

Ancient methane emitted to Arctic Ocean shelf waters is largely prevented from reaching the atmosphere.
Abstract
In response to warming climate, methane can be released to Arctic Ocean sediment and waters from thawing subsea permafrost and decomposing methane hydrates. However, it is unknown whether methane derived from this sediment storehouse of frozen ancient carbon reaches the atmosphere. We quantified the fraction of methane derived from ancient sources in shelf waters of the U.S. Beaufort Sea, a region that has both permafrost and methane hydrates and is experiencing significant warming. Although the radiocarbon-methane analyses indicate that ancient carbon is being mobilized and emitted as methane into shelf bottom waters, surprisingly, we find that methane in surface waters is principally derived from modern-aged carbon. We report that at and beyond approximately the 30-m isobath, ancient sources that dominate in deep waters contribute, at most, 10 ± 3% of the surface water methane. These results suggest that even if there is a heightened liberation of ancient carbon–sourced methane as climate change proceeds, oceanic oxidation and dispersion processes can strongly limit its emission to the atmosphere.
INTRODUCTION
Methane (CH4) emissions from Arctic Ocean shelf seas are anomalously large relative to those of the global mean ocean (1–4), but the source of these emissions remains largely unknown. Permafrost, which contains perennially frozen ancient carbon (C) (5), and CH4 hydrate, an ice-like form of CH4 that is principally ancient and older than surrounding sediment (6), are often invoked as likely sources because both constitute large C reservoirs and can be converted to CH4 gas as a result of warming climate. Although the global atmospheric CH4 inventory is increasing, arctic CH4 growth rates are comparable to or less than the global average (7) and appear to be derived mainly from biogenic sources (2, 8, 9). Ancient C stores, including arctic permafrost and hydrates, were recently determined to have contributed ≤19% of the CH4 released to the atmosphere during the Younger Dryas–Preboreal abrupt warming event (10), an analog to climate change today. Because of residual, fundamental unknowns about CH4 emissions from permafrost and hydrates, this potentially catastrophic climatological feedback has been absent from most Earth system models (5, 11).
Previous studies of CH4 dynamics in Arctic Ocean continental margins have measured atmospheric CH4 mole fractions ([CH4]), dissolved [CH4], and dissolved stable C isotopes (δ13C-CH4) to document emissions from the seafloor to the water column and from the water column to the atmosphere (1–4, 12–16). Because no study has conclusively fingerprinted the source of this CH4, it is unknown what fraction emitted to the atmosphere from the shallow arctic shelf seas is derived from ancient C sources. These ancient C CH4 sources are terrestrial and subsea permafrost via the biological transformation of thawed organic C (5), subsea permafrost–associated CH4 hydrates (6), and geologic CH4. Methane sources to seawater derived from modern-aged C include the atmosphere (17) and in situ production from more modern-aged substrates (12, 18).
Ancient and modern C–sourced CH4 can be readily distinguished with natural abundance 14C-CH4 measurements, as radioactive decay leaves ancient C sources substantially depleted in 14C with respect to modern C sources. Thermonuclear weapons and nuclear power generation have introduced anthropogenic 14C into atmospheric and oceanic CH4 (17, 19). We collected dissolved 14C-CH4 samples to test the hypotheses that (i) ancient C sources contribute CH4 to Arctic Ocean continental shelf waters and (ii) the contribution of ancient C sources to surface water and atmospheric CH4 in this environment diminishes as proximity to these sources decreases (that is, as water depth and distance from shore increase). Without newly developed techniques (Materials and Methods) (20), testing these hypotheses would not have been possible due to the challenge of collecting sufficient quantities of CH4 for natural abundance 14C-CH4 analysis in surface waters (1, 3, 4, 12, 13, 15, 16).
RESULTS AND DISCUSSION
The continental shelf offshore Prudhoe Bay, AK, in the U.S. Beaufort Sea was chosen as an ideal site to assess the input of ancient C–sourced CH4 to surface waters (Fig. 1A). Figure 1B illustrates the components of the Prudhoe Bay system schematically, including 14C measurements of dissolved CH4 and possible ancient and modern endmembers. The seaward extent of persistent subsea ice-bonded permafrost in this shelf sea, which was unglaciated land during the Late Pleistocene, has been determined from seismic reflection analysis (21) and verified with direct evidence from borehole well data (Fig. 1A) (22). Gas hydrates may occur within and beneath permafrost in this passive margin shelf (22) and may dissociate to release CH4 even after the permafrost matrix has thawed (6). Terrestrial peat and permafrost soils (5, 23, 24), including yedoma permafrost (25), are other potential sources of ancient CH4 delivered to the shelf by rivers [mainly the Colville and Mackenzie rivers (24)], coastal erosion, and submarine groundwater discharge (26) (Fig. 1B). Rates of both terrestrial permafrost degradation near the Colville River and erosion along the area’s permafrost-dominated coastline have been increasing in recent years (27, 28). Atmospheric CH4 in this system (and globally, as described above) has a 14C activity above modern because the atmosphere is both the site of natural 14C production and influenced by 14C-enriched CH4 produced by nuclear reactors (17). A second modern CH4 source in the system is in situ aerobic methanogenesis associated with the production and decomposition of phytoplankton biomass (12, 18), which we assume is similar to the measured 14C content of dissolved inorganic carbon (DIC) in surface waters (Fig. 1B). Anaerobic methanogenesis from the metabolism of recently fixed organic matter in sediment (29) is also a potential source of modern methane, but the substrate must be modern and not from one of the ancient C sources highlighted above. For this reason, we assume that this third potential modern CH4 source has a 14C content similar to that of DIC in surface waters (Fig. 1B).
