figure 3. ADAR expression increases FAK expression.

(A) Heat map of genes related to cellular movement differentially expressed in H358 scramble and ADAR KD cells (p = 0.0009). N = 2 biological replicates. (B) qRT-PCR of FAK mRNA relative to 18s in the indicated cells. *p<0.05, **p<0.01, and ***p<0.001 compared with scramble (Mann-Whitney test). Data are means ± SEM from three independent experiments. (C) Western blot of FAK in the indicated cells. N = 3 independent experiments. (D) Flag-tagged ADAR was stably transfected into H1975 ADAR KD cells. Western blot for the indicated proteins. N = 3 independent experiments. (E) The correlation of ADAR and FAK mRNA in advanced stages LUAD (TCGA cohort, N = 57, p=0.031). (F) Western blot of the indicated proteins in selected patient LUAD samples. N = 3 independent experiments. (G) Immunofluorescence staining cortactin in the indicated cells. Scale bar: 25μm. N = 3 independent experiments. (H) Phospho-cortactin in the indicated cells was detected by coimmunoprecipitation using an antibody against pan-phospho-tyrosine followed by immunoblotting with an antibody against cortactin. N = 3 independent experiments. (I) Western blot for phospho-paxillin Tyr118 and paxillin in the indicated cells. N = 3 independent experiments.
To experimentally address our hypothesis, we validated FAK expression in ADAR KD and scramble cells by qRT-PCR and Western blot. Consistent with our microarray data, FAK mRNA and protein were both greatly reduced in ADAR KD cells, compared to scramble control cells (Figure 3B and 3C). Moreover, “put-back” of ectopic Flag-tagged ADAR in H1975 ADAR KD cells rescued FAK protein (Figure 3D)—proving the specificity of ADAR on regulation of FAK expression. To investigate if ADAR increases FAK mRNA in human LUAD, we analyzed RNA-seq data in the TCGA-LUAD cohort (cbioportal.org). ADAR positively correlates with the mRNA expression of FAK in patients with advanced stage LUAD (stages III and IV) (Spearman r=0.29, Figure 3E). Additionally, protein analysis of selected human LUAD tumors showed that tumors with high ADAR also have high FAK (Figure 3F). These findings further strengthen our hypothesis that ADAR increases FAK expression.
To determine whether knockdown of ADAR also affects FAK signaling, we conducted immunofluorescence analysis for cortactin. Cortactin is a known downstream target of the FAK signaling pathway (23), and its localization at the peripheral edges of cells is a marker for actin-rich motility protrusions, such as lamellipodia and invadopodia (24). Moreover, phosphorylation of the tyrosine residues in cortactin by FAK (25) increases F-actin turnover and cell motility (26). We found cortactin was distributed throughout the cytoplasm in ADAR KD cells, whereas, in scramble cells it accumulated in the periphery (Figure 3G). To explore whether knockdown of ADAR leads to a reduction of phosphocortactin, we simultaneously performed an immunoprecipitation assay using an antibody against phosphotyrosine to pull down all phosphotyrosine proteins. Following detection with cortactin, we observed that knockdown of ADAR decreased phosphocortactin but not total cortactin expression (Figure 3H). Another downstream target of FAK is the focal adhesion protein paxillin which is phosphorylated by FAK on tyrosine 118 (Tyr118) (27). To further investigate the consequences of ADAR KD-induced decrease of FAK, we examined paxillin and phospho-paxillin. Knockdown of ADAR did not change total paxillin expression, but reduced phospho-paxillin (Figure 3I). Collectively, these data confirm that ADAR increases FAK expression and activity and reveal an association between ADAR-mediated cell migration and invasion and FAK signaling.
ADAR stabilizes FAK transcript in a RNA editing–dependent manner
Alteration of RNA stability is one of the mechanisms through which ADAR regulates gene expression (28). To determine whether ADAR increases FAK transcript through modulation of FAK RNA stability, we performed actinomycin D chase experiments in H358 and H1975 ADAR KD and scramble cells. The percentages of remaining FAK mRNA were statistically lower in both H1975 and H358 ADAR KD cells, compared with their scramble cells (Figure 4A). For both LUAD cells, FAK mRNA had a shorter half-life in ADAR KD cells than in scramble cells (Figure 4A).
Figure 4. ADAR stabilizes FAK transcript.
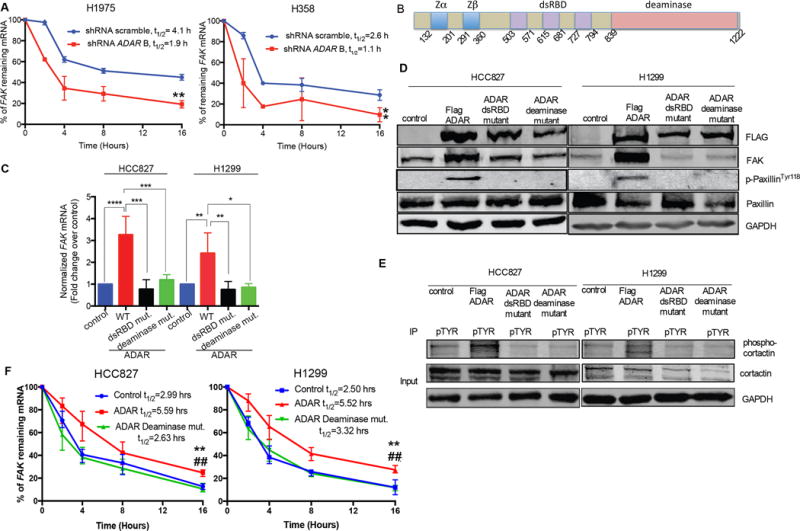
(A) Percentage of remaining FAK mRNA in the indicated cells following treatment with Actinomycin D. **p<0.01 compared with scramble (Wilcoxon rank sum test of area under the curve). Data are means ± SEM from three independent experiments. (B) Diagram of ADAR protein. Blue: Zα and Zβ domains, Purple: dsRBDs Red: deaminase domain. (C) qRT-PCR for FAK mRNA relative to 18s in HCC827 and H1299 cells transfected with ADAR wt, mutants or empty vector as control. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 compared with controls (Mann-Whitney test). Data are means ± SEM from three independent experiments. (D) Western blot for the indicated proteins in the indicated HCC827 and H1299 cells. N = 3 independent experiments. (E) Phosphocortactin in the indicated HCC827 and H1299 cells was detected by coimmunoprecipitation using an antibody against pan-phospho-tyrosine followed by immunoblotting with cortactin. N = 3 independent experiments. (F) Percentage of remaining FAK mRNA in the indicated HCC827 and H1299 cells following treatment with Actinomycin D. **p<0.01 compared with control. ##p < 0.01 compared with the deaminase mutant (Wilcoxon rank sum test of area under the curve). Data are means ± SEM from three independent experiments.
