

Abstract
Mammalian thioredoxin reductases (TrxRs) are selenocysteine-containing proteins (selenoproteins) that propel a large number of functions through reduction of several substrates including the active site disulfide of thioredoxins (Trxs). Well-known enzymatic systems that in turn are supported by Trxs and TrxRs include deoxyribonucleotide synthesis through ribonucleotide reductase, antioxidant defense through peroxiredoxins and methionine sulfoxide reductases, and redox modulation of a number of transcription factors. Although these functions may be essential for cells due to crucial roles in maintenance of cell viability and proliferation, findings during the last decade reveal that mammals have major redundancy in their cellular reductive systems. The synthesis of glutathione (GSH) and reductive functions of GSH-dependent pathways typically act in parallel with Trx-dependent pathways, with only one of these systems often being sufficient to support viability. Importantly, this does not imply that a modulation of the Trx system will remain without consequences, even when GSH-dependent pathways remain functional. As suggested by several recent findings, the Trx system in general and the TrxRs in particular, function as key regulators of signaling pathways. In this review article we will discuss findings that collectively suggest that modulation in mammalian systems of cytosolic TrxR1 (TXNRD1) or mitochondrial TrxR2 (TXNRD2) influence cell patterning and cellular stress responses. Effects of lower activities include increased adipogenesis, insulin responsiveness, glycogen accumulation, hyperproliferation, and distorted embryonic development, while increased activities correlate with decreased proliferation and extended lifespan, as well as worse cancer prognosis. The molecular mechanisms that underlie these diverse effects, involving regulation of protein phosphorylation cascades and of key transcription factors that guide cellular differentiation pathways, will be discussed. We conclude that the selenium-dependent oxidoreductases TrxR1 and TrxR2 should be considered as key components of signaling pathways that control cell differentiation and cellular stress responses.
Keywords: Thioredoxin, Thioredoxin Reductase, Cell Patterning, Redox Signaling
Graphical abstract
Cellular patterning programs and redox control
Redox regulation, i.e. regulatory oxidation-reduction modification of key proteins in cellular signaling pathways, is fundamental to life. Many diseases arise from imbalances in redox processes, including events of oxidative stress, hypoxia and ischemia-reperfusion injury. Importantly, redox processes are also essential parts of normal physiological events [1]. The two major reductive systems in mammalian cells are the thioredoxin (Trx) and glutathione (GSH) systems, which are generally complementary to each other in life supporting functions, but may differ in the context of modulated redox signaling pathways [2–7]. Indeed, these systems are well known to be able to modulate a large number of signaling pathways in the cytosol, nuclei, and mitochondria [4, 8–19], with major effects in cellular pathways of reduction and redox control [14, 19, 20].
Early insights suggesting both a robust redundancy between cytosolic disulfide reductase systems and their possible additional signaling roles in higher eukaryotes came with the onset of mouse genetic models having disruptions in these systems. First it was noted that germline genetic ablation of glutathione reductase (Gsr) had almost no measurable phenotypic impact on mice, and very curiously also did not substantially affect either intracellular levels or redox state of hepatic glutathione in the homozygous mutant animals, i.e. only a minor increase in the already very low intracellular glutathione disulfide levels were detected [21, 22]. This later observation suggested that mammalian cells had alternative means of sustaining pools of reduced GSH. These inferred systems were not resolved for more than another decade, after which it was revealed that both methionine-dependent de novo synthesis of GSH and cross-trafficking of reducing power from the cytosolic TrxR1/Trx1 system can participate in sustaining reduced GSH pools in the absence of Gsr [2, 23]. Further suggesting the GSH system might be of only minor importance in cell and organismal homeostasis, it had already been well established that mammalian cells as well as adult animals and humans were highly tolerant of chronic severe systemic GSH depletion by the drug buthionine sulfoximine (BSO) [24–28].
In contrast to these observations on the GSH system, homozygous germline knockouts of the genes encoding Trx1, Trx2, TrxR1, or TrxR2 were all found to result in embryonic lethality, suggesting that the cytosolic and mitochondrial Trx systems, unlike the GSH system, are critical for basal homeostasis in mammalian cells, at least during embryogenesis [29–32]. Interestingly however, none of the mouse thioredoxin system-knockout models showed simple zygotic arrest, as one might have expected for activities critical to basal cell survival or DNA replication. Rather, each homozygous mutation sustained early development and extensive cell proliferation, followed by a later lethal crisis. More detailed analyses of mouse embryos lacking TrxR1 verified that early TrxR1-null embryos underwent robust proliferative expansion and differentiation of early tissues, including trophectoderm and primitive endoderm, but arrested prior to gastrulation [33]. Single embryo transcriptome profiling and marker analyses revealed that mesodermal genes were not expressed and, indeed, there was no evidence of formation of either node or primitive streak in the mutant embryos [33]. These observations suggested that TrxR1-null embryos had no major basal cellular deficiencies, but rather had a patterning defect in which the intercellular signaling that establishes early embryonic patterning was affected [33].
The apparent normal proliferation and development of TrxR1-null embryonic trophectoderm and primitive endoderm was intriguing. One of our laboratories initiated a study in which a Cre-responsive conditional-null allele of the Txnrd1 gene (encoding TrxR1) was combined with a Cre-responsive dual-fluorescent reporter gene and inducible Cre expression systems in an attempt to use marked-mosaic analyses to identify cell types in vivo that absolutely required TrxR1 expression. We generated mosaic animals with marked TrxR1-null cells distributed throughout all organs and tissues of the fetal, juvenile, or adult body, and yet we were unable to identify any cell type that did not tolerate long-term disruption of TrxR1 (EES, unpublished data). To provide a convenient model system for studying the in vivo functions of TrxR1 in animals, we adopted a liver-specific model based on disruption of the conditional-null Txnrd1 allele coincident with hepatocyte differentiation using the classical AlbCre transgene [34, 35]. Ours and other groups also developed models lacking TrxR1 in different cell types including fibroblasts, neurons, heart, adipocytes, various cancer cells, and others [29, 32, 36–38]. Detailed studies on the TrxR1-null livers revealed that basal cell functions, including robust developmental and regenerative proliferation, were indeed sustained in the absence of TrxR1 [39, 40]. Intriguingly, the TrxR1-null livers also showed no measurable markers of oxidative stress, yet transcriptome analyses on these livers indicated that they had strong chronic induction of the Nrf2 oxidative stress response pathway [36, 41]. Similar findings were subsequently found using pharmacologic inhibition of TrxR1 [42]. These results suggested that TrxR1 might be a regulatory component on a signaling pathway that controls Nrf2 [10, 43]. In combination with the patterning defects in TrxR1-null embryos, these findings hinted that TrxR1 might play roles in modulating intracellular aspects of various signaling pathways.
In this article we shall specifically survey and discuss how the mammalian NADPH-dependent selenoprotein thioredoxin reductases TrxR1 and TrxR2 seem to modulate a large number of cellular processes that are important in regulation of cell patterning.
Mammalian thioredoxin reductases
Background and overall functions
The whole Trx system is NADPH dependent since it is propelled by NADPH-dependent oxidoreductases, mainly cytosolic TrxR1 (encoded by TXNRD1) and mitochondrial TrxR2 (encoded by TXNRD2). The system is also selenium-dependent since these enzymes are selenoproteins, i.e. they have a catalytic selenocysteine residue, and they are furthermore subject to splicing that results in variants of the enzymes differing at their N-terminal domains [14, 44–47]. It should be noted, considering cellular signaling pathways, that the normally cytosolic TrxR1 enzyme can also be found in the nucleus in the alternative splice-form of TXNRD1_v2, where it can modulate transcription factor activities, such as the estrogen receptors [14, 46]. The alternative splice-form of TXNRD1_v3 is targeted to cell membranes, where it co-localizes with membrane rafts and has the capacity to promote formation of filopodia [14, 48–50]. These splice variants of TrxR1 have not yet been examined at depth in relation to cellular signaling pathways, but should clearly also be specifically considered in this context. It should also be noted that mammalian TrxRs display very broad substrate specificities and reduce many low molecular weight compounds in addition to their primary Trx substrates [14]. Enzymatic activities with certain regulatory low molecular weight metabolites may hence, in theory, also be related to the impact of TrxR status on cellular patterning programs, but this possibility has yet been insufficiently studied.
Thioredoxins as primary TrxR substrates
The primary substrates of TrxR1 and TrxR2 are Trx1 and Trx2, respectively, and most actions of the mammalian thioredoxin system are likely to be carried out by the activities of Trx1 and Trx2 [12, 14, 51–55]. However, additional substrate proteins for TrxR1 such as the Trx-related protein of 14 kDa (TRP14, TXNDC17) are also likely to be important in relation to signaling events [56–61]. It should furthermore be noted that although major signaling aberrations can be seen upon genetic deletion of TrxR1 (the Txnrd1 gene in mice) or TrxR2 (the Txnrd2 gene in mice) these are not necessarily related to effects on Trx1 or Trx2 activities, respectively, because of significant crosstalk in redox pathways and specific effects on signaling depending upon more than solely general impairments of the cellular reduction capacity [8, 37, 40, 41, 62].
Ribonucleotide reductase
The antioxidant and redox-regulatory functions of the Trx system support cell growth and viability through a diverse set of mechanisms. Ribonucletide reductase, that catalyzes and controls the synthesis of DNA precursors, is an example of an important enzyme that receives reducing equivalents from the Trx system to maintain its function, although it can also be reduced by glutaredoxins [63–65], as in bacteria [66, 67]. Several redox active proteins can thus support ribonucleotide reductase, including Grx2 and its different splice variants [68]. Either TrxR or GSH can in turn reactivate Grx2 through reduction [64, 69, 70]. This redundancy in support of ribonucleotide reductase activity likely helps to explain the non-coherent expression and localization patterns of ribonucleotide reductase, Trx and TrxR in mammalian cells and tissues [71–73].
Peroxiredoxins
The peroxiredoxins (Prxs) are important antioxidant proteins that exist in different cellular compartments in high abundance. Their antioxidant functions and catalytic activities are in turn dependent upon direct reduction by Trxs. Prxs efficiently reduce H2O2 and are considered to be important antioxidant enzymes and also modulators of intracellular signaling through regulation of oxidative bursts [15, 18]. Inhibition of TrxR through compounds such as auranofin or isothiocyanates, has identified the mitochondrial Prx3 to be most susceptible peroxiredoxin to oxidation; time dependent analysis of the rate of oxidation during low dose TrxR1 inhibition showed that Prx3 was affected to a greater extent than cytosolic peroxiredoxins [74, 75]. The antioxidant function of Prx3, in the mitochondria, is supported by Trx2 and TrxR2 as well as a GSH-dependent peroxidase activity of Grx2 [76]. If Prx3 becomes overoxidized to its sulfinic acid form, the Trx system can no longer reactivate Prx3 but instead sulfiredoxin is required, which under such conditions becomes translocated from its natural location in the cytosol into the mitochondria [77]. The events of Prx3 overoxidation and sulfiredoxin being targeted to mitochondria are, interestingly, directly linked to the cellular mechanisms of circadian rhythm [78–80]. This poses the question whether activities or functions of the core players of the Trx system can also be directly linked with circadian rhythm. Some observations in Drosophila melanogaster [81] and plants [82] suggest this may be the case, but further studies of this topic are clearly needed.
Methionine sulfoxide reductases
The important Trx-dependent methionine sulfoxide reductase (Msr) enzymes repair methionine sulfoxide residues in proteins by reduction. Two MsrA and MsrB isoenzymes are specific for reduction of either methionine-S-sulfoxide or methionine-R-sulfoxide. Msrs are found in many organisms and, like TrxRs, some but not all Msrs are selenoproteins [83, 84]. In mammals, MsRB1 together with Mical enzymes controls actin polymerization through regulated methionine oxidation status [85]. Impaired Msr functions are related to degenerative disease processes and aging [83, 84]. For example, expression levels of both variants of mammalian methionine sulfoxide reductases decreased with increased age in rats, and also cytosolic Trx1 and TrxR1, which reduce Msrs, were found to be decreased [86]. Genetic deletion of MsrA increases oxidative stress, resulting in neurological defects and a reduced lifespan [87]. Selenium deficiency is known to decrease TrxR activity and combined with MsrA deficiency an increased oxidative damage is detected [88].