Fig. 1. Surface water 14C-CH4 data and potential CH4 endmembers in the U.S. Beaufort Sea shelf study area.
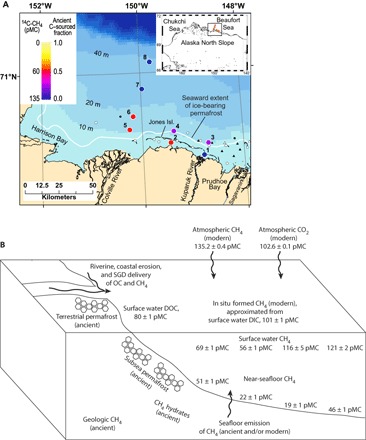
(A) Station map showing both the 14C-CH4 data in units of percent Modern Carbon (pMC), with the atmosphere in 1950 defined as 100 pMC (33, 34), as well as the calculated fraction of ancient C–sourced CH4 (fs) (Eqs. 1 to 5) in surface waters at each station. The white curve is the bulk sediment velocity contour (2000 m/s) used to delineate the seaward boundary of the sedimentary section that contains substantial (up to 29%) ice-bearing permafrost in the upper ~600 m (21). White circles and triangles respectively show boreholes (hundreds of meters deep) and geotechnical borings (<100 m) that contain permafrost based on an analysis of well logs and recovery of permafrost samples, respectively (22). Black circles and triangles respectively indicate no permafrost inferred or found in deep boreholes and geotechnical borings (22). (B) System schematic showing 14C values of dissolved CH4 (stations 5 to 8) and possible ancient and modern endmembers that were also measured here. SGD, submarine groundwater discharge; OC, organic carbon; DOC, dissolved organic carbon.
Although these disparate sources can contribute CH4 to the Beaufort Sea shelf (Fig. 1B), a plot of 14C-CH4 versus the reciprocal of molar [CH4], a so-called Keeling plot (30, 31), displays surprising linearity for a complex system (R2 = 0.75) (Fig. 2). The relationship is statistically significant (P < 0.01) and suggests that the observed (“obs”) system can be largely described as a mixture of modern background (“bkg”) and an ancient source (“s”); this result does not exclude the possibility that multiple sources of CH4 may contribute to the source and/or the background values, but it does suggest that potential CH4 endmembers can be linearly combined to establish a pseudo–two-component mixture
| (1) |
| (2) |
where “c” is [CH4] and “14C” is 14C-CH4 content. Combining and rearranging Eqs. 1 and 2 yields a linear equation (Eq. 3), whose y intercept indicates the 14C-CH4 content of the source (14Cs) when an infinite amount of source is added (Fig. 2B) (30).
| (3) |
Fig. 2. 14C-CH4 data from each station and Keeling plot analysis.
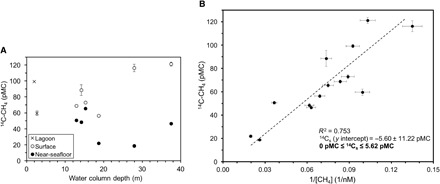
(A) Dissolved 14C-CH4 data for stations 1 to 8, plotted by the water depth of the station. The data include lagoon samples (×), surface samples (white circles), and near-seafloor samples (black circles). Error bars that are not visible are smaller than the markers. Uncertainty for 14C-CH4 data incorporates the collection, preparation, and measurement uncertainties (20). (B) A Keeling plot (Eq. 3) incorporating [CH4] and 14C-CH4 measurements from stations 1 to 8 suggests that the system can be viewed as a pseudo–two-component mixture and that the 14C-CH4 source signature (14Cs) likely ranges from 0 to 5.62 pMC.
Because the values of both 14Cobs and 1/cobs contain uncertainty, a standard Model I, linear least squares regression, is inappropriate to determine the y intercept; instead, a Model II, geometric mean regression, is often preferred (31, 32). This analysis is used here (Fig. 2B) and suggests that 14Cs equals −5.60 ± 11.22 percent Modern Carbon (pMC) relative to the 1950 atmosphere, which is defined as 100 pMC (33, 34). Negative values of pMC have no meaning, so 14Cs likely ranges from 0 to 5.62 pMC, indicating that ancient sources of CH4 (zero to low 14C content, <<100 pMC) (Fig. 1B) are being added to the background CH4 in these waters. Although this analysis cannot distinguish between different ancient sources of CH4, it does suggest that at least one, if not several, of the ancient sources is contributing CH4 to this region, confirming previous conjectures (5, 6, 13–16). The background CH4 to which these ancient sources are added is likely composed of more modern CH4 (≥100 pMC) from the atmosphere (135.2 ± 0.4 pMC; n = 3), in situ aerobic (water column) and anaerobic (sediment) methanogenesis (101 ± 1 pMC; n = 6), or some combination of the three (Fig. 1B).