While ADAR regulates RNA stability in either an RNA editing–dependent or –independent manner, RNA binding is essential for its RNA-editing activity (28). ADAR contains two Z DNA binding domains (blue), three double-stranded RNA binding domains (dsRBDs; purple), and the deaminase region (red) (Figure 4B). To discern whether ADAR-induced FAK stabilization depends on its deaminase activity we created (1) an ADAR RNA binding–deficient expression construct (dsRBD mutant) by mutating three lysine residues of the KKXXK motif to EAXXA in all three dsRBD regions and (2) a deaminase mutant through point mutation (E917A) (29, 30). Following stable expression of Flag-tagged ADAR in LUAD HCC827 and H1299 cells with low endogenous ADAR (Figure 1), we observed an increase in FAK transcript and protein, compared to vector control cells (Figure 4C and 4D). However, stable expression of either ADAR dsRBD mutant or the deaminase mutant failed to induce FAK expression (Figure 4C and 4D). Additionally, phosphorylation of paxillin (Figure 4D) and cortactin (Figure 4E) were increased only in cells with wild-type ADAR overexpression, not in ADAR mutant expressing cells (Figure 4D and 4E). To confirm that the deaminase function is required for ADAR-induced stabilization of FAK mRNA, we performed actinomycin D chase experiments using HCC827 and H1299 stable cells described above. Overexpression of ADAR significantly increased the percentages of remaining FAK mRNA and half-lives of FAK mRNA in both tested cell lines, compared to cells with either control or the deaminase mutant. (Figure 4F). Overexpression of ADAR deaminase mutant failed to alter the stability of FAK mRNA, compared to control (Figure 4F). These findings confirm that the deaminase activity of ADAR is critical for FAK transcript stabilization.
ADAR directly binds to and edits FAK RNA
To explore the functional ADAR-editing site(s) in FAK, we analyzed FAK RNA-seq. data in the TCGA LUAD cohort using the Integrated Genomic Viewer tool. We identified three putative functional ADAR-editing sites in FAK intronic regions by correlating with FAK mRNA expression (Figure S3A). Tumors with editing of each of the putative sites have significantly higher FAK mRNA expression, compared to unedited tumors (Figure 5A, Figure S3B and S3C). To examine if ADAR directly edits these sites in LUAD cells, we individually detected site-specific editing of these three sites in HCC827 and H1299 cells that stably express ADAR wild-type or mutants. Ectopic ADAR wild-type significantly increased the editing FAK only on site chr8:141,702,274 (Figure 5B and S3D), but not on the other two sites (Figure S3E and S3F). Neither ADAR dsRBD mutant nor deaminase mutant affected the editing FAK on all three sites (Figure 5B, Figure S3D, S3E, and S3F). These data indicate that site chr8:141,702,274 in the intron 26 of FAK is a functional ADAR-editing site in both LUAD patient specimens and cell lines, and the editing level of this site correlates with the expression of FAK mRNA in human LUAD.
Figure 5. ADAR stabilizes FAK through RNA-binding and editing.
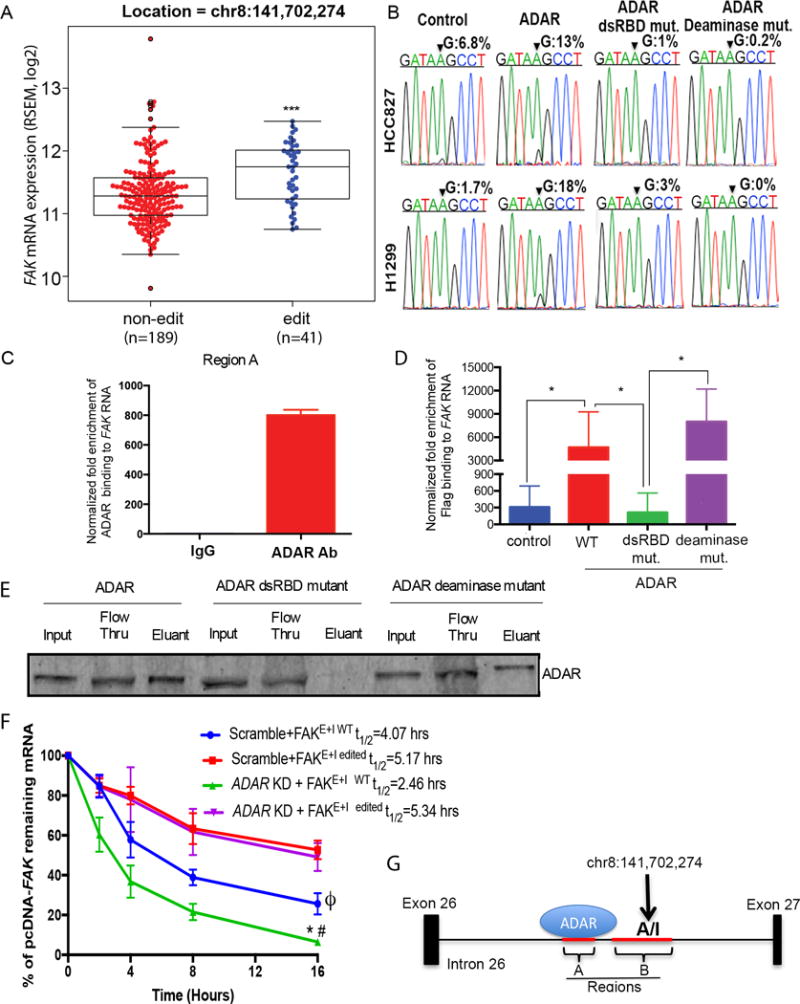
(A) FAK mRNA in tumors with (edit, N=41) or without (non-edit, N=189) A-to-I editing in the TCGA cohort. ***p<0.001, compared to non-edited tumors (Mann-Whitney test). (B) Chromatograms of FAK transcripts in the indicated cells. Arrow: the site chr8:141,702,274. The percentage of edited FAK detected by Sanger sequencing. N=3 biological replicates. (C) RIPs for region A in H1975 parental cells. Data are means ± SEM from three independent experiments. (D) RIPs for region A on the indicated H1299 stable cells. *p<0.05, comparing groups (Mann-Whitney test). Data are means ± SEM from three independent experiments. (E) RNA-protein interaction of in vitro dsRNA and ADARs. n = 3 independent experiments. (F) Percentage of remaining pcDNA-FAK mRNA in indicated H1975 cells following Actinomycin D treatment. *p<0.05 compared with FAKE+I WT in scramble (Blue); #p<0.05 compared with FAKE+I edited in ADAR KD (Purple); ϕp<0.05 compared with FAKE+I edited in scramble (Red) (Wilcoxon rank sum test of area under the curve). Data are means ± SEM from three independent experiments. (G) Schematic ADAR binding and editing FAK in the intron 26. Red lines: regions A and B used for RIP assays. Arrow: the editing site.
