Abstract
Low and disturbed blood flow drives the progression of arterial diseases including atherosclerosis and aneurysms. The endothelial response to flow and its interactions with recruited platelets and leukocytes determine disease progression. Here, we report widespread changes in alternative splicing of pre-mRNA in the flow-activated murine arterial endothelium in vivo. Alternative splicing was suppressed by depletion of platelets and macrophages recruited to the arterial endothelium under low and disturbed flow. Binding motifs for the Rbfox-family are enriched adjacent to many of the regulated exons. Endothelial deletion of Rbfox2, the only family member expressed in arterial endothelium, suppresses a subset of the changes in transcription and RNA splicing induced by low flow. Our data reveal an alternative splicing program activated by Rbfox2 in the endothelium on recruitment of platelets and macrophages and demonstrate its relevance in transcriptional responses during flow-driven vascular inflammation.
Research organism: Mouse
Introduction
Exposure of arterial endothelium to low and disturbed flow initiates a process of endothelial activation, resulting in increased expression of adhesion molecules and the rolling, adhesion and extravasation of leukocytes (Gimbrone and García-Cardeña, 2013; Ley et al., 2007). The level of shear stress to which endothelium is exposed is one of the best predictors of atherosclerosis and growth and rupture of aneurysms (Boussel et al., 2008; Cheng et al., 2006; Cicha et al., 2011; Xiang et al., 2011). Experiments with isolated endothelial cells exposed to altered flow in vitro revealed that increased laminar flow alone was sufficient to suppress endothelial apoptosis and expression of endothelial leukocyte recruitment molecules such as Icam1 (Nagel et al., 1994). Subsequent work revealed transcriptional programs induced by steady flow and suppressed by low and disturbed flow in vitro and in vivo (Dai et al., 2004). Many of these programs could be recapitulated by Erk5 activation and increased expression of the transcription factor Klf2 (Dekker et al., 2006; Lin et al., 2005; Parmar et al., 2006). Interfering with these programs increased markers of endothelial dysfunction and disease progression in atherosclerosis (Dekker et al., 2006; Lin et al., 2005; Parmar et al., 2006). Thus, exposure of arterial endothelium to low flow initiates a program of endothelial dysfunction characterized by transcriptional changes and alterations in the expression of leukocyte recruitment molecules.
Most work to date has focused on the primary endothelial response to low flow, rather than the chronic interactions with immune cells under low flow in vivo. However, chronic inflammation contributes to a wide variety of dangerous cardiovascular lesions, including the erosion and rupture of atherosclerotic plaques and aneurysms. Not surprisingly, inflammation is a clinical target in atherosclerosis (Ridker et al., 2012) and aortic aneurysm (Davis et al., 2014). The adaptive immune response is also being targeted, with good efficacy in pre-clinical models of atherosclerosis (Kimura et al., 2015). Historical perspectives on inflammation have typically focused on the leukocytes recruited in large numbers from the blood to the tissues, however, the endothelial lining of arterial vessels plays a critical role as a regulator of multiple steps in the cascade, including effects on immune cell recruitment, trafficking into tissues, and local activity (Carman and Martinelli, 2015; Galkina and Ley, 2009; Pober and Sessa, 2007). The endothelium, as the interface between recruited immune cells and tissues, and as a local regulator of the inflammatory process, is an attractive target for the regulation of local inflammation, potentially avoiding complications of systemic immunosuppressive agents.
Recently, we discovered that, in a chronic in vivo model of low flow, changes in alternative splicing of the extracellular matrix protein fibronectin (FN) prevented hemorrhagic rupture of the intima (Murphy and Hynes, 2014). Through alternative splicing, a single pre-mRNA produced from a transcribed gene can be spliced in different ways to generate multiple, sometimes hundreds, of various isoforms (Nilsen and Graveley, 2010; Wang and Burge, 2008). These new isoforms often lend completely novel interactions to proteins, resulting in interaction profiles often as divergent from the ‘canonical’ isoform as from other unrelated proteins (Yang et al., 2016). Interestingly, the splicing changes we observed in Fn, resulting in the incorporation of alternative exons EIIIA and EIIIB, could be blocked by the depletion of macrophages, which are recruited to the arterial endothelium under low flow (Murphy and Hynes, 2014). Our results suggested that regulation of alternative splicing might play an important role in the endothelial interaction with recruited immune cells and regulation of chronic arterial inflammation.
Here, we test the possibility that alternative splicing is broadly regulated in the arterial endothelium by low and disturbed flow, and hypothesize that alternative splicing events co-regulated with alternatively spliced Fn exons EIIIA and EIIIB may be important in coordinating the response of the arterial endothelium to low flow.
Results
Low flow induces a program of alternative splicing in the arterial intima
To determine the extent of alternative splicing changes in the arterial intima under low-flow conditions, we prepared pools of intimal RNA from the carotid arteries of mice exposed to low flow for 48 hr (Figure 1A). Control pools of RNA were prepared from mice subjected to a sham operation, or from the contralateral arteries of ligated mice, which were exposed to high flow. We detected 4766 differentially expressed genes between the low-flow carotid and the contralateral high-flow carotid (Padj <0.05 by DEseq2, N = 3 per group), which overlapped and extended previous experiments in this system and in vitro (Dai et al., 2004; Ni et al., 2010) (Figure 1B). We observed excellent correlation in Log2 fold-change between biological replicates (Pearson correlation of 0.82 for all genes, and 0.98 for genes found to be significantly regulated by low flow). Low flow, rather than high flow in the contralateral artery, was responsible for the majority of changes in mRNA levels, relative to the normal-flow, sham-operated controls (Figure 1—figure supplement 1). The flow-responsive signature included reduction in expression of the canonical flow-responsive genes Klf2 and Klf4 and we also detected an increase in signal from innate immune cells (CD45, F4/80, CD11b, Cxcr2; Figure 1—figure supplement 2), suggesting that such cells are recruited as early as 48 hr after the change in flow. However, transcripts from recruited or other contaminating cells represented a small portion of the total signal. This was assessed by the amount of eGFP (endothelial) mRNA copies versus tdTomato (all other cells) in Cdh5(PAC)Cre-ERT2; mT/mG mice, in which eGFP or tdTomato are driven from the same constitutive actin promoter in a cell-specific manner (Figure 1—figure supplement 3). By this measure, contaminating transcripts represented <1.5% of RNA analyzed.
Figure 1. Exposure of the arterial endothelium to low and disturbed flow induces a program of alternative splicing.
RNA from the arterial endothelium was isolated 48 hr after partial carotid ligation; from the low-flow side, the high-flow side, or sham-operated vessels. (A) Outline of splicing analysis. Three pools of mRNA from each condition were isolated by polyA and sequenced. (B) Plot showing the consistency of changes in gene expression in two independent biological comparisons of low-flow versus high-flow isolations. (C) Number of RNA splicing events of each category detected by MISO analysis as significantly different in two independent biological comparisons. Events were drawn from annotated databases (Burge, Tian) or from custom annotation of mapped splice junctions (novel). (D) Plot showing the changes in skipped-exon inclusion level (deltaPsi) between low-flow and high-flow isolations. The plot also indicates changes in transcription, highlighting exons in genes with a change in FPKM of more than 2-fold up or down. (E) Processes enriched in the genes with regulated skipped exons, relative to the entire set of genes expressed in the tissue with annotated skipped exons which were not significantly regulated. Padj = Adjusted P-value, BF = Bayes Factor; FPKM = Fragments Per Kilobase of transcript per Million mapped reads; CCDS = Consensus Coding DNA Sequence.

Figure 1—figure supplement 1. Gene expression changes between low-flow and high-flow intima are mainly due to low flow.

Figure 1—figure supplement 2. Hematopoietic cell recruitment within 48 hr of low and disturbed flow.

Figure 1—figure supplement 3. Enrichment of endothelial RNA demonstrated by lineage markers in mT/mG mice.

Figure 1—figure supplement 4. Splicing changes are primarily induced by low flow rather than high flow.

Figure 1—figure supplement 5. Arterial cells cultured in vitro replicate splicing changes observed under low and disturbed flow in vivo.

