

Supplemental Digital Content is available in the text.
Keywords: blood vessels, cadherin, cell differentiation, endothelial cells, polycomb-group proteins
Abstract
Rationale:
The mechanistic foundation of vascular maturation is still largely unknown. Several human pathologies are characterized by deregulated angiogenesis and unstable blood vessels. Solid tumors, for instance, get their nourishment from newly formed structurally abnormal vessels which present wide and irregular interendothelial junctions. Expression and clustering of the main endothelial-specific adherens junction protein, VEC (vascular endothelial cadherin), upregulate genes with key roles in endothelial differentiation and stability.
Objective:
We aim at understanding the molecular mechanisms through which VEC triggers the expression of a set of genes involved in endothelial differentiation and vascular stabilization.
Methods and Results:
We compared a VEC-null cell line with the same line reconstituted with VEC wild-type cDNA. VEC expression and clustering upregulated endothelial-specific genes with key roles in vascular stabilization including claudin-5, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and von Willebrand factor (vWf). Mechanistically, VEC exerts this effect by inhibiting polycomb protein activity on the specific gene promoters. This is achieved by preventing nuclear translocation of FoxO1 (Forkhead box protein O1) and β-catenin, which contribute to PRC2 (polycomb repressive complex-2) binding to promoter regions of claudin-5, VE-PTP, and vWf. VEC/β-catenin complex also sequesters a core subunit of PRC2 (Ezh2 [enhancer of zeste homolog 2]) at the cell membrane, preventing its nuclear translocation. Inhibition of Ezh2/VEC association increases Ezh2 recruitment to claudin-5, VE-PTP, and vWf promoters, causing gene downregulation. RNA sequencing comparison of VEC-null and VEC-positive cells suggested a more general role of VEC in activating endothelial genes and triggering a vascular stability-related gene expression program. In pathological angiogenesis of human ovarian carcinomas, reduced VEC expression paralleled decreased levels of claudin-5 and VE-PTP.
Conclusions:
These data extend the knowledge of polycomb-mediated regulation of gene expression to endothelial cell differentiation and vessel maturation. The identified mechanism opens novel therapeutic opportunities to modulate endothelial gene expression and induce vascular normalization through pharmacological inhibition of the polycomb-mediated repression system.
Several human pathological conditions are characterized by deregulated angiogenesis leading to the formation of unstable blood vessels.1 Abnormal angiogenesis is also a hallmark of cancer. Solid tumors get their nourishment from newly formed vessels, which, however, present several structural abnormalities such as wide and irregular interendothelial junctions.2
VEC (vascular endothelial cadherin), the main component of endothelial adherens junctions (AJs), plays a major role in the process of vessel maturation and stability. This molecule, specifically expressed by endothelial cells (ECs), mediates homophilic adhesion at cell–cell contacts regulating vascular permeability.3 VEC is indispensable for vascular maturation and inhibition of vascular regression.3 VEC clustering at AJs triggers intracellular signals inducing contact inhibition of cell growth, protection from apoptosis, cell polarity, and inhibition of migration.4
In previous work, we found that VEC expression and clustering at cell–cell contacts relieve the inhibitory effect of the FoxO1 (Forkhead box protein O1)/β-catenin complex on the expression of claudin-5, an endothelial-specific TJ (tight junction) protein,5 acting via the removal of a transcriptional repression mechanism.
The reversible nature of claudin-5 gene repression suggested the involvement of PcG (polycomb group) proteins and epigenetic mechanisms in VEC-mediated regulation of claudin-5 expression. Indeed, PcG proteins control the induction of reversible states of epigenetic silencing in most multicellular organisms, including humans, and regulate several developmental decisions by silencing genes involved in stem cell differentiation and specification of cellular identities.6,7 Their activity is exerted through 2 multiprotein complexes called PRC (polycomb repressive complex)1 and PRC2.6 PRC2 core subunits are Ezh (enhancer of zeste homolog)2, Suz (suppressor of zeste)12, Eed (embryonic ectoderm development), and Rbap (retinoblastoma-binding protein)48. Ezh2 is the catalytic subunit containing a SET [Su(var)3-9, enhancer of zeste and trithorax] domain, which harbors the active site for histone H3 trimethylation on lysine 27, although Suz12 and Eed association is needed for optimal enzymatic activity.8 According to the canonical model, H3K27me3 (histone H3 trimethylated on lysine 27) is a silencing histone mark promoting the recruitment of PRC1 selectively at genes that are targeted for repression. PRC1 includes Ring1B, which mediates monoubiquitylation of histone H2A on lysine 1199 blocking gene expression by multiple mechanisms such as chromatin compaction, inhibition of transcriptional initiation or elongation, recruitment of transcriptional inhibitors, and block of the binding of key activators.6,10
A fundamental step in PRC-mediated transcriptional repression is the ability of such complexes to be recruited specifically at target genes. In Drosophila, this is accomplished by polycomb response elements, clusters of DNA-binding sites for proteins that associate with PRC2 and PRC1.11 Mammalian polycomb response elements have not been identified yet, and many aspects of polycomb recruitment still remain obscure.
Here, we report that VEC-mediated inhibition of FoxO1/β-catenin nuclear accumulation triggers a transcriptional program of endothelial differentiation and maturation. PcG proteins exert an essential role in such program by targeting the promoters of key endothelial stability genes claudin-5, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and von Willebrand factor (vWf), causing their repression. VEC can inhibit such mechanism in multiple ways, by preventing polycomb binding to gene promoters through restriction of the nuclear localization of the FoxO1/β-catenin complex and by sequestering a fraction of Ezh2 at the plasma membrane.
These observations are supported by in vivo data in newborn mice treated with Ezh2 inhibitor UNC1999. Furthermore, dismantling of VEC clusters during pathological angiogenesis in human ovarian carcinomas is accompanied by the downregulation of Claudin-5 and VE-PTP.
These data highlight a key role of PcG proteins in the regulation of EC gene expression and open novel therapeutic opportunities to induce vascular normalization through pharmacological inhibition of the polycomb-mediated repression system.
Methods
Detailed Methods section is available in the Online Data Supplement.
The data that support the findings of this study are available from the corresponding author on reasonable request.
Results
VEC Clustering Triggers an Endothelial-Specific Transcription Program
To investigate whether VEC was able to upregulate other endothelial-specific genes besides claudin-5,5 we performed an RNA sequencing comparative analysis of a mouse VEC-null cell line (VEC-null) and the same line reconstituted with VEC wild-type cDNA (VEC-positive). Several genes were upregulated by VEC expression and clustering (Figure 1A). Taking advantage of previously published data on endothelial gene expression,12 we selected a list of endothelial genes whose expression was induced or repressed by VEC (Figure 1B; Online Tables I and II). Further validation of RNA sequencing data was performed by quantitative real-time polymerase chain reaction of VE-PTP,13 vWf,14 T-cell lymphoma invasion and metastasis-1 (Tiam1),15 LIM domain only-2 (Lmo2),16 signal transducer and activator of transcription-6 (Stat6)17 and Elk3 (ETS domain containing protein)18,19 among VEC-induced genes, hes-related family bHLH transcription factor with YRPW motif-1 (Hey1)20 and adrenomedullin (Adm)21 among VEC-repressed genes, and platelet/endothelial cell adhesion molecule-1 (Pecam1)22 and SRY (sex determining region Y)-box-18 (Sox18)19 as genes not influenced by VEC expression (Online Figures IA and IIA through IIC). Similar transcriptional changes were induced by VEC clustering when comparing human dermal microvascular ECs (HMEC-1) in sparse and confluent conditions (Online Figure IID through IIG). Interestingly, VEC also seemed to coordinate the expression of several genes promoting vascular stability, according DAVID Functional Annotation Clustering tool.23 For instance, we observed a general downregulation of genes involved in cell proliferation and sprouting angiogenesis, while extracellular matrix interaction and cell–cell adhesion were promoted (Figure 1C).
Figure 1.