We calculate the fraction of each dissolved CH4 sample that was derived from the ancient C source (fs) with an isotopic mass balance
| (4) |
| (5) |
where the radiocarbon content of each CH4 endmember is represented by the subscripts “h” (hydrate or geologic CH4; 0 pMC), “p” (permafrost CH4; 5.62 pMC), “a” (atmospheric CH4; 135.2 pMC), and “i” (in situ produced CH4; 101 pMC) (Fig. 1). Because this isotopic mass balance contains two equations and four unknowns (fh, fp, fa, and fi), we begin by defining fa and fi by systematically varying them from 0 to 1 in increments of 0.001, considering all possible combinations. Then, values of fh and fp are calculated using Eqs. 4 and 5 for each unique combination of fa and fi. When either fh or fp is determined to be less than 0 or greater than 1, all values are discarded for that linear combination. The resulting values of fh and fp are summed to more generally represent fs because 14Ch and 14Cp are assumed on the basis of the results of the Keeling plot (Fig. 2B) and not directly measured; the average and standard deviation of fs, fa, and fi are then calculated (Table 1 and Fig. 1A).
Table 1. Calculated fractions of ancient and modern C–sourced CH4 in each sample.
| Station |
Water depth (m) |
Distance offshore (km) |
Sample type |
Ancient C–sourced CH4 fraction, fs |
Atmospheric-sourced CH4 fraction, fa |
In situ produced CH4 fraction, fi |
| 1 | 2 | 3 | Lagoon | 0.18 ± 0.06 | 0.47 ± 0.18 | 0.35 ± 0.25 |
| 2 | 3 | 2 | Lagoon | 0.50 ± 0.04 | 0.23 ± 0.12 | 0.27 ± 0.17 |
| 3 | 14 | 12 | Surface | 0.26 ± 0.06 | 0.37 ± 0.18 | 0.37 ± 0.24 |
| Near-seafloor | 0.60 ± 0.04 | 0.18 ± 0.10 | 0.22 ± 0.13 | |||
| 4 | 15 | 10 | Surface | 0.39 ± 0.05 | 0.29 ± 0.15 | 0.33 ± 0.20 |
| Near-seafloor | 0.45 ± 0.05 | 0.25 ± 0.14 | 0.30 ± 0.18 | |||
| 5 | 13 | 18 | Surface | 0.42 ± 0.05 | 0.27 ± 0.14 | 0.31 ± 0.19 |
| Near-seafloor | 0.58 ± 0.04 | 0.19 ± 0.10 | 0.23 ± 0.14 | |||
| 6 | 19 | 27 | Surface | 0.53 ± 0.04 | 0.21 ± 0.12 | 0.26 ± 0.16 |
| Near-seafloor | 0.83 ± 0.02 | 0.07 ± 0.04 | 0.10 ± 0.06 | |||
| 7 | 28 | 48 | Surface | 0.10 ± 0.03 | 0.72 ± 0.10 | 0.18 ± 0.13 |
| Near-seafloor | 0.86 ± 0.02 | 0.06 ± 0.04 | 0.08 ± 0.05 | |||
| 8 | 38 | 69 | Surface | 0.07 ± 0.03 | 0.79 ± 0.07 | 0.14 ± 0.10 |
| Near-seafloor | 0.61 ± 0.03 | 0.17 ± 0.10 | 0.22 ± 0.13 |
In the back-barrier lagoon (stations 1 and 2), where sediment overlies intact subsea permafrost (Fig. 1A) (21, 22), just one “lagoon” 14C-CH4 sample was collected per station because of the shallow water depth (<3 m) (Fig. 2A). At each of the six deeper-water stations (stations 3 to 8), two 14C-CH4 samples were collected: a “surface” sample acquired at 2 m below the sea surface and a “near-seafloor” sample collected 3 to 8 m from the seafloor (table S1 and Fig. 2A).
The δ13C-CH4 and [CH4] data associated with each 14C-CH4 sample are presented in table S1. The average values for the surface samples [−58 ± 6‰, 11 ± 3 nmol/liter (nM); n = 6] are more enriched in 13C and have lower concentrations than those of the near-seafloor samples (−63 ± 6‰, 27 ± 15 nM; n = 6). These observations are also true of each station’s surface and near-seafloor pair (fig. S1). Because 12CH4 is oxidized faster than 13CH4, these trends support the traditional view of oceanic CH4 dynamics, in which CH4 is emitted from anoxic seafloor sediments and oxidized throughout its ascent in the water column (35).