Having established endothelial enriched RNA pools from the various flow conditions, we then asked whether alternative splicing in intimal endothelial cells was regulated by flow. We assessed levels of alternative exon inclusion, expressed as ‘percentage spliced in’ (Psi), in each condition and their differences using the MISO Bayesian statistical framework (Katz et al., 2010). We examined annotated events as well as novel events (see Materials and methods). Setting a threshold of significance at Bayes factor five in two biologically independent comparisons of low flow to high flow, we identified 768 regulated splicing events between known exons (skipped or mutually exclusive exons, retained introns, or alternative 5’ or 3’ splice sites) and over a thousand regulated changes to the last exon or 3’UTR (Figure 1C). As with the transcript level changes, the vast majority of the splicing changes were induced by low flow, rather than high flow, in comparison with sham-operated vessels (Figure 1—figure supplement 4). Approximately, 90% of the splicing changes observed in vivo were recapitulated (at least 50% in the same direction) in purified endothelial cells in vitro, supporting the endothelial regulation of these splicing events (Figure 1—figure supplement 5). It is not clear whether the 10% of events not similarly regulated in vitro reflect differences in the in vitro system or transcript variants from the ~1.5% contaminating mRNA.
Because the mechanisms of regulation of skipped exons are best understood, and because we are interested in alternative splicing events similar to the skipped exons in Fn, we focused on this subclass of alternative splicing events (Murphy and Hynes, 2014). Changes in skipped exons were consistent between completely independent biological sets, and not obviously correlated with increased or decreased abundance in the transcriptional levels of these genes between conditions (Figure 1D, each point indicates a regulated splicing event, colored points indicate events in genes with altered transcript levels). The majority of the skipped exons were as strongly conserved evolutionarily as constitutive exons in placental mammals, and much more so than introns (data not shown). Genes with regulated skipped exons had annotated functions in several categories (Figure 1E). Thus, altered flow results in changes in RNA splicing of exons coincident with, but independent of, transcriptional changes.
Platelets are required for regulation of a subset of flow-regulated skipped exons
We previously observed that monocyte recruitment was required for increased inclusion of Fn-EIIIA and EIIIB exons after 48 hr of low flow (Murphy and Hynes, 2014). Platelets and granulocytes are also recruited to arterial endothelium in regions of low and disturbed flow (Chèvre et al., 2014; Huo et al., 2003; Massberg et al., 2002). We depleted each of these recruited cell types using clodronate liposomes for macrophages (Murphy and Hynes, 2014), anti-GPIbα for platelets and anti-Gr1 for neutrophils (Labelle et al., 2014). Platelets were essential for the recruitment of both macrophages and neutrophils in our system (Figure 2—figure supplement 1). Platelet depletion resulted in suppression of Fn-EIIIA and –EIIIB relative to control treatments, and in widespread changes in a large number of other flow-regulated skipped exons (Figure 2—figure supplement 2). For the platelet-regulated skipped exons (about 1/3 of all flow-regulated skipped exons), platelet depletion causes low-flow arteries to cluster with high-flow arteries in their splicing profile rather than with IgG or untreated low-flow arteries (Figure 2A). Thus, platelets are necessary for the regulation of many of the low-flow regulated skipped exons we observed.
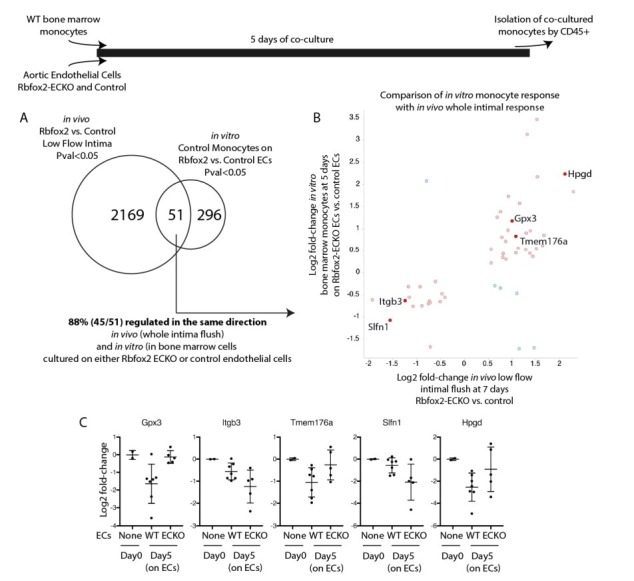
Figure 2. Platelets and macrophages regulate a subset of flow-regulated skipped-exon events.
(A) Effects of platelet depletion on splicing patterns in vivo (see also 2–2). Clustered heat map showing the change in skipped-exon inclusion frequency relative to high-flow contralateral arteries in biological replicates of untreated arteries (from data set created in Figure 1) or platelet-depleted (anti-GPIbα) or IgG control-treated (IgG) arteries. The data shown are for the platelet-dependent subset of all flow-regulated skipped exons (SE; 80/292). IgG and anti-GPIbα (low flow) are relative to average IgG (high flow). No antibody treatment in vitro and in vivo low flow are relative to average in vivo high flow. (B) Effects of platelet, macrophage and plasma addition to endothelial cells in vitro. (B1) Bar graphs showing in vitro changes in EIIIA inclusion frequency and Cd68 macrophage marker expression with the different treatments of conditionally immortalized aortic endothelial cells, relative to in vivo low-flow and high-flow samples. (B2) Plot showing the in vitro regulation of platelet-regulated skipped-exon events (40/80), in isolated and conditionally immortalized aortic endothelial cells. (B3) Clustered heat map, showing the change in skipped exon inclusion frequency in in vivo biological replicates of low-flow arteries with or without platelet depletion (in vivo low-flow+ IgG control, +GPIba) or conditionally immortalized aortic endothelial cells with the addition of the indicated cells or 10% plasma.

Figure 2—figure supplement 1. Hematopoietic cell depletion in aortic intima 48 hr after induction of low and disturbed flow.

Figure 2—figure supplement 2. In vivo depletions of individual hematopoietic cell populations and their effects on Fn-EIIIA and –EIIIB inclusion.

Figure 2—figure supplement 3. Isolation and conditional immortalization of aortic endothelial cells.

In vivo experiments were unable to discriminate between the effects of platelets and macrophages on the splicing response of endothelium, since platelet depletion suppressed macrophage recruitment. To address more directly the specific requirement for each blood cell type, we prepared a murine aortic endothelial cell line for in vitro co-culture experiments (Figure 2—figure supplement 3). We first examined EIIIA inclusion levels (Figure 2B1). Cultured cells in static conditions had higher baseline levels of EIIIA than high-flow arteries in vivo (compare bars 2 and 3). Co-culture with monocyte-derived macrophages, at similar stoichiometric ratios with the endothelial cells to those observed in vivo in low-flow arteries, did not further increase EIIIA inclusion. However, both platelets and plasma independently induced increased EIIIA inclusion, and exerted combinatorial effects, resulting in inclusion levels approximately 6-fold higher than in vivo levels when all three were added [EIIIA (from 15% to 80%) and EIIIB (from 11% to 60%)]. A broader examination of splicing changes, by RNA-seq, revealed that half (40 of 80) of the splicing events regulated by platelets in vivo were similarly regulated by a combination of platelets, macrophages and plasma in vitro (Figure 2B2). For this set of events, the addition of plasma, macrophages and platelets to cultured endothelial cells induced a low-flow-like change in splicing but the individual components - platelets, macrophages or plasma proteins - were unable to entirely recapitulate the potent effects of their combination (Figure 2B3). Thus, platelets alone were insufficient to induce low-flow regulated skipped exons, despite static in vitro conditions, and required both macrophages and plasma for maximal effect.
Together our results suggest that recruitment of both platelets and macrophages to the arterial endothelium is required for the flow-responsive splicing pattern in a large set of genes, including the Fn-EIIIA and EIIIB exons.
Binding motif for the splicing factor Rbfox2 is enriched in regulated vascular splicing events
We were particularly interested in the set of skipped exons induced in the endothelium in a platelet and macrophage-dependent manner, along with Fn-EIIIA and –EIIIB, and asked whether there might be a common upstream regulatory factor. Although analysis of transcriptional levels did not reveal obvious candidate splicing factors (Figure 3—figure supplement 1), the identity of relevant splicing regulators can in some cases be deduced by looking for enrichment of their RNA-binding motifs adjacent to regulated exons, where they typically bind to exert their effects (Shapiro et al., 2011).
To take an unbiased approach to the identification of regulatory splicing factors by flanking motifs, we developed an analysis pipeline using the ranking algorithm in GSEA and taking advantage of the recent development of a comprehensive database of RNA-protein binding preferences (CisBP-RNA) to provide the RNA-binding protein ‘finger-prints’ to be ranked (Ray et al., 2013). We confirmed that this pipeline was able to correctly identify known regulatory splicing factors from raw RNA-seq data from single-cell in vitro systems or complex in vivo tissues (Figure 3—figure supplement 2).
Having confirmed our pipeline, we then applied it to assess the regulation of skipped exons in the increasingly narrowly defined group regulated in vivo and in vitro by recruited platelets and macrophages (Figure 3A and B). Enrichment was judged relative to background sets created from known skipped exons expressed but not regulated in endothelial cells (see Materials and methods for details). The Rbfox family, of which Rbfox2 is known to regulate EIIIB (Underwood et al., 2005), stood out among this group, being enriched especially on the downstream side of exons with decreased inclusion and, to a lesser extent, on both the upstream and downstream side of exons with increased inclusion (Figure 3C–E).
Figure 3. Enrichment of an Rbfox motif in the platelet/macrophage-regulated subset identified in vivo and in vitro.
(A) Motif analysis pipeline, showing the number of potentially regulated skipped-exon events, and those passing each filter. (B) Events were separated into those with increased or decreased inclusion, and then the sequences of the upstream and downstream 200 bp flanking regions were isolated, resulting in four regions of analysis. 6-mer motifs in each region were assessed for enrichment relative to exons expressed in six matched background sets of skipped exons not regulated by flow. GSEA was used to identify splicing factor ‘fingerprints’ among these enriched motifs, using the motif-binding preference defined by the CisBP-RNA database (Ray et al.). (C) Example of enrichment of Rbfox binding motif in GSEA analysis of 85-unique RNA-binding protein signatures against flanking intron sequences of flow-regulated exons. Plot shows enrichment of Rbfox family motifs among the motifs most enriched above background in the downstream flanking region of exons regulated in vivo and in vitro (set iii). (D) Enrichment of the Rbfox2 motif in each of the sets of biologically defined regulated exons (i-iii) within the upstream and downstream flanking regions of exons with either increased or decreased inclusion. (E) Plot showing the average z-score enrichment of the top 5 Rbfox in vitro defined motifs in the flanking regions of the in vivo regulated exon set (80 SE) and the in vitro regulated subset (40 SE), relative to the six matched background sets (N = 6 per bar). (F) Transcript levels of Rbfox family of proteins in the carotid artery in vivo and in isolated aortic endothelial cells in vitro, with or without the deletion of Rbfox2 by Cdh5(PAC)-CreER (EC-KO). FPKM = Fragments Per Kilobase of transcript per Million mapped reads. FDR; false-discovery rate; n.f.; not found. p<0.0001 (****), p<0.01 (**).

