

Abstract
Purpose
Vimentin is an epithelial to mesenchymal transition (EMT) biomarker and intermediate filament protein that functions during cell migration to maintain structure and motility. Despite the abundance of clinical data linking vimentin to poor patient outcome, it is unclear if vimentin is required for metastasis or is a correlative biomarker. We developed a novel genetically engineered mouse model (GEMM) to probe vimentin in lung adenocarcinoma metastasis.
Experimental Design
We used the LSL-KrasG12D/Lkb1fl/fl/Vim−/− model (KLV−/−), which incorporates a whole-body knockout of vimentin and is derived from the Cre-dependent LSL-KrasG12D/Lkb1fl/fl model (KLV+/+). We compared the metastatic phenotypes of the GEMMs and analyzed primary tumors from the KLV models and lung adenocarcinoma patients to assess vimentin expression and function.
Results
Characterization of KLV+/+ and KLV−/− mice, show that while vimentin is not required for primary lung tumor growth, vimentin is required for metastasis and vimentin loss generates lower grade primary tumors. Interestingly, in the KLV+/+ mice, vimentin was not expressed in tumor cells but in cancer-associated fibroblasts (CAFs) surrounding collective invasion packs (CIPs) of epithelial tumor cells, with significantly less CIPs in KLV−/− mice. CIPs correlate with tumor grade, and are vimentin-negative and E-cadherin-positive, indicating a lack of cancer cell EMT. A similar heterotypic staining pattern was observed in human lung adenocarcinoma samples. In vitro studies show that vimentin is required for CAF motility to lead tumor cell invasion, supporting a vimentin-dependent model of collective invasion.
Conclusions
These data show that vimentin is required for lung adenocarcinoma metastasis by maintaining heterotypic tumor cell-CAF interactions during collective invasion.
Introduction
Epithelial to mesenchymal transition (EMT) has served as a guiding principle underlying the initiation and progression of metastatic disease (1). During EMT, epithelial cells from the primary site undergo genetic (2) and epigenetic (3) alterations that trigger cancer cell invasion. A hallmark of EMT is the loss of epithelial proteins, such as E-cadherin (4), and gain of mesenchymal proteins, such as vimentin(5). These events can be transient as cells transition through partial and reversible EMT states (6–9), and it is debated whether a full EMT is necessary for metastasis (10, 11).
Over half of all lung adenocarcinoma cases present with advanced metastatic disease which is associated with a 5-year survival rate of 4% (12). As the driving genomic aberrations of lung adenocarcinoma have become clearer, co-mutations in KRAS and LKB1 are recognized as major drivers of the disease (13, 14). The KRAS/LKB1 co-mutation has previously been modeled in a Cre-dependent LSL-KrasG12D, LKB1fl/fl genetically engineered mouse model (GEMM) (15) to investigate this co-mutation in the context of metabolism (16), therapeutic response (17, 18), and microenvironment remodeling (19). In this GEMM, primary lung tumors develop at 8 to 10 weeks with 52% of mice having metastatic disease to draining lymph nodes (15, 18). When Cre is delivered intranasally via lentiviral infection, 100% of primary tumors are lung adenocarcinomas that reproduce the stages of tumor progression from adenocarcinoma in situ to invasive disease (18).
To assess EMT in lung adenocarcinoma, and more specifically the role of the canonical mesenchymal marker vimentin, we generated an LSL-KrasG12D/Lkb1fl/fl/Vim−/− (KLV−/−) metastatic lung adenocarcinoma GEMM model, which contains a whole body vimentin knockout (20). Vimentin is an intermediate filament protein, which is associated with increased metastatic potential (21), high nuclear grade (22), and poor overall survival (23) across most solid tumor types including lung, prostate, and breast cancers (24–26). Since vimentin is integral for the structural integrity of the cell (27), lamellipodia formation (28), and adhesion signaling (29, 30), downregulating vimentin expression is sufficient to alter cell morphology in vitro (31) as well as inhibit cell motility and invasion (32).
The data presented here from GEMMs, human lung adenocarcinoma samples, and in vitro 3-D models show that vimentin is required for lung adenocarcinoma to metastasize at an early stage by acting in cancer associated fibroblasts (CAFs) that surround epithelial-like collective invasion packs (CIPs). These data support the concept that vimentin is critical for lung adenocarcinoma metastasis and could be a potential target for anti-metastatic therapies.