Effects of modulated activities of thioredoxin reductases on cellular patterning in mammalian cells and tissues
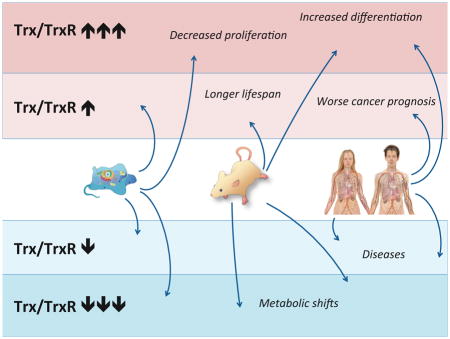
A large number of observations in recent years indicate that modulation of TrxR isoenzymes is typically linked with altered cellular phenotypes, with in many instances strong indications of casual relationships. In Table 1 and Figure 1 we have summarized an “A to Z” of a number of key observations in line with this notion, which shall here be discussed in further detail.
Table 1. The A to Z of Modulated Cell Patterning by Mammalian Thioredoxin Reductases.
This table lists selected key references from the literature that illustrate the effects of modulated expression of TrxR1 or TrxR2 on cell patterning. For a schematic illustration of these effects, see Figure 1 with the index letters as references.
| Index | Aberration | Cellular system | Effects on cell patterning | Comment | Ref. |
|---|---|---|---|---|---|
| Forced Overexpression of TrxR1 | |||||
| A | TrxR1 overexpression in Bovine arterial endothelial cells | TrxR1 overexpression is induced by Nrf2 activation and can enhancee TNFα-induced DNA binding activity of NF-κB and dependent gene expression | [206, 218] | ||
| B | TrxR1 and TrxR2 overexpression in HEK293 cells | Decreased cell proliferation and stimulation of differentiation | Expression of cell differentiation markers CK18, CK-Cam5.21 and BerEP4, decrease in GR activity and expression | [219] | |
| Forced Overexpression of TrxR2 | |||||
| B | TrxR1 and TrxR2 overexpression in HEK293 cells | Decreased cell proliferation and stimulation of differentiation | Expression of cell differentiation markers CK18, CK-Cam5.21 and BerEP4, decrease in GR activity and expression | [219] | |
| C | TrxR2 overexpression in Neuro2A, COS-7 and HeLa cells | Decreased growth rate | [220] | ||
| D | TrxR2A (alternative splicing variant) overexpression in HeLa and HEK293 cells | Expression induced apoptosis, decrease in cell proliferation of stable HEK293 cell lines. | Cleavage of procaspase-3 and PARP | [221] | |
| Endogenous Upregulation of TrxR1 | |||||
| E | 3T3-L1 | Differention of 3T3-L1 cells into adipocytes | Upregulation of TrxR1, Trx1, TrxR2 and Trx2 during differentiation | [105] | |
| F | Alternative splicing of TrxR1 in HEK293 cells | Regulation of ER signaling | Modulation of estrogen signaling | [46] | |
| G | Upregulation of TrxR1 in cancer | Upregulation of TrxR1 transcripts as well as activity correlates with worse cancer prognosis | Analyses of human data | [98–100] | |
| Endogenous Upregulation of TrxR2 | |||||
| E | 3T3-L1 | Differention of 3T3-L1 cells into adipocytes | Upregulation of TrxR1, Trx1, TrxR2 and Trx2 during differentiation | [105] | |
| H | Several model organisms | Overexpression of TrxR2 correlates with longer lifespan | Upregulation of TXNRD2 transcript and activity seen in long- lived long-lived species of rodents, primates, and | [106] | |
| Endogenous Downregulation of TrxR1 | |||||
| I | miR-23a and miR-23b dependent regulation of TrxR1 expression in C2C12 mouse myoblasts | miR-23a and miR-23b control skeletal myogenic differentiation through regulation of TrxR1 expression | Decreased expression of TrxR1 induced differentiation of C2C12 myoblasts and muscle satellite cells. Overexpression of TrxR1 delayed C2C12 myoblasts differentiation | [107] | |
| J | TrxR1 expression in a human patient study | TrxR1 and TrxR1- v.2,3,5 correlation with differentiation | TrxR1 and TrxR1-v.2,3,5 expression was correlated with differentiation in squamous carcinoma. | [108] | |
| K | Naturally occurring mutation in human patients | A homozygous Pro190Leu mutation correlates with generalized epilepsy | Found by whole-exome sequencing in a family with genetic generalized epilepsy | [109] | |
| Endogenous Downregulation of TrxR2 | |||||
| L | Rare mutations in the FAD-binding domain of the TNXRD2 gene (A59T and G375R) of human patients with dilated cardiomyopathy (DCM) | Heterozygous carriers of the two mutations mutations displayed impaired left ventricle function | However these mutations only contribute to disease burden since other DCM causing mutations have been described | [110] | |
| Knockout of TrxR1 | |||||
| M | Whole animal mouse model | No mesoderm formed. Disruption of gastrulation | No evidence of increased oxidative stress | [33] | |
| N | MEF −/− | Increased differentiation, lipogenesis | Strong effects on Akt signaling; possible to block with NAC | [37] | |
| O | MEF −/− | Increased apoptosis | TrxR1 is required for clearance of glucose derived H2O2 | [38] | |
| P | Hepatocyte specific disruption of Txnrd1 | Metabolic switch from lipogenic genes towards glycolysis, increased proliferation and hepatomegaly | Txnrd1-null livers were resistant to APAP- induced hepatotoxicity, replication is dependent on either at least one allele of Txnrd1 or sufficient GSH levels, and triple knockout of Txnrd1, Trx1 and GR yields pronounced hepatomegaly | [62, 111–113] | |
| Q | Neuronal specific Txnrd1 conditional knockout | Morphological alterations | Reduced proliferation of granule neurons resulting in thinner EGL in mutants (Postnatal day 7). Reduced weight, severe ataxia. Strongly disorganized development in anterior lobules (I–VI). Cereberal hypoplasia. Radial glial cells or granule cell precursors likely to be affected | [114] | |
| R | MEF TrxR1 −/− | Increase in PDGF induced proliferation | TrxR1 deficient cells display an increase in PTP1B oxidation corresponding to a decrease in activity and increase in PDGF induced proliferation | [58] | |
| Knockout of TrxR2 | |||||
| S | Whole mouse Txnrd2 knockout | Embryonic lethal at day 13 | Embryos were smaller and showed severe anemia. Apoptosis was increased in liver. Decreased proliferation of cardiomyocytes. Cardiac specific ablation of TrxR2 resulted in cardiomyopathy | [115] | |
| T | Heart specific Txnrd2 knockout | Increased heart to body weight, mitochondrial degeneration | Increased autophagy, reduced blood pressure, stabilization of HIF, decreased oxygen consumption and increased ROS | [116] | |
| U | MEF −/− subcutaneous tumor model | Decreased vascularization | Delay of angiogenic switch, impaired tumor angiogenesis. Increased prolyl hydroxylase (PHD2) levels with corresponding decrease in Hif1α. Decrease in VEGF-A levels. Increase in JNK activation | [121] | |
| V | Cardiomyocyte specific Txnrd1- or Txnrd2 deficient mice | Increased vulnerability to I/R induced injury. Increased mitochondrial ROS | [117] | ||
| X | Conditional B- and T- cell- specific inactivation of Txnrd2 | No effect on proliferation | [118] | ||
| Y | TrxR2 shRNA knockdown in Chondrogenic ATDC5 cells | TrxR2 deficiency enhance differentiation and apoptosis | Knockdown stimulates expression of Cartilage glycosaminoglycans (GAGs) and expression of (ECM) genes Collagen 2 and Aggrecan. Activation of Akt signaling and chondrocyte proliferation | [119] | |
| Z | Endothelium specific Txnrd2 knockout mice subjected to artery ligation | Impaired vascular remodeling processes in the model of femoral artery ligation | Endothelial deletion had no change in phenotype under normal conditions. However, artery ligation lead to increased vascular stiffness. Impaired EC vasodilation. The presence of global thrombi was seen in Txndr2 knockout animals suggesting a proaterogenic phenotype. Proinflammatory phenotype. Increase in leukocyte transmigration into tissues. Impaired sprouting of Txnrd2 KO cells in vitro. | [120] | |
Figure 1. Summary of Modulated Cell Patterning Effects Linked to Mammalian Thioredoxin Reductases.

This figure schematically summarizes key effects of forced overexpression, endogenous upregulation, endogenous downregulation or knockout of either TrxR1 or TrxR2 in cells, mice or human, as indicated. For further details and references, see the text and Table 1. The one-letter indices refer to the corresponding indices in Table 1.
Forced overexpression
With regards to cellular phenotypes triggered by forced overexpression of TrxR1 or TrxR2 it should be noted that because of highly inefficient and demanding synthesis machineries required for expression of selenoproteins in mammalian cells [89, 90], it is a difficult task to overexpress these proteins. Moreover, if the selenocysteine (Sec)-encoding UGA codon in the TXNRD1 or TXNRD2 transcripts leads to translational termination, instead of Sec insertion, the enzymes will be expressed as two-amino acid truncated forms ending with –Gly-Cys instead of their natural C-terminal end of –Gly-Cys-Sec-Gly. Such truncated forms of the enzymes are not inertly inactive, but interestingly retain redox activities as NADPH oxidases with pro-oxidant redox cycling capacity coupled to low molecular weight substrates, such as several quinone compounds including juglone. This pro-oxidant activity can become toxic to cells and these shortened forms of the enzyme were named SecTRAPs, for Selenium compromised thioredoxin reductase-derived apoptototic proteins [10, 91–95]. Formation of SecTRAPs in cancer cells during chemotherapy with alkylating agents can be beneficial in terms of therapeutic effects, while at least lower levels of SecTRAPs formed in normal cells may possibly trigger oxidative signaling pathways. Thus, the phenotypes observed upon forced expression of TrxR1 or TrxR2 may in fact be due to increased oxidative stress rather than increased reductive power, which may possibly explain some of the apparently paradoxical effects seen in such model systems [8]. In Table 1 and Figure 1 a number of observations are listed with effects upon cell proliferation after forced overexpression of either TrxR1 and TrxR2, which may hence possibly at least in part be due to increases in oxidative stress due to SecTRAP formation.
Endogenous overexpression
Several growth conditions and cell patterning events are related with endogenously increased expression levels of TrxR1 or TrxR2. The expression of TrxR1 is, interestingly, guided by a typical housekeeping-type promoter in combination with AU-rich elements in the mRNA that guide regulated mRNA stability, otherwise typically found in signaling proteins [19, 96]. Responding to oxidative stress, the TrxR1 transcript is also upregulated through Nrf2 [97] and the enzyme is highly overexpressed in several forms of cancer, where its high overexpression moreover correlates with poor prognosis [98–100]. Both the TXNRD1 and TXNRD2 genes furthermore give rise to several different splice variants, encoding TrxR1 or TrxR2 variants differing at their N-terminal ends, some of which may be dedicated players in cellular signaling pathways [44–49, 101, 102]. One of the variants of TrxR1, TXNRD1_v2, is targeted to the nucleus where it can modulate estrogen signaling by interactions with the estrogen receptor [46]. Another variant, TXNRD1_v3, is myristoylated and targeted to the cell membrane where it can trigger formation of filopodia and is possibly involved in receptor-linked or cell-to-cell mediated signaling events [103, 104]. The existence of these splice variants illustrates well that sole expression levels of TrxR1 (and its variants) can not be the only determinants of the effects of TrxR1 on cell patterning, but also compartmentalization effects and dedicated functions of specific splice variants. Such details in the mechanisms of TXNRD1- or TXNRD2-linked expression levels in relation to specific phenotypes are often overlooked, but should be considered for a full understanding of molecular mechanisms leading to any observed effects. Notwithstanding these aspects, it was found that both TrxR1 and TrxR2, as well as Trx1 and Trx2, are endogenously upregulated when 3T3-L1 cells differentiate into adipocytes [105]. This was an interesting finding since it was also shown that upon knockout of Txnrd1 in mouse embryonic fibroblasts this strongly promoted adipogenesis, which occurred in conjunction with potently augmented Akt and PPARγ activities [37].
With regards to endogenous upregulation of TrxR2 this seems to correlate with longer lifespan in several different model organisms [106]. The molecular mechanisms that may explain this interesting correlation are yet unknown.
Endogenous downregulation
Examples of studies correlating endogenous downregulation of TrxR1 or TrxR2 with cell patterning phenotypes are scarce. Recently one interesting study however discovered a control of TrxR1 expression by the small regulatory RNA species miR-23a and miR-23b, where downregulation of TrxR1 induced myogenic differentiation of mouse myoblasts [107]. It was also found that expression of some of the rarer splice variants of TrxR1 inversely correlates with differentiation in squamous carcinoma [108]. Surprisingly, a Pro190Leu mutation in TrxR1, as a result of a homogenous SNP variant that yields lower cellular activity of the enzyme, is linked to generalized epilepsy [109]. Heterozygous SNP variants in the TXNRD2 gene that lead to mutations in the FAD-binding domain of TrxR2 are instead linked to dilated cardiomyopathy [110]. These observations agree rather well with the phenotypes of mouse knockout models, showing that loss of Txnrd1 often promotes increased differentiation while loss of Txnrd2 mainly gives strong effects in heart development or hematopoiesis (see next).