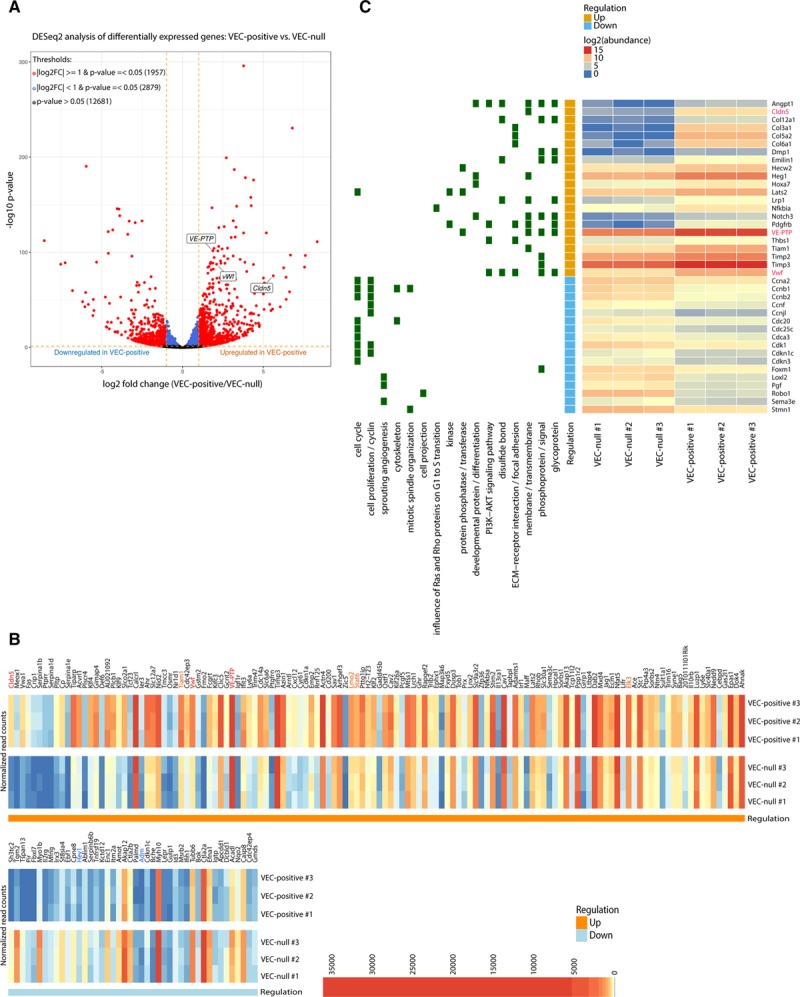
Transcriptome profile determined by VEC (vascular endothelial cadherin) expression and clustering. A, Volcano plot showing the magnitude of differential expression between VEC-positive and VEC-null endothelial cells (ECs). Each dot represents 1 gene with detectable expression in both cell types. The horizontal dashed line (orange) together with the vertical lines (orange) mark thresholds used (P value ≤0.05 and |log2FC| ≥1) to define a gene as differentially regulated in VEC-positive (red). Genes that only passed threshold P value ≤0.05 are depicted in blue. Dots representing claudin-5, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and von Willebrand factor (vWF) are labeled in the figure. B, Heat-map showing the expression pattern of significantly differentially expressed endothelial genes (P value ≤0.05 and |log2FC| ≥1) within and between biological replicates. Endothelial genes upregulated (red/orange) or downregulated (blue) in VEC-positive cells which were further investigated in this study are highlighted in the figure. Genes are displayed in decreasing |log2FC| order (left to right). C, Heat-map showing normalized abundance of significantly changing genes across all samples. Genes belonging to selected functionally enriched terms are highlighted in green on the left of the plot. Claudin-5, VE-PTP, and vWf genes are highlighted in red. In (B) and (C), VEC-positive numbers 1/2/3 represent biological replicates in VEC-positive cells, whereas VEC-null numbers 1/2/3 represent biological replicates in VEC-null cells.
Within VEC-upregulated endothelial genes, we selected the tyrosine phosphatase VE-PTP13 and the extracellular matrix protein vWf14 for further studies. These 2 genes were selected by virtue of their endothelial specificity and their described role in the induction of vascular stability.13,14 As reported for claudin-5,5 both genes were strongly upregulated by VEC expression and clustering (Online Figure IA). A mutant version of VEC composed of the cadherin cytoplasmic tail fused to the transmembrane and extracellular domains of IL-2 (interleukin-2) receptor α-chain (IL2-VEC),5 which is unable to cluster at cell–cell contacts, did not upregulate these genes, confirming the need of VEC clustering for this effect (Online Figure III). As for claudin-5,5 FoxO1 and β-catenin activity inhibited VE-PTP and vWf expression. Infection of confluent VEC-positive cells with an adenovirus encoding a constitutively active form of FoxO1 (FKHR-TM [Forkhead transcription factor triple mutant])24 significantly downregulated both VE-PTP and vWf expression (Online Figure IB). Similarly, increasing endogenous FoxO1 activity by LY294002-mediated inhibition of PI3K (phosphatidylinositol 3 kinase) reduced FoxO1-Ser256 (serine 256) phosphorylation level (Online Figure IC, upper) and downregulated both genes under study (Online Figure IC). β-Catenin associates with FoxO1 and stabilizes its binding to claudin-5 promoter.5 The expression of a stabilized version of β-catenin (ΔN-β-catenin)5 led to a marked downregulation of both VE-PTP and vWf genes (Online Figure ID), suggesting a regulatory mechanism similar to that of claudin-5. A promoter analysis spanning from 6000 bp upstream to 500 bp downstream of the transcription start site (TSS) of VE-PTP and vWf genes identified a series of paired Tcf (T-cell factor)/β-catenin/FoxO1-binding sites localized in 3 different regions on both promoters (Online Figure IE). Quantitative chromatin immunoprecipitation showed that FoxO1 binds all 3 regions in both promoters (Online Figure IF). Binding occurred only in confluent VEC-null and not in confluent VEC-positive cells, correlating with gene repression. Furthermore, β-catenin also bound all identified regions with different affinity (Online Figure IG), consistently with what previously reported.5 Thus, VEC upregulates the endothelial-specific genes vWf and VE-PTP through a mechanism similar to claudin-5 upregulation.
Claudin-5, VE-PTP, and vWf Are Polycomb Target Genes
We then aimed at clarifying how the FoxO1/β-catenin complex might act on claudin-5, VE-PTP, and vWf genes to induce their repression. Given the endothelial specificity of the identified genes, we hypothesized the possible involvement of PcG proteins, a group of transcriptional regulators involved in cell specification, which mediate reversible inhibition of transcription.6 We performed quantitative chromatin immunoprecipitation for PcG proteins on the TSS of claudin-5, VE-PTP, and vWf genes. Gene TSSs displayed higher enrichment in the components of both PRC2 (Ezh2 and Suz12) and PRC1 (Bmi1; B lymphoma Mo-MLV insertion region 1) in confluent VEC-null compared with VEC-positive cells (Figure 2A, top and lower left). The mark of Ezh2 enzymatic activity, H3K27me3, also showed a similar enrichment pattern (Figure 2A, lower right), and quantitative chromatin immunoprecipitation for total histone H3 showed comparable enrichments at the analyzed regions in VEC-null and VEC-positive cells (Online Figure IV), ruling out the possibility that the higher H3K27me3 signal in VEC-null cells could be because of widespread higher histone density. PcG-mediated repression is counteracted by the activity of TrxG (trithorax group) proteins.6 As expected, claudin-5, VE-PTP, and vWf TSSs showed a higher enrichment in H3K4me3, marker of TrxG activity and gene activation, in confluent VEC-positive cells than in VEC-null cells (Figure 2B, upper). RNA polymerase II phosphorylated on Ser5, a modification needed for the enzyme to escape the promoter and transcribe the gene, was also increased at the TSSs of genes in confluent VEC-positive cells (Figure 2B, lower).
Figure 2.
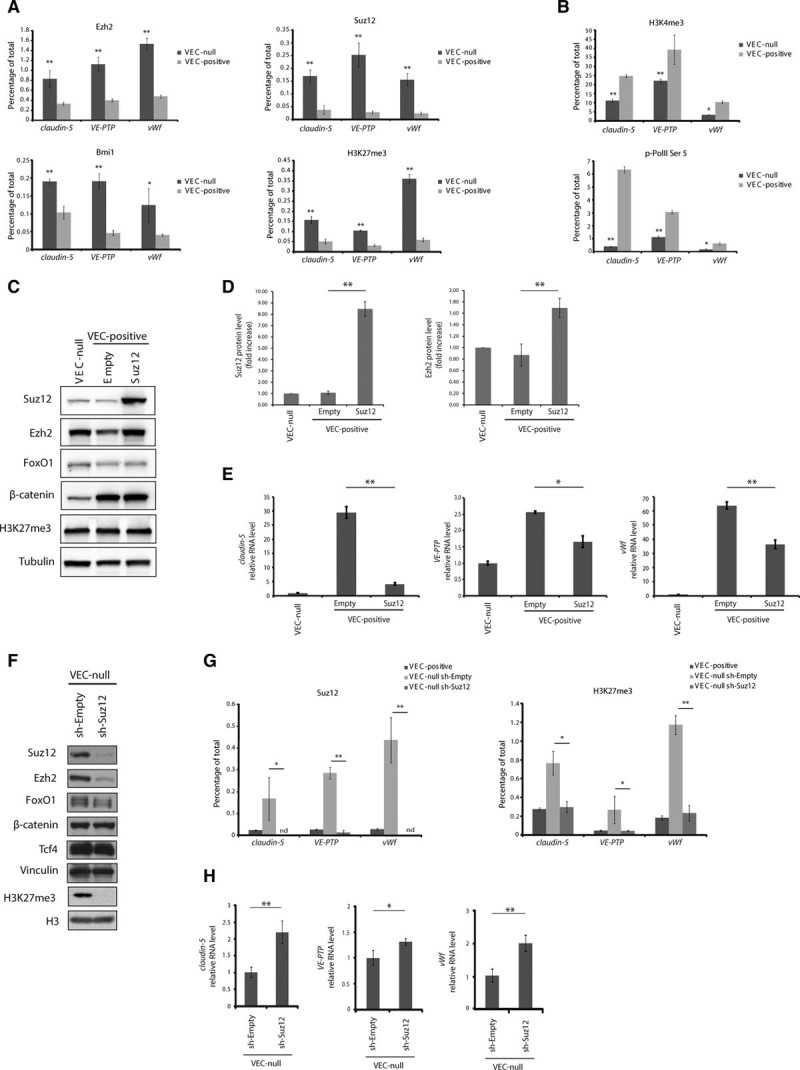
Claudin-5, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and von Willebrand factor (vWf) are polycomb targets. A, Quantitative real-time polymerase chain reaction (qRT-PCR) for the transcription start site (TSS) of claudin-5, VE-PTP, and vWf performed on endogenous Ezh (enhancer of zeste homolog)2-, Suz (suppressor of zeste)12-, Bmi1 (B lymphoma Mo-MLV insertion region 1)-, and H3K27me3 (histone H3 trimethylated on lysine 27)-bound chromatin immunoprecipitated from confluent VEC (vascular endothelial cadherin)-null and VEC-positive endothelial cells (ECs). B, qRT-PCR for the TSS of claudin-5, VE-PTP, and vWf performed on endogenous H3K4me3-bound and RNA polymerase II (p-PolII) Ser5–bound chromatin immunoprecipitated from confluent VEC-null and VEC-positive ECs. C, Western blot (WB) analysis of indicated proteins in extracts of confluent VEC-null and VEC-positive ECs upon Suz12 overexpression. D, Quantification of WB in (C). Suz12 and Ezh2 levels were normalized to tubulin. Columns are means±SEM of 3 independent experiments. E, qRT-PCR analysis of claudin-5, VE-PTP, and vWf expression in confluent VEC-null and VEC-positive ECs upon Suz12 overexpression. F, WB analysis of indicated proteins in extracts of confluent VEC-null ECs upon Suz12 knockdown (sh-Suz12). G, qRT-PCR for the TSS of claudin-5, VE-PTP, and vWf performed on endogenous Suz12- and H3K27me3-bound chromatin immunoprecipitated from confluent VEC-positive, VEC-null-sh-Empty, and VEC-null-sh-Suz12 ECs. H, qRT-PCR analysis of claudin-5, VE-PTP, and vWf expression in confluent VEC-null-sh-Empty and VEC-null-sh-Suz12 ECs. A, B, G, Levels of DNA are normalized to input, columns are means±SD of triplicates from a representative experiment. C, F, Tubulin and vinculin are the loading controls. E, H, Levels of mRNA are normalized to 18S; columns are means±SEM of triplicates from a representative experiment. In (A) and (B), *P<0.05; **P<0.01, t test VEC-null vs VEC-positive. In (G), *P<0.05; **P<0.01, t test VEC-null Sh-Empty vs VEC-null Sh-Suz12. In (D), (E), and (H), *P<0.05; **P<0.01, t test. nd indicates not detectable.
