

Abstract
This study investigated poly (ADP-ribose) polymerase-1 (PARP-1) activation in cultured human lens epithelial cells exposed to two levels of UVB light (312 nm peak wavelength), 0.014 and 0.14 J/cm2 (“low” and “high” dose, respectively). At the low dose, PARP-1 and poly (ADP-ribose) (PAR) polymers acted to repair DNA strand breaks rapidly with no subsequent major effects on either cell morphology or viability. However, following the high UVB dose, there was a dramatic second phase of PARP-1 activation, 90 min later, which included a sudden reappearance of DNA strand breaks, bursts of reactive oxygen species (ROS) formation both within the mitochondria and nucleus, a translocation of PAR from the nucleus to the mitochondria, and an ultimate 70% loss of cell viability occurring after 24 hrs. The results provide evidence for an important role for PARP-1 in protecting the human lens epithelium against low levels of UVB light, and possibly participating in the triggering of cell death following exposure to toxic levels of radiation.
Graphical Abstract
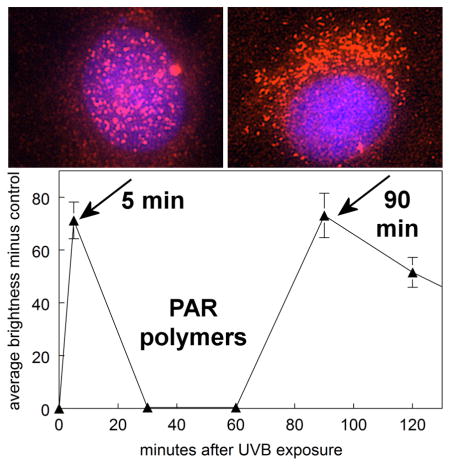
Five minutes after a 2.5 minute exposure of cultured human lens epithelial cells to 0.14 J/cm2 of UVB light, poly (ADP-ribose) (PAR) polymers were produced in the cell nucleus to assist in the repair of DNA, and the polymers then disappeared. Surprisingly, 90 minutes after the exposure, PAR polymers were produced once again, but this time the polymers appeared to travel out of the nucleus to the cell mitochondria, possibly to initiate cell death.
INTRODUCTION
Solar UV radiation has been linked epidemiologically with the formation of human cortical cataract (1–3). However, whether wavelengths of UVB or UVA light, or possibly both, may be responsible for the cataract is controversial (4). UVB light has a variety of lens epithelial targets, including DNA, mitochondria and the amino acid tryptophan, which is present in lens crystallins, enzymes and membrane proteins at high levels (5–7). In addition, UVA radiation can damage lens DNA and mitochondria through the generation of reactive oxygen species when it is absorbed by chromophores such as pyridine nucleotides and riboflavin (8–10). Whereas about 70% of solar UVA light striking the human cornea reaches the lens epithelium, only 1% of incident UVB radiation is ultimately absorbed by the lens (11, 12). However, following decades of exposure, even this relatively small amount of light can be damaging because of the high level of the UVB-absorber tryptophan present in the tissue (7). An even greater threat to lens transparency may be UV light entering from the side of the eye since this radiation can focus up to 20× stronger in the peripheral region of the lens on the nasal side (13–15). Since the lens periphery contains the germinative region of the tissue (16), which possesses a relatively high mitotic activity and slow rate of DNA repair (17–20), it is more susceptible to radiation-induced DNA damage. The fact that the nasal germinative region is typically the site of human cortical cataract (21–23) supports the belief that sunlight is indeed a major cause of this type of opacity.
Evidence exists to link DNA damage with the formation of human cortical cataract. Epithelia removed from the lenses of cataract patients prior to surgery showed a higher level of DNA damage (strand breaks) compared to similar tissue from human donor eyes, and the damage was significantly greater in cortical cataracts compared to nuclear and posterior subcapsular opacities (24). A similar investigation found increased amounts of DNA strand breaks present in lens epithelial cells and lymphocytes of patients with cortical, nuclear and posterior subcapsular cataracts, compared to controls (25). Results of earlier studies had suggested that genotoxic damage may be associated with the development of certain types of human cataracts (26, 27). In recent work, a major product of DNA oxidative damage, 8-oxo-7, 8-dihydroguanine (8-oxoG) (28), was detected at higher levels in lens epithelia from patients undergoing cataract surgery, compared to controls (29). Levels of mRNA coding for the enzyme responsible for repairing 8-oxoG, 8-oxoguanine-DNA glycosylase 1, were elevated in lens epithelia isolated from patients undergoing cataract surgery, and decreased in opaque regions of cortical cataracts (29, 30). Similarly, concentrations of the most abundant product of DNA oxidation, 8-hydroxy-2-deoxyguanosine (31), were elevated in various regions of cortical, nuclear and posterior subcapsular cataracts (32), as well as in leukocytes of cataract patients (33).
Repair of damaged DNA is a complex process involving a multitude of different proteins and cellular pathways, controlled by approximately 150 different genes in the human (34). Of 92 DNA repair genes detected in human central lens epithelium, 11 were reported to have changed expression in cataracts (35). One major DNA repair enzyme in the lens that becomes activated following oxidative challenge is poly(ADP-ribose) polymerase (PARP) (36–40). The PARP family consists of 17 proteins, of which the PARP-1 nuclear enzyme is the most abundant and most studied (41–43). PARP-1 catalyzes the synthesis of ADP-ribose units from donor NAD+ molecules to form large poly(ADP-ribose) (PAR) polymers, up to 200 units in length, that bind to target gene regulatory proteins and modulates their activities (44). PARP-1 performs an extraordinary number of key functions in the cell nucleus including the repair of DNA single and double strand breaks, which allows the cell to survive during times of oxidative challenge, as well as induction of cell death in the event of overactivation of the enzyme (45, 46). Low activities of PARP and elevated levels of 8-OHdG have been observed in the blood of patients with age-related cataract (47). Relatively few studies have explored UVB-induced activation of the PARP system in cultured cells (48–51). Here, we investigate cell survival/cell death functions of PARP-1 and PAR polymers in cultured human lens epithelial cells (LECs) following exposure to two levels of UVB radiation.
MATERIALS AND METHODS
Cell culture and exposure to UVB light
Experiments were conducted with an immortalized human lens epithelial cell (LEC) line, SRA 01/04 (52). The cells were grown in Dulbecco’s Modified Eagle Medium (MEM) with Earle’s salts, supplemented with 15% fetal bovine serum (FBS), 50 μg/ml gentamicin and 2.1 μg/ml amphotericin B contained in Fungizone®. Cultures were carried out at 37°C in 5% CO2, 95% air. FBS and Fungizone® were obtained from Gibco Life technologies (Grand Island, NY, USA). All other reagents were from Sigma-Aldrich (St. Louis, MO, USA), unless noted. After 6 days, when the cells had reached confluency in 100 mm plates, they were enzymatically removed from the plates 1 day prior to the experiment and transferred to either 60 mm plates (approximately 800,000 cells per plate), 4-well chamber slides (Thermo Fisher Scientific, Rochester, NY, U.S.A.; approximately 50,000 cells per well) or 96-well plates (Thermo Fisher Scientific; approximately 10,000 cells per well). On the day of the experiment, in preparation for UVB-irradiation, the cells were incubated for 2 hrs in MEM containing 1% FBS, and then for 30 min in serum-free medium. Prepared MEM was used within three weeks.