Knockout models
Although whole animal knockout models in mice with deletion of Txnrd1 or Txnrd2 are lethal, conditional knockouts show evident signs of major effects on cell patterning programs. Also analyses of the whole animal knockout of Txnrd1 revealed such effects, with no signs of increased oxidative stress but instead an almost complete lack of mesoderm formation [33]. When Txnrd1 is conditionally deleted in hepatocytes, the mice display larger livers presenting a striking metabolic switch towards glycogen accumulation together with very strong Nrf2 activation [62, 111–113]. Conditional knockout of Txnrd1 in neurons yield severe ataxia and disorganized brain development with cerebral hypoplasia [114]. Whole-animal knockout of Txnrd2 mainly gives lethal deficiencies in heart development and hematopoiesis [115], with major phenotypes also upon conditional knockout in heart [116] or cardiomyocytes [117], but no evident effect when deleted in B- and T-cells [118]. Lack of Txnrd2 in chondrocytes promotes proliferation, differentiation as well as apoptosis [119], while conditional knockout in endothelial cells displays a phenotype only under stressed conditions [120]. An interesting effect was found using Txnrd2 knockout mouse embryonic fibroblasts inoculated as subcutaneous tumors, which impaired the angiogenic switch as compared to controls [121].
In isolated Txnrd1 deficient mouse embryonic fibroblasts there is a major effect on cell differentiation with increased adipogenesis and insulin responsiveness [37]. It should be emphasized that in those TrxR1-deficient cells, Trx1 was still found to be reduced [37], which is likely to be explained by Trx1 reduction through GSH-dependent glutaredoxins [122]. These observations are noteworthy, as they suggest that effects of TrxR1 impairment on cellular phenotypes are not necessarily due to impaired Trx1 functions. Using the Txnrd1 deficient mouse embryonic fibroblasts it was however also shown that TrxR1 is an essential part for cellular antioxidant defense, but only in sparsely cultured cells stressed with high glucose and grown in medium lacking pyruvate or other antioxidants [38]. The same cells furthermore show increased responses to PDGF treatment due to increased inhibition of PTP1B [58], which again illustrates direct links of TrxR1 to cellular signaling pathways.
Molecular mechanisms by which thioredoxin reductases can control cellular patterning programs
With the several observations of disturbed cell patterning upon modulation of TrxR1 or TrxR2 activities, the question is by what molecular mechanisms these effects may arise. Here we shall discuss some of the possible mechanisms that could be involved and give references to key supporting observations in the literature. For a graphical summary of pathways that may be involved see Figure 2.
Figure 2. Cellular targets of mammalian TrxR1 and TrxR2.

This scheme briefly summarizes major downstream targets of the NADPH-dependent cytosolic enzyme TrxR1 (encoded by TXNRD1) and the mitochondrial TrxR2 (encoded by TXNRD2) through the activities of their principal substrates Trx1 and TRP14 (encoded by TXN and TXNDC17) in the cytosol and Trx2 (TXN2) in the mitochondria. The scheme summarizes substrates containing disulfides that are reduced as well as additional activities, as indicated. Note that this is a highly simplified scheme and that many additional targets exist that in turn may affect cell patterning. See text for further details.
Thioredoxins are well known as powerful and versatile protein disulfide reductases. Indeed, most of their actions are likely to be mediated mainly by catalytic reduction of disulfides in target proteins. This holds true for Trx-mediated reactivation of classical Trx substrates such as oxidized ribonucleotide reductase, peroxiredoxins, methionine sulfoxide reductases (where MsrB1 however forms a selenenylsulfide that is reduced by Trx), and many other target proteins for the thioredoxin system [14, 17, 51, 55], as also discussed above. However, additional target motif entities on protein Cys residues that may be reduced by Trx1, Trx2 and TRP14, likely to be involved in modulation of signaling pathways, include S-nitrosylated [12, 57, 123–126] or persulfidated [56, 127] motifs in target proteins. Also, in regulation of the activity of protein-tyrosine phosphatase-1B (PTP1B) the oxidized target seems to be a sulfenylamide motif that can be reduced by either Trx1 or TRP14 [128–130].
The activities of apoptosis signal regulating kinase 1 (ASK1) are also influenced by Trx1, yet by a distinct mechanism. Reduced Trx1 binds to ASK1 in an inhibitory manner, while oxidized Trx1 does not, thereby activating ASK1 under conditions where the active site motif of Trx1 becomes oxidized [16, 53, 131–133]. Trx2 regulates mitochondrial ASK1 in a similar manner [131]. The binding of reduced Trx1 to the Trx-interacting protein (TXNIP) can have a similar function; when Trx1 becomes oxidized it will release TXNIP, with TXNIP subsequently binding to the NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome that in turn triggers inflammatory signaling cascades [134–136]. Thus, it is not merely through potent disulfide reduction that the Trx system asserts modulatory roles in signaling, but also through more elaborate molecular interactions. The final outcome in signaling and modulation of cell patterning will depend upon a combination of the nature of the posttranslational modification that is targeted, the change in function incurred by the modification, the exact signaling role(s) of the target protein(s), and the overall cellular context in which modulation of such modifications occur. Considering the complexity of the web of signaling networks in a cell, it becomes obvious that although it is clear that the thioredoxin system modulates redox signaling pathways in cells, it seems almost impossible to fully predict the final impact on cellular phenotypes that may be the result of a particular modification in a particular cell. Notwithstanding this fact, we shall next discuss in further details a number of specific signaling systems that may be involved.
S-nitrosylation pathways
Nitrosylation of Cys residues in proteins, a process dependent upon regulated production of the gaseous signaling molecule nitrous oxide (NO) and its derivatives, is becoming recognized as a common mechanism for regulating protein functions in diverse signaling pathways [126, 137–140]. The human Trx1 active site has two Cys residues (Cys32 and Cys35) and in addition Trx1 has three structural Cys residues (Cys 62, Cys69 and Cys73), which can be nitrosylated. Potentially Cys73 of Trx1 might be involved in transnitrosylation reactions [124]. Also Trx2 can participate in transnitrosylation reactions, while during Fas-induced apoptosis, mitochondrial caspase-3 is activated by Trx2 dependent denitrosylation [125, 141, 142]. Trx2-mediated modulation of caspase-3 nitrosylation status can directly regulate the cellular phenotype of microglia [123]. In addition to Trx system-mediated denitrosylation activities, reduced lipoic acid is also able to carry out this reaction [143]. In this context it can be noted that lipoic acid is also a substrate of TrxR [144]. Several studies indicate that the Trx system is likely to modulate the balance between nitrosylation and denitrosylation reactions, and thereby control NO signaling pathways [125, 140, 145]. However, the exact mechanisms by which the Trx system regulates NO signaling is still unclear. Intriguingly, S-nitroso-glutathione (GSNO), which is spontaneously formed upon reaction of NO with GSH is a potent substrate for TrxR, with the reaction resulting in renewed liberation of GSH and NO [146]. GSNO itself was found as a positive regulator of Trx1 activity in rat pulmonary artery smooth muscle cells by lowering the expression levels of the Trx1-inhibitory protein Txnip [147], suggesting a highly intricate network of regulation. It should furthermore be noted that GSNO reductase (GSNOR, also known as class III alcohol dehydrogenase, ADH5) is a GSNO-reducing enzyme using NADH, which acts in parallell with the Trx system and NO synthases for regulation of nitrosylation pathways. In contrast to the Trx system, GSNOR can however not directly reduce nitrosylated proteins; thus it seems likely that the Trx system is the major cellular reducing system for nitrosylated Cys residues (so called SNO moieties) as found in proteins [12, 125, 148].
Protein phosphorylation cascades and PTP activities
Activation of tyrosine kinase phosphorylation cascades induces different cellular programs that control important events in cell fate determination, such as proliferation, migration and differentiation [149]. Protein phosphorylation cascades are mainly counteracted by the superfamily of protein tyrosine phosphatases (PTPs). Their activities are dependent upon conserved active site cysteine residues, which are sensitive to inhibition by oxidation. Inactivation of PTPs by oxidation is an important mechanism that is initiated by the onset of NADPH oxidase activity in response to growth factor receptor activation [150]. Local bursts of H2O2 thus become prerequisites for several pathways of protein phosphorylation cascades [151, 152]. Both PTPs and protein tyrosine kinases can be redox regulated and also modulated by the Trx system. Examples of such mechanisms shall here be further discussed.
The onset of lipolysis as stimulated by catecholamines or glucagon involves accumulation of cyclic adenosine monophosphate (cAMP), produced by adenylate cyclase, which initiates activation of cyclic AMP-dependent protein kinase (PKA) and triggers protein phosphorylation cascades [153]. Opposing metabolic signals are mediated through the actions of insulin that stimulate activation of lipogenesis. Interestingly, ligand dependent activation of the insulin receptor stimulates H2O2 production, which inactivates PKA by oxidation and formation of an intramolecular disulfide, which in turn is reduced and reactivated by TrxR1 and Trx1 [154]. In this context it is interesting to note that in yeast, the Trx1-dependent Prx1 orthologue Tsa1 is required for the increased lifespan seen upon caloric restriction, through interactions with PKA function [155]. Similar mechanisms of oxidative inactivation also takes occur with protein kinase C (PKC) isoforms, with reduction and reactivation being facilitated by TrxR and Trx [156, 157]. Intriguingly, it has been shown that PKC activity may in turn negatively regulate TrxR expression in human umbilical vein endothelial cells [158]. This might indicate a potential feedback loop of PKC signaling where TrxR and PKC activities counteract and balance each other.
The dual specificity phosphatase and tensin homolog, PTEN, is an important tumor suppressor that regulates cell proliferation, migration and survival. PTEN controls signaling through several growth factor receptors by dephosphorylating phosphatidylinositol 3,4,5-trisphosphate (PIP3), the product of phosphatidyl inositol 3-kinase (PIK). The active site cysteine (Cys124) of PTEN is readily oxidized during cellular responses as triggered by several stimuli, including growth factors, cytokines and hormones [159, 160]. The Trx system has been shown to effectively re-activate oxidized PTEN [129, 160], hence again regulating key intracellular signaling pathways that determine cellular fate.
Intriguingly, differential specificity has been identified for the Trx system with regards to reactivation of different PTPs. PTP1B, in contrast to SHP-2, displays an increase in its oxidation in MEF cells where TrxR1 is deleted [58]. This also correlates with an increased PDGF ligand-dependent phosphorylation of the PDGFRβ receptor site pY579–581, which is regulated by PTP1B. In contrast the phosphorylation site of SHP-2 was unaffected by the cellular TrxR1 status [58]. Differential sensitivity between PTP1B and SHP2 towards the Trx system was also confirmed by in vitro analyses [58, 161]. Recently, it was revealed that TrxR1 is also able to directly reduce a transient oxidation intermediate of PTP1B, potentially its sulfenic acid form [162], thus suggesting multiple modes of regulation of PTP1B by the Trx system. Indeed, TrxR1, Trx1 and interestingly also TRP14 are all potent regulators of PTP1B activity [128, 129, 161, 162]. This agrees well with the finding that the TrxR1 inhibitor auranofin inhibits cellular PTP activity and thereby augments signaling through src tyrosine kinases leading to ERK1/2 phosphorylation [163]. Similarly, TrxR-dependent pathways, but not GSH, can revive the activity of the dual specificity phosphatase Cdc25, which is a PTP that is rapidly oxidized by H2O2 [164].
Most growth factor receptor-linked signaling pathways, including those triggered by insulin, EGF, PDGF, FGF, Wnt, TGF or TNF, depend upon protein phosphorylation cascades. This is exemplified by the transmembrane receptor-tyrosine kinases (RTKs) linked to mitogen-activated protein kinase (MAPK) pathways [149, 165]. These pathways are also known to be redox modulated, i.e., altered in their signaling profiles by redox related events [166]. It is thereby not surprising that these pathways are also mediated by alterations in activities in proteins of the Trx system [61, 167]. More specifically, it seems clear that oxidative inactivation of PTPs is an important prerequisite for phosphorylation cascades to occur [130, 151, 168] and in this context the opposing PTP-activating capacity of the Trx system should be similarly important for inhibition of signaling [128, 129].