When we overexpressed the PRC2 member Suz12 in confluent VEC-positive cells using lentiviral-mediated gene delivery (Figure 2C), claudin-5, VE-PTP, and vWf expression was reduced (Figure 2E). PRC2 proteins are known to promote each other’s stability by physical interaction.8 Suz12 overexpression led to an almost 2-fold increase in Ezh2 protein level (Figure 2D). The promoter region of claudin-5 (3169 bp upstream of the coding sequence; Online Figure VA), comprising all 3 regions of paired Tcf/β-catenin/FoxO1-binding sites5 was cloned upstream of the firefly luciferase gene. As expected, transfection of this reporter in confluent VEC-null and VEC-positive cells showed higher luciferase activity in the presence of VEC expression and clustering (Online Figure VB). Suz12 overexpression in VEC-positive ECs significantly decreased luciferase expression (Online Figure VC). Lmo2 and Stat6 were also downregulated by Suz12 overexpression (Online Figure VIA), suggesting that the identified mechanism might regulate a wider set of endothelial genes. Hey1 expression was induced by Suz12 (Online Figure VIB), likely as a result of polycomb-mediated downregulation of a Hey1 gene repressor, while Pecam1 and Sox18 levels were not altered (Online Figure VIC). Conversely, Suz12 knockdown led to a marked decrease in Ezh2 and to the abrogation of detectable H3K27me3 levels (Figure 2F). FoxO1 protein levels were ≈30% lower on Suz12 knockdown (Figure 2F), although FoxO1 mRNA expression was unchanged (Online Figure VII). Suz12 knockdown abolished Suz12 and H3K27me3 signal at the TSSs of claudin-5, VE-PTP, and vWf genes (Figure 2G) causing a partial reactivation of their expression (Figure 2H). The incomplete rescue of expression might be because of the lack of specific gene activators missing in a VEC-null context or to PRC1 activity keeping genes partially repressed in the absence of PRC2.25 Furthermore, Suz12 knockdown induced an increase in claudin-5 promoter activity as assessed by luciferase reporter assay (Online Figure VD).
FoxO1/β-Catenin Enhance PcG Protein Binding to Claudin-5, VE-PTP, and vWf Promoters
We then hypothesized that the FoxO1/β-catenin complex could positively modulate polycomb interaction with the promoters of the identified genes. By coimmunoprecipitation, we found that constitutively active FKHR-TM or endogenous FoxO1 interact with Ezh2 (Figure 3A). Another member of PRC2, Suz12, also coimmunoprecipitated with FKHR-TM (Figure 3B). FKHR-TM overexpression in VEC-positive confluent cells increased PcG protein binding to the TSS of claudin-5, VE-PTP, and vWf genes, as shown by Ezh2 quantitative chromatin immunoprecipitation (Figure 3C, left). Consequently, H3K27me3 repressive histone mark was increased (Figure 3C, right). This is consistent with gene downregulation observed on FKHR-TM overexpression (Online Figure IB). No increase in Ezh2 protein levels was induced by FKHR-TM (Figure 3C inset), proving that the augmented Ezh2 enrichment was independent of protein upregulation. Conversely, FoxO1 knockdown strongly reduced Ezh2 recruitment at target sites in VEC-null cells to levels comparable to those detected in VEC-positive cells, or even lower (Figure 3D), in the absence of any Ezh2 downregulation (Figure 3D inset). Interestingly, this effect was dose dependent. SiRNA 2 was less efficient than siRNA 1 in knocking down FoxO1 (see FoxO1 long exp, Figure 3D inset), and this resulted in a weaker reduction of Ezh2 binding to gene TSSs.
Figure 3.
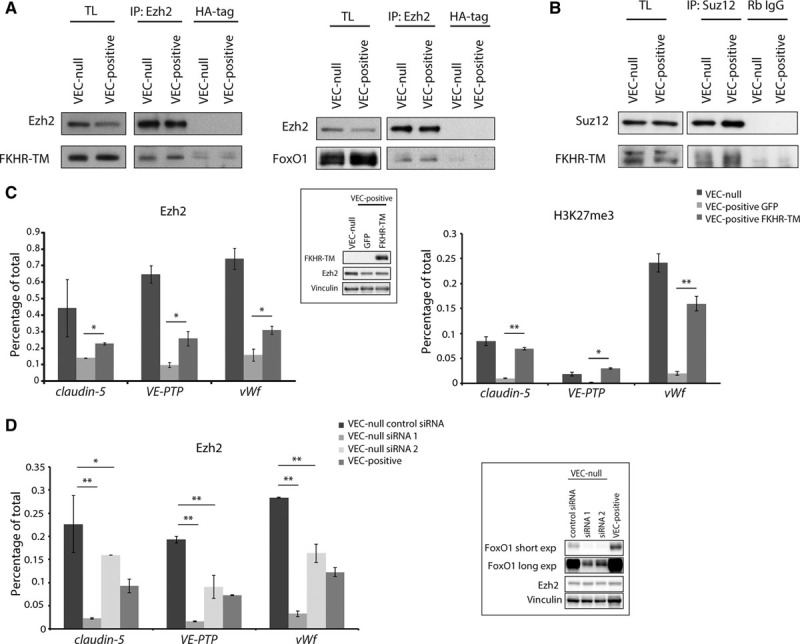
FoxO1 (Forkhead box protein O1) enhances PcG (polycomb group) protein association to claudin-5, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and von Willebrand factor (vWf) promoters. A, B, Coimmunoprecipitation and Western blot (WB) of endogenous Ezh (enhancer of zeste homolog)2 or Suz (suppressor of zeste)12 and endogenous FoxO1 or FKHR-TM (Forkhead transcription factor triple mutant) from extracts of confluent VEC (vascular endothelial cadherin)-null and VEC-positive endothelial cells (ECs) or the same cells types expressing FKHR-TM (myc-tagged). C, Quantitative real-time polymerase chain reaction (qRT-PCR) for the transcription start site (TSS) of claudin-5, VE-PTP, and vWf performed on endogenous Ezh2- and H3K27me3 (histone H3 trimethylated on lysine 27)-bound chromatin immunoprecipitated from confluent VEC-null and VEC-positive ECs expressing either FKHR-TM or GFP (green fluorescent protein; negative control). Inset: WB analysis of FKHR-TM and Ezh2. D, qRT-PCR for the TSS of claudin-5, VE-PTP, and vWf performed on endogenous Ezh2-bound chromatin immunoprecipitated from confluent VEC-positive or VEC-null ECs transfected with control siRNA or with 2 siRNAs targeting FoxO1 mRNA. Inset: WB analysis of FoxO1 and Ezh2. Two different film exposure timings are shown for FoxO1. C, D, Vinculin is the loading control. Levels of DNA are normalized to input, columns are means±SD of triplicates from a representative experiment. In (C), *P<0.05; **P<0.01, t test VEC-positive GFP vs VEC-positive FKHR-TM. In (D), *P<0.05; **P<0.01, t test VEC-null control siRNA vs VEC-null siRNA1 or VEC-null siRNA2. IP indicates immunoprecipitation; and TL, total cell lysate.
