

Abstract
The aqueous nature of complex coacervates provides a biologically-relevant context for various therapeutic applications. In this sense, biological applications demand a corresponding level of biocompatibility from the polyelectrolytes that participate in complex coacervation. Continued development with naturally-occuring polyelectrolytes such as heparin and chitosan underscore such aims. Herein, we design a synthetic polycation, in which betaine is conjugated to a biodegradable polyester backbone. Betaine is a naturally-occuring methylated amino acid that is ubiquitously present in human plasma. Inspired by its vast range of benefits – including but not limited to anti-inflammation, anti-cancer, anti-bacterial, anti-oxidant, protein stabilization, and cardiovascular health – we aim to impart additional functionality to a polycation for eventual use in a complex coacervate with heparin. We report on its in-vitro and in-vivo biocompatability, in-vitro and in-vivo effect on angiogenesis, in-vitro effect on microbial growth, and ability to form complex coacervates with heparin.
TOC image
Introduction
The self-assembly of materials can be characterized as a progression towards a global energy minimum. A central class of materials that undergo such transformations are complex coacervates. Driven by short-range1 or long-range electrostatic interactions2, complex coacervation is an associative phase separation phenomenon characterized by the aggregation of at least two polyelectrolytes into interpolymer complexes. Specifically, the release of counterions during the complexation of polyelectrolytes leads to a net gain in entropy3, 4, resulting in a dynamic equilibrium of a polyelectrolyte-rich coacervate and polyelectrolyte-poor supernatant phase. The relative simplicity of combining polyelectrolytes has given rise to a broad range of materials for complex coacervation, with applications that range from protocells5, 6 to microencapsulates of food or cosmetics7.
In particular, the substantial water content of complex coacervates8 has been capitalized on to facilitate the encapsulation of proteins9–11, small molecules12–14, and cells15 for various biological applications. These biological confines consequently limit the range of tenable polyelectrolytes to those of biocompatible nature. Not surprisingly then, a large number of complex coacervates take inspiration from naturally-occurring polyanions such as heparin16, 17 or hyaluronic acid18, 19. For example, heparin has been shown to bridge cationic sequences on proteins with their respective receptors20–22, rendering effective receptor-ligand interactions. The use of cationic polyelectrolytes for complex coacervation, however, requires more attention. While biological polysaccharides such as chitosan are regularly used18, 23, 24, synthetic cationic polymers are equally employed11, 16, 25–28, often for the capacity to formulate application-specific designs. Unfortunately, the scope of polycations for biological applications can be limited by their general toxicity to cells, necessitating biocompatible cationic polymers.
We have previously reported on an arginine-based polycation29, 30, poly(ethylene argininylaspartate diglyceride) (PEAD), which forms a biocompatible delivery vehicle upon complex coacervation with heparin. A number of derivative studies using PEAD have demonstrated an enhanced bioactivity of the encapsulated protein11, 16, 17, 31. Here, we aim to confer functionality to the cationic polymer itself through deliberate design, by replacing the arginine side group on poly(ethylene aspartate diglyceride) (PED) with betaine. Betaine is ubiquitously present in human plasma, primarily due to dietary intake as well as some endogenous level of synthesis in the liver and kidney32. Primarily understood as an osmolyte to protect cells from environmental stress33, betaine has numerous other chemical and physiological benefits, including but limited to: protein stabilization34, methyl donation to maintain liver and heart health35, supplement for the prevention of cancer36, reduction in angiogenesis and inflammation37, and anti-bacterial activity38. Given its numerous roles, we were motivated to assess its potential when grafted onto PED.
Herein, we report on a new polycation, poly(ethylene betainylaspartate diglyceride), or betaine-functionalized poly(ethylene aspartate diglyceride) (B-PED). The quaternary ammonium group on betaine imparts a positive charge to the polymer. We assess the in-vitro and in-vivo biocompatibility of the B-PED and explore its effect on angiogenesis and microbial activity. Finally, we examine its interaction with heparin and demonstrate its potential use as a polycation for complex coacervation.
Materials and Methods
Materials
Betaine and deuterium oxide (Sigma-Aldrich, St. Louis, MO), ethylene glycol diglycidyl ether (EGDE) (Pfaltz & Bauer, Waterbury, CT), t-Boc-aspartic acid (Boc-ASP-OH) (Bachem, Torrence, CA), 4-dimethylaminopyridine (DMAP) (Alfa Aesar, Ward Hill, MA), dimethylformamide (DMF), N-hydroxysuccinimide (NHS), N,N′-dicyclohexylcarbodiimide (DCC), and tetra-n-butylammonium bromide (TBAB) (Acros Organics, Geel, Belgium), heparin sodium USP (Scientific Protein Labs, Waunakee, WI), fluorescein-conjugated heparin, timentin, DMEM, and Quant-iT PicoGreen dsDNA kit (Thermo Fisher Scientific, Waltham, MA), Cultrex basement membrane extract (R&D Systems, Minneapolis, MN), Matrigel high concentration (Corning, Corning, NY), endothelial cell growth medium and BulletKit (Lonza, Basel, Switzerland), dialysis tubing (Spectrum Labs, Rancho Dominguez, CA), CellTiter-Blue cell viability kit (Promega, Madison WI), RFP-expressing human umbilical vein endothelial cells (gift from Dr. Steven R. Little), mouse anti-rat CD68 antibody (Millipore, Billerica, MA), rabbit anti-rat α-SMA antibody and mouse anti-rat CD31 antibody (Abcam, Cambridge, United Kingdom) were all used as received.