For UVB-irradiation, the cells were treated as described above and then rinsed with phosphate-buffered saline (PBS) and exposed to UVB light (from the bottom) in PBS (37°C, air) for 2.5 min using a Spectroline “medium wave” UV lamp (Model EB-160C, Spectronics Corp., Westbury, NY, USA) at a distance of 0.8 cm. Control cells were treated similarly, but not exposed to UVB light. UVB levels reaching the cells were determined with a radiometer (UVX Digital; San Gabriel, CA) equipped with a UVB sensor at 312 nm (model UVX-31). The UV beam had a wavelength spectral distribution of 270 – 390 nm with a peak at 312 nm (Fig. 1). The small amount of UVC radiation in the beam was removed using a Corning filter (0–53, 2 mm thick, Corning Co-Star, Corning, NY). The irradiance of the UVB light was either 0.09 or 0.9 mW/cm2, and the total fluences for the 2.5 min exposures were 0.014 J/cm2 (“low dose”) and 0.14 J/cm2 (“high dose”), respectively. Following UVB-exposure, cells were rinsed with PBS and cultured for various times in MEM containing 15% FBS. Photographs of control and UVB-treated cultured cells in 60 mm plates were taken using a Nikon Eclipse TS 100 microscope with an attached SPOT Idea camera and Spot 5.2 imaging software (SPOT Imaging, Sterling Heights, MI, U.S.A).
Figure 1.
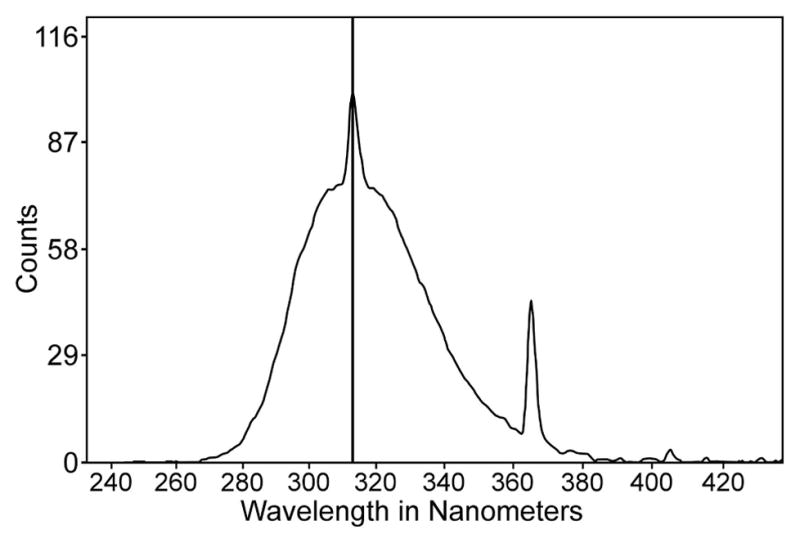
Spectral output of the UV lamp used in the study. Note the peak intensity at 312 nm wavelength. Light below 280 nm (UVC) was removed with a filter.
Cell viability
Viability of cells exposed to UVB light in 96-well plates was determined with use of an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Following UVB-exposure and subsequent culture, MEM containing 15% FBS was removed from the wells and replaced with 100 μl of MTT solution (5 mg MTT per ml MEM). After incubation of the cells for 2 hrs at 37°C, the MTT was removed and replaced with acidic organic solvent containing 90% isopropanol, 10% Triton X-100 and 1 mM HCL. Absorbance was read at a wavelength of 570 nm with a BioTek Epoch 2 Microplate Spectrophotometer (BioTek Instruments, Winooski, VT, U.S.A.), and the results graphed using Gen5 Data Analysis Software.
DNA strand break analysis
DNA strand breaks were detected in cultured LECs using a Roche TMR Red Cell Death Detection Kit (Roche Diagnostics Corporation, Indianapolis, IN, USA) following UV-exposure of the cells in 4-well chamber slides. TMR Red detects both single- and double-stranded DNA breaks that occur at early stages of apoptosis. Photographs were taken using a Zeiss Axio Imager .Z2 fluorescence microscope with an attached Axio Cam camera and Axio Vision software, version 4.8.2.
PARP-1, PAR and F-actin detection
Following a 2.5 min UVB-exposure of the cells in 4-well chamber slides and culture for various times, PARP-1 enzyme and PAR polymers were detected using immunocytochemistry. MEM containing 15% FBS was removed from the wells of the chamber slides and the cells were then washed once with PBS and fixed in 4% paraformaldehyde (PFA) for 30 minutes at room temperature. After fixation, the cells were washed 3× with PBS and incubated in 0.125% Triton X-100 for 15 minutes at room temperature. The Triton was removed, the cells washed 3× with PBS, and 5% Normal Goat Serum (NGS) added as blocking solution for 1 hr at room temperature. The NGS was removed and primary antibody for either PAR (PAR monoclonal antibody, Enzo Life Sciences, Farmingdale, NY or EMD Millipore, Billerica, MA; 1:400 dilution) or PARP-1 (PARP-1 monoclonal antibody, clone C2–10, Trevigen, Inc., Gaithersburg, MD, 1:400 dilution) was added and incubated overnight in the cold. The next day, the primary antibody was removed and the cells were washed 3× with PBS. Secondary antibody (Alexa Fluor 568 anti-mouse, 1:200 dilution) was added and incubated for 2 hours at room temperature. The secondary antibody was removed and the cells were washed 3× with PBS, and nuclei stained with 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) for 5 minutes at room temperature. The cells were washed 3× with PBS and the slides mounted with glycerol and photographed under a Zeiss fluorescence microscope as described above. In order to determine the composition of observed threadlike structures in UVB-damaged LECs, the fluorescent F-actin probe Rhodamine Phalloidin (Thermo Fisher Scientific; catalog number R415) was employed following the manufacturer’s instructions.
Reactive oxygen species and superoxide anion detection
Reactive oxygen species (ROS) were detected in cells exposed to UVB light in 4-well chamber slides using CellROX® Deep Red and CellROX® Green Reagents (Thermo Fisher Scientific) for detection of ROS (primarily H2O2) in the cell cytoplasm and nucleus, respectively. Following UVB-irradiation of the cells and subsequent incubation for various times, 1 μL each of the two reagents was added to each well containing 500 μL of MEM plus 15% FBS. After incubation for 30 minutes at 37°C, the mixture was removed from each well and the cells rinsed 3× with PBS and fixed in 4% PFA for 30 minutes at room temperature. The PFA was removed and the slides mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL, U.S.A.) and photographed under a Zeiss fluorescence microscope within 2 hours after addition of the reagents.