Next, to determine whether ADAR physically binds to FAK RNA endogenously, RNA immunoprecipitation (RIP) experiments were performed using H1975 cells, which have high endogenous ADAR expression (Figure 1). Two regions (A: chr8:141,702,900–141,703,155, and B: chr8:141,702,218–141,702,681) around the functional ADAR-editing site chr8:141,702,274 in the intron 26 of FAK, were amplified. Endogenous ADAR was found to bind FAK at region A (Figure 5C), but not region B (Figure S4). To determine if the functional dsRBDs are crucial for ADAR-FAK RNA binding, we performed RIP assays using H1299 cells that express ADAR wild-type, the dsRBDs mutant, or the deaminase mutant. To control for the effect of FAK transcript amount on the enrichment of ADAR-RNA binding, we normalized the enrichment of ADAR-RNA binding by FAK transcript in each corresponding cell line. There was increased ADAR binding to region A of FAK in cells expressing ADAR wild-type and the deaminase mutant, but not in those expressing the dsRBD mutant (Figure 5D). To determine if ADAR binds FAK RNA directly, we performed in vitro RNA-protein interaction assay using in vitro translated ADAR or its mutants, and in vitro transcribed double-stranded FAK RNA region A. ADAR and the deaminase mutant, not the dsRBD binding mutant, were able to bind the double-stranded FAK RNA (Figure 5E). Collectively these studies demonstrate that ADAR directly binds to FAK intronic region A endogenously through its functional dsRBDs.
To investigate whether ADAR-induced editing on site chr8:141,702,274 results in stabilization of FAK, we generated plasmids encoding FAK intronic regions A and B, as well as the adjacent exon (FAKE+I WT), and a T>C mutation was generated in plasmid FAK DNA on site chr8:141,702,274 (FAKE+I edited, Figure S5). We transfected either FAKE+I WT or FAKE+I edited into H1975 ADAR KD and scrambled cells and performed actinomycin D chase experiments. Consistent with our previous findings, the mRNA of FAKE+I WT had reduced stability in ADAR KD cells, compared to in scramble cells (Figure 5F). The mRNA of FAKE+I edited was significantly more stable than FAKE+I WT in scramble cells and ADAR KD cells, and ADAR KD failed to affect the mRNA stability of FAKE+I edited (Figure 5F). Collectively, our data suggests that ADAR binds to region A in intron 26 of FAK and edits FAK on an intronic site chr8:141,702,274 (Figure 5G), resulting in the stabilization and an increase of FAK mRNA.
ADAR-induced cell migration and invasion is FAK dependent
To assess whether knockdown of ADAR-induced repression of invasion is specifically mediated by the loss of FAK, Flag-tagged FAK was stably transfected in H358 and H1975 ADAR KD cells. Ectopic FAK partially rescued the invasion capability of ADAR KD cells in both LUAD cell lines (Figure 6A). To confirm that ectopic FAK reactivates downstream FAK signaling in ADAR KD cells, we evaluated the activity of the FAK downstream signaling target paxillin. Ectopic FAK rescued phospho-paxillin in ADAR KD cells without affecting paxillin (Figure 6B). Taken together, these findings suggest that FAK is a key mediator involved in ADAR-induced migration and invasion in LUAD.
Figure 6. ADAR-induced cell migration and invasion is FAK dependent.
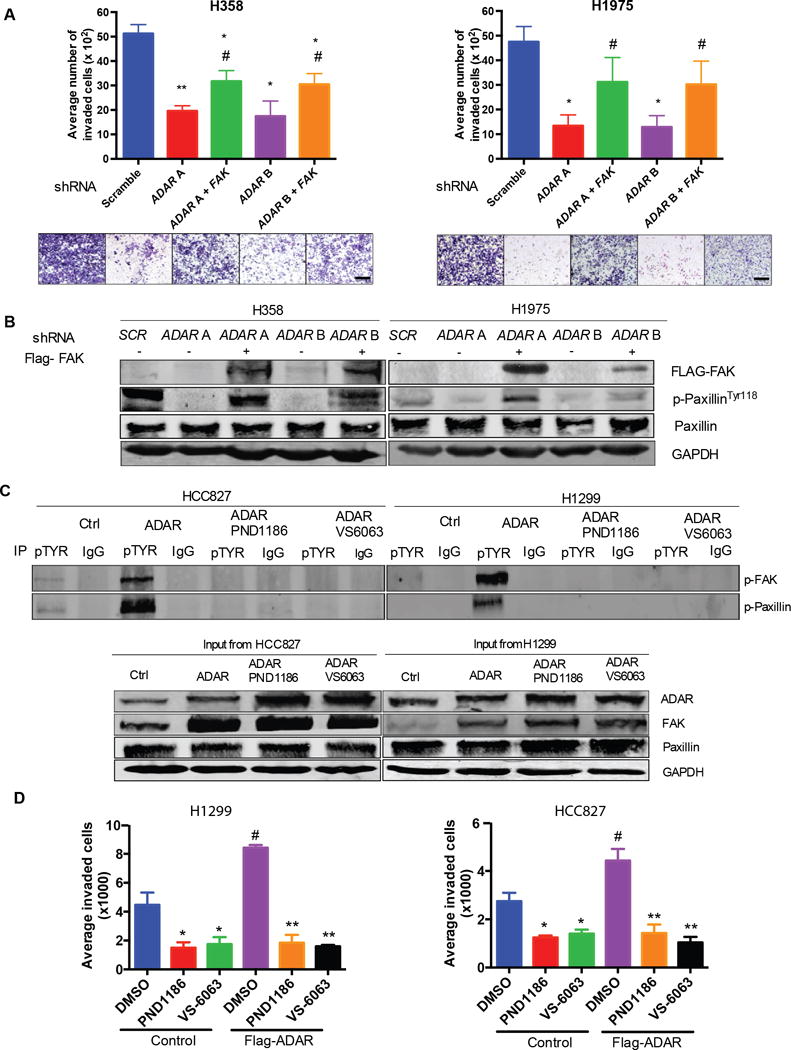
(A) Invasion assays of H358 and H1975 scramble, ADAR KD, and ADAR KD with Flag-FAK cells. #p<0.05, compared with corresponding ADAR KD cells. *p<0.05 and **p<0.01, compared with scramble (Mann-Whitney test). Data are means ± SEM from three independent experiments. Scale bar: 100μm (B) Western blot for the indicated proteins in H358 and H1975 scramble control, ADAR KD, and ADAR KD with Flag-FAK cells. N = 3 independent experiments. (C) Coimmunoprecipitation using antibody against pan-phospho-tyrosine in HCC827 and H1299 control or ADAR expressed cells treated with PND1186 or VS-6063 (2.5 μM) or DMSO for 72 h. Western blot for phospho-FAK and phospho-paxillin with antibody against FAK or paxillin. N = 3 independent experiments. (D) Invasion assays of HCC827 and H1299 control and ADAR expressing cells treated with FAK inhibitors PND1186 or VS-6063 (2.5 μM) or DMSO for 72 h. #p<0.05, compared with DMSO treated control cells. *p<0.05 and **p<0.01, compared with DMSO with the same vector (Mann-Whitney test). Data are means ± SEM from three independent experiments.