In sharp contrast, the values of fs computed from the 14C-CH4 data allow an entirely different interpretation of this system. The lagoon sample collected at station 1 is composed mainly of modern background CH4 (fs = 0.18 ± 0.06), whereas the sample collected from station 2 is of intermediate origin (fs = 0.50 ± 0.04), a roughly equivalent mixture of ancient C source and modern background. The mean value of fs in the near-seafloor samples ranges from 0.45 to 0.86 (n = 6), whereas the mean value of fs in the surface samples ranges from 0.07 to 0.53 (n = 6). The surface samples are all dominantly modern background CH4 except for the sample collected at station 6, which has an intermediate origin (fs = 0.53 ± 0.04).
At stations 3, 5, 7, and 8, CH4 in the near-seafloor sample is derived mainly from ancient C sources in contrast to CH4 derived mainly from modern background in the surface water sample. This decoupling is most evident at mid-outer shelf stations 7 and 8 (at water depths of 28 and 38 m, respectively), where little to no CH4 is sourced from ancient C in surface waters, whereas CH4 found near the seafloor is mainly sourced from ancient C (Table 1). These analyses suggest that (i) ancient C sources supply CH4 to shelf waters and (ii) ancient C sources contribute little to no CH4 to surface waters (and therefore to the atmosphere) with increasing water depth and thus confirms our hypotheses.
These results demonstrate that ancient C–sourced CH4 offshore Prudhoe Bay is largely not reaching the atmosphere beyond, approximately, the 30-m isobath. Our findings are consistent with other Arctic Ocean studies that have found CH4 removal processes to be highly efficient in sediment (36) and relatively shallow water columns (<100 m depth) (15, 16). The evidence of strong CH4 removal mechanisms operating in the Arctic from these studies suggests that an enhancement of ancient C mobilization due to climate change would not necessarily increase CH4 emission to the atmosphere from the Arctic Ocean. In addition to potential changes in the magnitude of CH4 sources in a warmer, increasingly ice-free Arctic Ocean (37), we must also consider that the rate of CH4 removal processes, such as aerobic CH4 oxidation by microorganisms in the water column (6, 35), could also change. Thus, to accurately constrain the mobilization of ancient C and the subsequent emission of CH4, we recommend that natural abundance 14C-CH4 analyses should be conducted in future studies of CH4 dynamics.
MATERIALS AND METHODS
Sample collection
Our study was carried out aboard the R/V Ukpik from 30 August to 5 September 2015, coincident with the period of the year that typically has the minimum extent of sea ice. Because the surface water [CH4] in the Prudhoe Bay area is lower than the limit of previous 14C-CH4 techniques (16 nM for a small sample accelerator mass spectrometry analysis) (38), a new dissolved 14C-CH4 sampling and preparation method was developed and used in this study (20). Using this method, seawater was continuously pumped onboard and the dissolved gases were continuously extracted from the water. In the Prudhoe Bay sample set, the average seawater sample volume was 32,000 ± 4000 liters (n = 14), and the average extracted gas volume was 350 ± 50 liters (n = 14). The extracted gas was compressed into a 2-liter cylinder for transport to the home laboratory, where it was prepared for 14C and stable isotope analyses. Although the cylinder is only pressurized to a maximum of 2100 psi, equivalent to 240 liters, it was necessary to extract 350 to 400 liters of gas to (i) flush the compressor pump and cylinder with sample and (ii) account for some small, unresolved loss of sample (that is, a leak) in the compression process.
Atmospheric CH4 for 14C-CH4 analyses was sampled in Utqiaġvik (formerly, Barrow), AK, on three separate days across 3 months (August to October 2015, bounding our cruise dates) and is reported as mean ± 1 SD (n = 3); the samples were collected when winds were coming from the north, so these measurements represent a circum-Arctic average, to some extent. Atmospheric CO2 for 14C-CO2 analyses was also sampled in Utqiaġvik, AK, on three separate days across 3 weeks (August to September 2015, bounding our cruise dates) and is reported as mean ± 1 SD (n = 3). DIC and DOC samples for 14C-DIC and 14C-DOC analyses were collected contemporaneously with 14C-CH4 sampling on our research cruise; these measurements are reported as the mean ± 1 SD of surface water samples (2 m depth) at stations 3 to 8 (n = 6).
A discrete vial for [CH4] analysis was collected at each sample collection depth using a single Niskin bottle following standardized procedures (39). In total, 16 samples were collected from the 14 sample collection depths because two duplicate vials were collected. Each sample was collected by transferring the seawater in the Niskin bottle to a 60-ml glass vial, which was flushed with seawater, filled, and sealed with a stopper and crimp cap. Then, a 10-ml gaseous headspace of ultrahigh-purity nitrogen was injected into each vial from a syringe while 10 ml of seawater from the vial was removed with a second syringe. Each sample was then sterilized with 25 μl of supersaturated mercuric chloride solution to prevent microbial perturbation of the original [CH4] and stored stopper side down to prevent any diffusion of headspace gas across the seal.