For detection of mitochondrial O2− in live LECs exposed to UVB light in 4-well chamber slides, a 1 μM solution of MitoSOX Red reagent (Thermo Fisher Scientific) was employed following the manufacturer’s instructions. After incubation for 10 min in the dark, the solution was removed and the cells counterstained with 5 μg/ml of Hoechst dye solution. The cells were washed 3× with warmed Dulbecco’s phosphate buffered saline plus magnesium and calcium (DPBS, Mediatech, Inc., Manassas, VA), and the slides mounted with a small amount of DPBS and a cover slip, and imaged with a Zeiss fluorescence microscope.
Fluorescent image analysis
Fluorescent images of cells were deconvoluted and background-subtracted using Nikon NIS Elements software. Quantitative densitometric scanning was conducted with use of ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2015). Cells were selected using the thresholding function and the average brightness level measured in the isolated cells. Averages, standard deviations and p-values were calculated with use of Excel software.
RESULTS
No distinct morphological effects were observed in cultured LECs at 1, 3 and 24 hr after a 2.5 min exposure to the low dose (0.014 J/cm2) of UVB light (Figs. 2b, c and d, respectively), compared to controls (Fig. 2a). Similarly, the high dose (0.14 J/cm2) of UVB light produced no damage after 1 and 3 hr (Figs. 2e and f). However, 24 hr after exposure to the high dose, significant changes in the cells were observed, including cell loss and formation of abnormal threadlike structures between cells (Fig. 2g), which stained for F-actin (Fig. 2h). Cells exposed to either the low or high UVB dose exhibited approximately 75% viability (MTT assay), compared to controls, after 1 and 3 hr of normal culture, and a similar result was obtained at the 24 hr mark for cells exposed to the low UVB dose (Fig. 3). In contrast, cell viability was observed to drop sharply to 30% 24 hr after exposure to the high UVB dose (Fig. 3; p<0.001). UVB-induced DNA strand-breaks were assessed using the TMR Red assay. For the low UVB dose, strand breaks were observed 5 min after UVB exposure (p<0.001), and were completely repaired within 30 min (Figs. 4a and c). For the high UVB dose, strand-breaks were observed immediately after UVB-exposure and at the 5 min mark (p<0.001), and were again repaired by 30 min (Figs. 4b and c). Surprisingly, DNA damage was seen to reappear for the high UVB dose, but not the low dose, at the 60 min (slight) and 90 min marks (p<0.001), and this second phase of damage was again completely repaired within 30 min (Figs. 4b and c). Similar results were obtained using the Comet assay (53) to detect DNA strand breaks (data not shown).
Figure 2.

Photographs of cultured human LECs at various times after exposure to either a low dose (0.09 mW/cm2) or high dose (0.9 mW/cm2) of UVB radiation for 2.5 min in 60 mm plates. a. 24 hr control; b. low dose UVB, 1 hr; c. low dose UVB, 3 hr; d. low dose UVB, 24 hr; e. high dose UVB, 1 hr; f. high dose UVB, 3 hr; g. high dose UVB, 24 hr; h. high dose UVB, 24 hr, stained for F-actin. Note the cell loss and abnormal threadlike structures between cells (arrows) in g and h. The results are representative of three experiments.
Figure 3.
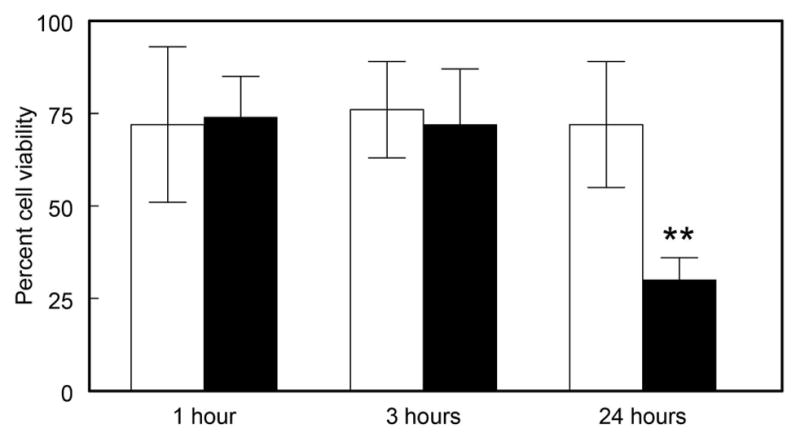
Viability of cultured human LECs at various times after exposure to either a low dose (0.09 mW/cm2) or high dose (0.9 mW/cm2) of UVB radiation for 2.5 min. Analysis was done using the MTT assay for cell viability. Results are expressed as percent of control cells not exposed to UVB light, means +/− S.D. for three experiments. Open bars: low UVB dose; solid bars: high UVB dose. **: p<0.01, relative to control.
Figure 4.

DNA strand-breaks (TMR Red assay) and DAPI staining in cultured human LEC’s at various times after exposure to either a low dose (0.09 mW/cm2) or high dose (0.9 mW/cm2) of UVB radiation for 2.5 min in 4-well chamber slides. a. low dose; b. high dose; c. densitometric fluorescence scanning of the cell nuclei; average cell nucleus brightness minus control for 6 to 24 cells, normalized per cell. Dashed line: low dose; solid line: high dose; n=3; error bars: S.D. (some error bars were smaller than the markers). ***: p<0.001, relative to control.
It was also of interest to investigate possible formation of ROS (primarily H2O2) in the cell cytoplasm and nucleus, as well as O2− in the mitochondria, at various times following UVB-exposure. A slight increase in ROS in both the cell cytoplasm and nucleus (red and green fluorescence, respectively) was observed immediately and 5 min after treatment with UVB-irradiation for both the low and high doses (Figs. 5a, b and c), but this had decreased to the control level by the 30 min mark. Levels of ROS remained at control values through 120 min for the low dose; however, in sharp contrast, for the high UVB dose, a dramatic increase in ROS reappeared at 90 min post UVB-exposure in both the cell cytoplasm and nucleus at levels that were 3× greater than those observed immediately after exposure (p<0.001; Figs. 5b and c). At 120 min after the high UVB dose, the level of ROS had decreased by 50%, compared to that seen at the 90 min mark (Figs. 5b and c). In contrast to the results for ROS, the first burst of mitochondrial O2− formation occurred primarily at 90 min after the high UVB dose, and this had decreased by approximately 60% 30 min later (p<0.001, Figs. 6a and b).
Figure 5.

Reactive oxygen species (ROS) detection (CellROX® Red and Green) in cultured human LEC’s at various times after exposure to either a low dose (0.09 mW/cm2) or high dose (0.9 mW/cm2) of UVB radiation for 2.5 min in 4-well chamber slides. Red fluorescence indicates ROS (mainly H2O2) in the cytoplasm. Green fluorescence indicates ROS (mainly H2O2) in the nucleus. a. low dose; b. high dose; c. densitometric fluorescence scanning of the cells; average cell brightness minus control for 6 to 40 cells, normalized per cell (red and green brightness added); Dashed line: low dose; solid line: high dose. n=3; error bars: S.D. ***: p<0.001, relative to control.
Figure 6.