We next sought to determine whether pharmacologic inhibition of FAK activity is able to affect ADAR-induced cell invasion. PND1186 and VS-6063 are specific FAK inhibitors that are currently in clinical trials for the treatment of solid tumors (clinicaltrials.org). Cell viability assays were conducted to establish the optimal doses of PND1186 and VS-6063. A concentration of 2.5 μM was chosen as it did not cause excessive cell death (Figure S6A and S6B) while still inhibiting phosphorylation of FAK and its downstream target paxillin (Figure 6C). Consistent with our previous findings, stable ectopic expression of ADAR resulted in increased invasion of both HCC827 and H1299 cells (Figure 6D). Treatment with either pharmacological FAK inhibitor reduced invasion in HCC827 and H1299 control cells. Moreover, treatment with either FAK inhibitor completely abrogated ectopic expression of ADAR-induced increases of cell invasion in both tested LUAD cells (Figure 6D). These findings support that pharmacological inhibition of FAK activity is able to block ADAR-induced increases in cell invasion.
Discussion
Advances in next-generation sequencing technology have led to the discovery of specific A-to-I RNA-editing events across different cancers, and LUAD is a cancer that is hyperedited (5). The “net” proportion of these overediting events significantly correlates with the expression of ADAR1 (ADAR), but not ADAR2 or ADAR3 (5). ADAR is located at chromosome 1q21, and this region is highly amplified in LUAD (31). It has been proposed that ADAR amplification is associated with NSCLC recurrence (12). Extending those studies, we now show that ADAR is amplified and overexpressed in multiple lung cancer cells and human lung cancer samples and that ADAR overexpression correlates with tumor recurrence and worse progression-free survival in early-stage LUAD. We also demonstrate that ADAR promotes cell migration and invasion in LUAD through stabilization of the FAK transcript in an RNA editing–dependent manner.
The precise role that ADAR-mediated RNA editing plays in the pathogenesis of cancer is increasingly being investigated. Reduced ADAR expression contributes to tumor growth and metastasis in brain tumors (32), as well as in metastatic melanoma through hypoediting of Alu repetitive elements or miRNA-455-5p (33). However, several tumor-specific A-to-I RNA-editing events in the coding regions in esophageal squamous cell carcinoma, such as AZIN1S367G and COPAI64V (34), are clearly associated with tumor progression and are related to increased ADAR expression. We found that knockdown of ADAR significantly inhibited cell migration and invasion in our LUAD cells. These functional discrepancies for ADAR in cancer might be related to tumor and/or target gene specificity. An alternative hypothesis is that the function of ADAR is dependent on tumor progression. Under certain pressures (such as cancer therapies) and natural selection, cancer cells dynamically alter their genetic architecture to contribute to tumor progression (35, 36). In this study, we observed that increased ADAR expression is associated with increased recurrence of LUAD and that reduction of ADAR by shRNA inhibits the mesenchymal formation of tumor cells and maintains an epithelial phenotype. Our data suggest that high ADAR expression in LUAD drives metastases in situ through induction of a mesenchymal phenotype. Therefore, in early-stage LUAD, amplification and overexpression of ADAR appear to be important oncogenic events that contribute to LUAD progression.
This study begins to unravel the mechanism(s) whereby ADAR enhances invasion and migration of LUAD cells—namely, through the stabilization of FAK. FAK overexpression is observed in more than 20% of solid tumors, including ovarian, breast, colorectal, and lung cancers, and is associated with poor prognosis (13). It is well-documented that FAK transcriptional activity is increased in cancer cells by NF-κB, Nanog, and Ago2 directly binding to the FAK promoter (37–39). In this study, we have identified posttranscriptional ADAR-induced RNA stabilization as a mechanism through which FAK is increased in cancer cells. ADAR edits FAK RNA and increases its stability, resulting in an increase of FAK expression and activation of FAK signaling.
Observed patterns of dysregulated A-to-I RNA editing in cancers (4–6) have shifted the focus in understanding how ADAR functions in cancer cells. For instance, ADAR editing of AZIN1 enhances hepatocellular carcinoma tumorigenesis and progression (34), and reduction of ADAR leads to regression of chronic myelogenous leukemia and pediatric astrocytomas (40, 41). Moreover, ADAR-catalyzed A-to-G changes are the most commonly identified RNA-DNA differences in lung cancer (42).
ADAR modulates gene expression in either an RNA editing–dependent or independent manner. Reports suggest that ADAR interacts with Dicer, a component of the RNA-induced silencing complex, or DCGR8 to mediate pre-miRNA processing, eventually leading to RNA destabilization (43, 44). Additionally, ADAR stabilizes RNA through interaction with an RNA-binding protein in B cells (HuR) (28). In this study, we have shown that ADAR stabilizes FAK RNA in an RNA editing-dependent manner. FAK RNA contains more than a thousand ADAR-editing sites, with the majority of these located in Alu repetitive elements. The editing of Alu sequences has several implications, such as “exonization” of intronic Alu sequences, retention of Alu sequences in the paraspeckles, suppression of interferon response, and heterochromatin formation, which has been reviewed (45). In this study, using the TCGA cohort and our experimental LUAD cell line model system, we identified an intronic site on chr8: 141,702,274 in FAK which is a functional ADAR-editing site that is directly related to FAK RNA stability. Given the differences in signaling pathways, genomic profiles, and tumor microenvironment between cultured cells and tumors (46, 47), there are likely to be other functional ADAR-editing sites in FAK. Overall, the mechanism(s) of ADAR’s editing on gene expression are evolving and more work is necessary to precisely determine how ADAR-induced intronic editing affects gene expression (48).
PND1186 and VS-6063 are orally bioavailable ATP-competitive small molecules that block FAK phosphorylation (49). PND1186 is currently in phase I clinical trials for nonhematologic tumors, whereas VS-6063 is in phase II clinical trials for KRAS-mutant NSCLC (clinicaltrials.gov) (50). Recently, the PF562271 FAK inhibitor was used in a preclinical high-grade mutant KRAS;INK4A/ARF–deficient LUAD mouse model and selectively promoted cell death by shutting down the dysregulated ERK/RHOA/FAK pathway (51). In this study, we show that inhibition of FAK activation by either PND1186 or VS-6063 abrogated ADAR-induced increases in invasion of LUAD HCC827 and H1299 cells, both of which are KRAS and INK4A/ARF wild-type. However, p-FAKY397 is also strongly or moderately expressed in 30% of KRASWT LUAD specimens (51), suggesting that alternative mechanisms to increase FAK activity (for example, ADAR overexpression, which occurs in ~30% of LUAD) may allow these tumors to be treated with FAK inhibitors. Other potential discrepancies between our work and prior studies include the use of different FAK inhibitors, different cells, and the biological function assays performed.