B-PED Synthesis
B-PED is synthesized according to conditions modified from those previously described29. Briefly, EGDE (1000 mg), Boc-ASP-OH (1338.8 mg), and TBAB (5 mg) were dissolved in 0.6 mL of DMF. The mixture was reacted at 120 °C under N2 for 20 minutes in a microwave reactor (Biotage, Uppsala, Sweden). The resulting intermediate polymer, poly(ethylene boc-aspartate diglyceride) (PED-boc) was solubilized in 2 mL DCM and precipitated into diethyl ether. t-Boc was removed via addition of 4:1 DCM:TFA ([TFA] = 2.5 mM). After 2 hours of stirring at room temperature, solvent was subsequently removed via a rotatory evaporator for 2 hours. Multiple precipitation steps in diethyl ether were used to remove excess reagents, DMF, and TFA. PED was then washed overnight in diethyl ether and dried under vacuum until further use. B-PED was prepared by combining PED (0.108 M of reactive sites), betaine (0.217 M), NHS (0.217 M), DCC (0.260 M), and DMAP (0.011 M) into DMF. The solution was stirred at 30 °C under N2 for 48 hours. An insoluble dicyclohexylurea by-product was removed via centrifugation and filtration (0.22 μm). B-PED was then purified via multiple precipitation steps in diethyl ether, washed in ethanol, and dialyzed against deionized water for 18 hours.
Characterization of B-PED
1H nuclear magnetic resonance spectroscopy (H-NMR) was performed on PED, B-PED, and betaine using deuterium oxide (D2O) as a solvent (Biospin Avance NMR, Bruker, Billerica, MA). Gel permeation chromatography (GPC) was used to assess the molecular weight of PED using DMF as the mobile and stationary phase (Viscotek VE2001); results were compared to polyethylene glycol calibration standards. Functional group assessment was carried out via Fourier transform infrared spectroscopy (FTIR) (Nicolet IR-100, Thermo, Waltham, MA).
Assessment of Cytotoxicity
In-vitro experiments were performed on NIH-3T3 cells (ATCC CRL-1658), and HUVECs (ATCC CRL-1730). NIH-3T3 cells and HUVECs were maintained in DMEM and endothelial basal media (EBM), respectively, and passaged using trypsin/EDTA upon confluence. Passage 4-6 cells were used for all experiments. For metabolic and proliferation assays, 7500 cells were seeded per 96 well and allowed to adhere overnight. Cells were then treated with varying concentrations of B-PED in the appropriate media (n=3). After 24 hours, cells were gently washed with DPBS three times and examined using CellTiter-Blue and PicoGreen kits, according to the manufacturer’s instructions. CellTiter-Blue and PicoGreen readouts were read on a microplate reader (SynergyMX, Biotek, Winooski, VT). In-vivo cytotoxicity experiments were carried out on Sprague-Dawley rats. 100 μL of saline or 10 mg/mL of B-PED in saline was subcutaneously injected into the dorsal area and explanted 1 week later (n=3). Tissues were fixed in 4% (w/v) PFA for 1 hour, followed by an overnight incubation step in 30% (w/v) sucrose. Samples were then embedded in OCT (Tissue-Tek), frozen on dry ice, and stored at −80 °C. OCT-embedded samples were sectioned at 10 μm thickness and stained via H&E staining and immunofluorescence (IF) staining. IF staining against CD68 was performed by cell permeabilization (0.2% triton-X 100), blocking with 5% (v/v) goat serum, incubation with mouse anti-rat CD68 antibody, and subsequent incubation with goat anti-mouse secondary antibody. H&E and IF images were captured on an inverted microscope (Eclipse Ti, Nikon).
Angiogenesis Assays
RFP-expressing HUVECs (Angio-Proteomie) were used for in-vitro angiogenic characterization. Briefly, basement membrane extract (reduced growth factor, Cultrex) was diluted to 10 mg/mL in sterile DPBS. 70 μL of the diluted basement membrane extract was added to each 96 well and allowed to gel at 37 °C for approximately 1 hour. 15,000 cells were then added to each well; EBM was used in the negative control while growth factor-supplemented EBM (EGM) was used for all other groups (n=4). After 12 hours of incubation, cells were examined every hour to determine the optimal end-point. At 15 hours post-seeding, cells were treated with Hoecst (1 μg/mL) for an additional 30 minutes, washed with DPBS, and imaged. To test the effect of B-PED on angiogenesis in-vivo, 1 mL of Matrigel (Corning) containing the appropriate treatment was injected into the caudal ventral area of Sprague-Dawley rats (negative: only Matrigel, positive: Matrigel + 1.5 μg FGF-2, B-PED: Matrigel + 1.5 μg FGF-2 + 10 mg B-PED). Matrigel plugs were extracted 10 days post-implantation and incubated at 37 °C for 30 minutes in a solution of 4% paraformaldehyde (w/v) and 0.5% glutaraldehyde (w/v). Plugs were then treated with 1 mg/mL sodium borohydride at 37 °C for 4 hours. Samples were embedded in OCT and processed for IF staining as described above.
Antimicrobial Assays
Staphylococcus Aureus were grown in 5 mL Tryptic Soy Broth (TSB) in a 15 mL falcon tube overnight at 37 °C. The next day, 200 μL of the overnight culture was added to 20 mL TSB in a 50 mL falcon tube and grown for 2 hours until the optical density reached 0.1. 100 μL of the Staphylococcus aureus were then transferred to a 96 well plate, after which different concentrations of PED or B-PED were added (n=3, final concentration: 5, 2.5, 1.25, 0.625, 0.313, 0.156 mg/mL). The 96 well plate was then incubated in a shaker at 37 °C for 5 hours; optical density measurements were carried out at 600 nm at 2, 3, and 5 hours post-treatment. Appropriate controls (positive control: 100 μg/mL timentin, negative control: TSB - no treatment) were utilized for this study.
Characterization of Complex Coacervates
Polymer solutions of B-PED and heparin were prepared at a concentration of 10 mg/mL in deionized water. After combining B-ED and heparin at varying mass ratios, the resulting complex coacervate was allowed to stabilize for one minute. Zeta potential measurements were then made using a folded capillary cell (Zetasizer Nano ZS, Malvern, Westborough, MA). To visualize coacervate behaviour, fluorescein-conjugated heparin was combined with unconjugated heparin at a 1:10 mass ratio. Finally, size measurements were taken via dynamic light scattering (DLS) to assess the effect of [NaCl] on coacervate hydrodynamic diameter; turbidity was also examined via absorbance measurements at 420 nm (SynergyMX, Biotek).