Mitochondrial O2− detection (MitoSOX Red reagent) and Hoechst nuclear staining in cultured human LEC’s at various times after exposure to a high dose (0.9 mW/cm2) of UVB radiation for 2.5 min in 4-well chamber slides. a. fluorescent images; b. densitometric fluorescence scanning of the cells; average cell brightness minus control for 12 to 46 cells, normalized per cell. n=3; error bars: S.D. (some error bars were smaller than the markers). ***: p<0.001, relative to control.
Immunocytochemical results indicated that PARP-1 enzyme is normally present in the nucleus of human LECs, and exposure of the cells to the high UVB dose did not produce a significant change in the level of the enzyme 5 and 90 mins following irradiation (Fig. 7a and b). In contrast, PAR polymers were observed to be absent in normal cells, but were rapidly produced following UVB-exposure at both the low and high doses (Figs. 8a, b and c). At both doses, levels of PAR increased at the 5 min mark (p<0.001), and then fell back to control values at 30 and 60 min. At the high UVB dose, but not the low dose, PAR polymers were seen to reappear at 90 min at a level equal to that observed at 5 min (p<0.001). Whereas the PAR polymers that formed soon after UVB-exposure rapidly disappeared (t1/2 of 8–12 min), the polymers that were produced at 90 min following the high UVB dose degraded much more slowly (t1/2 of nearly 60 min), and were still evident at 30 (p<0.001; Figs. 8b and c) and even 90 (p<0.001; Fig. 8c) min later. About 90% of the PAR present in the cells 5 min after exposure to the high UVB dose was located in the nucleus; however, at the 90 min mark, about 90% of the polymers appeared to have flowed out of the nucleus into the cytoplasm, apparently to reside within the mitochondria (Figs. 8b and 9).
Figure 7.

PARP-1 enzyme immunofluorescence and DAPI nuclear staining in cultured human LEC’s at 5 and 90 min after exposure to a high dose (0.9 mW/cm2) of UVB radiation for 2.5 min in 4-well chamber slides. a. the fluorescent images; b. densitometric fluorescence scanning of the cells; average cell nuclear brightness minus control for 27 to 41 cells. n=3; error bars: S.D. (some error bars were smaller than the markers).
Figure 8.

PAR polymer immunofluorescence and DAPI nuclear staining in cultured human LEC’s at various times after exposure to either a low dose (0.09 mW/cm2) or high dose (0.9 mW/cm2) of UVB radiation for 2.5 min in 4-well chamber slides. a. low dose; b. high dose; c. densitometric fluorescence scanning of the cells; average cell brightness minus control for 6 to 59 cells, normalized per cell. Dashed line: low dose; solid line: high dose; n=3; error bars: S.D. (some error bars were smaller than the markers). ***: p<0.001, relative to control.
Figure 9.

Higher magnification (b and d) of one cell each (shown in the rectangles of a and c) from the 5 min (a and b) and 90 min(c and d) merged results of the experiment conducted in Fig. 8b (PAR and DAPI following exposure to the high dose of UVB).
DISCUSSION
This study has demonstrated the presence of PARP-1 in the human LEC nucleus (Fig. 7), and documented its participation in the repair of oxidatively-damaged DNA following exposure of the cells to UVB radiation (Figs. 4 and 8). Similar findings have been reported for human lens cells challenged with H2O2 (40), and an earlier study showed that rabbit LECs treated with hyperbaric oxygen exhibited a doubling of PARP activity (37). Thus, PARP-1 appears to be a major defense against oxidative DNA damage in the lens epithelium, as has been reported for a variety of other tissues, including lung (54), brain (55), skin (56), liver (57), retina (58) and testes (59). PARP activation as a result of UVB-exposure has previously been reported to occur in human keratinocytes (50), and in both human (48) and mouse (49) fibroblasts. Although the PARP-1 enzyme was visible in the unstressed LEC (Fig. 7), PAR was not (Fig. 8). However, following oxidative stress and the formation of DNA strand breaks, the polymers were rapidly produced (Figs. 4 and 8). Similar results have been reported for a multitude of cell types and a variety of oxidative stresses including those caused by X-ray (60), H2O2 (61), UVB light (51, 49), lowered GSH level (55), and cigarette smoke (54).
Based on the maximum amount of solar UVB light reaching the human lens epithelium, 0.0037 J/cm2 in 1 hr (11), the two doses of UVB radiation employed in this study, 0.014 J/cm2 (“low dose”) and 0.14 J/cm2 (“high dose”), were equivalent to exposing cultured LECs to either 3.7 hrs or 37 hrs of sunlight, respectively, condensed into 2.5 min. However, another factor to be considered in assessing the physiological relevance of the study is the fact that the peak UVB wavelength of light employed, 312 nm (Fig. 1), is actually over 150 times less damaging to cultured LECs than 297 nm light (62), the lowest solar UVB wavelength that is able to pass through the human cornea to reach the lens (63, 12). In addition, sunlight striking the human eye from the side can focus on the lens up to 20× stronger than light entering from the front (13–15). The high UVB dose of this study was nearly identical to that used previously to induce PARP-1 activation in cultured mouse fibroblasts (49), but was 2 to 10-fold lower than the level required to activate PARP-1 in human keratinocytes (50, 51). It should also be noted that the exposure of LECs to UVB light in the current study was conducted in 20% oxygen (air), whereas the concentration of oxygen present in human aqueous humor in situ, close to the lens epithelium, has been reported to be <1% (64), possibly as a means of inhibiting the damaging effects of UVB (65). It cannot be ruled out that the use of a more physiological level of oxygen might diminish or even block the observed formation of UVB-induced ROS at 20% oxygen (Figs. 5 and 6).
Both the low and high UVB doses employed in the current study caused an approximate 25% decrease in LEC viability, 1 and 3 hr after the 2.5 min exposure (Fig. 3). This result may have been caused by effects of the light on non-confluent cells present in the 96 well plates during the UVB-exposure. Non-confluency is known to increase the susceptibility of cells to UV-induced damage (66). At 24 hr after the UVB exposure, although cell viability loss remained at about 25% for the low dose of UVB light, the observed loss increased substantially to 70% for the 10-fold higher dose (Fig. 3). It is well accepted that a mild oxidative stress will activate PARP-1 to facilitate cell survival, whereas a severe, stressful episode may lead to PARP-1-triggered cell death (55, 67). A similar phenomenon was observed in the current study in which the low UVB dose produced immediate DNA strand breaks and PAR polymer formation, with rapid repair of the DNA damage (Figs. 4a and c, 8a and c), and an absence of morphological cell damage after 24 hr (Fig. 2d). The high UVB dose also produced immediate DNA damage and repair, with coincident PAR formation (Figs. 4b and c, 8b and c); however, in this case, DNA damage and PAR formation remarkably reappeared at the 90 min mark and, after 24 hrs, the cells showed significant morphological damage with a 70% loss of cell viability (Figs. 2g and 3). This biphasic type of PARP-1 activation has also been observed in UVB-stressed mouse fibroblasts (49). In addition, a 4–8 hr delayed formation of PAR was seen in UVB-treated human keratocytes (51). The observed UVB-induced threadlike structures (Fig. 2g), which stained for F-actin (Fig. 2h), have also been reported to form in human LECs stressed by either peroxide (68) or UVA light (69). A previous study showed that an inhibition of PARP-1 activity in FHL124 LECs produced increased susceptibility to H2O2-induced DNA damage, along with lowered H2O2-induced cell death (40); however, a PARP-1 inhibitor was not employed in the current work.