Figure 3—figure supplement 1. No obvious candidate splice factors from differential expression under altered flow conditions.

Figure 3—figure supplement 2. Positive control sets to confirm the motif-based identification of regulated events in vitro and in vivo by their CisBP-RNA fingerprint in GSEA.

Figure 3—figure supplement 3. Conservation of Rbfox2 motifs enriched near flow-regulated skipped exons.

Rbfox2 motifs in the flanking regions of the regulated events were conserved, consistent with the conservation of the exons themselves (Figure 3—figure supplement 3). The Rbfox family contains three members, Rbfox1-3, with conserved RNA-binding motifs. To determine which of these might be most important in regulation of the EIIIA and EIIIB cohort of events, we examined their expression in vivo, and in in vitro purified endothelial cells. We found that only Rbfox2 was significantly expressed in arterial endothelial cells (Figure 3F).
Thus, bioinformatics analysis suggests Rbfox2 as a particularly important regulator of the skipped exon changes induced by platelet and macrophage recruitment to the arterial endothelium under low flow conditions.
Endothelial-specific deletion of Rbfox2 partially reverts flow-responsive skipping of vascular skipped exons induced by low flow in vivo
To test whether Rbfox2 is involved in the splicing response of the arterial endothelium to low flow, we genetically removed Rbfox2 in mice using an endothelial-specific and inducible Cre (Cdh5(PAC)-CreERT2) in combination with floxed Rbfox2 alleles (Gehman et al., 2012; Sörensen et al., 2009). This strategy resulted in efficient deletion of Rbfox2 without an increase in expression of the other Rbfox family members, 1 and 3 (Figure 3F; EC-KO).
To allow us to assess both the low-flow-responsive splicing patterns and the chronic transcriptional response of the flow-activated intima, we examined the effect of Rbfox2 deletion 7 days after the reduction in flow. Consistent with our previous observations on Fn-EIIIA and –EIIIB, we found that most splicing changes observed at 48 hr were also observed at 7 days (Murphy and Hynes, 2014). We found that Rbfox2 was important in the flow-responsive splicing change of many of the previously defined skipped exons, including Fn-EIIIB and –EIIIA to a lesser extent (Figure 4A, and Figure 4—source data 1). Altogether, Rbfox2 deletion suppressed flow-induced splicing changes of ~25% the skipped exons (Figure 4A, blue bars, right hand panel and 4B). This set of genes encodes extracellular matrix proteins (e.g. Fn1, Fbln2, Mfge8) and immune-regulatory proteins (e.g. Ikbkg, Gpr116, Ceacam1). Analysis of inclusion of specific exons is shown in Figure 4C demonstrating the loss of flow-responsive exons of Fbln2 on endothelial deletion of Rbfox2.
Figure 4. Endothelial Rbfox2 deletion affects many of the flow-regulated splicing events.
(A) Showing the percentage of each group of skipped exons regulated by endothelial deletion of Rbfox2 (BF >5) in any comparison and regulated similarly in both comparisons of Rbfox2 wt low-flow vs. Rbfox2 EC-KO low-flow – either consistent with the flow-induced change in splicing (red, ‘same’) or against the direction of the flow-induced change in splicing (blue, ‘reverted’). (B) Heat map showing the clustering of flow-regulated splicing events reverted by Rbfox2 deletion (67 of the 273 events consistently regulated between 48 hr and 7 days). 1 = C57 wild-type mice, 48 hr data set; 2 = C57 wild-type mice, IgG control 48 hr data set; 3 = Rbfox2 wt control (i.e. no Cre), 7 days data set; Rb EC-KO = Rbfox2 EC-KO 7 days data set. (C) Effect of endothelial Rbfox2 deletion on flow-mediated regulation of inclusion of Fbln2 alternative exons. (D) Plot shows the change in splicing of skipped exons (SE) following Rbfox2 deletion from carotid artery intima in vivo (under low flow) or in isolated primary aortic endothelial cells in vitro. SE from the Rbfox2 regulated set of events, in genes expressed >FPKM 1 in in vitro and in vivo sets. SE also regulated by a change in flow are highlighted.

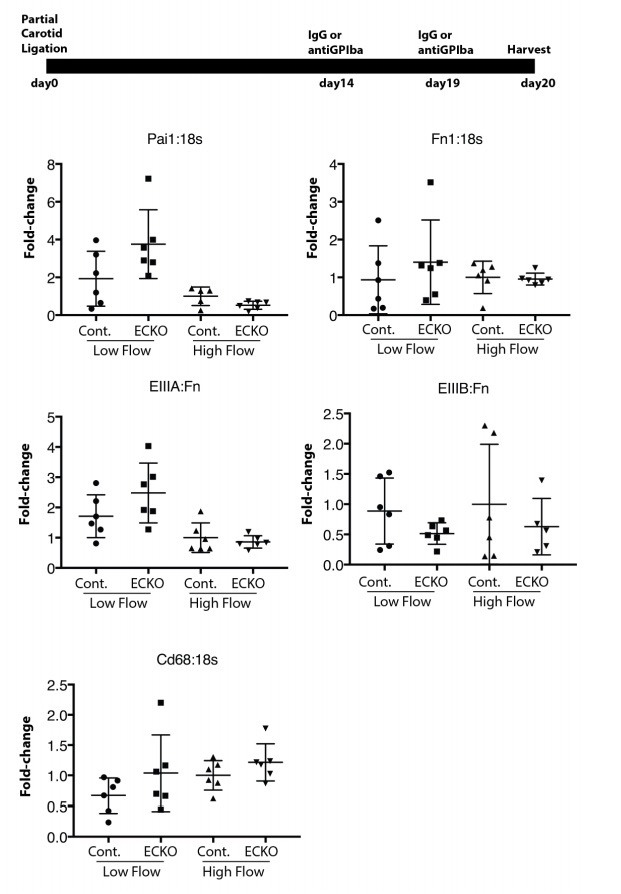
Figure 4—figure supplement 1. Rbfox2 deletion does not cause a reduction in markers of recruited platelets or macrophages.

Figure 4—figure supplement 2. Rbfox2 localization under serum stimulation in mouse aortic endothelial cells.