The [CH4] analyses were performed 2 months after the cruise in the home laboratory using an Agilent 6850 gas chromatograph with a flame ionization detector (GC-FID). The GC analysis of the headspace of each vial was performed in two consecutive runs. The [CH4] of the headspace was calculated by fitting the measured peak area to a four-point calibration curve created on the same day by analyzing a suite of CH4 gas standards {[CH4] = 0, 1, 10, and 100 parts per million (ppm)} that bound all of the measured values. The measured headspace [CH4] of each vial was translated to a dissolved [CH4] value (40) with knowledge of the sample incubator temperature and the salinity of the sampled seawater, the latter of which was measured with a water quality sonde in the field (YSI, 600R series). An uncertainty of 5.2% is associated with each measurement (39).
To evaluate the degree of CH4 saturation in the sampled seawater from the dissolved [CH4] data, it was necessary to calculate the [CH4] that would be found if each water sample had come to full equilibrium with the atmosphere (that is, the “equilibrium solubility”). The local atmosphere was sampled from bow air that was pumped to an onboard cavity ring-down spectrometer (CRDS; G2401, Picarro). The atmospheric [CH4] (2.000 ± 0.002 ppm; n = 79) was used along with the temperature- and salinity-dependent CH4 solubility (40) to calculate the CH4 equilibrium solubility of each sample. The degree of CH4 saturation is reported for all surface water samples in table S1. Samples that have CH4 concentrations greater than the seawater’s equilibrium solubility concentration have CH4 saturation values of >100% (that is, supersaturated), representing that the net flux of CH4 is from sea to air.
14C-CH4 and δ13C-CH4 sample preparation
The extracted gas cylinder samples were prepared for 14C-CH4 and δ13C-CH4 analyses on a newly developed shore-based vacuum line (20). From 15 collected samples, 17 samples were then prepared and analyzed for 14C-CH4 and δ13C-CH4, as two preparation duplicates were made by preparing a single extracted gas sample cylinder twice. Only 16 of these 17 prepared samples were analyzed (and discussed here) because a sample collected at one lagoon station (original station ID T5S29: 70.489°N, 149.114°W) was suspected to have been contaminated by carbon monoxide–C during the sample preparation process. The samples were prepared in a random order across 5 weeks. Vacuum line quality control assessments described by Sparrow and Kessler (20) were performed daily during the preparation period using gas standards with [CH4] of 0, 5, and 250 ppm.
The vacuum line technique achieves high-efficiency purification, oxidation, and collection of the sample CH4. The aliquots collected for the isotopic analyses are the CH4 oxidation products, CO2 and H2O, which are produced when the sample CH4 is oxidized on a heated platinized quartz wool catalyst. Although the gas sample volumes are large (≤240 liters), a high flow rate (2 liters/min) through the vacuum line allows multiple sample preparations per day. The total process blank of the procedure is small (5.0 μg of CH4-C), composing 1.2% of the average collected and prepared sample (424 ± 163 μg; n = 16). The 14C-CH4 blanks of the vacuum line have acceptably low radiocarbon content (0.22 ± 0.07 pMC; n = 8) relative to the 14C-dead (0 pMC) CH4 from which they are prepared, enabling radiocarbon dating of the dissolved CH4-C to the analytical limit of accelerator mass spectrometry (~50,000 years Before Present).
The 14C-CH4 data were analyzed and corrected for isotopic fractionation (33, 34) at the W. M. Keck Carbon Cycle Accelerator Mass Spectrometry (CCAMS) Laboratory at the University of California, Irvine. The uncertainties for 14C-CH4 data (both 14C-CH4 content and conventional 14C age of CH4) reported in Fig. 1A, fig. S1, and table S1 are calculated from the root mean square of the collection, preparation, and measurement uncertainties (20). Except for two smaller-sized samples (100 and 150 μg of CH4-C), δ13C-CH4 data were also analyzed at the Keck CCAMS facility to a precision of <0.1‰ relative to standards traceable to Pee Dee Belemnite using a Thermo Finnigan Delta Plus stable isotope ratio mass spectrometer (IRMS) with GasBench inlet. The δ13C-CH4 measurements for the two samples that had insufficient CH4-C for a separate IRMS aliquot were measured via CRDS (G2201-i, Picarro), analyzed directly from the sample cylinders; reported value is the 3-min average (n ≈ 120), and uncertainty is the standard error.