A unique finding of this study was the much slower rate of degradation of PAR observed for the second phase of UVB (high dose)-induced PARP-1 activation, compared to that for the first phase (Fig. 8c). This result is in marked contrast to previous reports indicating that PAR has a very short half-life due to its rapid degradation by poly(ADP-ribose) glycohydrolase (PARG) (70, 71). The observed PAR half-life of 8–12 min for the first phase of PARP-1 activation (Fig. 8c) compares favorably with the 6 min half-life reported for PAR degradation in UV-irradiated human fibroblasts (48). However, for the second phase of PAR formation in the current study, the t1/2 degradation rate increased to nearly 60 min (Fig. 8c), although DNA strand breaks in the second phase were completely repaired within 30 min (Figs. 4b and c). In addition, in the first phase after the high UVB dose (at 5 min post exposure), PAR polymers were formed and degraded within the cell nucleus (Figs. 8b, 9a and 9b). However, in the second phase (at 90 min post UVB exposure), PAR had flowed out of the nucleus into the cytoplasm, apparently to reside within the mitochondria (Figs. 8b, 9c and 9d).
It is not clear why PAR was degraded so slowly within the mitochondria following the second phase of UVB-induced (high dose) PARP-1 activation (Figs. 8b and c). The PAR degrading enzyme poly (ADP-ribose) glycohydrolase (PARG) has been shown previously to exist in the cytoplasm within the mitochondria of unstressed cells, and to translocate into the nucleus during oxidative stress-induced DNA damage (71, 70). Thus, it is possible that following the second phase of PARP-1 activation, the mitochondria may have been depleted of PARG activity, resulting in a longer PAR half-life. Indeed, it may have been essential for the level of PAR to remain high within the mitochondria following the high dose of UVB, triggering eventual loss of cell viability. It is known that a prolonged presence of PAR within a cell has deleterious consequences (72) since the polymers are toxic and represent a cellular death signal (73, 71). The >1.5 hr period that PAR was observed to be present within the mitochondria of the UVB (high dose)-treated cells (Fig. 8c) most likely acted as a trigger for future cell loss.
It is of interest to speculate on possible mechanisms for the UVB-induced cell death observed in this study, and how PARP-1 might have been involved. A previous investigation, employing the same line of LECs and identical UVB levels used in the current study, demonstrated the activation of two stress proteins associated with induction of apoptosis, caspase-3 and DFF 45 (74). Thus, it is likely that these proteins also contributed to cell death in this work. In addition, it is likely that the burst of PAR polymer formation observed at the 90 min mark for the high UVB dose (Figs. 8b and c) consumed intracellular NAD+ stores, leading to depletion of ATP and cell death (75), although energy depletion alone has been shown to not fully explain how PARP-1 activation kills cells (73). In certain cell types, over-activation of PARP-1 is known to induce a translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus to induce cell death (76); however, such a translocation was not observed in this study (unpublished results) or in a previous investigation that involved H2O2-challenged osteosarcoma cells (61). Exposure of human LECs to UVB light, at levels comparable to those of the current study, have been reported to cause loss of mitochondria (77) and decreased mitochondrial membrane potential (78, 79); however, these types of mitochondrial effects observed in oxidatively-stressed rat cerebral cortex neurons appeared to be independent of PARP-1 activation (55). A recent study showed that in oxidatively-stressed primary and SRA 01/04 LECs, there occurred a translocation of Parkin to depolarized mitochondria, resulting in the elimination of damaged mitochondria and a maintained level of LEC homeostasis (80). It should also be pointed out that the present studies were conducted with transformed SRA 01/04 LECs, which may exhibit altered apoptotic induction characteristics compared to non-transformed primary lens cells. A final limitation of the work was the fact that the LECs were necessarily cultured in the presence of serum, and growth factors contained within it, which the lens in situ is not exposed to. This may have affected survival of the cells following UVB exposure.
It appeared that the initial damage to LEC DNA, which occurred within 5 min after the 2.5 min UVB exposure at both the low and high doses (Fig. 4), was caused mainly by direct absorption of the radiation by DNA. A similar conclusion was reached in a previous study with UVB-treated mouse fibroblasts (49). Only slight amounts of ROS were detected in the cell cytoplasm or nucleus, soon after UVB exposure (Fig. 5), suggesting that DNA damage occurring during this period was due to direct UVB absorption. However, in the second phase of PARP-1 activation, occurring 90 min after the high UVB dose, bursts of ROS (primarily H2O2) in both the cytoplasm and nucleus (Figs. 5b and c), and O2− within the mitochondria (Fig. 6), were observed. The formation of these oxidants could not have been due directly to the 2.5 min UVB light exposure conducted 90 min earlier. It is likely that H2O2, produced within the mitochondria from the dismutation of O2−, was the cause of the nuclear DNA strand breaks occurring 90 min after the high UVB dose, and repaired within 30 min (Figs. 4b and c). Previous studies have also linked a delayed generation of H2O2 following UVB-treatment of cultured cells with DNA damage occurring hours after the exposure (49, 51).
It is difficult to ascertain the sequence of events occurring in the LECs at 90 min after the high UVB dose. At this time period, there existed damaged DNA in the nucleus (which was later repaired within 30 min) (Figs. 4b and c), H2O2 in both the cytoplasm and nucleus (Fig. 5b), PAR within the mitochondria (it is assumed that this PAR had flowed into the cytoplasm after being produced in the nucleus) (Figs. 8b and c, 9c and d), and O2− within the mitochondria (Fig. 6). One possible scenario is that PAR induced the observed burst of O2− within the mitochondria and, as outlined above, a subsequent dismutation of superoxide produced H2O2 that flowed from the mitochondria to the nucleus to damage DNA. It has been postulated previously that while PARP-1 activation is typically produced by an oxidative stress, such activation may also be able to generate ROS within a cell. PAR-induced formation of O2− was observed previously in high glucose-exposed human Schwann cells (81). In that 2005 study, the authors suggested that PAR would have been able to cause O2− production by a variety of mechanisms including a PAR-mediated upregulation of the major superoxide-generating enzyme, mitochondrial NADH oxidase. However, the above scenario does not explain how DNA strand breaks seen at 90 min were repaired within 30 min (Figs. 4b and c) if 90% of the PAR had already exited the nucleus (Figs. 9c and d). It is possible that the remaining 10% of PAR was able to assist in the repair, or it may have been accomplished by some other DNA repair system. PARP-1 activation has been demonstrated previously to result in a translocation of mitochondrial apoptosis-inducing factor (AIF) to the cell nucleus (76, 46). Although we observed both PAR (Figs. 9c and d) and AIF (fluorescence-labeled images not shown) residing within the mitochondria at 90 min after the high UVB dose, we did not see any significant translocation of AIF from the mitochondria to the nucleus.