Coexpression of Suz12 and FKHR-TM in VEC-positive confluent cells induced a stronger repression of claudin-5 than the expression of FKHR-TM alone (75.3% versus 53.1%; Online Figure VIIIA and VIIIB), further supporting the hypothesis that FoxO1 acts in concert with polycomb activity.
We then investigated whether β-catenin could interact with PcG protein complex. Coimmunoprecipitation experiments showed an interaction between β-catenin and both Ezh2 and Suz12 (Figure 4A). Surprisingly, we detected a strong Ezh2/β-catenin association in VEC-positive confluent cells that was unlikely to be connected to the analyzed recruitment mechanism (Figure 4A, left). The meaning of this interaction will be further explored in the next section.
Figure 4.
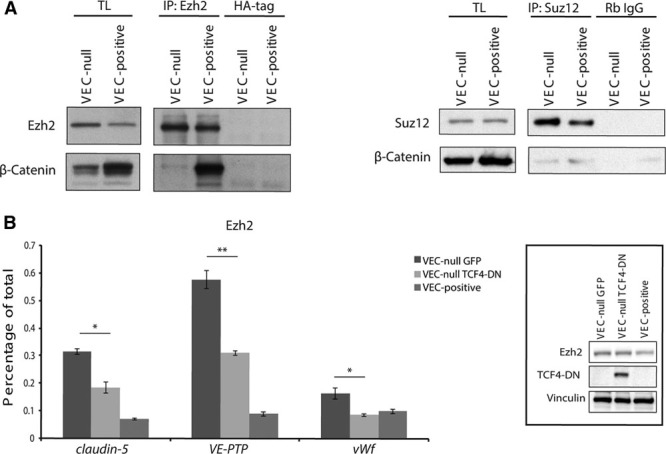
β-Catenin stabilizes polycomb/DNA interaction on claudin-5, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and von Willebrand factor (vWf) promoters. A, Coimmunoprecipitation and Western blot (WB) analysis of endogenous Ezh (enhancer of zeste homolog)2 or Suz (suppressor of zeste)12 and β-catenin from extracts of confluent VEC (vascular endothelial cadherin)-null and VEC-positive endothelial cells (ECs). B, Quantitative real-time polymerase chain reaction (qRT-PCR) for the TSS of claudin-5, VE-PTP, and vWf performed on endogenous Ezh2-bound chromatin immunoprecipitated from confluent VEC-positive and VEC-null ECs expressing dominant negative form of Tcf4 (TCF4-DN) or GFP (green fluorescent protein; negative control). Inset: WB analysis of TCF4-DN and Ezh2 in extracts from confluent VEC-positive and VEC-null ECs expressing TCF4-DN or control GFP. Vinculin is the loading control. *P<0.05; **P<0.01, t test VEC-null GFP vs VEC-null TCF4-DN. IP indicates immunoprecipitation; and TL, total cell lysate.
β-Catenin/FoxO1 association is known to stabilize FoxO1 binding to claudin-5 promoter. When TCF4-DN (dominant negative form of Tcf4), lacking the β-catenin–interacting region, was overexpressed in VEC-null cells, β-catenin/DNA interaction was abrogated,5 and Ezh2 binding to claudin-5, VE-PTP, and vWf promoters was strongly weakened (Figure 4B), suggesting a stabilizing role for β-catenin on PcG protein association to target sites.
VEC Associates With Ezh2 and Sequesters It at the Plasma Membrane
The strong Ezh2/β-catenin association detected in confluent VEC-positive cells (Figure 4A, left) suggested an alternative role for such interaction in this cell type. Indeed, Ezh2 coimmunoprecipitated with endogenous full-length VEC (Figure 5A, left), whereas no interaction was detected between Ezh2 and N-cadherin (Figure 5A, right). Ezh2–VEC interaction was confirmed in vivo in adult mice-derived whole lung extracts (Figure 5B). Biotinylation of cell surface proteins further proved that Ezh2 associates with VEC exposed on the cell membrane (Figure 5C). Moreover, junctional staining of Ezh2 was detected in confluent VEC-positive but not in VEC-null ECs (Figure 5D, arrow). Taken together these data demonstrate that the observed VEC–Ezh2 interaction sequesters the polycomb protein at the cell surface.
Figure 5.
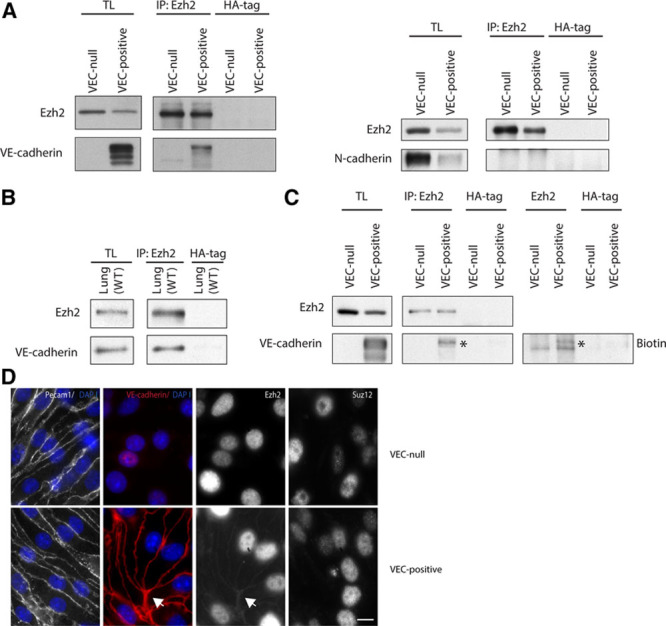
VEC (vascular endothelial cadherin) sequesters Ezh2 (enhancer of zeste homolog 2) at the plasma membrane. A, Coimmunoprecipitation and WB of endogenous Ezh2 and VEC or N-cadherin from extracts of confluent VEC-null and VEC-positive endothelial cells (ECs). B, Coimmunoprecipitation and Western blot (WB) of endogenous Ezh2 and VEC from wild-type (WT) murine whole lung extracts. C, Coimmunoprecipitation and WB of endogenous Ezh2 and VEC from extracts of confluent VEC-null and VEC-positive ECs after biotinylation of cell surface proteins. Asterisk highlights Ezh2-associated total and surface VEC bands. D, Immunofluorescence analysis of Ezh2 junctional localization (arrow) in confluent VEC-null and VEC-positive ECs. Junctional Suz (suppressor of zeste)12 was not detected. Platelet/endothelial cell adhesion molecule-1 (Pecam1) and VEC were used as junctional markers. Scale bar: 10 μm. IP indicates immunoprecipitation; and TL, total cell lysate.
To define whether Ezh2 interacts directly with VEC cytoplasmic tail and which Ezh2 domain is involved in such interaction, we designed a peptide array displaying 20-mers covering the entire amino acid sequence of mouse Ezh2 (746 amino acids). Each peptide spotted on the membrane overlapped the next one by 19 amino acids, resulting in a change of only a single amino acid per peptide and providing a high resolution in determining the interaction sites. Such peptide array was probed with glutathione S-transferase (GST)–tagged VEC cytoplasmic tail26 and free GST as control for nonspecific interactions (Online Figure IXA and IXB). Peptides corresponding to the spots displaying high signal in VEC cytoplasmic tail–probed array and no or low signal in GST-probed control (Online Figure IXB, red dots) were synthesized as biotinylated and used in streptavidin pull-down experiments to confirm protein interactions (Figure 6A). Peptides M6 and M10 corresponding to amino acids 450 to 469 and 454 to 473, respectively, were identified as the sites of Ezh2/VEC direct interaction (Figure 6A and 6B).
Figure 6.