AMPK and mTOR
Two proteins that play key roles in control of metabolic pathways in response to diverse growth conditions are the AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR). AMPK is activated in response to low ATP levels and helps to control growth, metabolism, autophagy and cell polarity [169]. Partly downstream of AMPK, mTOR is activated by nutrient-rich conditions and can in many aspects antagonize the cellular functions of AMPK [170]. The two pathways present significant crosstalk with each other and also show tissue- and organ-specific regulatory circuits and activities [171]. Interestingly, several activities in these pathways are modulated also by the Trx system. AMPK itself can be inhibited by oxidation through the formation of a disulfide, which is a substrate of Trx1; thereby Trx1 can be considered an essential cofactor for activation of AMPK [172]. It is in this context noteworthy that Akt is strongly upregulated upon Txnrd1 deletion [37], with Akt inhibiting AMPK and activating mTOR [170, 171]. However, in cardiomyocytes Trx1 was found to preserve the activities of mTOR by directly reducing a disulfide formed in mTOR upon oxidative stress [173]. Thus, it seems clear that the Trx system modulates both AMPK and mTOR, while the outcome of such modulation is likely to be both context- and cell type-dependent.
Regulation of transcription factor activities
Most if not all signaling pathways that alter cellular phenotypes converge upon transcription factors that ultimately regulate cell fate through modulated transcriptional programs. Also in this context the Trx system is important, as it directly regulates the activities of several transcription factors. Many transcription factors are redox regulated, including NF-κB, HIF, Nrf2, Oct-4, β-catenin, Notch, and c-Myc, with most of these being important regulators of development or cellular differentiation [4, 174, 175]. Examples of specific transcription factor that are modulated by the TrxR1 status are many, including the observations that TrxR1 is important for maturation of functional p53 [37, 176–178], may act as an important repressor of Nrf2 activation [10], and in the form of a specific splice variant affects estrogen receptor signaling in the nucleus [46]. The TrxR1 substrate TRP14 is a repressor of NFκB signaling [60, 61], and nuclear Trx1 modulates the activities of AP1 through Ref-1 [179], p21 through p53 [180], and Wnt/beta-catenin signaling through the Trx-like protein nucleoredoxin (NXN) [181, 182]. The Trx system thereby carries out specific roles in the nuclei that directly affect transcriptional responses to cellular signaling pathways, which in turn may have been initiated at the cell membrane or in the cytosol in processes that may also be modulated by the thioredoxin system, as discussed above. The complexity by which the Trx system may affect signaling pathways is thus extensive. Here we shall specifically discuss some of the key transcription factors known to be regulated by the Trx system.
Nrf2
Nrf2 is an important cap-n-collar BZip transcription factor that plays a predominant role in cellular stress responses across all metazoan phyla and in different cell types [183–187]. The mechanisms of Nrf2 activation are of specific interest in relation to TrxR1 modulation. During unstressed conditions the Kelch-like ECH-associated protein-1 (Keap1) in the cytosol targets Nrf2 for ubiqutination and rapid proteasomal degradation. Upon oxidative or electrophilic stress, however, Keap1 becomes modified and cannot release Nrf2 for degradation. As a consequence newly synthesized Nrf2 transits to the nucleus, where it heterodimerizes with small BZip Maf proteins. Nrf2/Maf heterodimers subsequently bind so called “antioxidant response elements” in the regulatory regions of genes encoding important proteins of the GSH and Trx systems as well as Phase-II and -III drug-metabolism enzymes [183–187]. Nrf2 also drives metabolic realignments to support these systems, including modulation of energy metabolism with augmented NADPH production [4, 188, 189]. It is in the context of this review article important to note that a wide range of conditions that lower cytosolic TrxR1 activities, including the use of inhibitors of the enzyme or when its activity is lowered by genetic means, typically leads to very strong Nrf2 activation [190].
Some evidence suggests that Nrf2 might also participate in normal developmental processes. For example, in intestinal stem cells of Drosophila melanogaster, constitutive Nrf2 activation sustains quiescence, in part via up-regulation of genes such as GCLC [191]. As an inducer of antioxidant defenses and detoxification, Nrf2 can also protect normal cells from oxidatively induced mutations that may lead to cancer initiation. However, Nrf2 activation in established cancer cells likely promotes cancer progression, by the resultant increased cellular protection against endogenous oxidative stress, cytotoxic immune responses, and therapeutic oxidative (e.g., in radiation therapy) or cytotoxic/electrophilic (e.g., in chemotherapies) challenges [183, 192]. Moreover, Nrf2 activation can accelerate malignant cell growth [193] and Nrf2 is indeed typically activated in many tumors [183, 194] and is critical for malignant progression of pancreatic, breast, and lung cancers [192, 195, 196].
HIF
The HIF1 transcription factor consists of two subunits, HIF1β (ARNT) and HIF1α [197]. During normoxia, a specific proline residue on HIF1α is hydroxylated by prolyl hydroxylase domain proteins (PHD), allowing the recognition and ubiquitination of HIF1α by the Von Hippel-Lindau (pVHL) protein followed by proteasomal degradation. Under hypoxic conditions (O2 below 3%), PHDs are inactivated by a shift from Fe3+ to Fe2+ in their active center and as a result, HIF1α becomes stabilized and translocates to the nucleus, where it together with HIF1β induces HIF target genes involved in e.g. the adaptation to hypoxia, angiogenesis, glucose transport, survival and invasion [198]. Interestingly, Trx1 strongly stimulates both the levels and transcriptional activities of HIF1 under normoxic as well as hypoxic conditions [199, 200]. Several pharmacological treatments that target the Trx system or redox states of cells hence affect HIF1 signaling, and there is also a significant crosstalk between HIF1, Nrf2 and NF-κB under such conditions [201].
NFkB
Nuclear transcription factor kappa-B (NF-κB) guides inflammatory pathways and cellular responses to inflammation, with intricate mechanisms in control of its activation [4, 202]. Under basal conditions, NF-κB is kept inactive in the cytosol by binding to I B, the inhibitor of NF-κB. Upon activation, a phosphorylation cascade results in degradation of I B and nuclear translocation of NF-κB [4, 202, 203]. Upon constitutively activated Wnt signaling, Rac1-driven H2O2 production by NADPH oxidase activation becomes required for NF-κB activation with initiation of colon tumorigenesis [204]. In the context of the Trx system, the alternative TrxR1 substrate TRP14 was initially discovered as a protein repressing NF-κB signaling through reduction of LC8 cytoplasmic dynein light chain [61]. Also Trx1 counteracts NF-κB but likely by other mechanisms than TRP14 not involving LC8 [205]. TrxR1, but not TrxR2, was shown to facilitate NF-κB signaling [206], but as those experiments were based upon forced overexpression of the selenoprotein it is also possible that formation of SecTRAPs contributed to the observed effects (see discussion above).
p53
The important tumor supressor p53 has long been known to be a redox sensitive transcription factor [207]. It was early shown that Trx1 augments p21 transactivation by p53, at the same time as Trx1 becomes targeted to the nucleus [180]. It was also shown in several studies that an intact TrxR1 activity is required for maturation and support of transcriptional activity of p53 [177, 178, 208, 209]. Since physiological functions of p53 include regulation of cell patterning during development and control of stem cell differentiation [210], it seems clear that effects on cell patterning upon Trx system aberrations may in part be due to modulated p53 function.
STAT3
STAT3 is a protein that mediates the expression of a variety of genes in response to cell stimuli and plays key roles in many cellular processes in cell patterning, moreover linked to NF signaling pathways [211]. With regards to STAT3 signaling in relation to the Trx system it is thus interesting to note that STAT3 activity upregulates TRP14 transcription, encoded by the TXNDC17 gene [212], and that thioredoxin interacting protein (TXNIP), an endogenous Trx1 inhibitor [213–215], can inactivate STAT3 [216]. The STAT3 regulation by H2O2 and its modulation by the Trx system, through several direct and indirect mechanisms via Prx2, Trx1, TRP14 and TXNIP [212, 216, 217], is another illustration of the intricate webs of highly complex signaling pathways that link the Trx system to cellular patterning programs.
Conclusions
Here we have discussed several key studies that indicate that the Trx system has important roles in control of cell patterning, in addition to its more known functions in support of reductive enzymes and antioxidant defense. We have focused upon studies showing effects on cell patterning by genetic manipulation of TrxR1 or TrxR2 expression in diverse model systems, and we have discussed the downstream effects and molecular mechanisms that may explain the observed phenotypes. It is safe to conclude that much of the details in these mechanisms are yet insufficiently understood, but it is just as evident that the Trx system should be considered as an important player in control of cell patterning processes.
Highlights.
The thioredoxin system is important for support of cellular reduction pathways and antioxidant defense
The thioredoxin system is also crucial in redox control of cellular signaling pathways
The major phenotypes triggered by aberrations in the thioredoxin system are altered cell patterning programs
This review discusses the possible molecular mechanisms underlying these observations
Acknowledgments
Funding to the authors from Karolinska Institutet, The Swedish Cancer Society, The Swedish Research Council, The Knut and Alice Wallenberg Foundations and The National Institutes of Health (grant R21AG055022) is thankfully acknowledged. The artists of the cell (File:Anima_cell_notext.svg), mouse (David Liao) and human diagrams (Human_body_diagrams) of Figure 1 as obtained from Wikimedia Commons are also acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312(5782):1882–3. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson S, Prigge JR, Talago EA, Arner ES, Schmidt EE. Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat Commun. 2015;6:6479. doi: 10.1038/ncomms7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deponte M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim Biophys Acta. 2013;1830(5):3217–66. doi: 10.1016/j.bbagen.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Brigelius-Flohe R, Flohe L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal. 2011;15(8):2335–81. doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toppo S, Flohe L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim Biophys Acta. 2009;1790(11):1486–500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780(11):1304–17. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Fernandes AP, Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal. 2004;6(1):63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 8.Lei XG, Zhu JH, Cheng WH, Bao Y, Ho YS, Reddi AR, Holmgren A, Arner ES. Paradoxical Roles of Antioxidant Enzymes: Basic Mechanisms and Health Implications. Physiol Rev. 2016;96(1):307–64. doi: 10.1152/physrev.00010.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye ZW, Zhang J, Townsend DM, Tew KD. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim Biophys Acta. 2015;1850(8):1607–21. doi: 10.1016/j.bbagen.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cebula M, Schmidt EE, Arner ES. TrxR1 as a Potent Regulator of the Nrf2-Keap1 Response System. Antioxid Redox Signal. 2015;23(10):823–53. doi: 10.1089/ars.2015.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groitl B, Jakob U. Thiol-based redox switches. Biochim Biophys Acta. 2014;1844(8):1335–43. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sengupta R, Holmgren A. Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antioxid Redox Signal. 2013;18(3):259–69. doi: 10.1089/ars.2012.4716. [DOI] [PubMed] [Google Scholar]