CONCLUSION
The results demonstrated two very different responses of PARP-1 activation in UVB-challenged human LECs. At a relatively low level of UVB stress, PARP-1 and PAR polymers acted to repair DNA strand breaks rapidly to ensure no subsequent major effects on either cell morphology or viability. However, exposure of the cells to a ten-fold higher level of UVB produced a dramatic second phase of PARP-1 activation, 90 min later, which included a sudden reappearance of DNA strand breaks, bursts of ROS formation both within the mitochondria and nucleus, a translocation of PAR from the nucleus to the mitochondria, and an ultimate 70% loss of cell viability occurring after 24 hrs. The results provide evidence for an important role for PARP-1 in protecting the human lens epithelium against low levels of UVB light, and possibly participating in the triggering of cell death following exposure to a toxic level of radiation.
Acknowledgments
The work was supported in part by NIH grant EY 02027(FJG), the Vision Research ROPARD Foundation, the Oakland University Center for Biomedical Research, and undergraduate student research awards provided by Oakland University. We thank Victor Leverenz for excellent technical assistance.
References
- 1.Cruickshanks KJ, Klein BE, Klein R. Ultraviolet light exposure and lens opacities: the Beaver Dam Eye Study. Am J Public Health. 1992;82:1658–62. doi: 10.2105/ajph.82.12.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor HR, West SK, Rosenthal FS, Munoz B, Newland HS, Abbey H, Emmett EA. Effect of ultraviolet radiation on cataract formation. N Engl J Med. 1988;319:1429–33. doi: 10.1056/NEJM198812013192201. [DOI] [PubMed] [Google Scholar]
- 3.West SK, Longstreth JD, Munoz BE, Pitcher HM, Duncan DD. Model of risk of cortical cataract in the US population with exposure to increased ultraviolet radiation due to stratospheric ozone depletion. Am J Epidemiol. 2005;162:1080–8. doi: 10.1093/aje/kwi329. [DOI] [PubMed] [Google Scholar]
- 4.Dillon J. Sunlight exposure and cataract. JAMA. 1999;281:229. author reply 230. [PubMed] [Google Scholar]
- 5.Hightower KR. The role of the lens epithelium in development of UV cataract. Curr Eye Res. 1995;14:71–8. doi: 10.3109/02713689508999916. [DOI] [PubMed] [Google Scholar]
- 6.Sinha RP, Hader DP. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci. 2002;1:225–36. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 7.Hightower K. A review of the evidence that ultraviolet radiation is a risk factor in cataractogenesis. Doc Ophthalmol. 1995;88:205–220. doi: 10.1007/BF01203675. [DOI] [PubMed] [Google Scholar]
- 8.Atherton SJ, Lambert C, Schultz J, Williams N, Zigman S. Fluorescence studies of lens epithelial cells and their constituents. Photochem Photobiol. 1999;70:823–8. [PubMed] [Google Scholar]
- 9.Tyrrell R. UVA (320–380nm) radiation as an oxidative stress. In: Sies H, editor. Oxidative stress: oxidants and antioxidants. Academic Press; San Diego, CA: 1991. pp. 57–83. [Google Scholar]
- 10.Wondrak GT, Jacobson MK, Jacobson EL. Endogenous UVA-photosensitizers: mediators of skin photodamage and novel targets for skin photoprotection. Photochem Photobiol Sci. 2006;5:215–37. doi: 10.1039/b504573h. [DOI] [PubMed] [Google Scholar]
- 11.Zigman S. Environmental near-UV radiation and cataracts. Optom Vis Sci. 1995;72:899–901. doi: 10.1097/00006324-199512000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Dillon J, Zheng L, Merriam JC, Gaillard ER. The optical properties of the anterior segment of the eye: implications for cortical cataract. Exp Eye Res. 1999;68:785–95. doi: 10.1006/exer.1999.0687. [DOI] [PubMed] [Google Scholar]
- 13.Kwok LS, Coroneo MT. Temporal and spatial growth patterns in the normal and cataractous human lens. Exp Eye Res. 2000;71:317–22. doi: 10.1006/exer.2000.0883. [DOI] [PubMed] [Google Scholar]
- 14.Coroneo MT, Muller-Stolzenburg NW, Ho A. Peripheral light focusing by the anterior eye and the ophthalmohelioses. Ophthalmic Surg. 1991;22:705–11. [PubMed] [Google Scholar]
- 15.Coroneo MT. Albedo concentration in the anterior eye: a phenomenon that locates some solar diseases. Ophthalmic Surg. 1990;21:60–6. [PubMed] [Google Scholar]
- 16.Rafferty NS. Lens Morphology. In: Maisel H, editor. The Ocular Lens: Structure, FUnction, and Pathology. Marcel Dekker, Inc; New York, New York: 1985. pp. 1–60. [Google Scholar]
- 17.Wolf N, Pendergrass W, Singh N, Swisshelm K, Schwartz J. Radiation cataracts: mechanisms involved in their long delayed occurrence but then rapid progression. Mol Vis. 2008;14:274–85. [PMC free article] [PubMed] [Google Scholar]
- 18.Pirie A, Flanders PH. Effect of x-rays on partially shielded lens of the rabbit. AMA Arch Ophthalmol. 1957;57:849–54. doi: 10.1001/archopht.1957.00930050863009. [DOI] [PubMed] [Google Scholar]
- 19.Markiewicz E, Barnard S, Haines J, Coster M, van Geel O, Wu W, Richards S, Ainsbury E, Rothkamm K, Bouffler S, Quinlan RA. Nonlinear ionizing radiation-induced changes in eye lens cell proliferation, cyclin D1 expression and lens shape. Open Biol. 2015;5:150011. doi: 10.1098/rsob.150011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Worgul BV, Rothstein H. Radiation cataract and mitosis. Opthalmic Res. 1975;7:21–32. [Google Scholar]
- 21.Schein OD, West S, Munoz B, Vitale S, Maguire M, Taylor HR, Bressler NM. Cortical lenticular opacification: distribution and location in a longitudinal study. Invest Ophthalmol Vis Sci. 1994;35:363–6. [PubMed] [Google Scholar]
- 22.Sasaki H, Kawakami Y, Ono M, Jonasson F, Shui YB, Cheng HM, Robman L, McCarty C, Chew SJ, Sasaki K. Localization of cortical cataract in subjects of diverse races and latitude. Invest Ophthalmol Vis Sci. 2003;44:4210–4. doi: 10.1167/iovs.01-1221. [DOI] [PubMed] [Google Scholar]
- 23.Abraham AG, Cox C, West S. The differential effect of ultraviolet light exposure on cataract rate across regions of the lens. Invest Ophthalmol Vis Sci. 2010;51:3919–23. doi: 10.1167/iovs.09-4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorte K, Sune P, Bhake A, Shivkumar VB, Gangane N, Basak A. Quantitative assessment of DNA damage directly in lens epithelial cells from senile cataract patients. Mol Vis. 2011;17:1–6. [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Wu J, Yang L, Zhu R, Yang M, Qin B, Shi H, Guan H. DNA damage in lens epithelial cells and peripheral lymphocytes from age-related cataract patients. Ophthalmic Res. 2014;51:124–8. doi: 10.1159/000356399. [DOI] [PubMed] [Google Scholar]