In addition to demonstrating that FAK plays an important role in mediating ADAR-induced increases in LUAD cell invasion, we found that the addition of FAK to ADAR KD cells was unable to completely rescue the invasion potential of the cells. Given that ADAR is involved in many cellular processes and FAK is only 1 of more than 27 genes related to cellular movement that are differentially expressed when ADAR is knocked down, it is likely that stabilization of FAK is not the only mechanism through which ADAR induces cell invasion. For example, RAC2 and RAC3 gene expression are also differentially expressed when ADAR is knocked down. Like FAK, these proteins are involved in cytoskeleton reorganization through promotion of actin assembly, resulting in lamellipodia and membrane ruffle formation (52).
In summary, we have highlighted the importance of ADAR amplification and overexpression in the pathogenesis of LUAD. By identifying FAK as a novel ADAR editing target, our work establishes a potential therapeutic strategy of targeting FAK to prevent metastasis in early-stage LUAD with high ADAR expression.
Materials and Methods
Cell culture, antibodies, and reagents
All cell lines used were obtained from American Type Culture Collection and tested for mycoplasma. Human LUAD cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA). Normal HBECs were grown in keratinocyte-SFM (Life Technologies, Grand Island, NY) containing 50 ug/mL bovine pituitary extract and 5 ng/mL epidermal growth factor. Human embryonic kidney cells (HEK293) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. The primary antibodies used were ADAR1 (ab88574, Abcam), cortactin (MAB6096, R&D Systems,), GAPDH (MAB374, EMD Millipore), FAK (sc-558, Santa Cruz Biotechnology), phosphotyrosine antibody (PY20, #03-7700, Thermo Scientific), FLAG-epitope (PA1-984B, Thermo Scientific), paxillin (AB32084, Abcam), and phospho-paxillin Tyr118 (MAB6164, R&D system). Collagen type IV (9007-34-5) and actinomycin D (50-76-0) were purchased from Sigma-Aldrich. Puromycin (A1113803) and Geneticin (10131027) were purchased from Life Technologies.
Droplet Digital PCR
Genomic DNA was extracted from LUAD cells using the Genomic DNA Purification Kit (ThermoFisher Scientific), in accordance with the manufacturer’s instructions. Droplet Digital PCR was performed using the QX200 system (Bio-Rad Laboratories) to determine ADAR copy number variation in LUAD cells; AP3B1 was used as the reference gene. Results were analyzed using QuantaSoft software (version 1.7, Bio-Rad Laboratories).
Analysis of existing microarray data
Microarray data, available on PrognoScan, from a study of Progression-free survival in patients with stage I and II LUAD—Okayama et al. (NCCRI cohort, N = 204) (20). The array was performed on the Affymetrix platform, with the probe (ID 201786_s_at). Progression-free survival was assessed using the Kaplan-Meier method and compared between high and low ADAR mRNA groups using the log-rank test. A cutoff of <8.8575 for low vs high ADAR mRNA was determined by maximally selected rank statistics (53) using the package maxstat in R 3.1.1 (R Core Development Team, Vienna, Austria).
Clinical specimens and immunohistochemical analysis
A clinically annotated tissue microarray containing 802 stage I LUAD specimens was created by the MSKCC Thoracic Surgery Service (54). Human LUAD specimens were obtained with written, informed consent and approval of the Human Investigations Committee at MSKCC. Immunohistochemical analysis was performed as described previously (55). The ADAR antibody was used at a dilution of 1:200. ADAR staining was scored by a pathologist on the basis of the average staining intensity of three tumor cores (0, no staining; 1, weak; 2, moderate, 3, strong), as previously described (56, 57). Cumulative incidence of recurrence (CIR) was defined as the time between surgery and locoregional recurrence or distant metastasis. A cutoff of ≤0.63 for low vs. high ADAR immunoreactivity score was determined by maximally selected rank statistics (53) using the package maxstat in R 3.1.1. The difference in CIR between ADAR low and high was determined by Gray’s test.
Plasmids and shRNA construction
ADAR pLKO.1 shRNA constructs TRN0000050788 (ADAR A) and TRNC0000050789 (ADAR B) were purchased from Sigma-Aldrich. The lentiviral plasmid encoding Flag-tagged ADAR (EX-Z3143-Lv101) was purchased from Genecopecia. To generate the ADAR RNA binding–deficient (dsRBD) mutant and the deaminase mutant (E912A), we used the site-directed mutagenesis kit (New England Biolabs, E0554S), in accordance with the manufacturer’s instructions. FAK intronic region chr8:141,702,243-chr8:141,703,155 (the intron 26) and the exon 27 was amplified from HBEC cDNA and cloned into pcDNA3.1+ (GenScript, FAKE+I WT) and a T>C mutation at site chr8:141,702,274 (FAKE+I edited) was generated by site-directed mutagenesis. The primer pairs used for mutations are listed in Table S. pWZL-Neo-Myr-Flag-PTK2 was a gift from William Hahn and Jean Zhao (Addgene plasmid #20610).
Virus production and infection
To produce virus, 10 μg of lentiviral (pReceiver or pLKO) or retroviral (pWZL-Neo Myr-Flag-PTK2) plasmid and 3 μg each of the packaging plasmid DNA (psPAX2 and pMD2.G for lentivirus, VSVG and Gag-Pol for retrovirus) were cotransfected into HEK293 cells by use of PolyFect reagent (Qiagen, 301107). NSCLC cell lines H358 and H1975 were plated into 100-mm culture dishes at 60% confluence the day before infection. The following day, 3 mL of lentivirus- or retrovirus-containing media was added to the cells, together with polybrene (8 μg/mL; Santa Cruz Biotechnology, sc-134220), for infection. For pLKO lentivirus infection, cells were selected by use of puromycin (2 μg/mL) for 2 weeks and screened by qPCR and Western blot for ADAR expression. To select cell lines that overexpress Flag-PTK2 or Flag-ADAR, geneticin (600 μg/mL) was used for 2 weeks, and the expression of Flag-tagged protein was screened using Western blot.
Total RNA isolation, qRT-PCR, and Sanger sequencing analyses of editing of FAK
Total RNA was isolated using TRIZOL reagent, in accordance with the manufacturer’s protocol (Invitrogen). To detect edited transcripts, we used the RNA-editing site-specific primer design strategy that is compatible with SYBR green qRT-PCR protocols (RESSq-PCR), as previously described (21).
For Sanger sequencing analyses, semiquantitative PCRs were performed using the pair of primers FAK274C forward and FAK274T reverse. The PCR products were purified using QIAquick PCR purification kit (Qiagen, 28106) and sent for Sanger sequencing using the primer FAK274T reverse at Genewiz (South Plainfield, NJ). The primers used are listed in Table S.