- 13.Kesarwani P, Murali AK, Al-Khami AA, Mehrotra S. Redox regulation of T-cell function: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2013;18(12):1497–534. doi: 10.1089/ars.2011.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnér ESJ. Focus on mammalian thioredoxin reductases – important selenoproteins with versatile functions. Biochim Biophys Acta. 2009;1790(6):495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 15.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45(5):549–61. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780(11):1325–36. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Lillig CH, Holmgren A. Thioredoxin and related molecules-from biology to health and disease. Antioxid Redox Signal. 2007;9(1):25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 18.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38(12):1543–52. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 19.Rundlöf A-K, Arnér ESJ. Regulation of the mammalian selenoprotein thioredoxin reductase 1 in relation to cellular phenotype, growth and signaling events. Antiox Redox Signal. 2004;6:41–52. doi: 10.1089/152308604771978336. [DOI] [PubMed] [Google Scholar]
- 20.Nordberg J, Arnér ESJ. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 21.Rogers LK, Tamura T, Rogers BJ, Welty SE, Hansen TN, Smith CV. Analyses of glutathione reductase hypomorphic mice indicate a genetic knockout. Toxicol Sci. 2004;82(2):367–73. doi: 10.1093/toxsci/kfh268. [DOI] [PubMed] [Google Scholar]
- 22.Rogers LK, Bates CM, Welty SE, Smith CV. Diquat induces renal proximal tubule injury in glutathione reductase-deficient mice. Toxicol Appl Pharmacol. 2006;217(3):289–98. doi: 10.1016/j.taap.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Prigge JR, Coppo L, Martin SS, Ogata F, Miller CG, Bruschwein MD, Orlicky DJ, Shearn CT, Kundert JA, Lytchier J, Herr AE, Mattsson A, Taylor MP, Gustafsson TN, Arner ESJ, Holmgren A, Schmidt EE. Hepatocyte Hyperproliferation upon Liver-Specific Co-disruption of Thioredoxin-1, Thioredoxin Reductase-1, and Glutathione Reductase. Cell reports. 2017;19(13):2771–2781. doi: 10.1016/j.celrep.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffith OW, Meister A. Glutathione: interorgan translocation, turnover, and metabolism. Proc Natl Acad Sci U S A. 1979;76(11):5606–10. doi: 10.1073/pnas.76.11.5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine) J Biol Chem. 1979;254(16):7558–60. [PubMed] [Google Scholar]
- 26.Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci U S A. 1985;82(14):4668–72. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meister A. New aspects of glutathione biochemistry and transport--selective alteration of glutathione metabolism. Nutrition reviews. 1984;42(12):397–410. doi: 10.1111/j.1753-4887.1984.tb02277.x. [DOI] [PubMed] [Google Scholar]
- 28.Meister A, Griffith OW, Novogrodsky A, Tate SS. New aspects of glutathione metabolism and translocation in mammals. Ciba Found Symp. 1979;(72):135–61. doi: 10.1002/9780470720554.ch9. [DOI] [PubMed] [Google Scholar]
- 29.Jakupoglu C, Przemeck GK, Schneider M, Moreno SG, Mayr N, Hatzopoulos AK, de Angelis MH, Wurst W, Bornkamm GW, Brielmeier M, Conrad M. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol. 2005;25(5):1980–8. doi: 10.1128/MCB.25.5.1980-1988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178(1):179–85. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 31.Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003;23(3):916–22. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conrad M. Transgenic mouse models for the vital selenoenzymes cytosolic thioredoxin reductase, mitochondrial thioredoxin reductase and glutathione peroxidase 4. Biochim Biophys Acta. 2009;1790(11):1575–85. doi: 10.1016/j.bbagen.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Bondareva AA, Capecchi MR, Iverson SV, Li Y, Lopez NI, Lucas O, Merrill GF, Prigge JR, Siders AM, Wakamiya M, Wallin SL, Schmidt EE. Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic Biol Med. 2007;43(6):911–23. doi: 10.1016/j.freeradbiomed.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274(1):305–15. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 35.Weisend CM, Kundert JA, Suvorova ES, Prigge JR, Schmidt EE. Cre activity in fetal albCre mouse hepatocytes: Utility for developmental studies. Genesis. 2009;47(12):789–92. doi: 10.1002/dvg.20568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suvorova ES, Lucas O, Weisend CM, Rollins MF, Merrill GF, Capecchi MR, Schmidt EE. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One. 2009;4(7):e6158. doi: 10.1371/journal.pone.0006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng X, Gimenez-Cassina A, Petrus P, Conrad M, Ryden M, Arner ES. Thioredoxin reductase 1 suppresses adipocyte differentiation and insulin responsiveness. Sci Rep. 2016;6:28080. doi: 10.1038/srep28080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng X, Mandal PK, Kaminskyy VO, Lindqvist A, Conrad M, Arner ES. Sec-containing TrxR1 is essential for self-sufficiency of cells by control of glucose-derived H2O2. Cell death & disease. 2014;5:e1235. doi: 10.1038/cddis.2014.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prigge JR, Eriksson S, Iverson SV, Meade TA, Capecchi MR, Arner ES, Schmidt EE. Hepatocyte DNA replication in growing liver requires either glutathione or a single allele of txnrd1. Free Radic Biol Med. 2012;52(4):803–10. doi: 10.1016/j.freeradbiomed.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rollins MF, van der Heide DM, Weisend CM, Kundert JA, Comstock KM, Suvorova ES, Capecchi MR, Merrill GF, Schmidt EE. Hepatocytes lacking thioredoxin reductase 1 have normal replicative potential during development and regeneration. J Cell Sci. 2010;123(Pt 14):2402–2412. doi: 10.1242/jcs.068106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iverson SV, Eriksson S, Xu J, Prigge JR, Talago EA, Meade TA, Meade ES, Capecchi MR, Arner ES, Schmidt EE. A Txnrd1-dependent metabolic switch alters hepatic lipogenesis, glycogen storage, and detoxification. Free Radic Biol Med. 2013;63:369–80. doi: 10.1016/j.freeradbiomed.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Locy ML, Rogers LK, Prigge JR, Schmidt EE, Arner ES, Tipple TE. Thioredoxin reductase inhibition elicits Nrf2-mediated responses in Clara cells: implications for oxidant-induced lung injury. Antioxid Redox Signal. 2012;17(10):1407–16. doi: 10.1089/ars.2011.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt EE. Interplay between cytosolic disulfide reductase systems and the Nrf2/Keap1 pathway. Biochem Soc Trans. 2015;43(4):632–8. doi: 10.1042/BST20150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su D, Gladyshev VN. Alternative splicing involving the thioredoxin reductase module in mammals: a glutaredoxin-containing thioredoxin reductase 1. Biochemistry. 2004;43(38):12177–88. doi: 10.1021/bi048478t. [DOI] [PubMed] [Google Scholar]
- 45.Rundlof AK, Janard M, Miranda-Vizuete A, Arner ES. Evidence for intriguingly complex transcription of human thioredoxin reductase 1. Free Radic Biol Med. 2004;36(5):641–56. doi: 10.1016/j.freeradbiomed.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 46.Damdimopoulos AE, Miranda-Vizuete A, Treuter E, Gustafsson JÅ, Spyrou G. An alternative splicing variant of the selenoprotein thioredoxin reductase is a modulator of estrogen signaling. J Biol Chem. 2004;279(37):38721–9. doi: 10.1074/jbc.M402753200. [DOI] [PubMed] [Google Scholar]
- 47.Sun QA, Zappacosta F, Factor VM, Wirth PJ, Hatfield DL, Gladyshev VN. Heterogeneity within animal thioredoxin reductases. Evidence for alternative first exon splicing. J Biol Chem. 2001;276:3106–3114. doi: 10.1074/jbc.M004750200. [DOI] [PubMed] [Google Scholar]
- 48.Cebula M, Moolla N, Capovilla A, Arner ES. The rare TXNRD1_v3 (“v3”) splice variant of human thioredoxin reductase 1 protein is targeted to membrane rafts by N-acylation and induces filopodia independently of its redox active site integrity. J Biol Chem. 2013;288(14):10002–11. doi: 10.1074/jbc.M112.445932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Damdimopoulou PE, Miranda-Vizuete A, Arner ES, Gustafsson JA, Damdimopoulos AE. The human thioredoxin reductase-1 splice variant TXNRD1_v3 is an atypical inducer of cytoplasmic filaments and cell membrane filopodia. Biochim Biophys Acta. 2009;1793(10):1588–96. doi: 10.1016/j.bbamcr.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 50.Dammeyer P, Damdimopoulos AE, Nordman T, Jimenez A, Miranda-Vizuete A, Arner ES. Induction of cell membrane protrusions by the N-terminal glutaredoxin domain of a rare splice variant of human thioredoxin reductase 1. J Biol Chem. 2008;283(5):2814–21. doi: 10.1074/jbc.M708939200. [DOI] [PubMed] [Google Scholar]
- 51.Gromer S, Urig S, Becker K. The thioredoxin system--from science to clinic. Med Res Rev. 2004;24(1):40–89. doi: 10.1002/med.10051. [DOI] [PubMed] [Google Scholar]
- 52.Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31(11):1287–312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 53.Nishiyama A, Masutani H, Nakamura H, Nishinaka Y, Yodoi J. Redox regulation by thioredoxin and thioredoxin-binding proteins. IUBMB Life. 2001;52(1–2):29–33. doi: 10.1080/15216540252774739. [DOI] [PubMed] [Google Scholar]
- 54.Nishinaka Y, Masutani H, Nakamura H, Yodoi J. Regulatory roles of thioredoxin in oxidative stress-induced cellular responses. Redox Rep. 2001;6(5):289–95. doi: 10.1179/135100001101536427. [DOI] [PubMed] [Google Scholar]
- 55.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267(20):6102–9. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 56.Doka E, Pader I, Biro A, Johansson K, Cheng Q, Ballago K, Prigge JR, Pastor-Flores D, Dick TP, Schmidt EE, Arner ESJ, Nagy P. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Science Advances. 2016;2(1):e1500968–e1500968. doi: 10.1126/sciadv.1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pader I, Sengupta R, Cebula M, Xu J, Lundberg JO, Holmgren A, Johansson K, Arner ES. Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc Natl Acad Sci U S A. 2014;111(19):6964–9. doi: 10.1073/pnas.1317320111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dagnell M, Frijhoff J, Pader I, Augsten M, Boivin B, Xu J, Mandal PK, Tonks NK, Hellberg C, Conrad M, Arner ES, Ostman A. Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGF-beta receptor tyrosine kinase signaling. Proc Natl Acad Sci U S A. 2013;110(33):13398–403. doi: 10.1073/pnas.1302891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woo JR, Kim SJ, Jeong W, Cho YH, Lee SC, Chung YJ, Rhee SG, Ryu SE. Structural basis of cellular redox regulation by human TRP14. J Biol Chem. 2004;279(46):48120–5. doi: 10.1074/jbc.M407079200. [DOI] [PubMed] [Google Scholar]
- 60.Jeong W, Yoon HW, Lee SR, Rhee SG. Identification and characterization of TRP14, a thioredoxin-related protein of 14 kDa. New insights into the specificity of thioredoxin function. J Biol Chem. 2004;279(5):3142–50. doi: 10.1074/jbc.M307932200. [DOI] [PubMed] [Google Scholar]
- 61.Jeong W, Chang TS, Boja ES, Fales HM, Rhee SG. Roles of TRP14, a thioredoxin-related protein in tumor necrosis factor-alpha signaling pathways. J Biol Chem. 2004;279(5):3151–9. doi: 10.1074/jbc.M307959200. [DOI] [PubMed] [Google Scholar]
- 62.Prigge JR, Eriksson S, Iverson SV, Meade TA, Capecchi MR, Arnér ESJ, Schmidt EE. Hepatocyte DNA replication in growing liver requires either glutathione or a single allele of txnrd1. Free Radic Biol Med. 2012;52:803–810. doi: 10.1016/j.freeradbiomed.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luthman M, Eriksson S, Holmgren A, Thelander L. Glutathione-dependent hydrogen donor system for calf thymus ribonucleoside-diphosphate reductase. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(5):2158–62. doi: 10.1073/pnas.76.5.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zahedi Avval F, Holmgren A. Molecular mechanisms of thioredoxin and glutaredoxin as hydrogen donors for Mammalian s phase ribonucleotide reductase. J Biol Chem. 2009;284(13):8233–40. doi: 10.1074/jbc.M809338200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 66.Holmgren A. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proceedings of the National Academy of Sciences of the United States of America. 1976;73(7):2275–9. doi: 10.1073/pnas.73.7.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holmgren A. Glutathione-dependent synthesis of deoxyribonucleotides. Purification and characterization of glutaredoxin from Escherichia coli. J Biol Chem. 1979;254(9):3664–3671. [PubMed] [Google Scholar]