Results and Discussions
Synthesis and Characterization of B-PED
B-PED synthesis is performed by grafting betaine onto PED, a previously-reported biodegradable ester29. Containing a primary amine group (Fig. 1A), PED is amenable to conjugation with a variety of carboxylic acid derivatives. Despite the low solubility of betaine in DMF, excess betaine in the presence of a catalyst pushes the reaction forward, yielding an amber-colored product after 48 hours (Supplementary Fig. 1). The resulting B-PED contains a quaternary ammonium group, which confers a permanent cationic charge to the polymer. Purification via multiple precipitation steps and membrane dialysis removes all solvents and impurities, as indicated by the 1H-NMR spectrum (Fig. 1B). Protons in the ethylene glycol region of B-PED continue to produce a multiplet between δ3.40 and δ3.90 (Fig. 1B, a). The conversion of the primary amine on aspartate to an amide bond results in a shift at 4.31 ppm, corresponding to the α proton of aspartate (Fig. 1B, b). In addition, protons of the trimethyl amine generate a shift at 3.24 ppm (Fig. 1B, c). Based on its NMR spectrum, B-PED has approximately 70% conjugation efficiency of betaine, yielding an extremely cationic polymer; zeta potential measurements of B-PED show a charge of 42.0 ± 2.3 mV (Supplementary Fig. 2). The molecular weight of B-PED was difficult to measure empirically due to the lack of a proper column for cationic polymers. Given the molecular weight of PED (Fig. 1C) and the conjugation efficiency of betaine onto PED, we estimate an MW of approximately 46.46 kDa. FTIR spectroscopy of B-PED further corroborates the conjugation of betaine onto PED (Fig. 1D). Clear differences are observed between PED and B-PED, in which absorptions at 1632 nm (“a”) and 1551 nm (“b”) correspond to amide I and II bonds, respectively. Furthermore, both betaine and B-PED show absorption at 1199 nm (“c”) and between 931 and 977 nm (“d”). Put together, these results demonstrate the successful synthesis of B-PED.
Fig. 1. B-PED Characterizaton.
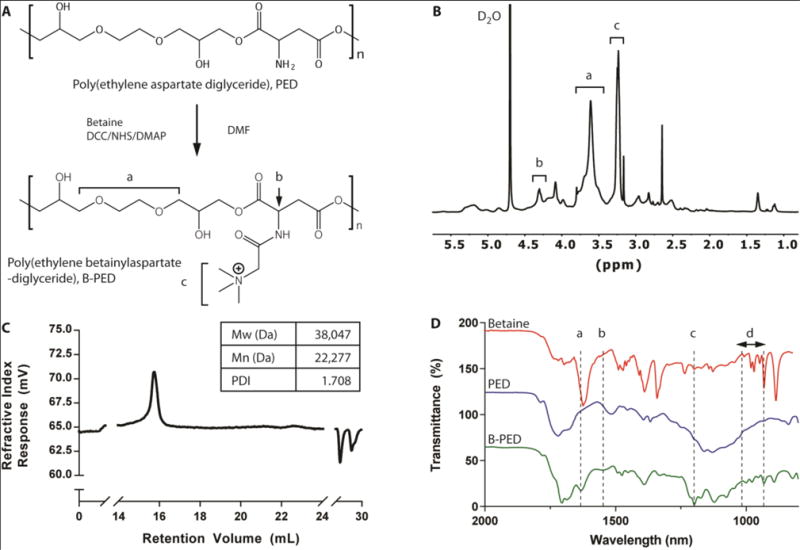
A. Betaine is grafted onto PED via an amide bond, yielding a polycation with a quaternary ammonium functional group. B. 1H NMR spectrum of B-PED; a (ethylene glycol), b (α proton on aspartate), c (trimethyl amine) (solvent peak: δ4.70). C. GPC of PED shows an MW of 38 kDa; B-PED is estimated to have an MW of ~46 kDa. D. FTIR spectrum reveals successful betaine conjugation onto PED; a,b: amide bond absorption; c,d: absorption peaks from betaine.
Biocompatibility of B-PED
Polycations such as poly(ethylenimine) and poly(L-lysine) generally exhibit poor biocompatibility, thereby limiting their widespread use. Given the relatively high conjugation efficiency of betaine onto PED, we examined the potential toxicity of B-PED on the metabolic activity and viability of NIH-3T3 fibroblasts and human umbilical vein endothelial cells (HUVECs). Both NIH-3T3 cells and HUVECs exhibit a corresponding increase in metabolic activity with [B-PED], peaking at 2.5 mg/mL (Fig. 2A). At 10 mg/mL, both cell types show metabolic activity that is not significantly different from that of untreated samples, though it should be noted that NIH-3T3 cells are trending towards significance (p=0.06). An examination of DNA quantity shows a similar effect, in which a peak response is observed for NIH-3T3 cells at 1.25 mg/mL (Fig. 2B). HUVECs, on the other hand, maintain a constant number of cells throughout, which may be more indicative of its longer doubling time. The increased level of metabolic and proliferative activity observed at low [B-PED] could be attributed to the protective effect of betaine on cells39, though less likely given the lack of free betaine to act as an osmolyte. A more likely explanation is that a betaine-induced increase of intracellular calcium40 leads to an upregulation of proliferative processes41. It should be noted that exogenously added betaine alone – concentrations of 1.25 mg/mL and above – results in an upregulation of metabolic activity for both cell types (Supplementary Fig. 3). Betaine-induced cellular toxicity shows a similar trend as cells treated with B-PED. On the other hand, treatment of NIH-3T3 cells with 10 mg/mL PED alone yields only 20% metabolic activity of untreated cells29. Put together, the slight toxicity of PED appears to be neutralized by the conjugation of betaine to its backbone.