Materials and Methods
Transgenic Mouse Model
KLV−/− mouse was generated by crossing the LSL-KrasG12D/LKB1fl/fl mouse (15) with the Vimentin−/− (20) mouse until a homozygous KLV−/− mouse was generated. KLV−/− mouse took approximately four generations over 7 months to create. KLV+/− mice were generated by crossing KLV+/+ mice with KLV−/− mice. All mice were housed and treated according to Institutional Animal Care and Use Committee protocol #2003525 by the Emory University Division of Animal Resources. Kras and Lkb1 mutant mice were purchased from The Jackson Laboratory. Vimentin null mice were obtained from the EMMA repository (20). Lentiviral Cre (1.2×10ˆ7 i.u.) was administered intranasally to mice 8-12 weeks old that were sedated with Avertin (20mg/mL working dilution). Infected mice were then monitored until tumorigenic symptoms (i.e., weight loss greater than 10%, respiratory distress, inactivity) presented. All mice infected with lentiviral Cre were monitored to a 25 weeks post infection endpoint and were sacrificed prior if they presented with clinical symptoms including weight loss, respiratory distress, and/or hunching. Upon sacrifice, lungs and mediastinal lymph nodes were harvested in either 10% formalin or flash frozen in OCT pending use.
Human Lung Adenocarcinoma Tissue
De-identified human lung adenocarcinoma tissue is a mixed cohort of samples from Emory and Wellstar tumor banks and Emory clinical formalin-fixed paraffin-embedded (FFPE) tumor specimens (IRB00009857). All Emory samples were sequenced using a workflow described (18). Lung adenocarcinoma staging was performed by a board certified thoracic pathologist according to the AJCC Cancer Staging Manual 7th Edition. Vimentin IHC was scored by a board certified thoracic pathologist with a 40× objective on a scale from 0-3 based on stain intensity and was stratified by stromal or tumor compartment. Q score was determined by the multiplication of stain intensity by percent of cells staining at that intensity for a given cell compartment (i.e., 30% of stromal cells at level 3 stain has Q score of 90).
Immunohistochemistry
Formalin-fixed paraffin embedded (FFPE) tissues were hydrated in a series of washes from xylenes to ethanol dilutions to water. Heat-mediated antigen retrieval was conducted with citrate buffer when necessary. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide. Samples were blocked with 2.5% Normal Goat Serum (Vector Labs) and incubated in primary antibody overnight in 1:1 NGS:PBS solution (Vimentin R28 Cell Signaling 1:300, alpha SMA Invitrogen 1:1000, Pro-SPC Santa Cruz FL-197 1:150, CD31 ab28364 1:50 CD3 ab16669 1:100). Samples were incubated in secondary antibody (Rabbit 1:200), biotin, and streptavidin (Vector Labs) according to kit protocol. Staining was developed using Peroxidase Substrate Kit DAB (Vector Labs). Proliferation was measured by staining for Ki67 (ab16667, 1:200) using IMMpact Kit (Vector Labs).
Immunofluorescence
Tissue
Tissue sections frozen in OCT Compound were sliced in 10μm sections and fixed in 100% acetone. Sections were stored at −80°C until used. Sections were blocked for 1 hour in 10% Normal Goat Serum in PBS. Primary antibodies were incubated in PBS overnight overnight for the following antibodies at indicated concentrations: vimentin R28 Cell Signaling 1:50, F4/80 abcam 1:100, FSP1 ab27957 1:200. FFPE tissues were hydrated as described above. Heat-mediated antigen retrieval was also conducted as above. A blocking buffer of 5% BSA in PBS was used. Primary antibodies were incubated at 4°C overnight in 1% BSA in PBS (FSP1 ab27957 1:200; E-cadherin BD Biosciences 610181 1:1000). Secondary antibodies were incubated on tissues for 1 hour (1:1000).
Cells
CAFs were fixed and stained as described (30). Primary antibodies were diluted in 5% normal goat serum (Vimentin R28 Cell Signaling 1:50) and added to cells on coverslips overnight at 4 °C. The coverslips were mounted on glass slides using Prolong diamond mounting medium (Invitrogen). The slides were imaged using Leica SP8 confocal microscope.
Spheroids
Spheroids embedded in matrigel were fixed with 4% paraformaldehyde for 20 minutes. Glycine rinse (TBS/glycine 130 mM NaCl, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 100 mM glycine) performed three times for 10 minutes at room temperature. Blocked in immunofluorescence (IF) buffer (130 mM NaCl, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 0.2% Triton X-100, 0.05% Tween-20) plus 3% BSA, 5% goat serum and 0.3% Triton X-100 for 2 hours at room temperature. Primary antibodies incubated overnight in blocking buffer (FSP1 1:250) at room temperature. IF buffer used for three 20 minute washes. Secondary antibody diluted 1:500 in blocking buffer (Phalloidin 488 1:40 and DAPI added) and incubated at room temperature overnight. Fluorescence was imaged on Confocal Leica SP8 microscope.