Since suppression of flow-responsive splicing was also impaired by depletion of platelets and macrophages, we asked whether recruitment of these cells was affected. However, we observed similar levels of macrophage markers (Cd68, F4/80) in the low-flow intimal flushes in the presence and absence of endothelial Rbfox2 (Figure 4—figure supplement 1), suggesting that recruitment of macrophages, and by extension the platelets that we had previously found to be required for macrophage recruitment, were not detectably affected by deletion of Rbfox2.
Consistent with a direct effect of Rbfox2 depletion on endothelial cells, we found a good correlation between the changes in splicing observed in vivo upon Rbfox2 deletion and those observed in vitro with Rbfox2 deletion in primary isolated aortic endothelial cells (Figure 4D). Notably, changes in splicing in vitro, in response to serum, did not correspond with an obvious alteration in the localization or abundance of Rbfox2 in the nucleus at the protein level (Figure 4—figure supplement 2).
Thus, Rbfox2 deletion in the endothelium prior to the induction of low flow suppresses a large portion of flow-responsive alternative splicing in endothelial cells, downstream of platelet and macrophage recruitment.
Endothelial deletion of Rbfox2 alters the intimal response to low flow
We predicted that changes in the splicing response of the arterial intima caused by Rbfox2 deletion would affect the response to low flow. To test this, we examined gene expression at the whole transcript level 7 days after the induction of low flow in Rbfox2 EC-KO mice and littermate controls, and in the isolated endothelial cells. We found that endothelial deletion of Rbfox2 significantly affected 641 genes in the carotid intima under low flow (DESeq2 p-adj <0.05) (Figure 5A). Of these 641 genes, 507 are part of the 4766 gene low-flow signature previously described (Figure 1). Clustering of expression changes in the Rbfox2-regulated subset of genes, relative to the contralateral artery of control mice, shows that deletion of Rbfox2 causes the low-flow arterial intima to resemble more closely the high-flow arterial intima (Figure 5A). Processes of cell proliferation and of antigen presentation were enriched among the differentially regulated genes (Figure 5B). RNA-seq was performed on pooled arteries, and quantitative PCR analysis of selected regulated genes in individual carotid arteries confirmed expression changes in individual animals (Figure 5C and D). These results indicate that Rbfox2 is essential for a portion of the endothelial response to low flow, regulating ~10% of the transcriptional response to low flow.
Figure 5. Endothelial Rbfox2 deletion suppresses low-flow transcript response in the arterial intima.
(A) Clustered heat map of the genes with adjusted p-values<0.05 shows the change in expression relative to contralateral controls at 7 days after the change in flow in the indicated genotypes (N = 641). (B) Enriched terms among the genes regulated by Rbfox2 deletion. (C) Volcano plot showing DESeq2 calculated p-values and log2 fold changes in genes expressed in the arterial intima at 7 days of arteries exposed to low and disturbed flow, with or without deletion of Rbfox2. Genes selected for qPCR in single arteries are shown. (D) Results of individual carotid artery qPCR for the genes indicated. Log2 fold-changes are relative to control, p values are from Mann-Whitney test (N = 14 control and N = 18 Rbfox2 EC-KO).

Figure 5—figure supplement 1. Rbfox2-dependent transcriptional response found in vivo is specific to the low-flow arterial intima.