Fig. 2. B-PED Biocompatibility.
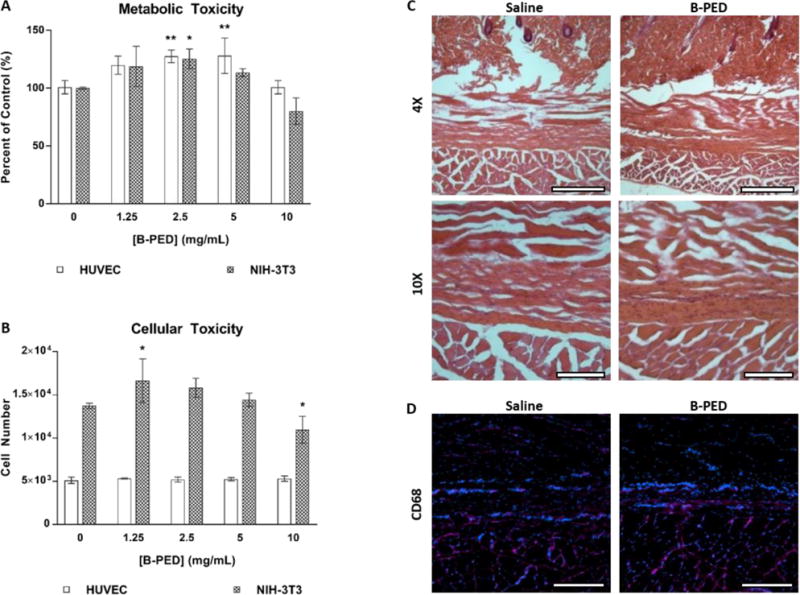
A. The effect of [B-PED] on HUVEC and NIH-3T3 cells is assessed via a metabolic and B. nucleic acid quantification assay (n=3, mean ± SD, *p<0.05, **p<0.01 compared to control). C. 100 μL of physiological saline or 10 mg/mL B-PED is subcutaneously injected into Sprague-Dawley rats. Tissues are harvested 1 week post-injection and visualized via H&E staining (scale bar, top: 100 μm, bottom: 500 μm). C. CD68 immunofluorescence staining shows minimal macrophage infiltration for both saline- and B-PED-treated rats (red: CD68, blue: nuclei, scale bar: 200 μm).
The biocompatibility of B-PED was then examined in-vivo, in which 100 μL of saline or B-PED (10 mg/mL) was injected subcutaneously in rats. H&E stains of tissues harvested 1 week post-injection show no significant difference in connective tissue and muscle morphology (Fig. 2C). Furthermore, no appreciable inflammation is observed. Finally, macrophage infiltration for B-PED – assessed via CD68 immunofluorescence – is minimal and comparable to rats receiving saline injections (Fig. 2D). Given the relative toxicity of poly(ethylenimine), in which cell viability is affected at concentrations as low as 0.01 mg/mL42, these results collectively demonstrate B-PED as a biocompatible polycation.
Effect of B-PED on Angiogenesis
Betaine has been shown to exhibit anti-inflammatory and anti-angiogenic effects via suppression of NF-κB and Akt signaling in-vitro43. Interestingly, we were unable to observe any anti-angiogenic effect of betaine on HUVEC tube formation (Supplementary Fig. 4). It should be noted that previous reports of betaine-induced anti-angiogenic behavior43 utilized increasing doses of betaine up to 1 mM; the lowest [betaine] tested in this study is 1.25 mg/mL (10.7 mM). Based on these comparisons, it appears that the anti-angiogenic effect of betaine is attenuated above a particular concentration. To assess how B-PED affects HUVEC tube formation, concentrations up to 10 mg/mL were tested based on cell viability results; HUVECs exhibit no change in cell metabolism or viability at 10 mg/mL compared to baseline levels (Fig. 2A, B). HUVECs cultured with basal medium (EBM) show an absence of tube formation while those cultured with growth factor-supplemented medium (EGM) develop well-connected tubes (Fig. 3A). Increasing concentrations of B-PED lead to a progressive inhibition of tube formation, in which 10 mg/mL yields a similar anti-angiogenic effect as HUVECs in EBM. Quantification of the fluorescent images indicates a significantly lower number of network-forming-loops for cells treated with 2.5 and 5 mg/mL of B-PED than those treated with EGM or 1.25 mg/mL B-PED (Fig. 3B). Treatment with 10 mg/mL of B-PED yields the smallest number of loops. While the number of junctions is also smallest for cells treated with 10 mg/mL of B-PED, there is no statistical difference among lower concentrations of B-PED (Fig. 3C). These in-vitro results demonstrate that betaine is effective in suppressing angiogenesis when grafted onto a polymer.
Fig. 3. B-PED Effect on Angiogenesis.