Multiphoton microscopy
Second harmonic generation (SHG) images of H&E sections were taken using a Zeiss Axio Examiner Z1 microscope with 20× water immersion objective (1.0 NA DIC [UV] VIS-IR) as described (33). The SHG signal was obtained using a band pass cube of 380–430nm. Images were taken with a Coherent Chameleon Verdi laser at 820 nm wavelength. Z-stack images were taken with a 1μm interval.
Western Blot
Lung tissue was harvested and flash frozen in liquid nitrogen. Frozen tissue was cut into 2mm pieces and ground in glass mortar and pestle in 1mL of lysis buffer TNES buffer (50 mM Tris pH 7.5, 100 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 1× Roche Complete Protease Inhibitors, 10 mM NaF, 1 mM NaVO4, 2 mM sodium pyrophosphate, and 2 mM β-glycerophosphate). Mixture was further homogenized with a sonicator. Lysate was centrifuged and supernatant was collected. Protein assay and western blot were performed as described (30). Primary antibody, diluted in either 5% BSA in TBST (Vimentin R28 Cell signaling 1:1000) or 5% milk in TBST (Tubulin Millipore MAB1864 1:20:000, GAPDH CST 1:30,000) was added to membrane overnight. Secondary antibodies were incubated at a concentration of 1:1000 in 5% milk in TBST for 1 hour.
Cell Culture
H460 lung cancer cell line was purchased from ATCC and subcultured in RPMI 1640 media supplemented with 10% FBS, 1% penicillin/streptomycin and 1% kanamycin. Human lung adenocarcinoma cancer-associated fibroblasts were purchased from Vitro Biopharma and subcultured in MSC Gro VitroPlus III, Low Serum media supplemented with 1% penicillin/streptomycin and 1% kanamycin. All cells were cultured in a humidified chamber at 37°C with 5% CO2. Experiments were performed with cells below passage 10. All cells were verified as mycoplasma-free on October 4, 2017 by DAPI staining.
Stable Cell Line Generation
For the production of stable vimentin knock-down cancer-associated fibroblasts, pLKO.1 vector and vimentin shRNA virus described previously (30) was added to cells 1:2 in media with 8ug/mL polybrene three times. Cells expressing the constructs were selected using Puromycin 2ug/mL treatment.
3D Spheroid Invasion Assay
Spheroids of H460s, CAFs, and H460/CAF 50/50 co-cultures were generated as described (18). Spheroids were collected and resuspended in 250u μL of 2.0mg/mL MatriGel (VWR) in 35mm Dish with 14mm bottom well (Cellvis) and a coverslip was attached with Vaseline. After 45min incubation at 37°C, 2mL of media was added. Images were taken at 0 and 24 hours on Olympus IX51 microscope. Spheroid invasion was quantified using ImageJ (NIH). For 3D immunofluorescence, spheroids were formed as described above. Each well of a 96 well flat-bottomed plate (Corning) was coated with 8mg/mL MatriGel. Spheroids were collected and resuspended in 50μL of 2.0mg/mL MatriGel. After a 45 minute incubation at 37°C, 100μL of media was added. Spheroids were allowed to invade for 48 hours.
PCR
Tissue was collected from candidate pups prior to weaning. Genomic DNA was extracted using Alkaline Lysis Buffer at 100°C for 45 minutes. A 40mM Tris-HCL Neutralization Buffer was then added and samples were stored at −20°C. PCR was performed using a GoTaq Flexi system from Promega. Samples were run on Biorad iCycler at recommended annealing temperatures.
Image Analysis
H&E and IHC stained tissue samples were imaged on the Zeiss Axioplan 2 widefield microscope or whole slides were scanned into ImageScope software. Immunofluorescence images were taken on the Confocal Leica SP8. All images were analyzed with ImageJ and Photoshop Elements. CellProfiler was used for fluorescent image quantification. Imaris software was used for vim/FSP1 colocalization analysis where a mask was generated for each marker and then a colocalization mask was generated from this. The 2D histogram was generated in Imaris by removing background levels of each marker. Pearson’s coefficient was generated from the histogram. Imaris was also used for CAF cell size analysis.
Tumor Histology
Stromal and tumor compartments in KLV mouse were identified by a board certified thoracic pathologist. KLV model tumor grading was performed as described (34).