Supplementary Material
Acknowledgments
We thank B. Kopplin, M. Fleming, and A. Schwartz of the R/V Ukpik for providing skillful assistance of our science in Prudhoe Bay. We are grateful to M. Leonte for running GC-FID hydrocarbon concentration analyses in the Kessler laboratory. K.J.S. thanks A. Andersson of Stockholm University for fruitful discussions. Funding: The National Science Foundation (PLR-1417149; awarded to J.D.K.) primarily supported this work with additional support provided by the U.S. Department of Energy (DE-FE0028980; awarded to J.D.K.). Atmospheric 14C-CH4 measurements were funded by NASA via the Jet Propulsion Laboratory (Earth Ventures project “Carbon in Arctic Reservoirs Vulnerability Experiment”) to the University of Colorado under contract 1424124. K.M.S. acknowledges support from the University of Minnesota Grant-in-Aid program. Author contributions: K.J.S. and J.D.K. led the study and drafted the manuscript, with contributions from C.D.R. Fieldwork in Prudhoe Bay was performed by K.J.S., J.D.K., F.G.-T., and K.M.S. Dissolved 14C-CH4 samples were prepared by K.J.S. and analyzed by J.R.S. Samples for 14C-DIC, 14C-DOC, atmospheric 14C-CH4, and atmospheric 14C-CO2 analyses were prepared by F.G.-T. and X.X., K.M.S., J.B.M. and S.J.L., and X.X., respectively. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. government. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The water column data are also available at doi:10.18739/A2B69R. Additional information is available from the authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaao4842/DC1
fig. S1. Dissolved CH4 concentration and isotopic data plotted by station depth.
table S1. Dissolved 14C-CH4, δ13C-CH4, and [CH4] data with relevant sample information.
REFERENCES AND NOTES
- 1.Thornton B. F., Geibel M. C., Crill P. M., Humborg C., Mörth C.-M., Methane fluxes from the sea to the atmosphere across the Siberian shelf seas. Geophys. Res. Lett. 43, 5869–5877 (2016). [Google Scholar]
- 2.Berchet A., Bousquet P., Pison I., Locatelli R., Chevallier F., Paris J.-D., Dlugokencky E. J., Laurila T., Hatakka J., Viisanen Y., Worthy D. E. J., Nisbet E., Fisher R., France J., Lowry D., Ivakhov V., Hermansen O., Atmospheric constraints on the methane emissions from the East Siberian Shelf. Atmos. Chem. Phys. 16, 4147–4157 (2016). [Google Scholar]
- 3.Lorenson T. D., Greinert J., Coffin R. B., Dissolved methane in the Beaufort Sea and the Arctic Ocean, 1992–2009; sources and atmospheric flux. Limnol. Oceanogr. 61, S300–S323 (2016). [Google Scholar]
- 4.Pohlman J. W., Greinert J., Ruppel C., Silyakova A., Vielstädte L., Casso M., Mienert J., Bünz S., Enhanced CO2 uptake at a shallow Arctic Ocean seep field overwhelms the positive warming potentional of emitted methane. Proc. Natl. Acad. Sci. U.S.A. 114, 5355–5360 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuur E. A. G., McGuire A. D., Schädel C., Grosse G., Harden J. W., Hayes D. J., Hugelius G., Koven C. D., Kuhry P., Lawrence D. M., Natali S. M., Olefeldt D., Romanovsky V. E., Schaefer K., Turetsky M. R., Treat C. C., Vonk J. E., Climate change and the permafrost carbon feedback. Nature 520, 171–179 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Ruppel C. D., Kessler J. D., The interaction of climate change and methane hydrates. Rev. Geophys. 55, 126–168 (2017). [Google Scholar]
- 7.Nisbet E. G., Dlugokencky E. J., Manning M. R., Lowry D., Fisher R. E., France J. L., Michel S. E., Miller J. B., White J. W. C., Vaughn B., Bousquet P., Pyle J. A., Warwick N. J., Cain M., Brownlow R., Zazzeri G., Lanoisellé M., Manning A. C., Gloor E., Worthy D. E. J., Brunke E.-G., Labuschagne C., Wolff E. W., Ganesan A. L., Rising atmospheric methane: 2007–2014 growth and isotopic shift. Global Biogeochem. Cycles 30, 1356–1370 (2016). [Google Scholar]
- 8.Fisher R. E., Sriskantharajah S., Lowry D., Lanoisellé M., Fowler C. M. R., James R. H., Hermansen O., Myhre C. L., Stohl A., Greinert J., Nisbet-Jones P. B. R., Mienert J., Nisbet E. G., Arctic methane sources: Isotopic evidence for atmospheric inputs. Geophys. Res. Lett. 38, L21803 (2011). [Google Scholar]