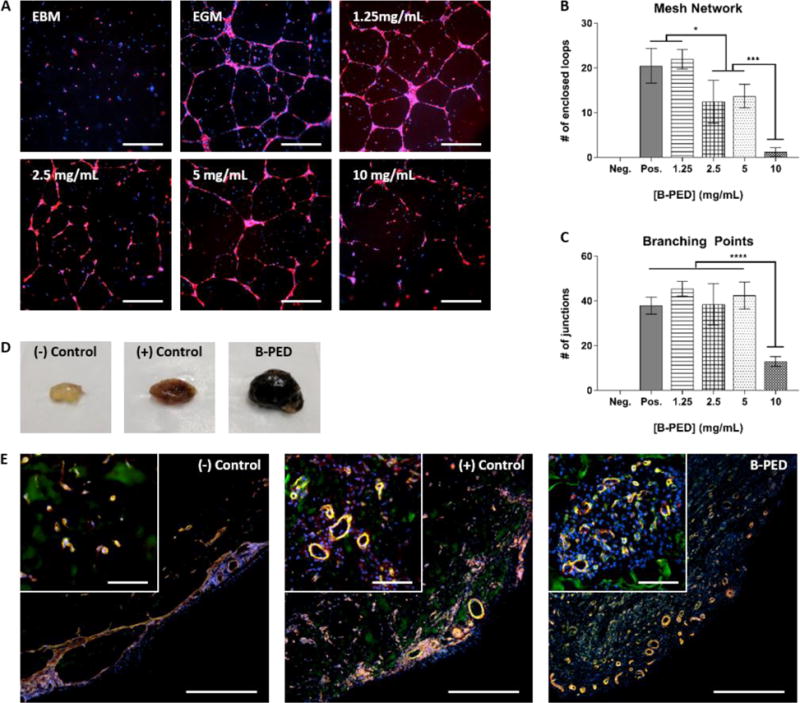
A. RFP-expressing HUVECs are cultured on basement membrane extract in the presence of varying [B-PED] (EBM: endothelial basal media, EGM: endothelial growth media). Cells are imaged 16 hours post-seeding (red: RFP, blue: nuclei, scale bar: 200 μm). B. Number of enclosed loops forming a mesh network and C. number of junctions are quantified from immunofluorescence images (n=4 wells, mean ± SD, *p<0.05, ***p<0.001, ****p<0.0001). D. Matrigel plugs are injected subcutaneously into the caudal ventral area of Sprague-Dawley rats (negative: Matrigel, positive: Matrigel + 1.5 μg FGF-2, B-PED: Matrigel + 1.5 μg FGF-2 + 10 mg B-PED). Plugs are harvested 10 days post-implantation. E. Immunofluorescence staining of Matrigel plug sections show varying levels of angiogenesis (scale bar: 500 μm, inset scale bar: 100 μm)
Interestingly, in-vivo application of B-PED in a rat Matrigel plug assay results in an entirely different response. Gross visualization of representative Matrigel plugs collected 10 days post-implantation are markedly different from one another (Fig. 3D), in which plugs without fibroblast growth factor 2 (FGF-2) are clear unlike those supplemented with FGF-2. In particular, the addition of B-PED (10 mg/mL) to FGF-2-supplemented Matrigel yield visibly larger and darker plugs. To better understand their cellular composition and blood vessel maturity, Matrigel plug explants were sectioned and assessed via double immunofluorescence staining against CD31 and α-SMA (Fig. 3E). Both FGF-2-supplemented plugs with or without B-PED show mature blood vessels, in which pericytes (α-SMA: red) are co-localized with endothelial cells (CD31: green). However, Matrigel plugs containing B-PED exhibit higher vessel density than even the positive control, which may explain their darker color at a gross macroscopic level. Additionally, a large number of non-endothelial cellular infiltrates (DAPI: blue) are observed in plugs supplemented with B-PED, which could explain its relatively large size.
Betaine has been shown to stabilize proteins34 and may have stabilized FGF-2 even in its grafted form. Given that monocyte/macrophage infiltration increases in the context of FGF-2-induced angiogenesis44, the CD31−/α-SMA− cellular infiltrates observed in plugs with B-PED could be macrophages. Furthermore, the pro-inflammatory environment promoted by macrophages has been reported to further activate FGF-2 and vascular endothelial growth factor (VEGF) secretion by endothelial cells44. In other words, although B-PED exhibits anti-angiogenic behavior in-vitro, its capacity to stabilize a large amount of FGF-2 (1.5 μg/plug/mouse) in a confined gel may have increased macrophage infiltrate and ultimately, angiogenesis, in-vivo. Finally, it should be noted that the average size of vessels in plugs containing B-PED are relatively small despite their high density. The inhibitory effect of betaine on cell migration43 could have carried over to B-PED, but further studies elucidating the mechanism of betaine-induced anti-angiogenesis are needed to explain this discrepancy.
Antimicrobial Property of B-PED
The prevention and treatment of bacterial infections is of particular importance in the field of medicine, in which the emergence of bacterial resistance poses additional challenges. Glycopeptide antibiotics are a class of antimicrobial agents for gram-positive bacteria and bind to bacterial cell wall precursors that contain (D-Ala-D-Ala), thereby inhibiting their cell wall synthesis45. Recently, vancomycin, a widely-used antibiotic, was systematically modified to include additional modes of antimicrobial activity independent of (D-Ala-D-Ala) binding38. Of particular interest was the inclusion of a quaternary ammonium group, which was found to induce cell wall permeability. Given that betaine contains the same functional group, we assessed the effect of B-PED on the growth of Staphylococcus aureus, a gram-positive bacteria predominant in skin infections and pneumonia.
The time-kill kinetics of B-PED at various concentrations demonstrate maximal anti-bacterial activity at 5 hours post-treatment (Fig. 4A). On the other hand, bacteria treated with timentin (positive control) show a response as early as 3 hours. Upon closer inspection at 5 hours post-treatment, we observe a unimodal dose response, in which B-PED shows an optimal concentration range for anti-bacterial activity (Fig. 4B). While lower and higher concentrations of B-PED appear to be ineffective in stemming Staphylococcus aureus growth, B-PED concentrations of 0.3125, 0.625, and 1.25 mg/mL show significantly lower levels of absorbance compared to that of untreated samples. As a control, Staphylococcus aureus were also incubated with PED, the backbone for B-PED. Increasing concentrations of PED have no effect on bacteria growth, thereby demonstrating that the anti-bacterial potency of B-PED is derived from the betaine side chain. It should be noted that the antimicrobial efficacy of B-PED exceeds or is comparable to other quaternary ammonium compound-based formulations. For instance, a cationic amphiphilic polycarbonate comprised of tetrametyl ammonium side groups shows activity against Staphylococcus aureus with a minimum inhibitory concentration (MIC) of 6.5 μM46. Similarly, the MIC90 of cetryltrimethylammonium bromide (CTAB), a commercially-available antiseptic, is 0.128 mg/mL47, or approximately 351 μM. B-PED shows an MIC of approximately 13.45 μM (0.625 mg/mL). On a side note, given that the mode of action of the quaternary ammonium group is contact-mediated membrane permeablization, the lack of anti-bacterial activity observed at higher B-PED concentrations may be due to some form of steric hindrance. Indeed, the molecular organization of cations within polycation aggregates has been shown to affect anti-bacterial activity, in which the size of aggregates and the number of active molecules that make up the aggregate play a determining role48. While we did not see [B-PED]-dependent aggregation (Supplementary Fig. 5), it is possible that the quaternary ammonium positive charge on B-PED is less accessible at higher [B-PED].