Microarray analysis
Total RNA was extracted from H358 shRNA scramble and shRNA ADAR B cells by use of the RNeasy kit (Qiagen), in accordance with the manufacturer’s instructions. Human HT-12 v4 Expression BeadChip (Illumina) was performed in duplicate by the Integrated Genomics Operation Core Facility at MSKCC. The raw data were extracted and analyzed using Partek Genomics Suite 6.6. Gene signal values were logarithm transformed and normalized using the quartile method (58). Array data were filtered using an adjusted p value < 0.01. Comparative analysis between shRNA scramble and shRNA ADAR B cells was performed on the basis of fold change in expression. Gene ontologic analysis was performed using Ingenuity Pathway Analysis software.
Western blot and coimmunoprecipitation
Western blotting was conducted as previously described (59). Primary antibodies were used at a dilution of 1:1000, and secondary antibodies (Licor, P/N 925-32210, P/N 925-32211) were used at a dilution of 1:20,000.
Blots were scanned using the Odyssey CLx machine (Licor). For immunoprecipitation, 500 μg of cell lysates were incubated with 1 μg of antibody against phosphotyrosine (PY20, Thermo Scientific) or control rabbit IgG. The presence of phosphocortactin was detected by immunoblot.
Immunofluorescence
Cells (2.5×104/100 mL) were plated in 4-well chamber slides, grown for 60 h, and fixed in 4% paraformaldehyde. After incubation with 1% bovine serum albumin in phosphate buffered saline Tween-20 for 30 min, rhodamine phalloidin (1:40, Life Technologies, R415) was added for F-actin detection for 30 min. For cortactin and FAK detection, antibodies were used at a dilution of 1:100 at 4°C overnight. Secondary antibodies, anti-rabbit Alexa Fluor 594 (Z25307) dye or anti-mouse Alexa Fluor 488 (Z25002), were use at a dilution of 1:2000 at room temperature for 1 h (Invitrogen). The slides were mounted with coverslips using UltraCruz Mounting Medium (Santa Cruz Biotechnology). Immunofluorescence images were taken on the Leica TCS SP5-II (upright stand) microscope with 40X oil 1.25 numeric aperture objective lens.
Soft agar colony formation assays
Soft agar assay for anchorage-independent growth was performed using 0.3% agarose with 5×103 suspended cells, which was plated on the top of a layer of 0.8% agar in a 6-well plate. Three weeks after initial plating, colonies were fixed, stained with 0.1% crystal violet, and counted.
Migration and invasion assays
Migration assays were performed using Boyden chambers (Corning, 353097), with 2.5×104 cells plated in each chamber. Cells were incubated for 24 h, fixed, and stained with 0.1% crystal violet. Invasion assays were performed by precoating the Boyden chambers with type IV collagen (0.25 mg/mL) in 0.25% acetic acid overnight. After incubation for 48 h, cells were fixed and stained with 0.1% crystal violet. Images were taken using the Olympus 1×71 microscope, and cells were counted using Image J software.
Cell viability assay and doubling time detection
Cells were seeded into 96-well plates at 2.5 × 103 cells per well. At days 0, 3, 5, and 7, cell viability was determined using the CellTiter-Glo luminescent cell viability assay kit (G7570, Promega) in accordance with the manufacturer’s instructions.
Cells were seeded into 6-well plates at a density of 5 × 103 cells/well, 5 × 104 cells/well, and 1 × 105 cells/well. Post-plating 48 hrs, cells were detached, and the trypan blue cell exclusion assay was used to calculate the number of cells. A countless automated cell counter (AMQAF1000, ThermoFisher Scientific) was used for cell counting. The doubling time was calculated using an online doubling time calculator (http://www.doubling-time.com/compute.php).
RNA stability assay
Cells were plated 24 h before treatment with 5 μg/mL actinomycin D. Treated cells were collected post-treatment 0, 2, 4, 8, and 16 hrs, and RNA was extracted. QRT-PCR was performed to assess RNA expression. Primers used to detect endogenous and exogenous FAK are listed in Table S. The percentage of remaining mRNA was normalized by the mRNA expression at day 0 in each group. The half-life of FAK mRNA was assessed using a one-phase exponential decay model, with 18s mRNA as normalization.
RNA immunoprecipitation (RIP)
RNA immunoprecipitation was performed using a Magna RNA-Binding Protein Immunoprecipitation Kit (EMD Millipore, 17-701). Before reverse transcription, DNAse digestion was performed on the precipitated RNA to remove any residual DNA. On the basis of the position of the functional ADAR-editing site in FAK RNA in the TCGA cohort, we used two putative ADAR-binding regions in the intron 26 of FAK RNA arbitrarily named regions A and B. These regions are located on chromosome 8, and their positions are as follows: A, 141,702,900–141,703,155 and B, 141,702,218–141,702,681. Regions A and B were pulled down by antibodies against ADAR1, FLAG-epitope, or negative-control IgG and amplified by qPCR after reverse transcription.
In vitro RNA-protein interaction
ADAR, ADAR dsRBD mutant, and ADAR deaminase mutant proteins were in vitro translated using the 1-Step Human Coupled IVT Kit (Thermo Scientific, 88882), in accordance with the manufacturer’s instructions. T7 promoter sequence was added by PCR to the 5′ and 3′ ends of FAK region A. Double-stranded RNA was in vitro transcribed using the Megascript T7 Kit (Ambion, AM1333) and purified using the phenol:chloroform extraction and isopropanol precipitation protocol. Double-stranded RNA was biotin-labeled using the RNA 3′ Desthiobiotinylation Kit (Thermo Scientific, 20163). Next, 100 μg of in vitro translated protein and 50 pmol labeled double-stranded RNA were mixed and put through the Magnetic RNA-Protein Pull Down Kit (Thermo Scientific, 20164), in accordance with the manufacturer’s instructions. Western blot was performed on the resulting eluent to assess for ADAR’s interaction with the double-stranded RNA.
FAK inhibitor treatment
H1299 and HCC827 cells (1×104/100 μL) were placed in 96-well plates for 24 h and then treated with the FAK inhibitors PND1186 (Selleckchem, SR-2156) and VS-6063 (Selleckchem, S7654) at varying concentrations (0.1, 1, 2.5, 5, and 10 μm) in 1% DMSO for 72 h. Cell viability was determined by cell titer glow (Promega, G7570). Invasion assays and Western blots were performed on cells treated with FAK inhibitors (2.5 μm) for 72 h.
Identification of editing sites in FAK in human LUAD
FAK editing sites were identified through a manually curated process where we compared mRNA sequenced reads for TCGA LUAD samples (N=230 patient) using the Integrated Genomic Viewer tool developed at the Broad Institute. Candidate editing locations were chosen when they exhibited a good concordance in T->C changes across the reference cell line and the patient samples. We then used the ‘mpileup’ tool from ‘samtools’ to interrogate the chosen positions across all the TCGA samples with available sequencing data, and we counted the number of reads with T->C changes at those chromosomal locations per sample.