In contrast with our observation of correlations between splicing patterns in vivo and in vitro, most transcriptional changes induced in the low-flow arterial intima were specific to the in vivo environment and were not observed in a comparison of isolated aortic endothelial cells or in the contralateral artery (Figure 5—figure supplement 1). In other systems, cytoplasmic Rbfox has been shown to have an important role in the regulation of transcript stability through the 3’UTR (Damianov et al., 2016; Lee et al., 2016). However, transcript levels regulated by Rbfox2 were not any more associated with characterized 3’ UTR binding by iClip-Seq (Lee et al., 2016) than those not regulated by Rbfox2 (iClip-Seq binding sites in 54/638 of the Rbfox2-regulated transcripts versus 1166/17130 of the transcripts not regulated by Rbfox2). Together, these data suggest that the consequences of endothelial Rbfox2 deletion on transcriptional changes involved cross-talk among cell types (e.g. effects of altered endothelial splicing on recruited immune cells) or other microenvironmental effects in the flow-activated intima in vivo not replicated in vitro.
Thus, Rbfox2 deletion affects skipped exon splicing patterns in endothelial cells, and results in the reversion of a large set of low-flow-induced gene expression changes in the arterial intima.
Discussion
Here, we report that recruitment of circulating hematopoietic cells to the arterial endothelium under low flow initiates an alternative splicing response regulated in part by Rbfox2. Profiling global transcriptional changes induced in the arterial endothelium by experimentally induced low flow, we show that hundreds of transcripts are regulated by alternative splicing. Focusing on skipped exons in particular, we find that one third of those affected by altered flow are dependent on recruited hematopoietic cells in vivo, particularly platelets. Rbfox-binding motifs are significantly enriched adjacent to these exons. Mechanistically, deletion of endothelial Rbfox2 reverts many of the flow-induced changes in splicing and suppresses ~10% of the flow-induced changes in transcription. Processes related to genes with changes in either splicing or transcription included cytoskeleton, cell adhesion, proliferation and antigen presentation suggesting that Rbfox2 may regulate these biological processes in the aortic intima downstream of hematopoietic cell recruitment.
Broad regulation of alternative splicing by flow in the arterial endothelium
Low flow exerts potent effects on the vasculature through the endothelium, which are achieved by effects on endothelial transcription, epigenetic regulation, and post-translational protein modifications and reorganization (Gimbrone and GarciaGarcía-CardenaCardeña, 2016). Our work reveals widespread changes in alternative splicing affecting a number of genes known to be critical in the pathways already implicated in the response to altered flow, inflammation, remodeling and flow-driven vascular disease. Examples include Pecam1, a component of the shear-sensing mechanism in the endothelium (Tzima et al., 2005), Yap1, a regulator of mechanical activation (Dupont et al., 2011) and Ikbkg (Nemo), a regulator of NFkappa-B signaling (Li et al., 1999). In previous work, differential splicing of Vcam1 was detected in IL-1-stimulated HUVECs (Cybulsky et al., 1991) and of Fn in TGFβ-induced liver sinusoidal endothelium (Chang et al., 2004). Exon arrays have also shown differential exon usage in HUVECs in response to hypoxia (Hang et al., 2009; Weigand et al., 2012). However, this hypoxic response appears to be very different from the responses we have observed, and less then 2% of the genes in which we detected alternative splicing overlap with those found in the hypoxic signature of HUVECs. It is possible that this is due to methods used (exon arrays versus our sequencing approach), cell type (HUVECs versus murine aortic endothelial cells), or pathway-specific responses. The latter is likely, since we observed no consistent regulation of hypoxia-induced genes (e.g. Vegfa) in the flow-responsive signature. Alternative splicing responses are distinct, similar to transcriptional responses (Graveley et al., 2011). Indeed, even within the alternative-splicing response to altered flow, we are able to detect a distinct response dependent on the recruitment of circulating hematopoietic cells.
Biological consequences of alternative splicing programs induced by low flow
We do not yet know the function of most of the splicing changes we have observed. Unlike mice deficient in both EIIIA and EIIIB, which exhibit an increased risk of arterial rupture under low flow, Rbfox2 EC-KO mice do not. This may be due to the lesser effect of Rbfox2 deletion on EIIIA than EIIIB in the Rbfox2 mutant mice (EIIIA inclusion was 50% in Rbfox2 EC-KO low-flow artery, versus 60% in WT low-flow artery, compared with 20% in high flow artery; while EIIIB inclusion was absent in all Rbfox2 EC-KO conditions). In previous work, we found that EIIIA-/- replicated much of the effect of the EIIIAB-/- mice; loss of EIIIB may have other consequences than loss of EIIIA (Murphy and Hynes, 2014).
Nevertheless, we have reason to believe that these splicing changes are biologically important. First, the alternatively spliced exons comprising this response, including Fn-EIIIA and -EIIIB, are highly conserved among all placental mammals. In fact ~80% of the flow-regulated skipped exons are as well conserved as consensus coding exons (CCDS). Conservation of sequence is not necessarily the same as conservation of regulation (Kalsotra et al., 2008; Li et al., 2015; Merkin et al., 2012; Pai et al., 2016; Yeo et al., 2005). Nevertheless, the conservation of regulatory regions, specifically the Rbfox2 motif, surrounding the skipped exons support conserved regulation as well. Second, deletion of a regulator of this splicing program, Rbfox2, affects the transcriptional response of the intima to low flow. Notably, both genes with Rbfox2-regulated transcript levels, and those with Rbfox2-regulated skipped exons were enriched in functions related to cytoskeletal remodeling, cell adhesion, and antigen presentation (Figure 1 and Figure 5). The particular genes regulated in each way (transcript abundance and splicing) did not directly overlap, suggesting that similar biological processes may be targeted through both transcriptional and post-transcriptional mechanisms in Rbfox2 EC-KO mice. Given the known activation of TLR4 signaling by EIIIA (Okamura et al., 2001), a key player in adaptive immunity, the potential impact on antigen presentation and the adaptive response warrants further research.
Regulation of alternative splicing in the aortic endothelium by platelets and macrophages
Much of the work to date on flow-induced changes in gene expression in the arterial endothelium has focused on the initial response to flow, which increases the transcriptional expression of adhesion receptors and chemokines involved in the recruitment of circulating immune cells (Conway and Schwartz, 2013; Ley et al., 2007). This has been a consequence of efforts to identify the first events leading to endothelial dysfunction under low flow. However, once circulating hematopoietic cells are recruited, they initiate ongoing interactions in the arterial wall likely to affect the ultimate outcome of flow-induced arterial injury (Pober and Sessa, 2007). Flow-driven vascular inflammation occurs as a chronic process, involving components of the innate and adaptive immune systems (Galkina and Ley, 2009; Pober and Sessa, 2007). Here, we show that recruitment of platelets and monocytes/macrophages in particular potently regulate alternative splicing in the arterial endothelium. Within this response, we observed alterations in a number of pathways that may impact ongoing interactions at the arterial wall. Thus, subsequent to the well-studied events leading to recruitment of immune cells, their interactions with the arterial endothelium appear to be affecting pathways that may help to shape the chronic inflammatory response.
Endothelial functions of Rbfox2
Rbfox2 is one of three Rbfox family members (Rbfox-1,-2 and -3). The Rbfox family of splice factors and the motif recognized by their RNA-binding domain are conserved throughout the vertebrates examined and also in C. elegans (Underwood et al., 2005). The motif which the Rbfox family recognizes was first identified adjacent to Fn-EIIIB (Huh and Hynes, 1993, 1994), which we find to be dependent on Rbfox2 in our system, consistent with previous work in other systems (Jangi et al., 2014). The Rbfox family has been shown to have important functions in heart (Gao et al., 2016), muscle (Singh et al., 2014), and brain development (Gehman et al., 2012) and EMT in cancer (Shapiro et al., 2011). De novo mutations in Rbfox2 have also been identified in congenital heart disease (Homsy et al., 2015), and alterations in Rbfox2 activity may be an early event in diabetes-induced heart pathology (Nutter et al., 2016). In both heart and brain, Rbfox2 has been shown to have a requirement in maintenance of tissue functions (Gao et al., 2016; Gehman et al., 2012). In contrast to the other tissues mentioned, Rbfox2 is the only member of the Rbfox family expressed in the arterial endothelium.
How Rbfox2 modulates splicing activity in this system remains unknown. We observe modest changes in expression of Rbfox2 (<20% increase in RNA in arterial endothelium exposed to low flow in vivo). Similar modest changes were observed in the in vitro system, in which we induced splicing changes with co-culture of platelets, monocytes and plasma. How else might Rbfox2 splicing activity be regulated? One possibility we examined is that upstream cues from platelet and monocyte recruitment alter sub-cellular localization of the Rbfox2 protein. However, we observed no obvious loss in nuclear Rbfox2 localization in vitro under conditions in which splicing changes are induced (Figure 4—figure supplement 2), suggesting that activity may be regulated in some other way. These data, together with the observation that deletion of Rbfox2 results in splicing alterations more widespread than the low-flow response alone (Figure 4D), suggest that the specificity of the endothelial splicing response is driven by other flow-responsive factors acting in an Rbfox2-dependent manner.
We have focused on the splicing functions of Rbfox2. However, in addition to affecting splicing patterns, Rbfox2 has also been reported to regulate gene expression patterns by other mechanisms. Splicing of Rbfox2 targets has been shown to regulate transcript stability (Jangi et al., 2014). Through recruitment of polycomb complexes to DNA, Rbfox2 may affect transcript expression (Wei et al., 2016). Rbfox2 may also stabilize transcripts directly by binding to the 3’UTR (Damianov et al., 2016; Lee et al., 2016), suppressing the formation of isoforms destined for nonsense-mediated decay (Jangi et al., 2014). However, as we observed no enrichment of known Rbfox-binding sites in the 3’UTR of regulated transcripts and no regulation of transcript levels in vitro which were regulated in vivo, we believe that many of the transcript level changes mediated by deletion of Rbfox2 are due to downstream or non-cell autonomous effects. Future work, examining the functional effects of individual splicing events identified will be important in teasing apart splicing-dependent and -independent effects.