Fig. 4. Antimicrobial Activity of B-PED.
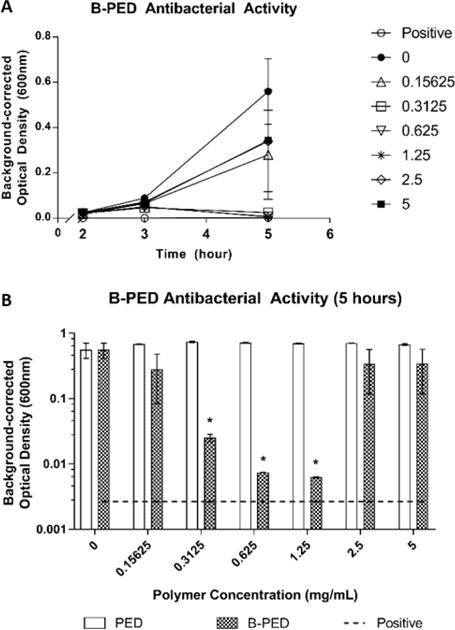
A. Staphylococcus aureus are cultured in the presence of varying [B-PED] and measured for absorbance to assess bacterial growth (positive: timentin, n=3, mean ± SEM). B. Antibacterial activity of PED is compared to B-PED 5 hours post-seeding (dashed line: positive control, n=3, mean ± SEM, *p<0.05 compared to negative control).
Complex Coacervation Behavior of B-PED
The conjugation of betaine onto PED yields a permanent positive charge per repeating unit via its quaternary ammonium functional group. Upon the addition of heparin, B-PED immediately self assembles into a complex coacervate, leading to liquid-liquid phase separation (Fig. 5A). The low interfacial energy between the coacervate and coexisting supernatant phase2 enables the coalescence of individual coacervate droplets into larger droplets over time. In the setting of a larger volume, an initially turbid solution of coacervate results in its sedimentation 24 hours post-mixing (Fig. 5B), which is easily resuspended by pipette agitation.
Fig. 5. Complex Coacervation of B-PED and Heparin.
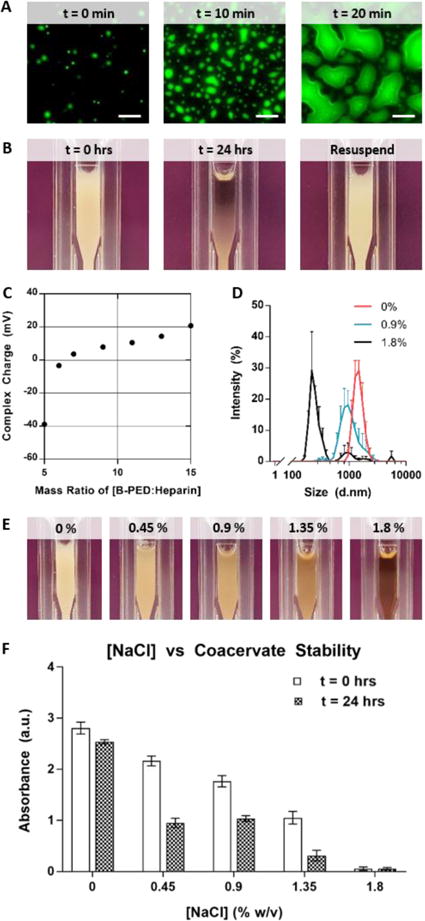
A. Fluorescein-labeled heparin is used to visualize complex coacervation behavior at a microscopic level (scale bar: 20 μm). B. Macroscopic observation shows the immediate formation of an off-white turbid solution, followed by a separation between the supernatant and coacervate phases within 24 hours; pipetting the solution resuspends the complex coacervate. C. Zeta potential measurements indicate a net neutral charge of B-PED/heparin complex coacervates is achieved at a mass ratio of 6.5:1 (B-PED:Heparin) (n=3, mean ± SD). D. Increasing [NaCl] decreases the hydrodynamic diameter of B-PED and heparin coacervates (n=3, mean ± SD). E. A visible reduction in turbidity is observed for increasing [NaCl] conditions (t=0 hrs). F. The absorbance of various solutions is measured (λ=420nm) at t = 0 hours and 24 hours (after resuspension) (n=3, mean ± SD).
The surface charge of B-PED and heparin coacervates is dependent on their relative ratio (Fig. 5C). As expected, a low B-PED:heparin mass ratio results in a negative zeta potential (−38.9 ± 0.5 mV). An increase in the B-PED:heparin ratio progressively increases the zeta potential, ultimately reaching an isoelectric point at approximately 6.5:1. At higher mass ratios, the excess B-PED acts to form positively-charged coacervates; the charge of B-PED alone is 42.0 ± 2.3 mV (Supplementary Fig. 2).