- 9.France J. L., Cain M., Fisher R. E., Lowry D., Allen G., O’Shea S. J., Illingworth S., Pyle J., Warwick N., Jones B. T., Gallagher M. W., Bower K., Le Breton M., Percival C., Muller J., Welpott A., Bauguitte S., George C., Hayman G. D., Manning A. J., Myhre C. L., Lanoisellé M., Nisbet E. G., Measurements of δ13C in CH4 and using particle dispersion modeling to characterize sources of Arctic methane within an air mass. J. Geophys. Res. Atmos. 121, 14257–14270 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrenko V. V., Smith A. M., Schaefer H., Riedel K., Brook E., Baggenstos D., Harth C., Hua Q., Buizert C., Schilt A., Fain X., Mitchell L., Bauska T., Orsi A., Weiss R. F., Severinghaus J. P., Minimal geological methane emissions during the Younger Dryas–Preboreal abrupt warming event. Nature 548, 443–446 (2017). [DOI] [PubMed] [Google Scholar]
- 11.James R. H., Bousquet P., Bussmann I., Haeckel M., Kipfer R., Leifer I., Niemann H., Ostrovsky I., Piskozub J., Rehder G., Treude T., Vielstädte L., Greinert J., Effects of climate change on methane emissions from seafloor sediments in the Arctic Ocean: A review. Limnol. Oceanogr. 61, S283–S299 (2016). [Google Scholar]
- 12.Damm E., Kiene R. P., Schwarz J., Falck E., Dieckmann G., Methane cycling in Arctic shelf water and its relationship with phytoplankton biomass and DMSP. Mar. Chem. 109, 45–59 (2008). [Google Scholar]
- 13.Westbrook G. K., Thatcher K. E., Rohling E. J., Piotrowski A. M., Pälike H., Osborne A. H., Nisbet E. G., Minshull T. A., Lanoisellé M., James R. H., Hühnerbach V., Green D., Fisher R. E., Crocker A. J., Chabert A., Bolton C., Beszczynska-Möller A., Berndt C., Aquilina A., Escape of methane gas from the seabed along the West Spitsbergen continental margin. Geophys. Res. Lett. 36, L15608 (2009). [Google Scholar]
- 14.Shakhova N., Semiletov I., Sergienko V., Lobkovsky L., Yusupov V., Salyuk A., Salomatin A., Chernykh D., Kosmach D., Panteleev G., Nicolsky D., Samarkin V., Joye S., Charkin A., Dudarev O., Meluzov A., Gustafsson O., The East Siberian Arctic Shelf: Towards further assessment of permafrost-related methane fluxes and role of sea ice. Philos. Trans. A Math. Phys. Eng. Sci. 373, 20140451 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graves C. A., Steinle L., Rehder G., Niemann H., Connelly D. P., Lowry D., Fisher R. E., Stott A. W., Sahling H., James R. H., Fluxes and fate of dissolved methane released at the seafloor at the landward limit of the gas hydrate stability zone offshore western Svalbard. J. Geophys. Res. Oceans 120, 6185–6201 (2015). [Google Scholar]
- 16.Myhre C. L., Ferré B., Platt S. M., Silyakova A., Hermansen O., Allen G., Pisso I., Schmidbauer N., Stohl A., Pitt J., Jansson P., Greinert J., Percival C., Fjaeraa A. M., O’Shea S. J., Gallagher M., Le Breton M., Bower K. N., Bauguitte S. J. B., Dalsøren S., Vadakkepuliyambatta S., Fisher R. E., Nisbet E. G., Lowry D., Myhre G., Pyle J. A., Cain M., Mienert J., Extensive release of methane from Arctic seabed west of Svalbard during summer 2014 does not influence the atmosphere. Geophys. Res. Lett. 43, 4624–4631 (2016). [Google Scholar]
- 17.Wahlen M., Tanaka N., Henry R., Deck B., Zeglen J., Vogel J. S., Southon J., Shemesh A., Fairbanks R., Broecker W., Carbon-14 in methane sources and in atmospheric methane: The contribution from fossil carbon. Science 245, 286–290 (1989). [DOI] [PubMed] [Google Scholar]
- 18.Repeta D. J., Ferrón S., Sosa O. A., Johnson C. G., Repeta L. D., Acker M., DeLong E. F., Karl D. M., Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nat. Geosci. 9, 884–887 (2016). [Google Scholar]
- 19.Kessler J. D., Reeburgh W. S., Valentine D. L., Kinnaman F. S., Peltzer E. T., Brewer P. G., Southon J., Tyler S. C., A survey of methane isotope abundance (14C, 13C, 2H) from five nearshore marine basins that reveals unusual radiocarbon levels in subsurface waters. J. Geophys. Res. 113, C12021 (2008). [Google Scholar]
- 20.Sparrow K. J., Kessler J. D., Efficient collection and preparation of methane from low concentration waters for natural abundance radiocarbon analysis. Limnol. Oceanogr. Meth. 15, 601–617 (2017). [Google Scholar]
- 21.Brothers L. L., Herman B. M., Hart P. E., Ruppel C. D., Subsea ice-bearing permafrost on the U.S. Beaufort Margin: 1. Minimum seaward extent defined from multichannel seismic reflection data. Geochem. Geophys. Geosyst. 17, 4354–4365 (2016). [Google Scholar]