- 26.Worgul BV, David J, Odrich S, Merriam GR, Jr, Medvedovsky C, Merriam JC, Trokel SL, Geard CR. Evidence of genotoxic damage in human cataractous lenses. Mutagenesis. 1991;6:495–9. doi: 10.1093/mutage/6.6.495. [DOI] [PubMed] [Google Scholar]
- 27.Kleiman NJ, Spector A. DNA single strand breaks in human lens epithelial cells from patients with cataract. Curr Eye Res. 1993;12:423–31. doi: 10.3109/02713689309024624. [DOI] [PubMed] [Google Scholar]
- 28.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–76. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 29.Kang L, Zhao W, Zhang G, Wu J, Guan H. Acetylated 8-oxoguanine DNA glycosylase 1 and its relationship with p300 and SIRT1 in lens epithelium cells from age-related cataract. Exp Eye Res. 2015;135:102–8. doi: 10.1016/j.exer.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Li F, Zhang G, Kang L, Qin B, Guan H. Altered DNA Methylation and Expression Profiles of 8-Oxoguanine DNA Glycosylase 1 in Lens Tissue from Age-related Cataract Patients. Curr Eye Res. 2015;40:815–21. doi: 10.3109/02713683.2014.957778. [DOI] [PubMed] [Google Scholar]
- 31.Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:120–39. doi: 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- 32.Xu B, Kang L, Zhang G, Wu J, Zhu R, Yang M, Guan H. The changes of 8-OHdG, hOGG1, APE1 and Pol beta in lenses of patients with age-related cataract. Curr Eye Res. 2015;40:378–85. doi: 10.3109/02713683.2014.924148. [DOI] [PubMed] [Google Scholar]
- 33.Ates O, Alp HH, Kocer I, Baykal O, Salman IA. Oxidative DNA damage in patients with cataract. Acta Ophthalmol. 2010;88:891–5. doi: 10.1111/j.1755-3768.2009.01583.x. [DOI] [PubMed] [Google Scholar]
- 34.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes, 2005. Mutat Res. 2005;577:275–83. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Li F, Wang Y, Zhang G, Zhou J, Yang L, Guan H. Expression and methylation of DNA repair genes in lens epithelium cells of age-related cataract. Mutat Res. 2014;766–767:31–6. doi: 10.1016/j.mrfmmm.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 36.Spector A, Kleiman NJ, Huang RR, Wang RR. Repair of H2O2-induced DNA damage in bovine lens epithelial cell cultures. Exp Eye Res. 1989;49:685–98. doi: 10.1016/s0014-4835(89)80063-6. [DOI] [PubMed] [Google Scholar]
- 37.Padgaonkar V, Giblin FJ, Reddan JR, Dziedzic DC. Hyperbaric oxygen inhibits the growth of cultured rabbit lens epithelial cells without affecting glutathione level. Exp Eye Res. 1993;56:443–52. doi: 10.1006/exer.1993.1057. [DOI] [PubMed] [Google Scholar]
- 38.Tamada Y, Fukiage C, Nakamura Y, Azuma M, Kim YH, Shearer TR. Evidence for apoptosis in the selenite rat model of cataract. Biochem Biophys Res Commun. 2000;275:300–6. doi: 10.1006/bbrc.2000.3298. [DOI] [PubMed] [Google Scholar]
- 39.Drel VR, Xu W, Zhang J, Kador PF, Ali TK, Shin J, Julius U, Slusher B, El-Remessy AB, Obrosova IG. Poly(ADP-ribose)polymerase inhibition counteracts cataract formation and early retinal changes in streptozotocin-diabetic rats. Invest Ophthalmol Vis Sci. 2009;50:1778–90. doi: 10.1167/iovs.08-2191. [DOI] [PubMed] [Google Scholar]
- 40.Smith AJ, Ball SS, Bowater RP, Wormstone IM. PARP-1 inhibition influences the oxidative stress response of the human lens. Redox Biol. 2016;8:354–62. doi: 10.1016/j.redox.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraus WL, Hottiger MO. PARP-1 and gene regulation: progress and puzzles. Mol Aspects Med. 2013;34:1109–23. doi: 10.1016/j.mam.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–28. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 43.Abbotts R, Wilson DM., 3rd Coordination of DNA single strand break repair. Free Radic Biol Med. 2017;107:228–244. doi: 10.1016/j.freeradbiomed.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–67. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 45.David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci (Landmark Ed) 2009;14:1116–28. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fatokun AA, V, Dawson L, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. 2014;171:2000–16. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cui NH, Qiao C, Chang XK, Wei L. Associations of PARP-1 variant rs1136410 with PARP activities, oxidative DNA damage, and the risk of age-related cataract in a Chinese Han population: A two-stage case-control analysis. Gene. 2017;600:70–76. doi: 10.1016/j.gene.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 48.Jacobson EL, Antol KM, Juarez-Salinas H, Jacobson MK. Poly(ADP-ribose) metabolism in ultraviolet irradiated human fibroblasts. J Biol Chem. 1983;258:103–7. [PubMed] [Google Scholar]
- 49.Vodenicharov MD, Ghodgaonkar MM, Halappanavar SS, Shah RG, Shah GM. Mechanism of early biphasic activation of poly(ADP-ribose) polymerase-1 in response to ultraviolet B radiation. J Cell Sci. 2005;118:589–99. doi: 10.1242/jcs.01636. [DOI] [PubMed] [Google Scholar]
- 50.Lakatos P, Szabo E, Hegedus C, Hasko G, Gergely P, Bai P, Virag L. 3-Aminobenzamide protects primary human keratinocytes from UV-induced cell death by a poly(ADP-ribosyl)ation independent mechanism. Biochim Biophys Acta. 2013;1833:743–51. doi: 10.1016/j.bbamcr.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 51.Chang H, Sander CS, Muller CS, Elsner P, Thiele JJ. Detection of poly(ADP-ribose) by immunocytochemistry: a sensitive new method for the early identification of UVB- and H2O2-induced apoptosis in keratinocytes. Biol Chem. 2002;383:703–8. doi: 10.1515/BC.2002.072. [DOI] [PubMed] [Google Scholar]
- 52.Ibaraki N, Chen SC, Lin LR, Okamoto H, Pipas JM, Reddy VN. Human lens epithelial cell line. Exp Eye Res. 1998;67:577–85. doi: 10.1006/exer.1998.0551. [DOI] [PubMed] [Google Scholar]
- 53.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–91. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 54.Kovacs K, Erdelyi K, Hegedus C, Lakatos P, Regdon Z, Bai P, Hasko G, Szabo E, Virag L. Poly(ADP-ribosyl)ation is a survival mechanism in cigarette smoke-induced and hydrogen peroxide-mediated cell death. Free Radic Biol Med. 2012;53:1680–8. doi: 10.1016/j.freeradbiomed.2012.08.579. [DOI] [PubMed] [Google Scholar]