Finally, the effect of a competing salt on coacervation was assessed via DLS measurements (Fig. 5D). In deionized water without NaCl (0% w/v), B-PED/heparin coacervates yield an average hydrodynamic diameter of 1477.7 ± 321.0 nm. The diameter slightly decreases to 1098.5 ± 343.8 nm in the presence of 0.9% NaCl. Further addition of NaCl to 1.8% w/v results in a non-uniform population of coacervates, with a primary peak diameter of 250.1 ± 24.9 nm. Apart from hydrodynamic diameter, the polydispersity index (PDI) of B-PED/heparin coacervates is relatively monodisperse for both 0% and 0.9% NaCl (Supplementary Table 1). On the other hand, the PDI of coacervates prepared in 1.8% NaCl is much higher and reflective of the salt-induced decrease in interfacial energy between phases. Macroscopic observation of B-PED/heparin coacervate solutions in different [NaCl] (0, 0.45, 0.9, 1.35, and 1.8% w/v) further corroborate the DLS data, in which increasing ionic strength progressively inhibits coacervate formation (Fig. 5E). After 24 hours, all groups show a clear supernatant phase, in which sedimented coacervate phase is visible for [NaCl] of 0, 0.45, and 0.9% (Supplementary Fig. 6); resuspension of the B-PED/heparin coacervate yields a turbid solution again. Though [NaCl] of 1.35% initially yields a turbid solution, the solution remains clear after 24 hours, even after re-agitation. Taken together, the critical salt resistance of the B-PED/heparin coacervates is between 0.9% and 1.35%. It should be noted that any coacervate-based therapy would be performed in physiological saline ([NaCl] = 0.9%) within 10 minutes of preparation. Furthermore, subcutaneous or intramuscular injections are traditionally used, in which the supernatant phase would quickly disperse to leave behind the coacervate phase.
Conclusions
We designed a betaine-functionalized polycation, B-PED, to capitalize on the various properties of betaine. Despite the high betaine content and corresponding cationic charge of B-PED, excellent in-vitro and in-vivo biocompatibility was observed. Depending on the type of cell examined, B-PED also appeared to act as a proliferation agent at certain concentrations. In addition, B-PED elicited a dose-responsive decrease in angiogenesis in-vitro. Interestingly, the presence of B-PED increased angiogenesis in-vivo as assessed by a rat Matrigel plug assay. The in vivo environment is far more complicated than a controlled cell culture environment; further mechanistic studies are required to explain the opposite effects. B-PED also exhibits an excellent unimodal antimicrobial response against Staphylococcus aureus. Finally, the strong positive charge of B-PED facilitates its complexation with heparin to form a stable coacervate. Given the anti-bacterial and pro-angiogenic activity of B-PED in the presence of FGF-2, we envision the potential utilization of B-PED/heparin coacervates for applications such as wound healing of the skin and open bone fracture.
Supplementary Material
Acknowledgments
We would like to thank Scientific Protein Laboratories, LLC for their generous donation of clinical grade heparin. We would also like to thank Dr. Steven Little for his generous donation of RFP-HUVECs. This work was supported by NIH grant # 5R01NR016436.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Kim S, Huang J, Lee Y, Dutta S, Yoo HY, Jung YM, Jho Y, Zeng H, Hwang DS. Proceedings of the National Academy of Sciences. 2016;113:E847–E853. doi: 10.1073/pnas.1521521113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qin J, Priftis D, Farina R, Perry SL, Leon L, Whitmer J, Hoffmann K, Tirrell M, de Pablo JJ. ACS Macro Letters. 2014;3:565–568. doi: 10.1021/mz500190w. [DOI] [PubMed] [Google Scholar]
- 3.Gummel J, Cousin F, Boué F. Journal of the American Chemical Society. 2007;129:5806–5807. doi: 10.1021/ja070414t. [DOI] [PubMed] [Google Scholar]
- 4.Priftis D, Laugel N, Tirrell M. Langmuir. 2012;28:15947–15957. doi: 10.1021/la302729r. [DOI] [PubMed] [Google Scholar]
- 5.Dora Tang TY, Rohaida Che Hak C, Thompson AJ, Kuimova MK, Williams DS, Perriman AW, Mann S. Nature Chemistry. 2014;6:527–533. doi: 10.1038/nchem.1921. [DOI] [PubMed] [Google Scholar]
- 6.Sokolova E, Spruijt E, Hansen MMK, Dubuc E, Groen J, Chokkalingam V, Piruska A, Heus HA, Huck WTS. Proceedings of the National Academy of Sciences. 2013;110:11692–11697. doi: 10.1073/pnas.1222321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakry AM, Abbas S, Ali B, Majeed H, Abouelwafa MY, Mousa A, Liang L. Comprehensive Reviews in Food Science and Food Safety. 2016;15:143–182. doi: 10.1111/1541-4337.12179. [DOI] [PubMed] [Google Scholar]
- 8.Spruijt E, Westphal AH, Borst JW, Cohen Stuart MA, van der Gucht J. Macromolecules. 2010;43:6476–6484. [Google Scholar]
- 9.Hwang DS, Waite JH, Tirrell M. Biomaterials. 2010;31:1080–1084. doi: 10.1016/j.biomaterials.2009.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishii S, Kaneko J, Nagasaki Y. Biomaterials. 2016;84:210–218. doi: 10.1016/j.biomaterials.2016.01.029. [DOI] [PubMed] [Google Scholar]
- 11.Chu H, Gao J, Chen CW, Huard J, Wang Y. Proceedings of the National Academy of Sciences. 2011;108:13444–13449. doi: 10.1073/pnas.1110121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pippa N, Kalinova R, Dimitrov I, Pispas S, Demetzos C. The Journal of Physical Chemistry B. 2015;119:6813–6819. doi: 10.1021/acs.jpcb.5b01664. [DOI] [PubMed] [Google Scholar]