Statistics
P-values of significance were obtained using a Fischer’s exact test, Chi square test, or unpaired t test when comparing two groups (ie KLV+/+ vs KLV−/− or metastatic vs non-metastatic). Data with more than two groups were analyzed by one-way ANOVA followed by Tukey’s multiple comparisons test. Analysis of vimentin IHC in patient samples was performed using Wilcoxon Rank Sum test. Kaplan-Meier curves were analyzed by Mantel-Cox and Gehan-Breslow-Wilcoxon tests.
Results
Generation of a KrasG12D/LKB1fl/fl/Vim−/− GEMM (KLV−/−)
Metastasis is well characterized in the LSL-KrasG12D/LKB1fl/fl (KLV+/+) model (15, 18), where approximately 52% of mice that develop primary lung tumors at 8-10 weeks also develop metastasis to mediastinal lymph nodes at 12-16 weeks (18). A whole-body vimentin−/− mouse (20) was crossed to this KLV+/+ mouse to test the hypothesis that vimentin is necessary for metastatic disease. Through this cross a novel LSL-KrasG12D,LKB1fl/fl,Vim−/− (KLV−/−) mouse was generated (Supplemental Figure 1A) that lacks vimentin expression throughout and has the LSL-KrasG12D,LKB1fl/fl conditionally mutated in the lungs via intranasal delivery of lentiviral Cre recombinase. Disruption of the vimentin allele and lack of protein expression in the KLV−/− mouse was validated by RT-PCR, western blotting, and immunofluorescence (Supplemental Figure 1B–D). Disruption of one vimentin allele in the heterozygous KLV+/− mouse did not greatly reduce vimentin protein expression (Supplemental Figure 1C, D). These results demonstrate the development of a novel KLV−/− GEMM to probe the role of vimentin in lung adenocarcinoma metastasis.
KLV+/+ and KLV−/− mice have a similar primary tumor burden
KLV+/+ and KLV−/− were infected intranasally with Cre lentivirus and a comparison of tumor burden between the two genotypes (either at 25 weeks or before due to morbidity) revealed no significant differences in primary tumor initiation or growth (Table I and Figure 1A). In both genotypes, 100% of primary tumors were lung adenocarcinomas with no significant difference in tumor incidence (Table 1). Both KLV+/+ and KLV−/− mice generated primary lung tumors (Figure 1A) without a significant difference in lung weight (Figure 1B) or tumor multiplicity (number of tumor foci per H&E slice) (Figure 1C). Tumor histology was similar across genotypes with both KLV+/+ and KLV−/− mice developing tumors with and without invasive fronts (Figure 1D). Proliferative index using Ki-67 staining was also not significantly different between the two genotypes (Figure 1E, F). Based on these data, we conclude that vimentin loss does not impact primary lung tumor initiation or growth. A Kaplan-Meier curve to clinical symptoms shows that KLV−/− mice have decreased survival compared to KLV+/+ (Supplemental Figure 2).
Table 1.
Infection rate and histology of lentiviral-Cre treated mice of both KLV+/+ and KLV−/− mice
| Genotype | Primary Tumor Rate | Adenocarcinoma Histology |
|---|---|---|
| KLV+/+ | 17/26 (66%) | 17/17 (100%) |
|
|
||
| KLV−/− | 47/70 (67%) | 47/47 (100%) |
|
|
||
Figure 1. KLV+/+ and KLV−/− mice have a similar primary tumor burden.

(A) Representative images of PBS control, KLV+/+ and KLV−/− lungs (scale = 50mm). (B) Graphs showing weight of PBS control, KLV+/+ and KLV−/− lungs. Statistical significance was determined by one-way ANOVA and Tukey’s multiple comparisons test (p<0.05). (C) Tumor multiplicity of KLV+/+ and KLV−/− mice quantified as the number of tumor foci per H&E lung slice. Statistical significance was determined by unpaired t test (p<0.05). (D) Representative H&E sections of KLV+/+ and KLV−/− primary tumors (scale = 200μm). (E) IHC of Ki67 in KLV+/+ and KLV−/− primary tumors. (F) Quantification of percent of Ki67 positive tumor cells. Statistical significance was determined by unpaired t test (p<0.05).