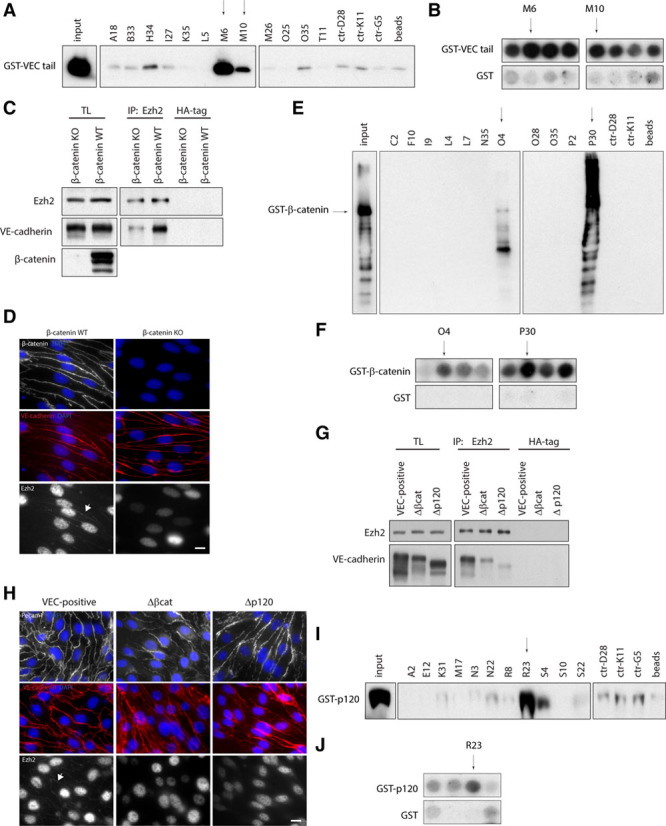
Analysis of Ezh2 (enhancer of zeste homolog 2) interaction with VEC (vascular endothelial cadherin) junctional complex. A, Streptavidin pull-down of selected biotinylated Ezh2 peptides and GST-tagged VEC cytoplasmic tail. GST-VEC cytoplasmic tail (400 ng) was loaded as input. Peptides displaying no interaction in peptide array were used as controls. Arrows indicate peptides showing positive signal. B, Regions of peptide array in the Online Figure IXA and IXB corresponding to selected peptides in (A). C, Coimmunoprecipitation and Western blot (WB) of Ezh2 and VEC from extracts of confluent β-catenin knockout (KO) and β-catenin wild-type (WT) endothelial cells (ECs). D, Immunofluorescence analysis of Ezh2 (arrow) and β-catenin junctional localization in confluent β-catenin KO and β-catenin WT ECs. VE-cadherin (red) was used as junctional marker. E, Streptavidin pull-down of selected biotinylated Ezh2 peptides and GST-tagged β-catenin. GST-β-catenin (400 ng) was loaded as input. Peptides displaying no interaction in peptide array were used as controls. Arrows indicate peptides showing positive signal. F, Regions of peptide array in the Online Figure IXA and IXC corresponding to selected peptides in (E). G, Coimmunoprecipitation and WB of Ezh2 and VEC from extracts of confluent VEC-positive, Δβcat, and Δp120 ECs. H, Immunofluorescence analysis of Ezh2 junctional localization (arrow) in confluent VEC-positive, Δβcat, and Δp120 ECs. VEC and platelet/endothelial cell adhesion molecule-1 (Pecam1) were used as junctional markers. I, Streptavidin pull-down of selected biotinylated Ezh2 peptides and GST-tagged p120-catenin. GST-p120-catenin (300 ng) was loaded as input. Peptides displaying no interaction in peptide array were used as controls. Arrow indicates peptide showing positive signal. J, Regions of peptide array in the Online Figure IXA and IXD corresponding to selected peptides in (I). In (D) and (H), scale bar: 10 μm. GST indicates glutathione S-transferase; HA, human influenza hemagglutinin; IP, immunoprecipitation; and TL, total cell lysate.
We then investigated the role of VEC cytoplasmic partner β-catenin in such interaction. Ezh2/VEC association was reduced in a β-catenin–null EC line (β-catenin knockout) compared with its wild-type counterpart (β-catenin wild type; Figure 6C and 6D). When VEC-null cells were reconstituted with a truncated mutant of VEC lacking the β-catenin–binding domain (Δβcat),5 thus unable to sequester β-catenin at the membrane, VEC displayed a marked reduction of Ezh2 binding (Figure 6G and 6H). These results strongly suggested that β-catenin association to VEC cytoplasmic tail is required for optimal Ezh2 junctional recruitment. Ezh2 peptide array was probed with GST-tagged β-catenin (Online Figure IXC). Biotinylated peptide pull-down with peptides corresponding to the spots displaying high signal in β-catenin–probed array and no or low signal in GST-probed control (Online Figure IXC, red dots) confirmed Ezh2 interaction with β-catenin at amino acids 522 to 541 and 585 to 604, corresponding to peptides O4 and P30 (Figure 6E and 6F).
Ezh2 also associated with another constituent of VEC cytoplasmic junctional complex, p120-catenin (Online Figure X). VEC-null cells reconstituted with a mutant version of VEC lacking the juxtamembrane p120-catenin binding region showed a marked reduction of VEC/Ezh2 interaction (Figure 6G and 6H), suggesting a role for p120-catenin in such association. Ezh2 peptide array was probed with GST-tagged p120-catenin, and the identified putative interacting peptides (Online Figure IXD, red dots) were selected for validation by biotinylated peptide pull-down (Figure 6I). Amino acids 652 to 671, corresponding to peptide R23, were identified as Ezh2 interaction site with p120-catenin (Figure 6I and 6J).
Overall, these data prove that Ezh2 interacts with VEC junctional complex at multiple sites. Whether the identified interactions correspond to different junctional pools of Ezh2 or a single complex is formed is a matter of future investigation.
Inhibition of Ezh2/VEC Interaction Downregulates Claudin-5, VE-PTP, and vWf Expression
To understand whether Ezh2 sequestration at AJs impacts the expression of claudin-5, VE-PTP, and vWf, we aimed at inhibiting Ezh2/VEC and Ezh2/β-catenin interactions. Selected Ezh2 peptides M6-M10 (VEC-binding sites) and O4-P30 (β-catenin–binding sites) were synthesized in tandem with transactivator of transcription (TAT) of human immunodeficiency virus, to allow peptide entry into the cell. Such TAT-conjugated peptides are expected to bind VEC or β-catenin engaging the domains needed for Ezh2 interaction, thus acting in a dominant negative fashion to inhibit Ezh2 junctional recruitment. VEC-positive cells were treated from subconfluent state throughout the period of time needed to establish AJs to saturate Ezh2-binding sites before the polycomb protein could localize at cell–cell contacts. Treatment with peptides TAT-P30 and TAT-M10 reduced VEC/Ezh2 association by 28% and 45.3%, respectively (Figure 7A). This caused an increase in Ezh2 recruitment to the TSS of claudin-5, VE-PTP, and vWf (Figure 7B), which, in the case of TAT-M10 treatment, corresponded to a significant repression of gene expression in comparison to treatment with a nonbinding TAT-control peptide (Figure 7C). The lack of gene downregulation after TAT-P30 treatment might be because of interference of this peptide with the correct assembly of the FoxO1/β-catenin/PRC2 complex at gene promoters.
Figure 7.
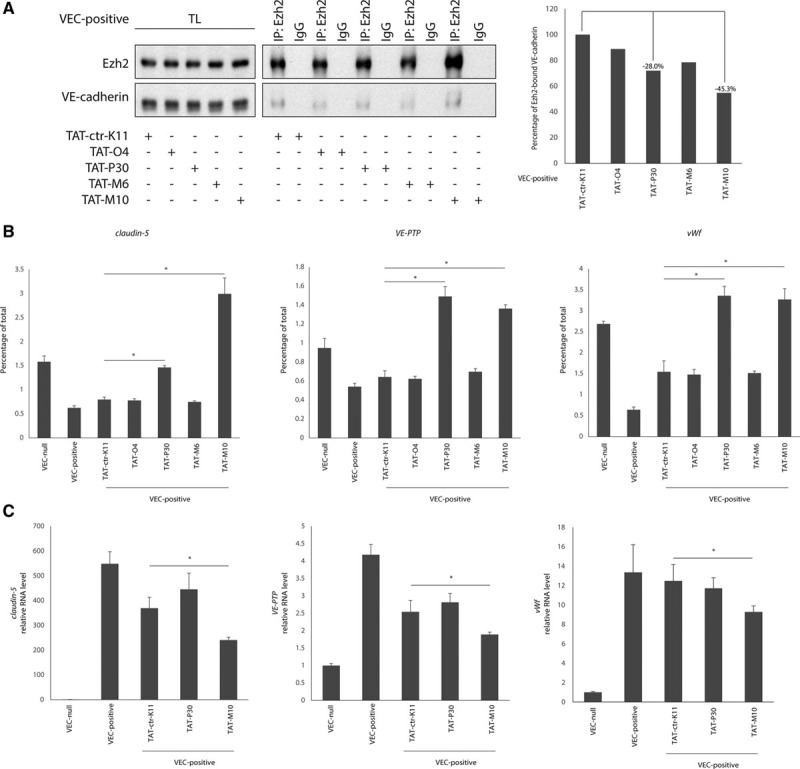
Inhibition of Ezh2 (enhancer of zeste homolog 2)/VEC (vascular endothelial cadherin) interaction causes claudin-5, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and von Willebrand factor (vWf) downregulation. A, Coimmunoprecipitation and Western blot (WB) of endogenous Ezh2 and VEC from extracts of VEC-positive endothelial cells (ECs) treated with VEC-binding transactivator of transcription (TAT)-M6 and TAT-M10 peptides, β-catenin–binding TAT-O4 and TAT-P30 peptides or nonbinding TAT-ctr-K11 peptide as control (left). Quantification of coprecipitated VEC protein normalized on precipitated Ezh2 level (right). B, Quantitative real-time polymerase chain reaction (qRT-PCR) for the transcription start site (TSS) of claudin-5, VE-PTP, and vWf performed on endogenous Ezh2-bound chromatin immunoprecipitated from VEC-positive ECs treated with VEC-binding TAT-M6 and TAT-M10 peptides, β-catenin–binding TAT-O4 and TAT-P30 peptides or nonbinding TAT-ctr-K11 peptide as control. Levels of DNA are normalized to input; columns are means±SD of triplicates from a representative experiment. *P<0.01, t test TAT-ctr-K11 vs TAT-P30 or TAT-M10 treatment. C, qRT-PCR analysis of claudin-5, VE-PTP, and vWf expression in VEC-positive ECs treated with VEC-binding TAT-M10 peptide, β-catenin–binding TAT-P30 peptide or nonbinding TAT-ctr-K11 peptide as control. Levels of mRNA are normalized to GAPDH; columns are means±SEM of triplicates from a representative experiment. *P<0.01, t test TAT-ctr-K11 vs. TAT-M10 treatment. IP indicates immunoprecipitation; and TL, total cell lysate.
Polycomb Activity Correlates With Claudin-5, VE-PTP, and vWf Repression In Vivo
To verify whether the inhibition of PRC2 activity could enhance the expression of the identified endothelial genes in vivo, we analyzed the effect of Ezh2/Ezh1 inhibition in mouse pups by pharmacological treatment with UNC1999. As reported in Figure 8A, drug administration increased VE-PTP and vWf expression in lung ECs, but was unable to increase claudin-5 expression over physiological levels.