Statistical analysis
The results of all experiments represent the mean ± SD of at least three separate experiments. All statistical tests were two-sided and performed with R 3.3.1 (R Core Team, Vienna, Austria) using the clinfun and coin packages. Statistical significance is defined as p<0.05. No formal multiple-testing adjustments were conducted.
Supplementary Material
Fig. S1. The genetic alteration of ADAR in TCGA cohorts.
Fig. S2. The impact of ADAR KD on the downstream targets and cell viability.
Fig. S3 The editing status of FAK in the TCGA cohort.
Fig. S4. ADAR does not bind to region B in FAK.
Fig. S5. Comparison of DNA sequences of pcDNA-FAKE+I WT and edited.
Fig. S6. Cell viability after treatment with FAK inhibitors.
Table S. PCR primers.
One sentence summary.
RNA binding and editing are required for ADAR-mediated stabilization of FAK RNA, which promotes lung adenocarcinoma cell migration and invasion.
Acknowledgments
Funding: This work was supported by grants R01 CA136705 (to D.R.J.), R01 CA192399 (to M.W.M.), R01 CA132580 (to M.W.M.), and U54 CA137788 (to P.S.A.). This work was also supported, in part, by NIH/NCI Cancer Center Support Grant P30 CA008748.
Footnotes
Author contributions: E.M.A. and S.D. designed and performed the experiments, analyzed the data, and wrote the paper. Y.L. helped with experimental designs, data analysis, and writing of the paper. P.S.A provided lung adenocarcinoma tissue microarray samples and data. K.S.T. assisted with statistical analyses. N.C., S.D., M.W.M., F.S.V., and N.D.S. assisted with writing and preparation of the manuscript. D.R.J provided conceptual design of the study, supervision, analysis of the data, and writing of the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The Illumina microarray data have been submitted to the Gene Expression Omnibus (GEO [http://www.ncbi.nlm.nih.gov/geo]) under accession no. GSE93035.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18:349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 3.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fumagalli D, Gacquer D, Rothe F, Lefort A, Libert F, Brown D, Kheddoumi N, Shlien A, Konopka T, Salgado R, Larsimont D, Polyak K, Willard-Gallo K, Desmedt C, Piccart M, Abramowicz M, Campbell PJ, Sotiriou C, Detours V. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep. 2015;13:277–289. doi: 10.1016/j.celrep.2015.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HM, Eterovic AK, Yuan Y, Li J, Nair N, Minelli R, Tsang YH, Cheung LW, Jeong KJ, Roszik J, Ju Z, Woodman SE, Lu Y, Scott KL, Li JB, Mills GB, Liang H. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell. 2015;28:515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015;13:267–276. doi: 10.1016/j.celrep.2015.08.080. [DOI] [PubMed] [Google Scholar]
- 7.Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–1098. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- 8.Wagner RW, Smith JE, Cooperman BS, Nishikura K. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc Natl Acad Sci U S A. 1989;86:2647–2651. doi: 10.1073/pnas.86.8.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CX, Cho DSC, Wang QD, Lai F, Carter KC, Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. Rna-a Publication of the Rna Society. 2000;6:755–767. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galeano F, Tomaselli S, Locatelli F, Gallo A. A-to-I RNA editing: the “ADAR” side of human cancer. Semin Cell Dev Biol. 2012;23:244–250. doi: 10.1016/j.semcdb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Slotkin W, Nishikura K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013;5:105. doi: 10.1186/gm508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anadon C, Guil S, Simo-Riudalbas L, Moutinho C, Setien F, Martinez-Cardus A, Moran S, Villanueva A, Calaf M, Vidal A, Lazo PA, Zondervan I, Savola S, Kohno T, Yokota J, de Pouplana LR, Esteller M. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene. 2015 doi: 10.1038/onc.2015.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer and Metastasis Reviews. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63:610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, Spencer KS, Cheresh DA, Schlaepfer DD. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–767. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 18.Michael KE, Dumbauld DW, Burns KL, Hanks SK, Garcia AJ. Focal adhesion kinase modulates cell adhesion strengthening via integrin activation. Mol Biol Cell. 2009;20:2508–2519. doi: 10.1091/mbc.E08-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, Takenoshita S, Gotoh N, Mizuno H, Sarai A, Kawano S, Yamaguchi R, Miyano S, Yokota J. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 21.Crews LA, Jiang Q, Zipeto MA, Lazzari E, Court AC, Ali S, Barrett CL, Frazer KA, Jamieson CH. An RNA editing fingerprint of cancer stem cell reprogramming. J Transl Med. 2015;13:52. doi: 10.1186/s12967-014-0370-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev. 2004;14:92–101. doi: 10.1016/j.gde.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Tehrani S, Tomasevic N, Weed S, Sakowicz R, Cooper JA. Src phosphorylation of cortactin enhances actin assembly. Proc Natl Acad Sci U S A. 2007;104:11933–11938. doi: 10.1073/pnas.0701077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene. 1999;18:4440–4449. doi: 10.1038/sj.onc.1202827. [DOI] [PubMed] [Google Scholar]
- 25.Tomar A, Lawson C, Ghassemian M, Schlaepfer DD. Cortactin as a Target for FAK in the Regulation of Focal Adhesion Dynamics. Plos One. 2012;7 doi: 10.1371/journal.pone.0044041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacGrath SM, Koleske AJ. Cortactin in cell migration and cancer at a glance. Journal of Cell Science. 2012;125:1621–1626. doi: 10.1242/jcs.093781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellis SL, Miller JT, Turner CE. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J Biol Chem. 1995;270:17437–17441. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- 28.Wang IX, So E, Devlin JL, Zhao Y, Wu M, Cheung VG. ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep. 2013;5:849–860. doi: 10.1016/j.celrep.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, Freund S, Bycroft M, St Johnston D, Varani G. RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Devarakonda SHK, Waqar SN, Guebert K, Maggi LB, Carpenter D, Ozenberger B, Govindan R, Morgensztern D. Characteristics of 1q amplification in adenocarcinoma of the lung (LUAD) Journal of Clinical Oncology. 2014;32 [Google Scholar]