Vimentin null mice have reduced metastatic disease and less focal invasion
The KLV+/+ model develops robust primary lung tumors and metastasis to the mediastinal lymph node (15, 18). We leveraged this phenotype to investigate how the vimentin loss affects the metastatic potential of Kras/Lkb1 co-mutated lung adenocarcinomas. Vimentin loss in KLV−/− mice results in 54% less mediastinal lymph nodes metastasis compared to KLV+/+ mice (Figure 2A, B). In mice with clinical symptoms (respiratory distress, weight loss, hunching), vimentin loss was associated with significantly less metastatic disease to the draining lymph nodes compared to KLV+/+ mice (Figure 2C). In addition to decreased incidence of lymph node metastasis, vimentin loss in the KLV−/− is associated with significantly less invasive foci than KLV+/+ mice tumors as measured by the number of invasive fields (defined as a field with focal invasion) per mouse (Figure 2D).
Figure 2. Vimentin depletion inhibits invasion and metastasis.

(A) Representative H&E images of lung draining lymph nodes of LV-Cre infected mice (scale= 20μm). (B) Metastatic incidence by KLV genotype. Statistical significance was determined by Fisher’s exact test (p<0.05). (C) Metastatic incidence of mice sacrificed prior to 25-week endpoint due to lung cancer symptoms. Statistical significance was determined by Fisher’s exact test (p<0.05). (D) Quantification of invasive fields per mouse by genotype. Statistical significance was determined by unpaired t test (p<0.05). (E) KLV tumor grading by genotype. Statistical significance was determined by Chi-square test (p<0.05).
To assess the impact of vimentin loss on tumor progression, primary tumors of KLV+/+ and KLV−/− were graded on a scale of 1 to 4 with grade 1 tumors classified as atypical adenomatous hyperplasia and grade 4 tumors demonstrating stromal reaction in addition to irregular mitoses and invasive borders along lymphatic vessels. Higher tumor grade are also associated with more invasive and metastatic tumors (34). Based on this analysis, the KLV+/+ mice developed significantly more high-grade tumors than KLV−/− mice (Figure 2E) further supporting the claim that vimentin loss reduces metastatic disease and tumor progression. Taken together, these data show that vimentin is required for efficient metastasis in Kras/Lkb1 co-mutated lung adenocarcinoma.
Vimentin is required for collective invasion pack (CIP) formation
To determine vimentin localization within the primary tumor, vimentin immunohistochemistry (IHC) was performed on KLV+/+ primary tumor samples (Figure 3A). The vimentin antibody was validated for specificity by IHC in vimentin null tissue and by western blot (Supplemental Figure 3). To our surprise, in KLV+/+ samples, vimentin expression was absent in tumor cells but present in the stromal niche often surrounding tumor cells that appear to have budded off from the primary tumor. We previously termed these tumor cell histologies collective invasion packs (CIPs) (18). CIPs are surrounded by vimentin-positive, elongated stromal cells that resemble fibroblasts in morphology (Figure 3A).
Figure 3. Vimentin is required for collective invasion pack (CIP) formation.

(A) Immunohistochemical staining of vimentin in KLV+/+ mice with serial H&E sections. Black arrows mark CIPs (10× scale =100μm, 40× =20μm). (B) Representative Pro-SPC stained sections of KLV+/+ and KLV−/− primary tumors with CIPs marked by black arrows. (C) Quantification of number of cells per CIP in both KLV+/+ and KLV−/− mice. Statistical significance determined by unpaired t test (p>0.05). (D) Quantification of CIPs per invasive field in KLV+/+ and KLV−/− mice. Statistical significance determined by unpaired t test (p<0.05) (E) CIPs per invasive field stratified by genotype and tumor grade. Statistical significance determined by ANOVA and Tukey’s multiple comparisons test (p<0.05) (F) Quantification of CIPs per invasive field stratified by metastatic phenotype in KLV models (p<0.05). (G) Quantification of invasive fields by metastatic phenotype in KLV models Statistical significance determined by unpaired t test (p<0.05). (H) Immunofluorescence of fibronectin in KLV primary tumor samples at the invasive front, emerging from the primary tumor, and at site distant from the primary tumor. Yellow arrows mark CIPs with invasive frontmarked by dashed line (scale=50μm). (I) Second harmonic generation imaging of collagen in KLV samples at the invasive front and surrounding CIPs in both primary tumor and metastatic lymph node. Yellow arrows mark CIPs with invasive front marked by dashed line (scale= 50μm).