- 55.Diaz-Hernandez JI, Moncada S, Bolanos JP, Almeida A. Poly(ADP-ribose) polymerase-1 protects neurons against apoptosis induced by oxidative stress. Cell Death Differ. 2007;14:1211–21. doi: 10.1038/sj.cdd.4402117. [DOI] [PubMed] [Google Scholar]
- 56.Ding W, Liu W, Cooper KL, Qin XJ, de Souza Bergo PL, Hudson LG, Liu KJ. Inhibition of poly(ADP-ribose) polymerase-1 by arsenite interferes with repair of oxidative DNA damage. J Biol Chem. 2009;284:6809–17. doi: 10.1074/jbc.M805566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pang J, Gong H, Xi C, Fan W, Dai Y, Zhang TM. Poly(ADP-ribose) polymerase 1 is involved in glucose toxicity through SIRT1 modulation in HepG2 hepatocytes. J Cell Biochem. 2011;112:299–306. doi: 10.1002/jcb.22919. [DOI] [PubMed] [Google Scholar]
- 58.Martin-Oliva D, Martin-Guerrero SM, Matia-Gonzalez AM, Ferrer-Martin RM, Martin-Estebane M, Carrasco MC, Sierra A, Marin-Teva JL, Calvente R, Navascues J, Cuadros MA. DNA damage, poly(ADP-Ribose) polymerase activation, and phosphorylated histone H2AX expression during postnatal retina development in C57BL/6 mouse. Invest Ophthalmol Vis Sci. 2015;56:1301–9. doi: 10.1167/iovs.14-15828. [DOI] [PubMed] [Google Scholar]
- 59.Kilarkaje N, Al-Hussaini H, Al-Bader MM. Diabetes-induced DNA damage and apoptosis are associated with poly (ADP ribose) polymerase 1 inhibition in the rat testis. Eur J Pharmacol. 2014;737:29–40. doi: 10.1016/j.ejphar.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 60.Benjamin RC, Gill DM. ADP-ribosylation in mammalian cell ghosts. Dependence of poly(ADP-ribose) synthesis on strand breakage in DNA. J Biol Chem. 1980;255:10493–501. [PubMed] [Google Scholar]
- 61.Robaszkiewicz A, Erdelyi K, Kovacs K, Kovacs I, Bai P, Rajnavolgyi E, Virag L. Hydrogen peroxide-induced poly(ADP-ribosyl)ation regulates osteogenic differentiation-associated cell death. Free Radic Biol Med. 2012;53:1552–64. doi: 10.1016/j.freeradbiomed.2012.08.567. [DOI] [PubMed] [Google Scholar]
- 62.Andley UP, Lewis RM, Reddan JR, Kochevar IE. Action spectrum for cytotoxicity in the UVA- and UVB-wavelength region in cultured lens epithelial cells. Invest Ophthalmol Vis Sci. 1994;35:367–73. [PubMed] [Google Scholar]
- 63.Kinsey VE. Spectral transmission of the eye to ultraviolet radiations. Arch Ophthal. 1948;39:508–13. doi: 10.1001/archopht.1948.00900020516005. [DOI] [PubMed] [Google Scholar]
- 64.Siegfried CJ, Shui YB, Holekamp NM, Bai F, Beebe DC. Oxygen distribution in the human eye: relevance to the etiology of open-angle glaucoma after vitrectomy. Invest Ophthalmol Vis Sci. 2010;51:5731–8. doi: 10.1167/iovs.10-5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eaton JW. UV-mediated cataractogenesis: a radical perspective. Doc Ophthalmol. 1994;88:233–42. doi: 10.1007/BF01203677. [DOI] [PubMed] [Google Scholar]
- 66.Carvalho H, da Costa RM, Chigancas V, Weinlich R, Brumatti G, Amarante-Mendes GP, Sarasin A, Menck CF. Effect of cell confluence on ultraviolet light apoptotic responses in DNA repair deficient cells. Mutat Res. 2003;544:159–66. doi: 10.1016/j.mrrev.2003.06.004. [DOI] [PubMed] [Google Scholar]
- 67.Virag L. Structure and function of poly(ADP-ribose) polymerase-1: role in oxidative stress-related pathologies. Curr Vasc Pharmacol. 2005;3:209–14. doi: 10.2174/1570161054368625. [DOI] [PubMed] [Google Scholar]
- 68.Hawse JR, V, Padgaonkar A, Leverenz VR, Pelliccia SE, Kantorow M, Giblin FJ. The role of metallothionein IIa in defending lens epithelial cells against cadmium and TBHP induced oxidative stress. Mol Vis. 2006;12:342–9. [PMC free article] [PubMed] [Google Scholar]
- 69.Padgaonkar VA, V, Leverenz R, Bhat AV, Pelliccia SE, Giblin FJ. Thioredoxin reductase activity may be more important than GSH level in protecting human lens epithelial cells against UVA light. Photochem Photobiol. 2015;91:387–96. doi: 10.1111/php.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Poitras MF, Koh DW, Yu SW, Andrabi SA, Mandir AS, Poirier GG, Dawson VL, Dawson TM. Spatial and functional relationship between poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in the brain. Neuroscience. 2007;148:198–211. doi: 10.1016/j.neuroscience.2007.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haince JF, Ouellet ME, McDonald D, Hendzel MJ, Poirier GG. Dynamic relocation of poly(ADP-ribose) glycohydrolase isoforms during radiation-induced DNA damage. Biochim Biophys Acta. 2006;1763:226–37. doi: 10.1016/j.bbamcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 72.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A. 2004;101:17699–704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–13. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Long AC, Colitz CM, Bomser JA. Apoptotic and necrotic mechanisms of stress-induced human lens epithelial cell death. Exp Biol Med (Maywood) 2004;229:1072–80. doi: 10.1177/153537020422901012. [DOI] [PubMed] [Google Scholar]
- 75.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 76.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–63. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 77.Youn HY, McCanna DJ, Sivak JG, Jones LW. In vitro ultraviolet-induced damage in human corneal, lens, and retinal pigment epithelial cells. Mol Vis. 2011;17:237–46. [PMC free article] [PubMed] [Google Scholar]
- 78.Liu L, Yu R, Shi Y, Dai Y, Zeng Z, Guo X, Ji Q, Wang G, Zhong J. Transduced protein transduction domain linked HSP27 protected LECs against UVB radiation-induced damage. Exp Eye Res. 2014;120:36–42. doi: 10.1016/j.exer.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 79.Du Y, Guo D, Wu Q, Shi J, Liu D, Bi H. Protective effects of appropriate Zn(2+) levels against UVB radiation-induced damage in human lens epithelial cells in vitro. J Biol Inorg Chem. 2016;21:213–26. doi: 10.1007/s00775-015-1324-9. [DOI] [PubMed] [Google Scholar]
- 80.Brennan L, Khoury J, Kantorow M. Parkin elimination of mitochondria is important for maintenance of lens epithelial cell ROS levels and survival upon oxidative stress exposure. Biochim Biophys Acta. 2017;1863:21–32. doi: 10.1016/j.bbadis.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Obrosova IG, V, Drel R, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, Yorek MA. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54:3435–41. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]