- 32.Maas S, Patt S, Schrey M, Rich A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci U S A. 2001;98:14687–14692. doi: 10.1073/pnas.251531398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shoshan E, Mobley AK, Braeuer RR, Kamiya T, Huang L, Vasquez ME, Salameh A, Lee HJ, Kim SJ, Ivan C, Velazquez-Torres G, Nip KM, Zhu K, Brooks D, Jones SJ, Birol I, Mosqueda M, Wen YY, Eterovic AK, Sood AK, Hwu P, Gershenwald JE, Robertson AG, Calin GA, Markel G, Fidler IJ, Bar-Eli M. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol. 2015;17:311–321. doi: 10.1038/ncb3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, Liu M, Yuan YF, Fu L, Kong KL, Qi L, Li Y, Zhang N, Tong AH, Kwong DL, Man K, Lo CM, Lok S, Tenen DG, Guan XY. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19:209–216. doi: 10.1038/nm.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, Shah RH, Huynh T, Mino-Kenudson M, Sgroi D, Isakoff S, Thabet A, Elamine L, Solit DB, Lowe SW, Quadt C, Peters M, Derti A, Schegel R, Huang A, Mardis ER, Berger MF, Baselga J, Scaltriti M. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature. 2015;518:240–244. doi: 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merlo LMF, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nature Reviews Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 37.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111–125. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Ho BT, Olson G, Figel S, Gelman I, Cance WG, Golubovskaya VM. Nanog Increases Focal Adhesion Kinase (FAK) Promoter Activity and Expression and Directly Binds to FAK Protein to Be Phosphorylated. Journal of Biological Chemistry. 2012;287:18656–18673. doi: 10.1074/jbc.M111.322883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng N, Li YD, Han ZG. Argonaute2 Promotes Tumor Metastasis by Way of Up-regulating Focal Adhesion Kinase Expression in Hepatocellular Carcinoma. Hepatology. 2013;57:1906–1918. doi: 10.1002/hep.26202. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, Rusert JM, Dao KH, Morris SR, Goldstein LS, Marra MA, Frazer KA, Jamieson CH. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2013;110:1041–1046. doi: 10.1073/pnas.1213021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cenci C, Barzotti R, Galeano F, Corbelli S, Rota R, Massimi L, Di Rocco C, O’Connell MA, Gallo A. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem. 2008;283:7251–7260. doi: 10.1074/jbc.M708316200. [DOI] [PubMed] [Google Scholar]
- 42.Liu J, Lee W, Jiang Z, Chen Z, Jhunjhunwala S, Haverty PM, Gnad F, Guan Y, Gilbert HN, Stinson J, Klijn C, Guillory J, Bhatt D, Vartanian S, Walter K, Chan J, Holcomb T, Dijkgraaf P, Johnson S, Koeman J, Minna JD, Gazdar AF, Stern HM, Hoeflich KP, Wu TD, Settleman J, de Sauvage FJ, Gentleman RC, Neve RM, Stokoe D, Modrusan Z, Seshagiri S, Shames DS, Zhang Z. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res. 2012;22:2315–2327. doi: 10.1101/gr.140988.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bahn JH, Ahn J, Lin XZ, Zhang Q, Lee JH, Civelek M, Xiao XS. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nature Communications. 2015;6 doi: 10.1038/ncomms7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153:575–589. doi: 10.1016/j.cell.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nature Reviews Molecular Cell Biology. 2016;17:83–96. doi: 10.1038/nrm.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ertel A, Verghese A, Byers SW, Ochs M, Tozeren A. Pathway-specific differences between tumor cell lines and normal and tumor tissue cells. Mol Cancer. 2006;5:55. doi: 10.1186/1476-4598-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stein WD, Litman T, Fojo T, Bates SE. A Serial Analysis of Gene Expression (SAGE) database analysis of chemosensitivity: comparing solid tumors with cell lines and comparing solid tumors from different tissue origins. Cancer Res. 2004;64:2805–2816. doi: 10.1158/0008-5472.can-03-3383. [DOI] [PubMed] [Google Scholar]
- 48.Daniel C, Lagergren J, Ohman M. RNA editing of non-coding RNA and its role in gene regulation. Biochimie. 2015;117:22–27. doi: 10.1016/j.biochi.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 49.Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacology & Therapeutics. 2015;146:132–149. doi: 10.1016/j.pharmthera.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Gerber DE, Ramalingam SS, Morgensztern D, Kelly RJ, Burns TF, Lopez-Chavez A, Spigel DR, Wehbe AM, Sorensen R, Weaver DT, Horobin J, Keegan M, Scaglioni PP, Camidge DR, Pls V. A phase 2 study of defactinib (VS-6063), a cancer stem cell inhibitor that acts through inhibition of focal adhesion kinase (FAK), in patients with KRAS-mutant non-small cell lung cancer. Journal of Clinical Oncology. 2014;32 [Google Scholar]
- 51.Konstantinidou G, Ramadori G, Torti F, Kangasniemi K, Ramirez RE, Cai YR, Behrens C, Dellinger MT, Brekken RA, Wistuba II, Heguy A, Teruya-Feldstein J, Scaglioni PP. RHOA-FAK Is a Required Signaling Axis for the Maintenance of KRAS-Driven Lung Adenocarcinomas. Cancer Discovery. 2013;3:444–457. doi: 10.1158/2159-8290.CD-12-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The Small Gtp-Binding Protein Rac Regulates Growth-Factor Induced Membrane Ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 53.Lausen BS. M. Maximally selected rank statistics. Biometrics. 1992;48:73–85. [Google Scholar]
- 54.Kadota K, Eguchi T, Villena-Vargas J, Woo KM, Sima CS, Jones DR, Travis WD, Adusumilli PS. Nuclear estrogen receptor-alpha expression is an independent predictor of recurrence in male patients with pT1aN0 lung adenocarcinomas, and correlates with regulatory T-cell infiltration. Oncotarget. 2015;6:27505–27518. doi: 10.18632/oncotarget.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y, Mayo MW, Xiao A, Hall EH, Amin EB, Kadota K, Adusumilli PS, Jones DR. Loss of BRMS1 promotes a mesenchymal phenotype through NF-kappaB-dependent regulation of Twist1. Mol Cell Biol. 2015;35:303–317. doi: 10.1128/MCB.00869-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung GG, Provost E, Kielhorn EP, Charette LA, Smith BL, Rimm DL. Tissue microarray analysis of beta-catenin in colorectal cancer shows nuclear phospho-beta-catenin is associated with a better prognosis. Clin Cancer Res. 2001;7:4013–4020. [PubMed] [Google Scholar]
- 57.O’Shannessy DJ, Yu G, Smale R, Fu YS, Singhal S, Thiel RP, Somers EB, Vachani A. Folate receptor alpha expression in lung cancer: diagnostic and prognostic significance. Oncotarget. 2012;3:414–425. doi: 10.18632/oncotarget.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 59.Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol. 2006;26:8683–8696. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The genetic alteration of ADAR in TCGA cohorts.
Fig. S2. The impact of ADAR KD on the downstream targets and cell viability.
Fig. S3 The editing status of FAK in the TCGA cohort.
Fig. S4. ADAR does not bind to region B in FAK.
Fig. S5. Comparison of DNA sequences of pcDNA-FAKE+I WT and edited.
Fig. S6. Cell viability after treatment with FAK inhibitors.
Table S. PCR primers.