CIPs are positive for the lung adenocarcinoma marker pro-SPC (35) (Figure 3B), indicating their type II pneumocyte origin, retain epithelial morphology (non-elongated), and express E-cadherin (18), while lacking vimentin protein expression (Figure 3A). This pattern suggests that tumor cells within CIPs retain homotypic cell contacts and are likely not undergoing a traditional EMT. CIPs range in size from 5-25 cells (Figure 3C) and are found within this range in KLV+/+ and KLV−/− primary tumor samples. CIP density in each genotype was assessed and these data show that KLV−/− mice have significantly less CIPs per invasive field than KLV+/+ mice overall (Figure 3D). When CIP density is stratified by tumor grade (Figure 3E), high-grade KLV−/− tumors have significantly less CIPs than high-grade KLV+/+ tumors indicating that vimentin is required to maintain CIP density in invading tumors. Further, these data indicate that within the KLV models, the predominant role of vimentin lies within stromal cells rather than tumor cells.
To determine the role of CIPs in tumor progression and metastasis, CIP density and invasive area was stratified by extent of disease or tumor stage. These data demonstrate that CIPs are found in both early (non-metastatic) and late (metastatic) stage KLV tumors (Figures 3F, G) with significantly higher CIP density and invasion in late stage tumors. To probe CIP biology, immunofluorescence of extracellular matrix (ECM) components was performed. Fibronectin staining reveals that cancer cells themselves are negative for fibronectin, but fibronectin is expressed in the regions surrounding emerging CIPs at the invasive front of the primary tumor in both KLV+/+ and KLV−/− samples (Figure 3H, Supplemental Figure 4A). Interestingly, fibronectin expression is lost in CIPs farther away from the primary tumor, suggesting that fibronectin may only be important for early invasion events (Figure 3H).
Second harmonic generation imaging was performed to assess the collagen ECM which showed high levels of aligned collagen at the invasive front of the primary tumor and surrounding CIPs in the KLV models (Figure 3I, Supplemental Figure 4B). This aligned collagen persisted at metastatic lymph nodes, indicating that collagen alignment and secretion is maintained at the distant metastatic site (Figure 3I). To probe cancer invasion and EMT, immunofluorescence of TGFβ1 ligand was performed in primary tumor samples (Supplemental Figure 4C). High expression was observed in areas of focal invasion at or near the primary tumor indicating TGFβ1 is promoting cancer invasion and progression in the KLV models without inducing some canonical EMT markers.
Vimentin+ cancer-associated fibroblasts surround CIPs
To characterize vimentin positive cells surrounding CIPs, FSP1 (Fibroblast Specific Protein 1) and alpha-Smooth Muscle Actin (α-SMA) were used to mark cancer-associated fibroblasts (CAFs; (36). Dual labeling of vimentin and FSP1 in KLV+/+ primary tumor samples demonstrates that vim+ stromal cells are also FSP1+ (Figure 4A). FSP1+/vim+ cells are randomly localized within the primary tumor (Figure 4A left) but surround CIPs at sites of focal invasion (Figure 4A right). Quantitative analysis of FSP1+/vim+ cells surrounding CIPs show co-localization between vimentin and FSP1 (Pearson coefficient = 0.59; Figure 4B). IHC of serial sections show that vim+ cells are also α-SMA+ (Supplemental Figure 5A). These data strongly suggest that vim+ cells are cancer-associated fibroblasts (CAFs). FSP1+ CAFs are found at the invasive fronts of both KLV+/+ and KLV−/− primary tumors (Supplemental Figure 5B); however, the significantly greater extent of focal invasion in KLV+/+ tumors (Figure 3D) indicates increased CAF recruitment in this model.
Figure 4. Vimentin+ cancer-associated fibroblasts surround CIPs.

(A) Immunofluorescence of vimentin (488) and FSP1 (555) on primary tumors KLV+/+ mice (scale=50μm) (B) Colocalization of vimentin and FSP1 staining (scale=10μm). (C) Quantification of percent of mice of each genotype with CAFs stratified by metastatic or non-metastatic phenotypes. (D) Immunohistochemical staining of vimentin in the metastatic and non-metastatic lymph nodes (scale bars 10× =100μm, 40× =20μm)
Since many tumor-promoting components of the tumor microenvironment express vimentin, several members of the microenvironment were probed within the KLV models, including macrophages, inflammatory cells, and vascular cells. Immunofluorescence of tumor-associated macrophages (TAMs) with F4/80 in KLV+/+ and KLV−/− primary tumor samples show no significant difference in macrophage recruitment (Supplemental Figure 6). IHC of a Pan T-cell marker (CD3) and endothelial marker (CD31) also demonstrated no difference in recruitment of these factors (Supplemental Figure 7A, B). Taken together, these data indicate vimentin loss does not impede the recruitment of vascular or inflammatory cells to the primary tumor microenvironment.