- 22.Ruppel C. D., Herman B. M., Brothers L. L., Hart P. E., Subsea ice-bearing permafrost on the U.S. Beaufort Margin: 2. Borehole constraints. Geochem. Geophys. Geosyst. 17, 4333–4353 (2016). [Google Scholar]
- 23.Anthony K. M. W., Anthony P., Grosse G., Chanton J., Geologic methane seeps along boundaries of Arctic permafrost thaw and melting glaciers. Nat. Geosci. 5, 419–426 (2012). [Google Scholar]
- 24.Schreiner K. M., Bianchi T. S., Eglinton T. I., Allison M. A., Hanna A. J. M., Sources of terrigenous inputs to surface sediments of the Colville River Delta and Simpson’s Lagoon, Beaufort Sea, Alaska. J. Geophys. Res. Biogeosci. 118, 808–824 (2013). [Google Scholar]
- 25.Schreiner K. M., Bianchi T. S., Rosenheim B. E., Evidence for permafrost thaw and transport from an Alaskan North Slope watershed. Geophys. Res. Lett. 41, 3117–3126 (2014). [Google Scholar]
- 26.Lecher A. L., Kessler J., Sparrow K., Garcia-Tigreros Kodovska F., Dimova N., Murray J., Tulaczyk S., Paytan A., Methane transport through submarine groundwater discharge to the North Pacific and Arctic Ocean at two Alaskan sites. Limnol. Oceanogr. 61, S344–S355 (2016). [Google Scholar]
- 27.Jorgenson M. T., Shur Y. L., Pullman E. R., Abrupt increase in permafrost degradation in Arctic Alaska. Geophys. Res. Lett. 33, L02503 (2006). [Google Scholar]
- 28.Jones B. M., Arp C. D., Jorgenson M. T., Hinkel K. M., Schmutz J. A., Flint P. L., Increase in the rate and uniformity of coastline erosion in Arctic Alaska. Geophys. Res. Lett. 36, L03503 (2009). [Google Scholar]
- 29.Crill P. M., Martens C. S., Methane production from bicarbonate and acetate in an anoxic marine sediment. Geochim. Cosmochim. Acta 50, 2089–2097 (1986). [Google Scholar]
- 30.Keeling C. D., The concentration and isotopic abundances of atmospheric carbon dioxide in rural areas. Geochim. Cosmochim. Acta 13, 322–334 (1958). [Google Scholar]
- 31.Pataki D. E., Ehleringer J. R., Flanagan L. B., Yakir D., Bowling D. R., Still C. J., Buchmann N., Kaplan J. O., Berry J. A., The application and interpretation of Keeling plots in terrestrial carbon cycle research. Global Biogeochem. Cycles 17, 1022 (2003). [Google Scholar]
- 32.Miller J. B., Tans P. P., Calculating isotopic fractionation from atmospheric measurements at various scales. Tellus B 55, 207–214 (2003). [Google Scholar]
- 33.Stuiver M., Polach H. A., Discussion reporting of 14C data. Radiocarbon 19, 355–363 (1977). [Google Scholar]
- 34.Fahrni S. M., Southon J. R., Santos G. M, Palstra S. W. L., Meijer H. A. J., Xu X., Reassessment of the 13C/12C and 14C/12C isotopic fractionation ratio and its impact on high-precision radiocarbon dating. Geochim. Cosmochim. Acta 213, 330–345 (2017). [Google Scholar]
- 35.Leonte M., Kessler J. D., Kellermann M. Y., Arrington E. C., Valentine D. L., Sylva S. P., Rapid rates of aerobic methane oxidation at the feather edge of gas hydrate stability in the waters of Hudson Canyon, US Atlantic Margin. Geochim. Cosmochim. Acta 204, 375–387 (2017). [Google Scholar]
- 36.Overduin P. P., Liebner S., Knoblauch C., Günther F., Wetterich S., Schirrmeister L., Hubberten H.-W., Grigoriev M. N., Methane oxidation following submarine permafrost degradation: Measurements from a central Laptev Sea shelf borehole. J. Geophys. Res. Biogeosci. 120, 965–978 (2015). [Google Scholar]
- 37.Stroeve J. C., Markus T., Boisvert L., Miller J., Barrett A., Changes in Arctic melt season and implications for sea ice loss. Geophys. Res. Lett. 41, 1216–1225 (2014). [Google Scholar]
- 38.Kessler J. D., Reeburgh W. S., Preparation of natural methane samples for stable isotope and radiocarbon analysis. Limnol. Oceanogr. Meth. 3, 408–418 (2005). [Google Scholar]
- 39.Weinstein A., Navarrete L., Ruppel C., Weber T. C., Leonte M., Kellermann M. Y., Arrington E. C., Valentine D. L., Scranton M. I., Kessler J. D., Determining the flux of methane into Hudson Canyon at the edge of methane clathrate hydrate stability. Geochem. Geophys. Geosyst. 17, 3882–3892 (2016). [Google Scholar]
- 40.Wiesenburg D. A., Guinasso N. L. Jr, Equilibrium solubilities of methane, carbon monoxide, and hydrogen in water and sea water. J. Chem. Eng. Data 24, 356–360 (1979). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaao4842/DC1
fig. S1. Dissolved CH4 concentration and isotopic data plotted by station depth.
table S1. Dissolved 14C-CH4, δ13C-CH4, and [CH4] data with relevant sample information.