Figure 8.
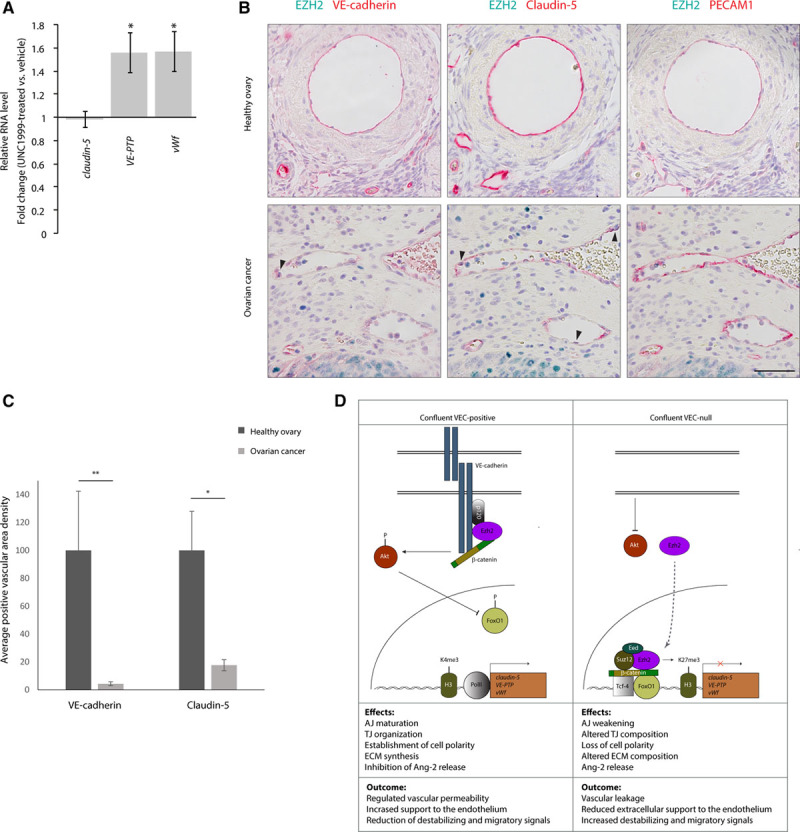
Ezh2 (enhancer of zeste homolog 2) activity correlates with claudin-5, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and von Willebrand factor (vWf) repression in vivo. A, Quantitative real-time polymerase chain reaction (qRT-PCR) analysis of claudin-5, VE-PTP, and vWf expression in lungs of vehicle- or UNC1999-treated pups (P6). Data are represented as fold change of UNC1999 treated vs vehicle and are means±SD from at least 4 mice per group. Gene expression was normalized to VEC (vascular endothelial cadherin) expression. B, Immunohistochemistry (IHC) staining of VEC, Claudin-5, platelet/endothelial cell adhesion molecule-1 (PECAM1; red) and EZH2 (green) expression in serial sections of human healthy ovary (upper) or serous surface papillary ovarian carcinoma (lower). Black arrowheads point to tumor vessel endothelial cells (ECs) expressing high levels of EZH2. Scale bar: 50 μm. C, Quantification of IHC stainings in (B). For VEC and claudin-5, areas of specific signal, divided by the total measured area, were normalized to the corresponding values of PECAM1 staining. Columns are means±SEM (n=3 healthy ovaries; 4 ovarian carcinomas; at least 3 fields per sample). D, Suggested model for the regulation of claudin-5, VE-PTP, and vWf genes. Clustered VEC recruits β-catenin and activates Akt leading to FoxO1 (Forkhead box protein O1) phosphorylation and inhibition.5 Furthermore, Ezh2 is sequestered at the cell membrane by association with VEC cytoplasmic tail (left). These mechanisms allow gene activation by impeding the recruitment of PcG (polycomb group) proteins to gene promoters. Claudin-5 expression allows the correct organization of tight junctions (TJs) and regulation of vessel permeability.5,27 VE-PTP regulates adherens junction (AJ) maturation28 and VEGFR2 (vascular endothelial growth factor receptor-2) activity,13,29 whereas vWf contributes to extracellular matrix (ECM) formation and inhibits Ang (angiopoietin)-2 release.14,30 These effects are likely to contribute to vessel stabilization and prevent vascular leakage. In (A) and (C), *P<0.05; **P<0.01, t test.
EZH2 overexpression is frequent in tumor cells and in vessels of human epithelial ovarian cancers and is associated with poor prognosis.31 Vessels of human healthy ovarian tissue presented very low EZH2 expression, while VEC, Claudin-5, and PECAM1 were clearly detectable (Figure 8B, upper). In contrast, in tumor vasculature, high nuclear EZH2 staining paralleled a significant reduction in VEC and Claudin-5, whereas PECAM1 expression was unaltered (Figure 8B, lower, and 8C). EC-associated vWf staining was highly variable in both healthy ovarian tissue and tumor samples, preventing a quantitative evaluation of its expression. VE-PTP staining quantification also presented problems of antibody specificity. Nonetheless, a reduction of VE-PTP signal could be appreciated in tumor vessels (Online Figure XI).
Discussion
In this study, we describe a novel mechanism through which VEC expression and clustering upregulate endothelial-specific genes and contribute to endothelial differentiation and stability. We found that VEC engagement at cell–cell contacts acts by inhibiting PcG protein–mediated epigenetic modifications of EC chromatin. PcG proteins have been implicated in several developmental processes,7,8 and Ezh2 activity was claimed to be involved in the regulation of genes important in tumor angiogenesis and Kaposi Sarcoma.31 Furthermore, Ezh2 was reported to play a role in maintaining vascular integrity during embryonic development.32
However, these studies mainly focused on the effects of Ezh2 inhibition without investigating in detail the mechanisms of polycomb regulation in ECs. We observed that VEC expression and clustering were able to trigger a gene expression program contributing to endothelial differentiation. Some of these endothelial genes seemed to be regulated by polycomb activity, thus pointing to a possible central role of VEC/polycomb crosstalk in orchestrating endothelial specification.
Within the set of genes upregulated by VEC, we selected endothelial-specific claudin-5, VE-PTP, and vWf. Claudin-5 is a major component of endothelial TJs and determines size and charge selectivity of endothelial paracellular permeability.4,5,27,33
VE-PTP exerts different context-specific activities including associating with VEC and reducing its tyrosine phosphorylation.34 Absence of VE-PTP impairs AJ maturation, inducing vessel destabilization and increased permeability.28,35 VE-PTP associates with Tie-2 receptor modulating Ang (angiopoietin) signaling36 and interacts with VEGFR2 (vascular endothelial growth factor receptor-2) limiting its signaling activity.13,29
vWf is a crucial component of EC extracellular matrix that provides mechanical support to the EC monolayer30 and limits the release of vessel-destabilizing Ang-2 from endothelial Weibel–Palade bodies.14 Consistent with our data, a larger pool of vWf is present in confluent ECs compared with subconfluent condition.37
Although our analysis was mainly focused on these 3 genes, other genes regulated by VEC are involved in inhibition of cell proliferation, decrease of sprouting angiogenesis, promotion of cell adhesion, and production of extracellular matrix, suggesting a broader role of VEC expression and clustering in inducing vessel stabilization.
We previously found5 that VEC expression derepresses the endothelial-specific gene claudin-5 by restraining the activity of FoxO1 and β-catenin.33 Here, we show that nuclear FoxO1 and β-catenin increase polycomb targeting to the promoter regions of a set of endothelial genes. We observed that induction of claudin-5,5 VE-PTP, and vWf relies on VEC capacity to activate the PI3K/AKT pathway, leading to FoxO1 inactivation,4 and to sequester β-catenin at the cell membrane. The absence of VEC clustering prevented the expression of this set of genes. The work presented here confirms and extends what previously observed for claudin-55 and further underlines the role of FoxO1 as a key transcription factor in endothelial differentiation and homeostasis.38 This is in agreement with a recent report highlighting a key role of Akt1 activation and FoxO inhibition in stabilizing the endothelial barrier and preventing vascular leakage.39
Little is known about the molecular mechanisms of FoxO1-mediated gene repression. We found that FoxO1 physically associates with PcG proteins and, through still unknown mechanisms, contributes to their association to a selected set of endothelial gene promoters inducing chromatin conformational changes and gene inhibition. β-Catenin takes part in such multiprotein complex and stabilizes PcG protein binding to DNA, as TCF4-DN–mediated abrogation of β-catenin/DNA binding destabilizes Ezh2/promoter interaction.
PcG proteins have been previously reported to have atypical extranuclear localizations.40,41 We show here that VEC also reduces polycomb nuclear activity by sequestering Ezh2 at the plasma membrane in a β-catenin–dependent and p-120-catenin–dependent manner. Ezh2 interaction with VEC was confirmed in vivo in mouse lung extracts in the absence of any crosslinking reaction, whereas no association was detected between Ezh2 and the other major classical endothelial cadherin, N-cadherin, likely as a result of the reduced binding of this adhesion molecule to p120-catenin.42 Recently, EZH2 has been shown to interact with β-catenin in liver cancer stem cells43 through its N-terminal domain (amino acids 1–334). This was defined by domain mapping assays using EZH2 truncation mutants. Our analysis identified the Ezh2 sites of direct interaction with β-catenin in the domain comprised between amino acids 522 and 604. Deletion of domain 1 to 334 might alter the protein structural integrity, affecting EZH2/β-catenin interaction at downstream residues.