To determine the relationship between CAF recruitment and metastatic disease, the presence or absence of CAFs was stratified by genotype and metastatic phenotype. These results show that 100% of metastatic mice contained CAFs at the primary tumor site (Figure 4C) but less than 50% of non-metastatic mice had CAFs recruited to the primary tumor in both KLV+/+ and KLV−/− genotypes. Vimentin IHC identified that CAFs are also found surrounding CIPs at the metastatic lymph nodes demonstrating that CAFs are important components of the tumor microenvironment at the secondary site (Figure 4D) and perhaps travel with tumor cells to the lymph node. These data suggest that CAF recruitment contributes to tumor metastasis by promoting CIP formation and facilitating metastasis to the secondary site.
Vimentin regulates CAF invasion and stroma-cancer cell crosstalk
To probe the mechanisms of the heterotypic tumor cell-CAF interactions and CAF recruitment found in the KLV models, in vitro studies were performed. Stable knockdown of vimentin was achieved in human lung adenocarcinoma CAFs via shRNA (Figure 5A, B). CAFs lacking vimentin (shVIM19 and shVIM22) were significantly smaller than pLKO.1 vector control (Figure 5C, D). To test the hypothesis that vimentin depletion inhibits CAF invasion in the KLV models, 3D spheroid invasion assays were performed with pLKO.1 and shVIM CAFs embedded in MatriGel (Figure 5E). Results show that shVIM CAFs invaded significantly less than pLKO.1 CAFs (Figure 5F), supporting the hypothesis that CAF motility is impaired with vimentin loss in the KLV models.
Figure 5. Vimentin regulates CAF invasion and stroma-cancer cell crosstalk.

(A) Western blot demonstrating generation of shVIM CAFs with two different shRNA clones. (B) Immunofluorescence of vimentin in pLKO.1 andshVIM19 CAFs (scale=50μm). (C) Immunofluorescence of actin and vimentin in pLKO.1 and shVIM CAFs demonstrating cell size (10× scale= 100μm, 20× scale = 100μm). (D) Quantification of cell area of pLKO.1 and shVIM CAFs. Significance determined by unpaired t-test (p<0.05). (E) Representative images of spheroid invasion assay with pLKO.1 and shVIM CAFs in matrigel (scale = 100μm). (F) Quantification of invasive area in spheroid invasion assay. Significance determined by unpaired t-test (p<0.05). (G) Representative images ofcoculture spheroid invasion assay with H460 lung cancer line alone or in combination with pLKO.1 or shVIMCAFs (scale = 100μm). (H) Circularity of invasive areas in representative trial (3 total trials) of coculture spheroids with pLKO.1(n=6) or shVIM (n=3) CAFs. Value of 1= perfect circle. Significance determined by unpaired t-test (p<0.05). (I) Quantification of invasive chains in representative coculture spheroids with pLKO.1 or shVIM CAFs. Significance determined by unpaired t-test (p<0.05). (J) Representative images of immunofluorescence of FSP1and phalloidin in coculture spheroids. Yellow arrows mark CAFs positive for cytoplasmic FSP1 leading invasive chain of H460s (scale = 100μm).
Co-culture 3D spheroid invasion assays were then performed to assess heterotypic cancer cell-CAF interactions (Figure 5G). Invasion analysis revealed that the addition of CAFs to spheroids of H460s (lung adenocarcinoma cell line) decreases circularity, a surrogate for collective invasion branching (i.e., increased circularity means less collective invasion packs; Supplemental Figure 8). Co-culture spheroids with shVIM CAFs exhibit significantly higher circularity (decreased branching) compared to those with pLKO.1 CAFs (Figure 5H). These same co-culture spheroids show significantly less invasive chains (37) per spheroid with shVIM CAFs compared to pLKO.1 CAFs (Figure 5I). These data suggest impairment in shVIM CAFs to drive collective invasion. Immunofluorescence of FSP1 and actin in co-culture spheroids (Figure 5J) show that CAFs facilitate collective invasion by leading chains of H460s into the surrounding ECM. CAFs lacking vimentin expression lose the ability to lead cancer cells out of the spheroid as evidenced by the decrease in invasive branches with CAFs leading the chain (Figure 5H). Together, these data indicate that heterotypic in vitro 3D invasion is vimentin-dependent and consistent with the in vivo GEMM results.