- 13.Kuo CH, Leon L, Chung EJ, Huang RT, Sontag TJ, Reardon CA, Getz GS, Tirrell M, Fang Y. Journal of Materials Chemistry B. 2014;2:8142–8153. doi: 10.1039/C4TB00977K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vehlow D, Schmidt R, Gebert A, Siebert M, Lips KS, Müller M. Nanomaterials. 2016;6:53. doi: 10.3390/nano6030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baruch L, Machluf M. Biopolymers. 2006;82:570–579. doi: 10.1002/bip.20509. [DOI] [PubMed] [Google Scholar]
- 16.Chen WC, Lee BG, Park DW, Kim K, Chu H, Kim K, Huard J, Wang Y. Biomaterials. 2015;72:138–151. doi: 10.1016/j.biomaterials.2015.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson NR, Wang Y. Journal of Controlled Release. 2013;166:124–129. doi: 10.1016/j.jconrel.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayitmazer AB, Koksal AF, Kilic Iyilik E. Soft Matter. 2015;11:8605–8612. doi: 10.1039/c5sm01829c. [DOI] [PubMed] [Google Scholar]
- 19.Miller DR, Das S, Huang KY, Han S, Israelachvili JN, Waite JH. ACS Biomaterials Science & Engineering. 2015;1:1121–1128. doi: 10.1021/acsbiomaterials.5b00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pellegrini L, Burke DF, von Delft F, Mulloy B, Blundell TL. Nature. 2000;407:1029–1034. doi: 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- 21.Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD, Leahy DJ. Proceedings of the National Academy of Sciences. 2007;104:6152–6157. doi: 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teran M, Nugent MA. The Journal of Biological Chemistry. 2015;290:16451–16462. doi: 10.1074/jbc.M114.627372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de la Torre PM, Enobakhare Y, Torrado G, Torrado S. Biomaterials. 2003;24:1499–1506. doi: 10.1016/s0142-9612(02)00512-4. [DOI] [PubMed] [Google Scholar]
- 24.Butstraen C, Salaün F. Carbohydrate Polymers. 2014;99:608–616. doi: 10.1016/j.carbpol.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Salehi A, Desai PS, Li J, Steele CA, Larson RG. Macromolecules. 2015;48:400–409. [Google Scholar]
- 26.Hao Y, Zhang M, He J, Ni P. Langmuir. 2012;28:6448–6460. doi: 10.1021/la300208n. [DOI] [PubMed] [Google Scholar]
- 27.Trubetskoy VS, Wong SC, Subbotin V, Budker VG, Loomis A, Hagstrom JE, Wolff JA. Gene Therapy. 2003;10:261–271. doi: 10.1038/sj.gt.3301888. [DOI] [PubMed] [Google Scholar]
- 28.Meng X, Perry SL, Schiffman JD. ACS Macro Letters. 2017;6:505–511. doi: 10.1021/acsmacrolett.7b00173. [DOI] [PubMed] [Google Scholar]
- 29.Chu H, Gao J, Wang Y. Biotechnology Progress. 2012;28:257–264. doi: 10.1002/btpr.728. [DOI] [PubMed] [Google Scholar]
- 30.Zern BJ, Chu H, Osunkoya AO, Gao J, Wang Y. Advanced Functional Materials. 2011;21:434–440. doi: 10.1002/adfm.201000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Awada HK, Long DW, Wang Z, Hwang MP, Kim K, Wang Y. Biomaterials. 2017;125:65–80. doi: 10.1016/j.biomaterials.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Day CR, Kempson SA. Biochimica et Biophysica Acta (BBA) - General Subjects. 2016;1860:1098–1106. doi: 10.1016/j.bbagen.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Burg MB, Ferraris JD. The Journal of Biological Chemistry. 2008;283:7309–7313. doi: 10.1074/jbc.R700042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar N, Kishore N. Biophysical Chemistry. 2014;189:16–24. doi: 10.1016/j.bpc.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Lever M, Slow S. Clinical Biochemistry. 2010;43:732–744. doi: 10.1016/j.clinbiochem.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Sun S, Li X, Ren A, Du M, Du H, Shu Y, Zhu L, Wang W. Scientific Reports. 2016;6:35547. doi: 10.1038/srep35547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Detopoulou P, Panagiotakos DB, Antonopoulou S, Pitsavos C, Stefanadis C. The American Journal of Clinical Nutrition. 2008;87:424–430. doi: 10.1093/ajcn/87.2.424. [DOI] [PubMed] [Google Scholar]
- 38.Okano A, Isley NA, Boger DL. Proceedings of the National Academy of Sciences. 2017;114:E5052–E5061. doi: 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petronini PG, De Angelis EM, Borghetti P, Borghetti AF, Wheeler KP. Biochemical Journal. 1992;282:69–73. doi: 10.1042/bj2820069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villa I, Senesi P, Montesano A, Ferraretto A, Vacante F, Spinello A, Bottani M, Bolamperti S, Rubinacci A, Luzi L, Terruzzi I. Journal of Translational Medicine. 2017;15:132. doi: 10.1186/s12967-017-1233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munaron L, Antoniotti S, Lovisolo D. Journal of Cellular and Molecular Medicine. 2004;8:161–168. doi: 10.1111/j.1582-4934.2004.tb00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. Biomaterials. 2003;24:1121–1131. doi: 10.1016/s0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- 43.Yi EY, Kim YJ. International Journal of Oncology. 2012;41:1879–1885. doi: 10.3892/ijo.2012.1616. [DOI] [PubMed] [Google Scholar]
- 44.Andres G, Leali D, Mitola S, Coltrini D, Camozzi M, Corsini M, Belleri M, Hirsch E, Schwendener RA, Christofori G, Alcami A, Presta M. Journal of Cellular and Molecular Medicine. 2009;13:2083–2108. doi: 10.1111/j.1582-4934.2008.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.James RC, Pierce JG, Okano A, Xie J, Boger DL. ACS Chemical Biology. 2012;7:797–804. doi: 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nederberg F, Zhang Y, Tan JPK, Xu K, Wang H, Yang C, Gao S, Guo XD, Fukushima K, Li L, Hedrick JL, Yang YY. Nature Chemistry. 2011;3:409–414. doi: 10.1038/nchem.1012. [DOI] [PubMed] [Google Scholar]
- 47.Wu G, Yang Q, Long M, Guo L, Li B, Meng Y, Zhang A, Wang H, Liu S, Zou L. Journal of Antibiotics. 2015;68:661–665. doi: 10.1038/ja.2015.51. [DOI] [PubMed] [Google Scholar]
- 48.Kanazawa A, Ikeda T, Endo T. Journal of Applied Bacteriology. 1995;78:55–60. doi: 10.1111/j.1365-2672.1995.tb01673.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.