- 68.Hudemann C, Lonn ME, Godoy JR, Avval FZ, Capani F, Holmgren A, Lillig CH. Identification, Expression Pattern, and Characterization of Mouse Glutaredoxin 2 Isoforms. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2068. [DOI] [PubMed] [Google Scholar]
- 69.Hudemann C, Lonn ME, Godoy JR, Zahedi Avval F, Capani F, Holmgren A, Lillig CH. Identification, expression pattern, and characterization of mouse glutaredoxin 2 isoforms. Antioxid Redox Signal. 2009;11(1):1–14. doi: 10.1089/ars.2008.2068. [DOI] [PubMed] [Google Scholar]
- 70.Johansson C, Lillig CH, Holmgren A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J Biol Chem. 2004;279(9):7537–43. doi: 10.1074/jbc.M312719200. [DOI] [PubMed] [Google Scholar]
- 71.Hansson H-A, Holmgren A, Rozell B, Stemme S. Localization of thioredoxin, thioredoxin reductase and ribonucleotide reductase in cells: immunohistochemical aspects. In: Holmgren A, et al., editors. Thioredoxin and Glutaredoxin Systems: Structure and Function. Raven Press; New York: 1986. pp. 177–187. [Google Scholar]
- 72.Hansson HA, Rozell B, Stemme S, Engstr:om Y, Thelander L, Holmgren A. Different cellular distribution of thioredoxin and subunit M1 of ribonucleotide reductase in rat tissues. Exp Cell Res. 1986;163(2):363–9. doi: 10.1016/0014-4827(86)90067-4. [DOI] [PubMed] [Google Scholar]
- 73.Hansson H-A, Holmgren A, Norstedt G, Rozell B. Changes in the distribution of insulin-like growth factor I, thioredoxin, thioredoxin reductase and ribonucleotide reductase during the development of the retina. Exp Eye Res. 1989;48:411–420. doi: 10.1016/s0014-4835(89)80009-0. [DOI] [PubMed] [Google Scholar]
- 74.Brown KK, Eriksson SE, Arner ES, Hampton MB. Mitochondrial peroxiredoxin 3 is rapidly oxidized in cells treated with isothiocyanates. Free Radic Biol Med. 2008;45(4):494–502. doi: 10.1016/j.freeradbiomed.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 75.Cox AG, Brown KK, Arner ES, Hampton MB. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem Pharmacol. 2008 doi: 10.1016/j.bcp.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 76.Fernando MR, Lechner JM, Lofgren S, Gladyshev VN, Lou MF. Mitochondrial thioltransferase (glutaredoxin 2) has GSH-dependent and thioredoxin reductase-dependent peroxidase activities in vitro and in lens epithelial cells. Faseb J. 2006;20(14):2645–7. doi: 10.1096/fj.06-5919fje. [DOI] [PubMed] [Google Scholar]
- 77.Noh YH, Baek JY, Jeong W, Rhee SG, Chang TS. Sulfiredoxin Translocation into Mitochondria Plays a Crucial Role in Reducing Hyperoxidized Peroxiredoxin III. J Biol Chem. 2009;284(13):8470–7. doi: 10.1074/jbc.M808981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rhee SG, Kil IS. Mitochondrial H2O2 signaling is controlled by the concerted action of peroxiredoxin III and sulfiredoxin: Linking mitochondrial function to circadian rhythm. Free Radic Biol Med. 2016;100:73–80. doi: 10.1016/j.freeradbiomed.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 79.Kil IS, Ryu KW, Lee SK, Kim JY, Chu SY, Kim JH, Park S, Rhee SG. Circadian Oscillation of Sulfiredoxin in the Mitochondria. Mol Cell. 2015;59(4):651–63. doi: 10.1016/j.molcel.2015.06.031. [DOI] [PubMed] [Google Scholar]
- 80.Kil IS, Lee SK, Ryu KW, Woo HA, Hu MC, Bae SH, Rhee SG. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol Cell. 2012;46(5):584–94. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 81.Huang Y, Ainsley JA, Reijmers LG, Jackson FR. Translational profiling of clock cells reveals circadianly synchronized protein synthesis. PLoS Biol. 2013;11(11):e1001703. doi: 10.1371/journal.pbio.1001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Barajas-Lopez JD, Serrato AJ, Cazalis R, Meyer Y, Chueca A, Reichheld JP, Sahrawy M. Circadian regulation of chloroplastic f and m thioredoxins through control of the CCA1 transcription factor. J Exp Bot. 2011;62(6):2039–51. doi: 10.1093/jxb/erq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oien DB, Moskovitz J. Substrates of the methionine sulfoxide reductase system and their physiological relevance. Curr Top Dev Biol. 2008;80:93–133. doi: 10.1016/S0070-2153(07)80003-2. [DOI] [PubMed] [Google Scholar]
- 84.Kim HY, Gladyshev VN. Methionine sulfoxide reductases: selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem J. 2007;407(3):321–9. doi: 10.1042/BJ20070929. [DOI] [PubMed] [Google Scholar]
- 85.Lee BC, Peterfi Z, Hoffmann FW, Moore RE, Kaya A, Avanesov A, Tarrago L, Zhou Y, Weerapana E, Fomenko DE, Hoffmann PR, Gladyshev VN. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol Cell. 2013;51(3):397–404. doi: 10.1016/j.molcel.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vinokur V, Grinberg L, Berenshtein E, Gross M, Moskovitz J, Reznick AZ, Chevion M, Eliashar R. Methionine-centered redox cycle in organs of the aero-digestive tract of young and old rats. Biogerontology. 2008 doi: 10.1007/s10522-008-9152-8. [DOI] [PubMed] [Google Scholar]
- 87.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci USA. 2001;98:12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moskovitz J. Prolonged selenium-deficient diet in MsrA knockout mice causes enhanced oxidative modification to proteins and affects the levels of antioxidant enzymes in a tissue-specific manner. Free Radic Res. 2007;41(2):162–71. doi: 10.1080/10715760600978823. [DOI] [PubMed] [Google Scholar]
- 89.Fixsen SM, Howard MT. Processive selenocysteine incorporation during synthesis of eukaryotic selenoproteins. J Mol Biol. 2010;399(3):385–96. doi: 10.1016/j.jmb.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stoytcheva Z, Tujebajeva RM, Harney JW, Berry MJ. Efficient incorporation of multiple selenocysteines involves an inefficient decoding step serving as a potential translational checkpoint and ribosome bottleneck. Mol Cell Biol. 2006;26(24):9177–84. doi: 10.1128/MCB.00856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu J, Cheng Q, Arner ES. Details in the catalytic mechanism of mammalian thioredoxin reductase 1 revealed using point mutations and juglone-coupled enzyme activities. Free Radic Biol Med. 2016;94:110–20. doi: 10.1016/j.freeradbiomed.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 92.Xu J, Eriksson SE, Cebula M, Sandalova T, Hedstrom E, Pader I, Cheng Q, Myers CR, Antholine WE, Nagy P, Hellman U, Selivanova G, Lindqvist Y, Arner ES. The conserved Trp114 residue of thioredoxin reductase 1 has a redox sensor-like function triggering oligomerization and crosslinking upon oxidative stress related to cell death. Cell Death Dis. 2015;6:e1616. doi: 10.1038/cddis.2014.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheng Q, Antholine WE, Myers JM, Kalyanaraman B, Arnér ESJ, Myers CR. The selenium-independent inherent pro-oxidant NADPH oxidase activity of mammalian thioredoxin reductase and its selenium-dependent direct peroxidase activities. J Biol Chem. 2010;285(28):21708–23. doi: 10.1074/jbc.M110.117259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anestål K, Prast-Nielsen S, Cenas N, Arnér ESJ. Cell Death by SecTRAPs – Thioredoxin Reductase as a Prooxidant Killer of Cells. PLoS ONE. 2008;3(4):e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arner ESJ. Selenium compromised thioredoxin reductase-derived apoptotic proteins (SecTRAPs): Potent killers of cancer cells. Free Radical Research. 2006;40:S51–S51. [Google Scholar]
- 96.Rundlöf A-K, Carlsten M, Arnér ESJ. The core promoter of human thioredoxin reductase 1: Cloning, transcriptional activity and Oct-1, Sp1 and Sp3 binding reveal a housekeeping-type promoter for the ARE-regulated gene. J Biol Chem. 2001;276:30542–30551. doi: 10.1074/jbc.M101452200. [DOI] [PubMed] [Google Scholar]
- 97.Hintze KJ, Wald KA, Zeng H, Jeffery EH, Finley JW. Thioredoxin reductase in human hepatoma cells is transcriptionally regulated by sulforaphane and other electrophiles via an antioxidant response element. J Nutr. 2003;133(9):2721–7. doi: 10.1093/jn/133.9.2721. [DOI] [PubMed] [Google Scholar]
- 98.Leone A, Roca MS, Ciardiello C, Costantini S, Budillon A. Oxidative Stress Gene Expression Profile Correlates with Cancer Patient Poor Prognosis: Identification of Crucial Pathways Might Select Novel Therapeutic Approaches. Oxidative Medicine and Cellular Longevity. 2017;2017:18. doi: 10.1155/2017/2597581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bhatia M, McGrath KL, Di Trapani G, Charoentong P, Shah F, King MM, Clarke FM, Tonissen KF. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016;8:68–78. doi: 10.1016/j.redox.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cadenas C, Franckenstein D, Schmidt M, Gehrmann M, Hermes M, Geppert B, Schormann W, Maccoux LJ, Schug M, Schumann A, Wilhelm C, Freis E, Ickstadt K, Rahnenfuhrer J, Baumbach JI, Sickmann A, Hengstler JG. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010;12(3):R44. doi: 10.1186/bcr2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dammeyer P, Damdimopoulos A, Nordman T, Jimenez A, Miranda-Vizuete A, Arnér ESJ. Induction of cell membrane protrusions by the N-terminal glutaredoxin domain of a rare splice variant of human thioredoxin reductase 1. J Biol Chem. 2007;283(5):2814–2821. doi: 10.1074/jbc.M708939200. [DOI] [PubMed] [Google Scholar]
- 102.Osborne SA, Tonissen KF. Genomic organisation and alternative splicing of mouse and human thioredoxin reductase 1 genes. BMC Genomics. 2001;2(1):10. doi: 10.1186/1471-2164-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cebula M, Moolla N, Capovilla A, Arner ESJ. The Rare TXNRD1_v3 (“v3”) Splice Variant of Human Thioredoxin Reductase 1 Protein Is Targeted to Membrane Rafts by N-Acylation and Induces Filopodia Independently of Its Redox Active Site Integrity. Journal of Biological Chemistry. 2013;288(14):10002–10011. doi: 10.1074/jbc.M112.445932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Damdimopoulou PE, Miranda-Vizuete A, Arner ESJ, Gustafsson J-A, Damdimopoulos AE. The human thioredoxin reductase-1 splice variant TXNRD1_v3 is an atypical inducer of cytoplasmic filaments and cell membrane filopodia. Biochimica Et Biophysica Acta-Molecular Cell Research. 2009;1793(10):1588–1596. doi: 10.1016/j.bbamcr.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 105.Rajalin AM, Micoogullari M, Sies H, Steinbrenner H. Upregulation of the thioredoxin-dependent redox system during differentiation of 3T3-L1 cells to adipocytes. Biol Chem. 2014;395(6):667–77. doi: 10.1515/hsz-2014-0102. [DOI] [PubMed] [Google Scholar]
- 106.Pickering AM, Lehr M, Gendron CM, Pletcher SD, Miller RA. Mitochondrial thioredoxin reductase 2 is elevated in long-lived primate as well as rodent species and extends fly mean lifespan. Aging Cell. 2017;16(4):683–692. doi: 10.1111/acel.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mercatelli N, Fittipaldi S, De Paola E, Dimauro I, Paronetto MP, Jackson MJ, Caporossi D. MiR-23-TrxR1 as a novel molecular axis in skeletal muscle differentiation. Sci Rep. 2017;7(1):7219. doi: 10.1038/s41598-017-07575-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fernandes AP, Capitanio A, Selenius M, Brodin O, Rundlof AK, Bjornstedt M. Expression profiles of thioredoxin family proteins in human lung cancer tissue: correlation with proliferation and differentiation. Histopathology. 2009;55(3):313–20. doi: 10.1111/j.1365-2559.2009.03381.x. [DOI] [PubMed] [Google Scholar]
- 109.Kudin AP, Baron G, Zsurka G, Hampel KG, Elger CE, Grote A, Weber Y, Lerche H, Thiele H, Nuernberg P, Schulz H, Ruppert A-K, Sander T, Cheng Q, Arner ESJ, Schomburg L, Seeher S, Fradejas-Villar N, Schweizer U, Kunz WS. Homozygous mutation in TXNRD1 is associated with genetic generalized epilepsy. Free Radical Biology and Medicine. 2017;106:270–277. doi: 10.1016/j.freeradbiomed.2017.02.040. [DOI] [PubMed] [Google Scholar]
- 110.Sibbing D, Pfeufer A, Perisic T, Mannes AM, Fritz-Wolf K, Unwin S, Sinner MF, Gieger C, Gloeckner CJ, Wichmann HE, Kremmer E, Schafer Z, Walch A, Hinterseer M, Nabauer M, Kaab S, Kastrati A, Schomig A, Meitinger T, Bornkamm GW, Conrad M, von Beckerath N. Mutations in the mitochondrial thioredoxin reductase gene TXNRD2 cause dilated cardiomyopathy. Eur Heart J. 2011;32(9):1121–33. doi: 10.1093/eurheartj/ehq507. [DOI] [PubMed] [Google Scholar]