Ezh2/VEC association was shown to play a functional role in regulating gene expression. Inhibition of such interaction, and likely the consequent release of Ezh2 protein in the cytoplasm, increased its recruitment to claudin-5, VE-PTP, and vWf gene promoters causing their downregulation. The strong increase of Ezh2 recruitment at gene promoters, however, did not parallel a similarly strong repression of gene expression.
This was likely because of reduced FoxO1 and β-catenin nuclear abundancy in the confluent VEC-positive condition, impeding the correct assembly of the nuclear repressive complex.
Thus, we propose that an event happening at cell–cell contacts, that is, VEC clustering, triggers and orchestrates a network of pathways and epigenetic events, regulating several aspects of endothelial homeostasis and vascular stability (Figure 8D).
Pharmacological inhibition of Ezh2/Ezh1 led to the upregulation of VE-PTP and vWf in mouse pups, confirming that these genes are dependent on polycomb activity in vivo. Claudin-5 was not significantly upregulated by such treatment, likely because its expression is unable to increase over a physiological threshold. Additional studies are needed for understanding the kinetics of expression of this gene in the presence or absence of polycombs in different vascular regions.
Deregulated angiogenesis is a characteristic of several human pathologies and is a hallmark of cancer. Tumor vessels present altered endothelial cell–cell junctions, are hyperpermeable, have low pericyte coverage, and altered basement membrane composition and stiffness. Tumor vessel stabilization by increased VEC has been shown to reduce tumor metastasis.44,45 Ezh2 expression is increased in tumor-associated ECs, and this has been linked to increased vascular density and reduced vessel maturation.31 Our results show that reduced VEC and increased EZH2 nuclear staining in human ovarian carcinoma vessels parallel the downregulation of Claudin-5 and VE-PTP, suggesting a possible alteration of the identified signaling mechanism.
In conclusion, this work introduces a novel role for VEC in the regulation of polycomb activity and, consequently, EC differentiation and vascular maturation. The available genetic and pharmacological approaches to inhibit polycomb function will be instrumental to identify the role of this mechanism in different pathological conditions of the vascular system and to investigate new therapeutic opportunities to induce vessel normalization in cancer.4
Acknowledgments
We thank Luca Ferrarini for help in bioinformatic analysis, Djamil A. Damry for scientific input, and Emanuele Martini and Dario Parazzoli for imaging analysis. The anti–vascular endothelial-protein tyrosine phosphatase (VE-PTP; hPTPb1-8) antibody and FKHR-TM (Forkhead transcription factor triple mutant) adenovirus were kind gifts of Dietmar Vestweber and Christopher Daly, respectively.
Sources of Funding
This work was supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC001057), the UK Medical Research Council (FC001057), and the Wellcome Trust (FC001057); by Associazione Italiana per la Ricerca sul Cancro (AIRC; investigator grant [IG] 16683), AIRC IG2016 18683 and Special Program Molecular Clinical Oncology 5x1000 to AGIMM (AIRC Gruppo Italiano Malattie Mieloproliferative; 10005); by the European Research Council (project EC-ERC-VEPC contract 742922); by ITN (initial training networks) BtRAIN grant 675619; by CARIPLO Foundation (2008.2463) and TELETHON (GGP14149). Research was also supported by the grants R01CA218600, R01GM122749, and R01HD088626 (to J. Jin) from the US National Institutes of Health. Sequencing was performed by the SNP&SEQ Technology platform in Uppsala. The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory. The SNP&SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation. E. Dejana is supported by the Swedish Science Council and the Knut and Alice Wallenberg Foundation.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- Adm
- adrenomedullin
- AJs
- adherens junctions
- Ang
- angiopoietin
- ECs
- endothelial cells
- Eed
- embryonic ectoderm development
- Ezh2
- enhancer of zeste homolog 2
- H3K27me3
- histone H3 trimethylated on lysine 27
- Hey1
- Hes-related family bHLH transcription factor with YRPW motif-1
- HMEC-1
- human dermal microvascular endothelial cells-1
- IL
- interleukin
- Lmo2
- LIM domain only-2
- PcG
- polycomb group
- Pecam1
- platelet/endothelial cell adhesion molecule-1
- PI3K
- phosphatidylinositol 3 kinase
- PRC
- polycomb repressive complex
- Rbap48
- retinoblastoma-binding protein 48
- Sox18
- SRY (sex determining region Y)-box-18
- Stat6
- signal transducer and activator of transcription-6
- Suz12
- suppressor of zeste 12
- TAT
- transactivator of transcription
- Tcf
- T-cell factor
- TCF4-DN
- dominant negative form of Tcf4
- Tiam1
- T-cell lymphoma invasion and metastasis-1
- TJ
- tight junction
- TrxG
- trithorax group
- TSS
- transcription start site
- VEC
- vascular endothelial cadherin
- VEGFR2
- vascular endothelial growth factor receptor-2
- VE-PTP
- vascular endothelial-protein tyrosine phosphatase
- vWf
- von Willebrand factor
In November 2017, the average time from submission to first decision for all original research papers submitted to Circulation Research was 11.99 days.
M.F. Morini and C. Giampietro contributed equally to this study.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.117.312392/-/DC1.
Novelty and Significance
What Is Known?
Pathological conditions such as inflammation, diabetic retinopathy, and age-related macular degeneration are characterized by deregulated angiogenesis and unstable blood vessels.
Solid tumors receive nutrients and oxygen from newly formed vessels that display structural abnormalities such as lack of hierarchy, abnormal lumen, altered endothelial cell–cell junctions, and poor control of permeability.
VEC (vascular endothelial cadherin) and its clustering at cell–cell adherens junctions upregulate claudin-5, key component of endothelial tight junctions regulating vessel permeability, via inhibition of FoxO1 (Forkhead box protein O1) and β-catenin nuclear translocation.
What New Information Does This Article Contribute?
VEC expression and clustering upregulate a wide set of genes involved in endothelial differentiation and vascular stabilization, such as vascular endothelial-protein tyrosine phosphatase (VE-PTP) and von Willebrand factor (vWf) along with previously identified claudin-5.
Claudin-5, VE-PTP, and vWF are expressed via inhibition of polycomb protein binding to their promoters by preventing the nuclear accumulation of FoxO1 and β-catenin, which associate with PRC2 (polycomb repressive complex-2) and promote its localization at target genes.
VEC sequesters Ezh2 (enhancer of zeste homolog 2), a key component of PRC2, at the plasma membrane in a β-catenin–dependent and p120-catenin–dependent manner, further reducing polycomb nuclear activity.
Solid tumors and other human pathologies display abnormal, destabilized blood vessels, with wide and irregular endothelial cell–cell junctions. These leaky vessels favor metastatic dissemination of tumor cells, cause hemorrhages, and reduce delivery of therapeutic agents. We found that VEC is able to coordinate the expression of genes involved in endothelial differentiation and vascular stabilization. In particular, VEC expression and clustering upregulate stability-related genes claudin-5, VE-PTP, and vWF by preventing polycomb protein binding to their promoters via inhibition of FoxO1/β-catenin nuclear localization and sequestration of PRC2 component Ezh2 at the plasma membrane. This study shows that adherens junction establishment influences chromatin organization by modulating polycomb activity and that cadherin-mediated recruitment of a polycomb protein can influence gene expression. This mechanism depends on cadherin type, as no Ezh2/N-cadherin interaction is observed. Moreover, the work highlights a new role for FoxO1/β-catenin complex in localizing polycombs at target endothelial gene promoters. In vessels of human ovarian carcinomas, downregulation of VEC parallels reduced Claudin-5 and VE-PTP expression, pointing at a possible involvement of the identified mechanism. The data presented here suggest therapeutic opportunities to induce vascular normalization through inhibition of the polycomb-mediated repression system in pathological states characterized by vascular leakiness and fragility.