In conclusion, we report that hundreds of splicing changes are regulated in endothelial cells exposed to an acute reduction in blood flow, many of which were dependent on recruitment of platelets and monocytes to the arterial wall and impact key pathways in endothelial cell biology. Rbfox2 emerged as an important regulator in this system, and its depletion resulted in a partial suppression of transcriptional responses characteristic of low-flow-induced endothelial activation. Thus, Fn is only one of many genes affected by splicing changes in the activated vascular endothelium. Alterations in Fn splicing contribute to a variety of vascular diseases with an inflammatory component; stroke (Dhanesha et al., 2015), atherosclerosis (Tan et al., 2004), myocardial infarction (Arslan et al., 2011), organ transplant and other fibroses (Bhattacharyya et al., 2014; Booth et al., 2012). Therefore, further investigation of the other alternative splicing events we have identified here may uncover novel regulatory mechanisms in chronic inflammatory disease.
Materials and methods
Key resources table.
| Reagent type (species) or resource | Designation | Source or reference | Identifiers |
|---|---|---|---|
| antibody | anti-GP1bα | Emfret | RRID:AB_2721041 |
| antibody | anti-Gr1 | Biolegend | RRID:AB_467731 |
| antibody | anti-Rbfox2 | Bethyl Labs | RRID:AB_609476 |
| commercial assay or kit | SMARTer Universal Low Input RNA Kit | Clontech | |
| commercial assay or kit | RNAeasy microcolumns | Qiagen | |
| software, algorithm | RSEM v.1.2.15 | GitHub | RRID:SCR_013027 |
| software, algorithm | MISO v.0.4.9 | GitHub | RRID:SCR_003124 |
| software, algorithm | STAR 2.5.1b | GitHub | RRID:SCR_015899 |
| software, algorithm | Tophat 2.0.6 | GitHub | RRID:SCR_013035 |
| software, algorithm | Bowtie2 v. 2.0.5 | GitHub | RRID:SCR_005476 |
| software, algorithm | GSEA | http://www.gsea-msigdb.org/gsea/index.jsp | RRID:SCR_003199 |
| strain, strain background (M. musculus) | Rbfox2lox/lox | Jackson Lab | RRID:IMSR_JAX:014090 |
| strain, strain background (M. musculus) | Rosa26-mTmG | Jackson Lab | RRID:IMSR_JAX:007676 |
| strain, strain background (M. musculus) | Cdh5(PAC)-CreERT2 | Jackson Lab | RRID:IMSR_TAC:13073 |
| strain, strain background (M. musculus) | C57BL/6J | Jackson Lab | RRID:IMSR_JAX:000664 |
Flow alterations
Partial carotid ligations were performed as previously described (Ni et al., 2010), with minor modifications (Murphy and Hynes, 2014). Briefly, the distal branches of the left carotid artery were identified in mice anesthetized with isoflurane. The left external carotid, internal carotid, and occipital artery were ligated with 9–0 Ethilon suture, leaving only the superior thyroid artery intact. Sham operations consisted of the same carotid dissection and encircling with suture, except that the vessels were not tied off. High-resolution ultrasound, using the VisualSonics Vevo 770, was performed at the experimental endpoint (2–7 days after partial carotid ligation) to confirm vessel patency.
RNA-isolation and sequencing
Isolation of intimal RNA was performed as previously described from the carotid intima (Murphy and Hynes, 2014; Ni et al., 2010). Briefly, vessels were isolated and imaged from euthanized mice, flushed with PBS solution and then 150 µL Trizol. RNA from cultured aortic endothelial cells was isolated after one week in culture by sorting 1–2K endothelial cells (by CD31+ and Icam2+) from BD Aria directly into cold Trizol solution. RNA from all samples was isolated with Qiagen RNAeasy microcolumns. Pools of RNA were created and then DNAse-treated in solution, before concentrating on RNAeasy columns with elution in minimal volume. RNA quality was assessed by Agilent Bioanalyzer or Advanced Analytical, library preparation was performed using the Clontech SMARTer Universal Low Input RNA Kit, and samples were sequenced on Illumina HiSeq 2000 or NextSeq 500 using 80 bp paired-end (PE) reads (triplicates of low-flow, sham and high-flow), 100 bp PE reads (re-sequencing of triplicates of low-flow, sham and high-flow, and cultured aortic endothelial cells) or 150 bp PE reads (hematopoeitic cell depletions with controls and endothelial Rbfox2 deletion with controls in vivo and in vitro). Base calls were performed using the Offline Base Caller (Illumina) v. 1.9.4 and reads mapping to different genomic features were tallied and read densities were compared for each sample.
Read-mapping and transcript analysis
For experiments with alteration of flow (triplicates of low-flow, sham and high-flow), reads were trimmed to 80 bp and mapped with Tophat 2.0.6 and Bowtie2 v. 2.0.5, allowing the detection of novel junctions, with options -p 8 --read-edit-dist 4 --min-intron-length 10 --read-mismatches 4 --max-intron-length 1000000 --read-realign-edit-dist 0 --mate-std-dev 30 --segment-length 20 --library-type fr-firststrand -r 50, guided (-G) by an mm9-based refseq gtf file [https://www.ncbi.nlm.nih.gov/pubmed/23618408].
For experiments with alteration of flow with or without depletion of hematopoeitic cells or deletion of Rbfox2, 150 bp reads were mapped with STAR 2.5.1b and options --runThreadN 8 --runMode alignReads --outFilterType BySJout --outFilterMultimapNmax 20 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --alignIntronMin 10 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --outSAMtype BAM SortedByCoordinate --quantMode TranscriptomeSAM [https://www.ncbi.nlm.nih.gov/pubmed/23104886].
RSEM v.1.2.15 (options --paired-end --calc-ci -bam -p 8 ) was used to calculate transcript levels [https://www.ncbi.nlm.nih.gov/pubmed/21816040], and DESeq2 v1.10.1 in the R statistical environment v3.2.3 was used to calculate differential transcript levels using the DEseq() integrated command with default parameters, which estimates library sizes and dispersions and fits a negative binomial generalized linear model for fold-changes and Wald statistics estimation using a 2-condition design. [https://www.ncbi.nlm.nih.gov/pubmed/25516281].
For eGFP and Tomato analysis, a custom index containing eGFP and tdTomato sequences was created, and bowtie was used to align reads to this index using options --best --strata -m 1 –q. The number of reads mapping to eGFP and to tdTomato were counted, and % tdTomato reads determined [tdTomato reads / (tdTomato+ eGFP reads) *100%].
Splicing analysis
Positive control data sets for regulation of splicing by Rbfox2 in vitro or by Mbnl in vivo were taken from published data, and run through our own informatics pipeline (Jangi et al., 2014; Wang et al., 2015).
MISO v.0.4.9 was used to determine differential splicing regulation. The SE, A3SS, A5SS, RI and MXE event index was derived from the MISO splicing events database (mm9 version 2). Briefly, this set of events was derived by considering all transcripts annotated in Ensembl genes, knownGenes (UCSC) and RefSeq genes (as of June 2013). The ALE and TandemUTR events index was derived from the MISO database compiled from published data, made available on the MISO website (Hoque et al., 2013). Additional novel SE, A3SS, A5SS and MXE events were detected using a method developed by Paul Boutz. Identification and classification of novel alternative splicing events was performed as described in Boutz et al. (2015). Briefly, mapped splice junctions were filtered for minimal transcript level and classified as SE, A3SS, A5SS, or MXE without reference to annotated exonic loci. This set of novel events was then used to generate a novel index, which was passed through the same MISO pipeline as all other indices.
Selection of subgroups of regulated exons
Flow-regulated: BF >5 in two completely independent biological comparisons of low flow versus high flow, where the difference between high flow and low flow is >2 x the difference between replicate high-flow conditions.
Flow-regulated and endothelial
Of the events above, at least 50% of the difference between low-flow in vivo and high flow in vivo must be recapitulated in differences between FACs-purified mouse arterial cells in culture and high-flow cells in vivo.
Flow-regulated and endothelial and platelet-regulated
Of the events above, if platelet depletion reverted flow-induced splicing changes by >50% in two completely independent biological comparisons.
Flow-regulated and endothelial and platelet-regulated and in vitro regulated
Of the events above, if the change in splicing induced in isolated aortic endothelial cells by the addition of platelets, macrophages and plasma was >50% of the flow-regulated change in splicing.
Rbfox2-regulated
If change in inclusion with Rbfox2 deletion was BF >5 in any of three comparisons, in vivo low-flow Rbfox2 control vs. Rbfox2 EC-KO artery at 7 days, in vivo high-flow Rbfox2 control vs. Rbfox2 EC-KO artery at 7 days, or in vitro Rbfox2 control vs. Rbfox2-deleted aortic endothelial cells, and consistently changed in the same direction in all in vivo comparisons under low flow between Rbfox2 control and Rbfox2 EC-KO arteries. In this set, Rbfox2 control arteries have floxed alleles but no Cre activity.
Identification of enriched splice factor motifs adjacent to regulated skipped exons
To perform motif enrichment analysis, we compared endothelial alternative skipped exons regulated by blood cells in vivo and in vitro (the foreground) with skipped exons expressed by endothelial cells, but not consistently regulated under the same conditions (the background). In general, the foreground events were more often poised at an intermediate Psi level (neither complete inclusion or exclusion). They also tended to be expressed at a higher level than background events. Thus, we created background sets in which we normalized each of these parameters independently (data not shown). We looked for motifs enriched either upstream (3’splice site, or 3’SS) or downstream (5’ splice site, or 5’SS) of the regulated exons. Since the direction of change in exon inclusion, increased dPsi or decreased dPsi, has been correlated with the location of splice factor binding, we further separated the foreground into exons with increased (+dPsi) or decreased (-dPsi) inclusion. Intronic sequences adjacent to the 3’SS and the 5’SS of skipped exons with increased or decreased inclusion were isolated from mm9 sequence data using Bedtools 2.16.1 (getfasta –s), and enrichment was calculated based on enrichment of 6-mer or 7-mer motifs in the foreground versus the background sets, split across 10 GC-normalized bins, iterated multiple times.
From the lists of enriched motifs we obtained, we aimed to identify the likely RNA-binding proteins. To do this in an unbiased manner, we used the CisBP-RNA data set, which includes 154 unique motifs and 373 total murine RNA-binding proteins (Ray et al., 2013). We modified the ranking algorithm GSEA, typically used to identify transcriptional signatures, to identify RNA-binding protein signatures. A Kolmogorov-Smirnov statistic embedded in GSEA was used to identify which of these RNA-binding-protein motif sets was most abundant at the leading edge of the ranked enrichment set (Subramanian et al., 2005).
Depletion of innate immune cells