- 111.Prigge JR, Coppo L, Martin SS, Ogata F, Miller CG, Bruschwein MD, Orlicky DJ, Shearn CT, Kundert JA, Lytchier J, Herr AE, Mattsson A, Taylor MP, Gustafsson TN, Arner ESJ, Holmgren A, Schmidt EE. Hepatocyte Hyperproliferation upon Liver-Specific Co-disruption of Thioredoxin-1, Thioredoxin Reductase-1, and Glutathione Reductase. Cell Reports. 2017;19(13):2771–2781. doi: 10.1016/j.celrep.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eriksson S, Prigge JR, Talago EA, Arner ESJ, Schmidt EE. Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nature Communications. 2015;6 doi: 10.1038/ncomms7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Iverson SV, Eriksson S, Xu J, Prigge JR, Talago EA, Meade TA, Meade ES, Capecchi MR, Arner ESJ, Schmidt EE. A Txnrd1-dependent metabolic switch alters hepatic lipogenesis, glycogen storage, and detoxification. Free Radical Biology and Medicine. 2013;63:369–380. doi: 10.1016/j.freeradbiomed.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Soerensen J, Jakupoglu C, Beck H, Forster H, Schmidt J, Schmahl W, Schweizer U, Conrad M, Brielmeier M. The role of thioredoxin reductases in brain development. PLoS ONE. 2008;3(3):e1813. doi: 10.1371/journal.pone.0001813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F, Schmahl W, Chien KR, Wurst W, Bornkamm GW, Brielmeier M. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol. 2004;24(21):9414–23. doi: 10.1128/MCB.24.21.9414-9423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kiermayer C, Northrup E, Schrewe A, Walch A, de Angelis MH, Schoensiegel F, Zischka H, Prehn C, Adamski J, Bekeredjian R, Ivandic B, Kupatt C, Brielmeier M. Heart-Specific Knockout of the Mitochondrial Thioredoxin Reductase (Txnrd2) Induces Metabolic and Contractile Dysfunction in the Aging Myocardium. J Am Heart Assoc. 2015;4(7) doi: 10.1161/JAHA.115.002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Horstkotte J, Perisic T, Schneider M, Lange P, Schroeder M, Kiermayer C, Hinkel R, Ziegler T, Mandal PK, David R, Schulz S, Schmitt S, Widder J, Sinowatz F, Becker BF, Bauersachs J, Naebauer M, Franz WM, Jeremias I, Brielmeier M, Zischka H, Conrad M, Kupatt C. Mitochondrial thioredoxin reductase is essential for early postischemic myocardial protection. Circulation. 2011;124(25):2892–902. doi: 10.1161/CIRCULATIONAHA.111.059253. [DOI] [PubMed] [Google Scholar]
- 118.Geisberger R, Kiermayer C, Homig C, Conrad M, Schmidt J, Zimber-Strobl U, Brielmeier M. B- and T-cell-specific inactivation of thioredoxin reductase 2 does not impair lymphocyte development and maintenance. Biol Chem. 2007;388(10):1083–90. doi: 10.1515/BC.2007.131. [DOI] [PubMed] [Google Scholar]
- 119.Yan J, Xu J, Fei Y, Jiang C, Zhu W, Han Y, Lu S. TrxR2 deficiencies promote chondrogenic differentiation and induce apoptosis of chondrocytes through mitochondrial reactive oxygen species. Exp Cell Res. 2016;344(1):67–75. doi: 10.1016/j.yexcr.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 120.Kirsch J, Schneider H, Pagel JI, Rehberg M, Singer M, Hellfritsch J, Chillo O, Schubert KM, Qiu J, Pogoda K, Kameritsch P, Uhl B, Pircher J, Deindl E, Muller S, Kirchner T, Pohl U, Conrad M, Beck H. Endothelial Dysfunction, and A Prothrombotic, Proinflammatory Phenotype Is Caused by Loss of Mitochondrial Thioredoxin Reductase in Endothelium. Arterioscler Thromb Vasc Biol. 2016;36(9):1891–9. doi: 10.1161/ATVBAHA.116.307843. [DOI] [PubMed] [Google Scholar]
- 121.Hellfritsch J, Kirsch J, Schneider M, Fluege T, Wortmann M, Frijhoff J, Dagnell M, Fey T, Esposito I, Kolle P, Pogoda K, Angeli JP, Ingold I, Kuhlencordt P, Ostman A, Pohl U, Conrad M, Beck H. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid Redox Signal. 2015;22(11):938–50. doi: 10.1089/ars.2014.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Du Y, Zhang H, Zhang X, Lu J, Holmgren A. Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. J Biol Chem. 2013;288(45):32241–7. doi: 10.1074/jbc.M113.495150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shen X, Burguillos MA, Osman AM, Frijhoff J, Carrillo-Jimenez A, Kanatani S, Augsten M, Saidi D, Rodhe J, Kavanagh E, Rongvaux A, Rraklli V, Nyman U, Holmberg J, Ostman A, Flavell RA, Barragan A, Venero JL, Blomgren K, Joseph B. Glioma-induced inhibition of caspase-3 in microglia promotes a tumor-supportive phenotype. Nat Immunol. 2016;17(11):1282–1290. doi: 10.1038/ni.3545. [DOI] [PubMed] [Google Scholar]
- 124.Hashemy SI, Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J Biol Chem. 2008;283(32):21890–8. doi: 10.1074/jbc.M801047200. [DOI] [PubMed] [Google Scholar]
- 125.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320(5879):1050–4. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tannenbaum SR, White FM. Regulation and specificity of S-nitrosylation and denitrosylation. ACS Chem Biol. 2006;1(10):615–8. doi: 10.1021/cb600439h. [DOI] [PubMed] [Google Scholar]
- 127.Wedmann R, Onderka C, Wei S, Szijarto IA, Miljkovic JL, Mitrovic A, Lange M, Savitsky S, Yadav PK, Torregrossa R, Harrer EG, Harrer T, Ishii I, Gollasch M, Wood ME, Galardon E, Xian M, Whiteman M, Banerjee R, Filipovic MR. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem Sci. 2016;7(5):3414–3426. doi: 10.1039/c5sc04818d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dagnell M, Frijhoff J, Pader I, Augsten M, Boivin B, Xu J, Mandal PK, Tonks NK, Hellberg C, Conrad M, Arnér ESJ, Östman A. Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGFβ-receptor tyrosine kinase signaling. Proc Nat’l Acad Sci USA. 2013;110:13398–13403. doi: 10.1073/pnas.1302891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schwertassek U, Haque A, Krishnan N, Greiner R, Weingarten L, Dick TP, Tonks NK. Reactivation of oxidized PTP1B and PTEN by thioredoxin 1. FEBS J. 2014;281(16):3545–58. doi: 10.1111/febs.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423(6941):769–73. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 131.Lim PL, Liu J, Go ML, Boelsterli UA. The mitochondrial superoxide/thioredoxin-2/Ask1 signaling pathway is critically involved in troglitazone-induced cell injury to human hepatocytes. Toxicol Sci. 2008;101(2):341–9. doi: 10.1093/toxsci/kfm273. [DOI] [PubMed] [Google Scholar]
- 132.Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, Rouis M. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307. doi: 10.1016/j.redox.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim SJ, Lee SM. NLRP3 inflammasome activation in D-galactosamine and lipopolysaccharide-induced acute liver failure: role of heme oxygenase-1. Free Radic Biol Med. 2013;65:997–1004. doi: 10.1016/j.freeradbiomed.2013.08.178. [DOI] [PubMed] [Google Scholar]
- 136.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 137.Mannick JB, Schonhoff CM. Measurement of protein S-nitrosylation during cell signaling. Methods Enzymol. 2008;440:231–42. doi: 10.1016/S0076-6879(07)00814-2. [DOI] [PubMed] [Google Scholar]
- 138.Sun J, Steenbergen C, Murphy E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid Redox Signal. 2006;8(9–10):1693–705. doi: 10.1089/ars.2006.8.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6(2):150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 140.Holmgren A. Biochemistry. SNO removal. Science. 2008;320(5879):1019–20. doi: 10.1126/science.1159246. [DOI] [PubMed] [Google Scholar]
- 141.Mitchell DA, Morton SU, Fernhoff NB, Marletta MA. Thioredoxin is required for S-nitrosation of procaspase-3 and the inhibition of apoptosis in Jurkat cells. Proc Natl Acad Sci U S A. 2007;104(28):11609–14. doi: 10.1073/pnas.0704898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1(3):154–8. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 143.Stoyanovsky DA, Tyurina YY, Tyurin VA, Anand D, Mandavia DN, Gius D, Ivanova J, Pitt B, Billiar TR, Kagan VE. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J Am Chem Soc. 2005;127(45):15815–23. doi: 10.1021/ja0529135. [DOI] [PubMed] [Google Scholar]
- 144.Arnér ESJ, Nordberg J, Holmgren A. Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem Biophys Res Commun. 1996;225:268–274. doi: 10.1006/bbrc.1996.1165. [DOI] [PubMed] [Google Scholar]
- 145.Sengupta R, Ryter SW, Zuckerbraun BS, Tzeng E, Billiar TR, Stoyanovsky DA. Thioredoxin catalyzes the denitrosation of low-molecular mass and protein S-nitrosothiols. Biochemistry. 2007;46(28):8472–83. doi: 10.1021/bi700449x. [DOI] [PubMed] [Google Scholar]
- 146.Nikitovic D, Holmgren A. S-Nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem. 1996;271:19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 147.Schulze PC, Liu H, Choe E, Yoshioka J, Shalev A, Bloch KD, Lee RT. Nitric oxide-dependent suppression of thioredoxin-interacting protein expression enhances thioredoxin activity. Arterioscler Thromb Vasc Biol. 2006;26(12):2666–72. doi: 10.1161/01.ATV.0000248914.21018.f1. [DOI] [PubMed] [Google Scholar]
- 148.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10(10):721–32. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 149.Katz M, Amit I, Yarden Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta. 2007;1773(8):1161–76. doi: 10.1016/j.bbamcr.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63(1):218–42. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 151.Cho SH, Lee CH, Ahn Y, Kim H, Ahn CY, Yang KS, Lee SR. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS lett. 2004;560(1–3):7–13. doi: 10.1016/s0014-5793(04)00112-7. [DOI] [PubMed] [Google Scholar]
- 152.Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 153.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr. 2007;27:79–101. doi: 10.1146/annurev.nutr.27.061406.093734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.de Pina MZ, Vazquez-Meza H, Pardo JP, Rendon JL, Villalobos-Molina R, Riveros-Rosas H, Pina E. Signaling the signal, cyclic AMP-dependent protein kinase inhibition by insulin-formed H2O2 and reactivation by thioredoxin. J Biol Chem. 2008;283(18):12373–86. doi: 10.1074/jbc.M706832200. [DOI] [PubMed] [Google Scholar]
- 155.Molin M, Yang J, Hanzen S, Toledano MB, Labarre J, Nystrom T. Life span extension and H(2)O(2) resistance elicited by caloric restriction require the peroxiredoxin Tsa1 in Saccharomyces cerevisiae. Mol Cell. 2011;43(5):823–33. doi: 10.1016/j.molcel.2011.07.027. [DOI] [PubMed] [Google Scholar]
- 156.Gundimeda U, Schiffman JE, Chhabra D, Wong J, Wu A, Gopalakrishna R. Locally generated methylseleninic acid induces specific inactivation of protein kinase C isoenzymes: Relevance to selenium-induced apoptosis in prostate cancer cells. J Biol Chem. 2008 doi: 10.1074/jbc.M807007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kahlos K, Zhang J, Block ER, Patel JM. Thioredoxin restores nitric oxide-induced inhibition of protein kinase C activity in lung endothelial cells. Mol Cell Biochem. 2003;254(1–2):47–54. doi: 10.1023/a:1027380828645. [DOI] [PubMed] [Google Scholar]
- 158.Anema SM, Walker SW, Howie AF, Arthur JR, Nicol F, Beckett GJ. Thioredoxin reductase is the major selenoprotein expressed in human umbilical-vein endothelial cells and is regulated by protein kinase C. Biochem J. 1999;342:111–117. [PMC free article] [PubMed] [Google Scholar]
- 159.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101(47):16419–24. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277(23):20336–42. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 161.Parsons ZD, Gates KS. Thiol-dependent recovery of catalytic activity from oxidized protein tyrosine phosphatases. Biochemistry. 2013;52(37):6412–23. doi: 10.1021/bi400451m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dagnell M, Pace PE, Cheng Q, Frijhoff J, Ostman A, Arner ESJ, Hampton MB, Winterbourn CC. Thioredoxin reductase 1 and NADPH directly protect protein tyrosine phosphatase 1B from inactivation during H2O2 exposure. J Biol Chem. 2017;292(35):14371–14380. doi: 10.1074/jbc.M117.793745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shinozaki Y, Koizumi S, Ohno Y, Nagao T, Inoue K. Extracellular ATP counteracts the ERK1/2-mediated death-promoting signaling cascades in astrocytes. Glia. 2006;54(6):606–18. doi: 10.1002/glia.20408. [DOI] [PubMed] [Google Scholar]