References
- 1.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–427. doi: 10.1038/nrd3455. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 2.Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003;163:1801–1815. doi: 10.1016/S0002-9440(10)63540-7. doi: 10.1016/S0002-9440(10)63540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26:441–454. doi: 10.1016/j.devcel.2013.08.020. doi: 10.1016/j.devcel.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 4.Dejana E, Giampietro C. Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol. 2012;19:218–223. doi: 10.1097/MOH.0b013e3283523e1c. doi: 10.1097/MOH.0b013e3283523e1c. [DOI] [PubMed] [Google Scholar]
- 5.Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- 6.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 7.Conway E, Healy E, Bracken AP. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol. 2015;37:42–48. doi: 10.1016/j.ceb.2015.10.003. doi: 10.1016/j.ceb.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061–4071. doi: 10.1038/sj.emboj.7600402. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morey L, Helin K. Polycomb group protein-mediated repression of transcription. Trends Biochem Sci. 2010;35:323–332. doi: 10.1016/j.tibs.2010.02.009. doi: 10.1016/j.tibs.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Simon JA, Kingston RE. Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell. 2013;49:808–824. doi: 10.1016/j.molcel.2013.02.013. doi: 10.1016/j.molcel.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuettengruber B, Cavalli G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development. 2009;136:3531–3542. doi: 10.1242/dev.033902. doi: 10.1242/dev.033902. [DOI] [PubMed] [Google Scholar]
- 12.Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010;5:e13741. doi: 10.1371/journal.pone.0013741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Küppers V, Vockel M, Nottebaum AF, Vestweber D. Phosphatases and kinases as regulators of the endothelial barrier function. Cell Tissue Res. 2014;355:577–586. doi: 10.1007/s00441-014-1812-1. doi: 10.1007/s00441-014-1812-1. [DOI] [PubMed] [Google Scholar]
- 14.Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TA, Sutton RE, Payne EM, Haskard DO, Hughes AD, Cutler DF, Laffan MA, Randi AM. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117:1071–1080. doi: 10.1182/blood-2010-01-264507. doi: 10.1182/blood-2010-01-264507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lampugnani MG, Zanetti A, Breviario F, Balconi G, Orsenigo F, Corada M, Spagnuolo R, Betson M, Braga V, Dejana E. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell. 2002;13:1175–1189. doi: 10.1091/mbc.01-07-0368. doi: 10.1091/mbc.01-07-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coma S, Allard-Ratick M, Akino T, van Meeteren LA, Mammoto A, Klagsbrun M. GATA2 and Lmo2 control angiogenesis and lymphangiogenesis via direct transcriptional regulation of neuropilin-2. Angiogenesis. 2013;16:939–952. doi: 10.1007/s10456-013-9370-9. doi: 10.1007/s10456-013-9370-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalmasso AP, Goldish D, Benson BA, Tsai AK, Wasiluk KR, Vercellotti GM. Interleukin-4 induces up-regulation of endothelial cell claudin-5 through activation of FoxO1: role in protection from complement-mediated injury. J Biol Chem. 2014;289:838–847. doi: 10.1074/jbc.M113.455766. doi: 10.1074/jbc.M113.455766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dejana E, Taddei A, Randi AM. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim Biophys Acta. 2007;1775:298–312. doi: 10.1016/j.bbcan.2007.05.003. doi: 10.1016/j.bbcan.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Fontijn RD, Volger OL, Fledderus JO, Reijerkerk A, de Vries HE, Horrevoets AJ. SOX-18 controls endothelial-specific claudin-5 gene expression and barrier function. Am J Physiol Heart Circ Physiol. 2008;294:H891–H900. doi: 10.1152/ajpheart.01248.2007. doi: 10.1152/ajpheart.01248.2007. [DOI] [PubMed] [Google Scholar]
- 20.Kofler NM, Shawber CJ, Kangsamaksin T, Reed HO, Galatioto J, Kitajewski J. Notch signaling in developmental and tumor angiogenesis. Genes Cancer. 2011;2:1106–1116. doi: 10.1177/1947601911423030. doi: 10.1177/1947601911423030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang TH, Chu TY. Repression of miR-126 and upregulation of adrenomedullin in the stromal endothelium by cancer-stromal cross talks confers angiogenesis of cervical cancer. Oncogene. 2014;33:3636–3647. doi: 10.1038/onc.2013.335. doi: 10.1038/onc.2013.335. [DOI] [PubMed] [Google Scholar]
- 22.Nourshargh S, Krombach F, Dejana E. The role of JAM-A and PECAM-1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J Leukoc Biol. 2006;80:714–718. doi: 10.1189/jlb.1105645. doi: 10.1189/jlb.1105645. [DOI] [PubMed] [Google Scholar]
- 23.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 24.Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, Lai KM, Lin HC, Ioffe E, Yancopoulos GD, Rudge JS. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev. 2004;18:1060–1071. doi: 10.1101/gad.1189704. doi: 10.1101/gad.1189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010;24:265–276. doi: 10.1101/gad.544410. doi: 10.1101/gad.544410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giampietro C, Disanza A, Bravi L, Barrios-Rodiles M, Corada M, Frittoli E, Savorani C, Lampugnani MG, Boggetti B, Niessen C, Wrana JL, Scita G, Dejana E. The actin-binding protein EPS8 binds VE-cadherin and modulates YAP localization and signaling. J Cell Biol. 2015;211:1177–1192. doi: 10.1083/jcb.201501089. doi: 10.1083/jcb.201501089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carra S, Foglia E, Cermenati S, Bresciani E, Giampietro C, Lora Lamia C, Dejana E, Beltrame M, Cotelli F. Ve-ptp modulates vascular integrity by promoting adherens junction maturation. PLoS One. 2012;7:e51245. doi: 10.1371/journal.pone.0051245. doi: 10.1371/journal.pone.0051245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayashi M, Majumdar A, Li X, et al. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun. 2013;4:1672. doi: 10.1038/ncomms2683. doi: 10.1038/ncomms2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruggeri ZM. The role of von Willebrand factor in thrombus formation. Thromb Res. 2007;120(suppl 1):S5–S9. doi: 10.1016/j.thromres.2007.03.011. doi: 10.1016/j.thromres.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu C, Han HD, Mangala LS, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18:185–197. doi: 10.1016/j.ccr.2010.06.016. doi: 10.1016/j.ccr.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delgado-Olguín P, Dang LT, He D, Thomas S, Chi L, Sukonnik T, Khyzha N, Dobenecker MW, Fish JE, Bruneau BG. Ezh2-mediated repression of a transcriptional pathway upstream of Mmp9 maintains integrity of the developing vasculature. Development. 2014;141:4610–4617. doi: 10.1242/dev.112607. doi: 10.1242/dev.112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beard RS, Jr, Haines RJ, Wu KY, Reynolds JJ, Davis SM, Elliott JE, Malinin NL, Chatterjee V, Cha BJ, Wu MH, Yuan SY. Non-muscle Mlck is required for β-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1β-mediated barrier dysfunction in brain endothelial cells. J Cell Sci. 2014;127:1840–1853. doi: 10.1242/jcs.144550. doi: 10.1242/jcs.144550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, Golding M, Shima DT, Deutsch U, Vestweber D. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. 2002;21:4885–4895. doi: 10.1093/emboj/cdf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, Filippova K, Lyck R, Engelhardt B, Kamenyeva O, Bixel MG, Butz S, Vestweber D. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med. 2008;205:2929–2945. doi: 10.1084/jem.20080406. doi: 10.1084/jem.20080406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saharinen P, Eklund L, Miettinen J, Wirkkala R, Anisimov A, Winderlich M, Nottebaum A, Vestweber D, Deutsch U, Koh GY, Olsen BR, Alitalo K. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol. 2008;10:527–537. doi: 10.1038/ncb1715. doi: 10.1038/ncb1715. [DOI] [PubMed] [Google Scholar]
- 37.Howell GJ, Herbert SP, Smith JM, Mittar S, Ewan LC, Mohammed M, Hunter AR, Simpson N, Turner AJ, Zachary I, Walker JH, Ponnambalam S. Endothelial cell confluence regulates Weibel-Palade body formation. Mol Membr Biol. 2004;21:413–421. doi: 10.1080/09687860400011571. doi: 10.1080/09687860400011571. [DOI] [PubMed] [Google Scholar]
- 38.Wilhelm K, Happel K, Eelen G, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529:216–220. doi: 10.1038/nature16498. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao F, Artham S, Sabbineni H, Al-Azayzih A, Peng XD, Hay N, Adams RH, Byzova TV, Somanath PR. Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover. Cell Mol Life Sci. 2016;73:3917–3933. doi: 10.1007/s00018-016-2232-z. doi: 10.1007/s00018-016-2232-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Witte V, Laffert B, Rosorius O, Lischka P, Blume K, Galler G, Stilper A, Willbold D, D’Aloja P, Sixt M, Kolanus J, Ott M, Kolanus W, Schuler G, Baur AS. HIV-1 Nef mimics an integrin receptor signal that recruits the polycomb group protein Eed to the plasma membrane. Mol Cell. 2004;13:179–190. doi: 10.1016/s1097-2765(04)00004-8. [DOI] [PubMed] [Google Scholar]
- 41.Su IH, Dobenecker MW, Dickinson E, Oser M, Basavaraj A, Marqueron R, Viale A, Reinberg D, Wülfing C, Tarakhovsky A. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell. 2005;121:425–436. doi: 10.1016/j.cell.2005.02.029. doi: 10.1016/j.cell.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 42.Giampietro C, Taddei A, Corada M, Sarra-Ferraris GM, Alcalay M, Cavallaro U, Orsenigo F, Lampugnani MG, Dejana E. Overlapping and divergent signaling pathways of N-cadherin and VE-cadherin in endothelial cells. Blood. 2012;119:2159–2170. doi: 10.1182/blood-2011-09-381012. doi: 10.1182/blood-2011-09-381012. [DOI] [PubMed] [Google Scholar]
- 43.Zhu P, Wang Y, Huang G, Ye B, Liu B, Wu J, Du Y, He L, Fan Z. lnc-β-Catm elicits EZH2-dependent β-catenin stabilization and sustains liver CSC self-renewal. Nat Struct Mol Biol. 2016;23:631–639. doi: 10.1038/nsmb.3235. doi: 10.1038/nsmb.3235. [DOI] [PubMed] [Google Scholar]
- 44.Mazzone M, Dettori D, de Oliveira RL, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Y, Ting KK, Li J, et al. Targeting vascular endothelial-cadherin in tumor-associated blood vessels promotes T-cell-mediated immunotherapy. Cancer Res. 2017;77:4434–4447. doi: 10.1158/0008-5472.CAN-16-3129. doi: 10.1158/0008-5472.CAN-16-3129. [DOI] [PubMed] [Google Scholar]