For platelet depletion experiments, mice were injected intraperitoneally with a single dose of platelet-depleting antibody (anti-GP1bα; 50 µg per mouse; R300; Emfret; RRID:AB_2721041) or IgG control at the time of partial carotid ligation. To deplete Ly6G+ cells, anti-Gr1 (RB6-8C5; BioLegend; RRID:AB_467731) or IgG controls were administered by intraperitoneal injection (25 µg per mouse) 24 hr prior to partial carotid ligation. To deplete macrophages, mice were injected by tail vein with 150 µL clodronate liposomes or PBS liposome controls at the time of partial carotid ligation. Clodronate liposomes or PBS liposomes were purchased from ClodronateLiposomes.com (Kruisweg 59, 2011 LB Haarlem, The Netherlands).
Mice
Male C57BL/6J mice from Jackson Laboratories were used at 6–7 weeks of age for experiments on the regulation of transcription and splicing in the carotid intima in wild-type mice, or in wild-type mice with the depletion of various hematopoietic cells.
For the endothelial deletion of Rbfox2, Cdh5(PAC)-CreERT2, Rosa26-mTmG and Rbfox2lox/lox mice have been previously described (Gehman et al., 2012; Muzumdar et al., 2007; Wang et al., 2010) RRID:IMSR_JAX:014090, RRID:IMSR_JAX:007676, RRID:IMSR_TAC:13073. They were intercrossed to create the Rbfox2 EC-KO mice (Cdh5(PAC)-CreERT2; Rosa26-mTmG; Rbfox2lox/lox) and littermate controls (Rosa26-mTmG and Rbfox2lox/lox) used here. Mice used were between 2 and 7 months of age in paired groups of males and females.
All mice were housed and handled in accordance with protocols approved by the Massachusetts Institute of Technology Division of Comparative Medicine.
Aortic endothelial cell isolation
Mouse aortic endothelial cells were isolated following previously described methods (Kobayashi et al., 2005), with modifications. Briefly, mice were perfused via the left ventricle with PBS, the aorta was isolated and filled with 2% collagenase II (Worthington) in serum-free DMEM, and then closed at the ends with 7–0 suture. The vessel was digested for 30–45 min in 10% FBS at 37C, and endothelial cells were flushed out into EC culture medium, and onto a collagen I-coated plate. EC culture medium consisted of DMEM with 10% FBS and 10 mg/mL endothelial growth supplement (ECGS, Biomedical Technologies) with primocin (InvivoGen). Typical isolations of ~1000 cells expanded to ~10,000–50,000 cells. Cells were then FACs-sorted (BD Aria) on endothelial markers, eGFP for Cdh5(PAC)-CreERT2; Rosa26-mTmG on Pecam+ and Icam2+ for Cdh5(PAC)-CreERT2; Rosa26-mTmG; Rbfox2lox/lox mice and littermate controls.
Aortic endothelial cell immortalization
After purification by FACs, endothelial cells were immortalized by lentiviral TetOn-Sv40T. The construct was developed from a version of pTRIPZ (Clontech) with removal of the puro-selection cassette. SV40 T antigen was inserted into the tet-regulated region of the lentivirus from pBabe-SV40T (Zhao et al., 2003). After infection, cells were expanded in EC media with 2 µg/mL Doxycycline (Dox).
Aortic endothelial cell co-culture
Cells derived from Cdh5(PAC)-CreERT2; Rosa26-mTmG, tamoxifen-treated in vivo, and then isolated by FACs purification on eGFP, expanded and immortalized by TetOn-SV40T were trypsinized and split in DMEM into new dishes, without Dox to turn off SV40T. After culture for 24 hr in DMEM, combinations of platelets, bone-marrow-derived monocytes, and/or 10% plasma were added for 48 hr. Cells in plate were then imaged and lysed with Trizol for RNA extraction.
Platelets were isolated from the platelet-rich plasma (PRP) of C57BL/6J mouse blood by collection in ACD buffer. They were spun down at 500xG for 7 min with isolation of the upper PRP, from which a platelet pellet was isolated after being spun down at 2800xG for 5 min and resuspended in PIPES buffer. ~20 million platelets (in 6 µL PIPES) were added to each confluent well of endothelial cells in the 24-well plate. Plasma was taken from C57BL/6J mouse blood collected in EDTA and spun down at 2000xG for 15 min at 4°C. 10% plasma was added to DMEM in 24-well plates. Bone-marrow-derived monocytes were isolated from the femurs of C57BL/6J mice, flushed with 10% FBS DMEM (+2 mM EDTA) into collection tube with 10 mL syringe and 25Ga needle, and then purified using an EasySep Mouse Monocyte Isolation Kit (by negative selection, depleting Cd3+, Cd45R+, Cd117+, Ly6G+, Nk1.1+, Siglec F+ cells). ~10K monocytes were added to each well of the 24-well plate.
Aortic endothelial cell immunofluorescence
Cells (TetOn-Sv40, eGFP+ from Cdh5(PAC)CreERT2; mT/mG mice were cultured on poly-L-lysine treated coverslips and treated or not with 10% serum (FBS) for 48 hr. Coverslips and cells were washed with phosphate buffered saline (PBS) and fixed with ice cold methanol. Rbfox2 protein was stained by anti-Rbfox2 (Bethyl Labs A300-864A, 1:1000; RRID:AB_609476), with goat anti-rabbit Alexa594 secondary. Block was 10% normal goat serum in PBS with 0.1% tritonX-100.
Acknowledgements
We thank members of the Hynes lab and other labs in the Koch Institute for advice and discussions, particularly Mohini Jiangi for discussions of Rbfox2 functions and John Lamar (Albany Medical Center) for critical reading. We thank the Swanson Biotechnology Center at the Koch Institute/MIT, especially the Applied Therapeutics and Whole Animal Imaging Facility, the Hope Babette Tang (1983) Histology Facility, the Microscopy Facility, the Barbara K Ostrom (1978) Bioinformatics and Computing Facility, and Scott Malstrom, Denise Crowley, Eliza Vasile and Charlie Whittaker for technical support. The authors wish to dedicate this paper to the memory of Officer Sean Collier for his caring service to the MIT community.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Patrick A Murphy, Email: pamurphy@uchc.edu.
Richard O Hynes, Email: rohynes@mit.edu.
Douglas L Black, University of California, Los Angeles, United States.
Funding Information
This paper was supported by the following grants:
National Heart, Lung, and Blood Institute F32-HL110484 to Patrick A Murphy.
National Cancer Institute P30-CA14051 to Vincent L Butty.
Howard Hughes Medical Institute Investigator Award to Richard O Hynes.
National Heart, Lung, and Blood Institute K99/R00-HL125727 to Patrick A Murphy.
National Heart, Lung, and Blood Institute PO1-HL66105 to Patrick A Murphy.
National Institute of General Medical Sciences R01-GM034277 to Phillip A Sharp.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Resources, Data curation, Software, Formal analysis, Funding acquisition, Validation, Investigation, Visualization, Methodology, Writing—original draft, Project administration, Writing—review and editing.
Data curation, Software, Formal analysis, Supervision, Methodology, Writing—review and editing.
Data curation, Software, Formal analysis, Investigation, Methodology, Writing—review and editing.
Investigation.
Investigation.
Resources, Software, Funding acquisition, Writing—review and editing.
Conceptualization, Methodology.
Conceptualization, Resources, Supervision, Funding acquisition, Methodology, Project administration, Writing—review and editing.
Ethics
Animal experimentation: All mice were housed and handled in accordance with protocols approved by the Massachusetts Institute of Technology Committee on Animal Care (CAC) protocol (0415-033-18). All surgery was performed under isoflurane anesthesia with post-operative analgesia.
Additional files
Major datasets
The following dataset was generated:
Murphy PA, author; Butty VL, author; Boutz PL, author; Begum S, author; Sharp PA, author; Burge CB, author; Hynes RO, author. Alternative RNA Splicing in the Endothelium Mediated in Part by Rbfox2 Regulates the Arterial Response to Low Flow. 2017 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101826 Publicly available at the NCBI Gene Expression Omnibus (accession no: GSE101826)
The following previously published datasets were used:
Wang ET, author; Cody NA, author; Jog S, author; Biancolella M, author; Wang TT, author; Treacy DJ, author; Luo S, author; Schroth GP, author; Housman DE, author; Reddy S, author; Lécuyer E, author; Burge CB, author. Transcriptome-wide Regulation of Splicing and mRNA Localization by Muscleblind Proteins. 2012 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39911 Publicly available at the NCBI Gene Expression Omnibus (accession no: GSE39911)
Jangi M, author; Boutz PL, author; Paul P, author; Sharp PA, author. Rbfox2 controls autoregulation in RNA binding protein networks. 2014 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54794 Publicly available at the NCBI Gene Expression Omnibus (accession no: GSE54794)
References
- Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circulation Research. 2011;108:582–592. doi: 10.1161/CIRCRESAHA.110.224428. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, White ES, Varga J. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Science Translational Medicine. 2014;6:232ra50. doi: 10.1126/scitranslmed.3008264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth AJ, Wood SC, Cornett AM, Dreffs AA, Lu G, Muro AF, White ES, Bishop DK. Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. The Journal of Pathology. 2012;226:609–618. doi: 10.1002/path.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussel L, Rayz V, McCulloch C, Martin A, Acevedo-Bolton G, Lawton M, Higashida R, Smith WS, Young WL, Saloner D. Aneurysm growth occurs at region of low wall shear stress: patient-specific correlation of hemodynamics and growth in a longitudinal study. Stroke. 2008;39:2997–3002. doi: 10.1161/STROKEAHA.108.521617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Bhutkar A, Sharp PA. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes & Development. 2015;29:63–80. doi: 10.1101/gad.247361.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Martinelli R. T lymphocyte-endothelial interactions: emerging understanding of trafficking and antigen-specific immunity. Frontiers in Immunology. 2015;6:603. doi: 10.3389/fimmu.2015.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang ML, Chen JC, Alonso CR, Kornblihtt AR, Bissell DM. Regulation of fibronectin splicing in sinusoidal endothelial cells from normal or injured liver. PNAS. 2004;101:18093–18098. doi: 10.1073/pnas.0408439102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- Chèvre R, González-Granado JM, Megens RT, Sreeramkumar V, Silvestre-Roig C, Molina-Sánchez P, Weber C, Soehnlein O, Hidalgo A, Andrés V. High-resolution imaging of intravascular atherogenic inflammation in live mice. Circulation Research. 2014;114:770–779. doi: 10.1161/CIRCRESAHA.114.302590. [DOI] [PubMed] [Google Scholar]
- Cicha I, Wörner A, Urschel K, Beronov K, Goppelt-Struebe M, Verhoeven E, Daniel WG, Garlichs CD. Carotid plaque vulnerability: a positive feedback between hemodynamic and biochemical mechanisms. Stroke. 2011;42:3502–3510. doi: 10.1161/STROKEAHA.111.627265. [DOI] [PubMed] [Google Scholar]
- Conway DE, Schwartz MA. Flow-dependent cellular mechanotransduction in atherosclerosis. Journal of Cell Science. 2013;126:5101–5109. doi: 10.1242/jcs.138313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulsky MI, Fries JW, Williams AJ, Sultan P, Davis VM, Gimbrone MA, Collins T. Alternative splicing of human VCAM-1 in activated vascular endothelium. The American Journal of Pathology. 1991;138:815–820. [PMC free article] [PubMed] [Google Scholar]
- Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, García-Cardeña G, Gimbrone MA. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. PNAS. 2004;101:14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damianov A, Ying Y, Lin CH, Lee JA, Tran D, Vashisht AA, Bahrami-Samani E, Xing Y, Martin KC, Wohlschlegel JA, Black DL. Rbfox proteins regulate splicing as part of a large multiprotein complex LASR. Cell. 2016;165:606–619. doi: 10.1016/j.cell.2016.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Rateri DL, Daugherty A. Mechanisms of aortic aneurysm formation: translating preclinical studies into clinical therapies. Heart. 2014;100:1498–1505. doi: 10.1136/heartjnl-2014-305648. [DOI] [PubMed] [Google Scholar]
- Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–4363. doi: 10.1182/blood-2005-08-3465. [DOI] [PubMed] [Google Scholar]
- Dhanesha N, Ahmad A, Prakash P, Doddapattar P, Lentz SR, Chauhan AK. Genetic ablation of extra domain A of fibronectin in hypercholesterolemic mice improves stroke outcome by reducing thrombo-inflammation. Circulation. 2015;132:2237–2247. doi: 10.1161/CIRCULATIONAHA.115.016540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*) Annual Review of Immunology. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Ren S, Lee JH, Qiu J, Chapski DJ, Rau CD, Zhou Y, Abdellatif M, Nakano A, Vondriska TM, Xiao X, Fu XD, Chen JN, Wang Y. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. Journal of Clinical Investigation. 2016;126:195–206. doi: 10.1172/JCI84015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Meera P, Stoilov P, Shiue L, O'Brien JE, Meisler MH, Ares M, Otis TS, Black DL. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes & Development. 2012;26:445–460. doi: 10.1101/gad.182477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbrone MA, García-Cardeña G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovascular Pathology. 2013;22:9–15. doi: 10.1016/j.carpath.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circulation Research. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, Brown JB, Cherbas L, Davis CA, Dobin A, Li R, Lin W, Malone JH, Mattiuzzo NR, Miller D, Sturgill D, Tuch BB, Zaleski C, Zhang D, Blanchette M, Dudoit S, Eads B, Green RE, Hammonds A, Jiang L, Kapranov P, Langton L, Perrimon N, Sandler JE, Wan KH, Willingham A, Zhang Y, Zou Y, Andrews J, Bickel PJ, Brenner SE, Brent MR, Cherbas P, Gingeras TR, Hoskins RA, Kaufman TC, Oliver B, Celniker SE. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang X, Li P, Li Z, Qu W, Yu Y, Li H, Shen Z, Zheng H, Gao Y, Wu Y, Deng M, Sun Z, Zhang C. Transcription and splicing regulation in human umbilical vein endothelial cells under hypoxic stress conditions by exon array. BMC Genomics. 2009;10:126. doi: 10.1186/1471-2164-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, Jin SC, Deanfield J, Giardini A, Porter GA, Kim R, Bilguvar K, López-Giráldez F, Tikhonova I, Mane S, Romano-Adesman A, Qi H, Vardarajan B, Ma L, Daly M, Roberts AE, Russell MW, Mital S, Newburger JW, Gaynor JW, Breitbart RE, Iossifov I, Ronemus M, Sanders SJ, Kaltman JR, Seidman JG, Brueckner M, Gelb BD, Goldmuntz E, Lifton RP, Seidman CE, Chung WK. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, Tian B. Analysis of alternative cleavage and polyadenylation by 3' region extraction and deep sequencing. Nature Methods. 2013;10:133–139. doi: 10.1038/nmeth.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Hynes RO. Elements regulating an alternatively spliced exon of the rat fibronectin gene. Molecular and Cellular Biology. 1993;13:5301–5314. doi: 10.1128/MCB.13.9.5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Hynes RO. Regulation of alternative pre-mRNA splicing by a novel repeated hexanucleotide element. Genes & Development. 1994;8:1561–1574. doi: 10.1101/gad.8.13.1561. [DOI] [PubMed] [Google Scholar]
- Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nature Medicine. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- Jangi M, Boutz PL, Paul P, Sharp PA. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes & Development. 2014;28:637–651. doi: 10.1101/gad.235770.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. PNAS. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nature Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Tse K, Sette A, Ley K. Vaccination to modulate atherosclerosis. Autoimmunity. 2015;48:152–160. doi: 10.3109/08916934.2014.1003641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T. A simple method of isolating mouse aortic endothelial cells. Journal of Atherosclerosis and Thrombosis. 2005;12:138–142. doi: 10.5551/jat.12.138. [DOI] [PubMed] [Google Scholar]
- Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. PNAS. 2014;111:E3053–E3061. doi: 10.1073/pnas.1411082111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Damianov A, Lin CH, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, Martin KC. Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron. 2016;89:113–128. doi: 10.1016/j.neuron.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature Reviews Immunology. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Li Y, Kang J, Friedman J, Tarassishin L, Ye J, Kovalenko A, Wallach D, Horwitz MS. Identification of a cell protein (FIP-3) as a modulator of NF-kappaB activity and as a target of an adenovirus inhibitor of tumor necrosis factor alpha-induced apoptosis. PNAS. 1999;96:1042–1047. doi: 10.1073/pnas.96.3.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Sanchez-Pulido L, Haerty W, Ponting CP. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Research. 2015;25:1–13. doi: 10.1101/gr.181990.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Balasubramanian V, García-Cardeña G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circulation Research. 2005;96:e48–e57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- Massberg S, Brand K, Grüner S, Page S, Müller E, Müller I, Bergmeier W, Richter T, Lorenz M, Konrad I, Nieswandt B, Gawaz M. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. The Journal of Experimental Medicine. 2002;196:887–896. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PA, Hynes RO. Alternative splicing of endothelial fibronectin is induced by disturbed hemodynamics and protects against hemorrhage of the vessel wall. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014;34:2042–2050. doi: 10.1161/ATVBAHA.114.303879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nagel T, Resnick N, Atkinson WJ, Dewey CF, Gimbrone MA. Shear stress selectively upregulates intercellular adhesion molecule-1 expression in cultured human vascular endothelial cells. Journal of Clinical Investigation. 1994;94:885–891. doi: 10.1172/JCI117410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, Visvader JE, Jo H. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood. 2010;116:e66–e73. doi: 10.1182/blood-2010-04-278192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, Lozano MJ, Abass IJ, Ijaz T, Brasier AR, Garg NJ, Wehrens XHT, Yeo GW, Kuyumcu-Martinez MN. Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Reports. 2016;15:2200–2213. doi: 10.1016/j.celrep.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF. The extra domain A of fibronectin activates Toll-like receptor 4. Journal of Biological Chemistry. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- Pai AA, Baharian G, Pagé Sabourin A, Brinkworth JF, Nédélec Y, Foley JW, Grenier JC, Siddle KJ, Dumaine A, Yotova V, Johnson ZP, Lanford RE, Burge CB, Barreiro LB. Widespread Shortening of 3' Untranslated Regions and Increased Exon Inclusion Are Evolutionarily Conserved Features of Innate Immune Responses to Infection. PLoS Genetics. 2016;12:e1006338. doi: 10.1371/journal.pgen.1006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, García-Cardeña G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. Journal of Clinical Investigation. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nature Reviews Immunology. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, Na H, Irimia M, Matzat LH, Dale RK, Smith SA, Yarosh CA, Kelly SM, Nabet B, Mecenas D, Li W, Laishram RS, Qiao M, Lipshitz HD, Piano F, Corbett AH, Carstens RP, Frey BJ, Anderson RA, Lynch KW, Penalva LO, Lei EP, Fraser AG, Blencowe BJ, Morris QD, Hughes TR. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, Thuren T, CANTOS Pilot Investigative Group Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genetics. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Xia Z, Bland CS, Kalsotra A, Scavuzzo MA, Curk T, Ule J, Li W, Cooper TA. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Molecular Cell. 2014;55:592–603. doi: 10.1016/j.molcel.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sörensen I, Adams RH, Gossler A. DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood. 2009;113:5680–5688. doi: 10.1182/blood-2008-08-174508. [DOI] [PubMed] [Google Scholar]
- Tan MH, Sun Z, Opitz SL, Schmidt TE, Peters JH, George EL. Deletion of the alternatively spliced fibronectin EIIIA domain in mice reduces atherosclerosis. Blood. 2004;104:11–18. doi: 10.1182/blood-2003-09-3363. [DOI] [PubMed] [Google Scholar]
- Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Molecular and Cellular Biology. 2005;25:10005–10016. doi: 10.1128/MCB.25.22.10005-10016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, Barberis A, Benjamin LE, Mäkinen T, Nobes CD, Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, Lambert NJ, Freese P, Saxena T, Cooper TA, Burge CB. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Research. 2015;25:858–871. doi: 10.1101/gr.184390.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Xiao R, Chen L, Cui H, Zhou Y, Xue Y, Hu J, Zhou B, Tsutsui T, Qiu J, Li H, Tang L, Fu XD. RBFox2 binds nascent RNA to globally regulate polycomb complex 2 targeting in mammalian genomes. Molecular Cell. 2016;62:875–889. doi: 10.1016/j.molcel.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigand JE, Boeckel JN, Gellert P, Dimmeler S. Hypoxia-induced alternative splicing in endothelial cells. PLoS One. 2012;7:e42697. doi: 10.1371/journal.pone.0042697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J, Natarajan SK, Tremmel M, Ma D, Mocco J, Hopkins LN, Siddiqui AH, Levy EI, Meng H. Hemodynamic-morphologic discriminants for intracranial aneurysm rupture. Stroke. 2011;42:144–152. doi: 10.1161/STROKEAHA.110.592923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Coulombe-Huntington J, Kang S, Sheynkman GM, Hao T, Richardson A, Sun S, Yang F, Shen YA, Murray RR, Spirohn K, Begg BE, Duran-Frigola M, MacWilliams A, Pevzner SJ, Zhong Q, Trigg SA, Tam S, Ghamsari L, Sahni N, Yi S, Rodriguez MD, Balcha D, Tan G, Costanzo M, Andrews B, Boone C, Zhou XJ, Salehi-Ashtiani K, Charloteaux B, Chen AA, Calderwood MA, Aloy P, Roth FP, Hill DE, Iakoucheva LM, Xia Y, Vidal M. Widespread expansion of protein interaction capabilities by alternative splicing. Cell. 2016;164:805–817. doi: 10.1016/j.cell.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GW, Van Nostrand E, Holste D, Poggio T, Burge CB. Identification and analysis of alternative splicing events conserved in human and mouse. PNAS. 2005;102:2850–2855. doi: 10.1073/pnas.0409742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–495. doi: 10.1016/S1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]