- 164.Sohn J, Rudolph J. Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry. 2003;42(34):10060–70. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 165.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80(2):179–85. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 166.Liu Y, He C. A review of redox signaling and the control of MAP kinase pathway in plants. Redox Biol. 2016;11:192–204. doi: 10.1016/j.redox.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Latimer HR, Veal EA. Peroxiredoxins in Regulation of MAPK Signalling Pathways; Sensors and Barriers to Signal Transduction. Mol Cells. 2016;39(1):40–5. doi: 10.14348/molcells.2016.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ostman A, Frijhoff J, Sandin A, Bohmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150(4):345–56. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 169.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400. doi: 10.1146/annurev-pharmtox-010611-134537. [DOI] [PubMed] [Google Scholar]
- 171.Xu J, Ji J, Yan XH. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr. 2012;52(5):373–81. doi: 10.1080/10408398.2010.500245. [DOI] [PubMed] [Google Scholar]
- 172.Shao D, Oka S, Liu T, Zhai P, Ago T, Sciarretta S, Li H, Sadoshima J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014;19(2):232–45. doi: 10.1016/j.cmet.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Oka SI, Hirata T, Suzuki W, Naito D, Chen Y, Chin A, Yaginuma H, Saito T, Nagarajan N, Zhai P, Bhat S, Schesing K, Shao D, Hirabayashi Y, Yodoi J, Sciarretta S, Sadoshima J. Thioredoxin-1 maintains mTOR function during oxidative stress in cardiomyocytes. J Biol Chem. 2017 doi: 10.1074/jbc.M117.807735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ufer C, Wang CC, Borchert A, Heydeck D, Kuhn H. Redox control in mammalian embryo development. Antioxid Redox Signal. 2010;13(6):833–75. doi: 10.1089/ars.2009.3044. [DOI] [PubMed] [Google Scholar]
- 175.Ding S, Li C, Cheng N, Cui X, Xu X, Zhou G. Redox Regulation in Cancer Stem Cells. Oxidative medicine and cellular longevity. 2015;2015:750798. doi: 10.1155/2015/750798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Maillet A, Pervaiz S. Redox regulation of p53, redox effectors regulated by p53: a subtle balance. Antioxid Redox Signal. 2011;16(11):1285–94. doi: 10.1089/ars.2011.4434. [DOI] [PubMed] [Google Scholar]
- 177.Cassidy PB, Edes K, Nelson CC, Parsawar K, Fitzpatrick FA, Moos PJ. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis. 2006;27(12):2538–49. doi: 10.1093/carcin/bgl111. [DOI] [PubMed] [Google Scholar]
- 178.Moos PJ, Edes K, Cassidy P, Massuda E, Fitzpatrick FA. Electrophilic prostaglandins and lipid aldehydes repress redox-sensitive transcription factors p53 and hypoxia-inducible factor by impairing the selenoprotein thioredoxin reductase. J Biol Chem. 2003;278(2):745–50. doi: 10.1074/jbc.M211134200. [DOI] [PubMed] [Google Scholar]
- 179.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci USA. 1997;(94):3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ueno M, Masutani H, Arai RJ, Yamauchi A, Hirota K, Sakai T, Inamoto T, Yamaoka Y, Yodoi J, Nikaido T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J Biol Chem. 1999;274:35809–35815. doi: 10.1074/jbc.274.50.35809. [DOI] [PubMed] [Google Scholar]
- 181.Funato Y, Miki H. Nucleoredoxin, a novel thioredoxin family member involved in cell growth and differentiation. Antioxid Redox Signal. 2007;9(8):1035–57. doi: 10.1089/ars.2007.1550. [DOI] [PubMed] [Google Scholar]
- 182.Kurooka H, Kato K, Minoguchi S, Takahashi Y, Ikeda J, Habu S, Osawa N, Buchberg AM, Moriwaki K, Shisa H, Honjo T. Cloning and characterization of the nucleoredoxin gene that encodes a novel nuclear protein related to thioredoxin. Genomics. 1997;39:331–339. doi: 10.1006/geno.1996.4493. [DOI] [PubMed] [Google Scholar]
- 183.Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. 2012;2:200. doi: 10.3389/fonc.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Higgins LG, Hayes JD. The cap’n’collar transcription factor Nrf2 mediates both intrinsic resistance to environmental stressors and an adaptive response elicited by chemopreventive agents that determines susceptibility to electrophilic xenobiotics. Chem Biol Interact. 2011;192(1–2):37–45. doi: 10.1016/j.cbi.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 185.Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008;74(13):1526–39. doi: 10.1055/s-0028-1088302. [DOI] [PubMed] [Google Scholar]
- 186.Osburn WO, Kensler TW. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat Res. 2008;659(1–2):31–9. doi: 10.1016/j.mrrev.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38(4):769–89. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 188.Arner ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochimica et biophysica acta. 2009;1790(6):495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 189.Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicological sciences: an official journal of the Society of Toxicology. 2004;82(1):308–17. doi: 10.1093/toxsci/kfh231. [DOI] [PubMed] [Google Scholar]
- 190.Cebula M, Schmidt EE, Arner ESJ. TrxR1 as a Potent Regulator of the Nrf2-Keap1 Response System. Antioxidants & Redox Signaling. 2015;23(10):823–853. doi: 10.1089/ars.2015.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox Regulation by Keap1 and Nrf2 Controls Intestinal Stem Cell Proliferation in Drosophila. Cell stem cell. 2011;8(2):188–199. doi: 10.1016/j.stem.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Ohlund D, Wright K, Filippini D, Lee EJ, Da Silva B, Schoepfer C, Wilkinson JE, Buscaglia JM, DeNicola GM, Tiriac H, Hammell M, Crawford HC, Schmidt EE, Thompson CB, Pappin DJ, Sonenberg N, Tuveson DA. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell. 2016;166(4):963–976. doi: 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Satoh H, Moriguchi T, Saigusa D, Baird L, Yu L, Rokutan H, Igarashi K, Ebina M, Shibata T, Yamamoto M. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer research. 2016;76(10):3088–3096. doi: 10.1158/0008-5472.CAN-15-1584. [DOI] [PubMed] [Google Scholar]
- 194.Ganan-Gomez I, Wei Y, Yang H, Boyano-Adanez MC, Garcia-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med. 2013;65:750–64. doi: 10.1016/j.freeradbiomed.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 195.Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez-Rivera FJ, Subbaraj L, Martinez B, Bronson RT, Prigge JR, Schmidt EE, Thomas CJ, Goparaju C, Davies A, Dolgalev I, Heguy A, Allaj V, Poirier JT, Moreira AL, Rudin CM, Pass HI, Vander Heiden MG, Jacks T, Papagiannakopoulos T. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23(11):1362–1368. doi: 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z, Li WY, Cescon DW, Li YT, Molyneux S, Penrod N, Lupien M, Schmidt EE, Stambolic V, Gauthier ML, Mak TW. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc Natl Acad Sci U S A. 2014;111(12):4472–7. doi: 10.1073/pnas.1324136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang GL, Semenza GL. Purification and Characterization of Hypoxia-Inducible Factor-1. Journal of Biological Chemistry. 1995;270(3):1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 198.Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J. 2012;31(11):2448–60. doi: 10.1038/emboj.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Welsh SJ, Bellamy WT, Briehl MM, Powis G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002;62(17):5089–95. [PubMed] [Google Scholar]
- 200.Kim WJ, Cho H, Lee SW, Kim YJ, Kim KW. Antisense-thioredoxin inhibits angiogenesis via pVHL-mediated hypoxia-inducible factor-1alpha degradation. Int J Oncol. 2005;26(4):1049–52. [PubMed] [Google Scholar]
- 201.Johansson K, Cebula M, Rengby O, Dreij K, Carlstrom KE, Sigmundsson K, Piehl F, Arner ES. Cross Talk in HEK293 Cells Between Nrf2, HIF, and NF-kappaB Activities upon Challenges with Redox Therapeutics Characterized with Single-Cell Resolution. Antioxid Redox Signal. 2015 Nov 11; doi: 10.1089/ars.2015.6419. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18(18):2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 204.Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D, Timpson P, Vidal M, Murray GI, Greten FR, Anderson KI, Sansom OJ. ROS Production and NF-kappa B Activation Triggered by RAC1 Facilitate WNT-Driven Intestinal Stem Cell Proliferation and Colorectal Cancer Initiation. Cell stem cell. 2013;12(6):761–773. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wu XL, Li X, Li Y, Kong LP, Fang JL, Zhou XS, Li M, Jia JJ, Bai J. The overexpression of Thioredoxin-1 suppressing inflammation induced by methamphetamine in spleen. Drug Alcohol Depend. 2016;159:66–71. doi: 10.1016/j.drugalcdep.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 206.Sakurai A, Yuasa K, Shoji Y, Himeno S, Tsujimoto M, Kunimoto M, Imura N, Hara S. Overexpression of thioredoxin reductase 1 regulates NF-kappa B activation. J Cell Physiol. 2004;198(1):22–30. doi: 10.1002/jcp.10377. [DOI] [PubMed] [Google Scholar]
- 207.Sun Y, Oberley LW. Redox regulation of transcriptional activators. Free Radic Biol Med. 1996;21(3):335–48. doi: 10.1016/0891-5849(96)00109-8. [DOI] [PubMed] [Google Scholar]
- 208.Ma X, Hu J, Lindner DJ, Kalvakolanu DV. Mutational analysis of human thioredoxin reductase 1. Effects on p53-mediated gene expression and interferon and retinoic acid-induced cell death. J Biol Chem. 2002;277:22460–22468. doi: 10.1074/jbc.M202286200. [DOI] [PubMed] [Google Scholar]
- 209.Hu J, Ma X, Lindner DJ, Karra S, Hofmann ER, Reddy SP, Kalvakolanu DV. Modulation of p53 dependent gene expression and cell death through thioredoxin-thioredoxin reductase by the Interferon-Retinoid combination. Oncogene. 2001;20:4235–4248. doi: 10.1038/sj.onc.1204585. [DOI] [PubMed] [Google Scholar]
- 210.Molchadsky A, Rivlin N, Brosh R, Rotter V, Sarig R. p53 is balancing development, differentiation and de-differentiation to assure cancer prevention. Carcinogenesis. 2010;31(9):1501–8. doi: 10.1093/carcin/bgq101. [DOI] [PubMed] [Google Scholar]
- 211.Fan Y, Mao R, Yang J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013;4(3):176–85. doi: 10.1007/s13238-013-2084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhang Z, Wang A, Li H, Zhi H, Lu F. STAT3-dependent TXNDC17 expression mediates Taxol resistance through inducing autophagy in human colorectal cancer cells. Gene. 2016;584(1):75–82. doi: 10.1016/j.gene.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 213.Chong CR, Chan WP, Nguyen TH, Liu S, Procter NE, Ngo DT, Sverdlov AL, Chirkov YY, Horowitz JD. Thioredoxin-interacting protein: pathophysiology and emerging pharmacotherapeutics in cardiovascular disease and diabetes. Cardiovasc Drugs Ther. 2014;28(4):347–60. doi: 10.1007/s10557-014-6538-5. [DOI] [PubMed] [Google Scholar]
- 214.Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H, Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:514. doi: 10.3389/fimmu.2013.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Spindel ON, World C, Berk BC. Thioredoxin interacting protein: redox dependent and independent regulatory mechanisms. Antioxid Redox Signal. 2012;16(6):587–96. doi: 10.1089/ars.2011.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Xu G, Chen J, Jing G, Shalev A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat Med. 2013;19(9):1141–6. doi: 10.1038/nm.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, Dick TP. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol. 2015;11(1):64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 218.Sakurai A, Nishimoto M, Himeno S, Imura N, Tsujimoto M, Kunimoto M, Hara S. Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. J Cell Physiol. 2005;203(3):529–37. doi: 10.1002/jcp.20246. [DOI] [PubMed] [Google Scholar]
- 219.Nalvarte I, Damdimopoulos AE, Nystom C, Nordman T, Miranda-Vizuete A, Olsson JM, Eriksson L, Bjornstedt M, Arner ES, Spyrou G. Overexpression of enzymatically active human cytosolic and mitochondrial thioredoxin reductase in HEK-293 cells. Effect on cell growth and differentiation. J Biol Chem. 2004;279(52):54510–7. doi: 10.1074/jbc.M408494200. [DOI] [PubMed] [Google Scholar]
- 220.Patenaude A, Ven Murthy MR, Mirault ME. Mitochondrial thioredoxin system: effects of TrxR2 overexpression on redox balance, cell growtha, and apoptosis. J Biol Chem. 2004;279(26):27302–14. doi: 10.1074/jbc.M402496200. [DOI] [PubMed] [Google Scholar]
- 221.Chang EY, Son SK, Ko HS, Baek SH, Kim JH, Kim JR. Induction of apoptosis by the overexpression of an alternative splicing variant of mitochondrial thioredoxin reductase. Free Radic Biol Med. 2005;39(12):1666–75. doi: 10.1016/j.freeradbiomed.2005.08.008. [DOI] [PubMed] [Google Scholar]