Vimentin is expressed in cancer-associated fibroblasts of lung cancer patient CIPs which lack EMT in tumor cells
To analyze the clinical relevance of the CIP phenotype found in the KLV models, vimentin staining in human lung adenocarcinoma patients with varying genetic subtypes was performed to assess vimentin localization, invasive histology, and EMT (Figure 6A). A similar pattern to the KLV+/+ mouse samples was observed in human tissues, where the vast majority of patient samples (25 of 26) expressed vimentin in the mesenchymal stroma and not within the collectively invading cancer cells (Figure 6A). Vimentin stain intensity was scored and was significantly higher in stromal cells compared to tumor cells across the majority of samples, independent of driver mutation (Figure 6B). Furthermore, there were no observable differences in vimentin stromal Q score (product of stain intensity and percent of vimentin-positive cells in stromal compartment) based upon genetic subtype (Supplemental Figure 9).
Figure 6. Vimentin is expressed in cancer-associated fibroblasts of lung cancer patient CIPs which lack EMT in tumor cells.
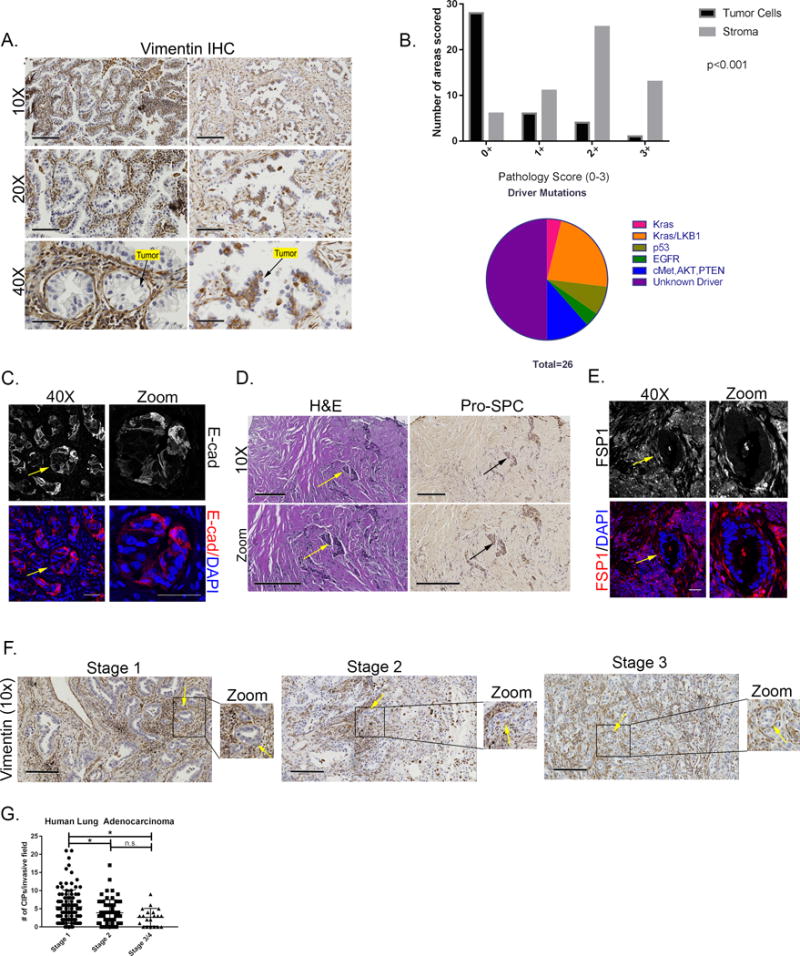
(A) Representative images of patient primary tumor samples stained for vimentin by immunohistochemistry (scale bars 10× =200μm, 20× =100μm, 40× =50μm). (B) Quantification of vimentin staining in tumor and stromal compartments. Pie chart of driver mutations in patient samples stained for vimentin. Statistical analysis was conducted using SAS Version 9.4. For the ordinal variable, frequency and average rank are calculated and presented. The univariate association of staining and different group, i.e., Tumor cell and Stroma was detect by Wilcoxon Rank Sum test. (D) Representative images of E-cadherin immunofluorescence of human lung adenocarcinoma sample marking collective invasion packs of tumor cells (40× = 50μm) (D) H&E and SPC staining of adenocarcinoma samples.
CIPs marked with arrows (scale =200μm). (E) Immunofluorescence of FSP1 on Kras/LKB1 patient sample. CIP marked with an arrow (scale=50μm). (F) Representative images of human lung adenocarcinoma samples stained for vimentin at stages 1-3. (G) CIPs per invasive field by stage in human lung adenocarcinoma samples. Statistical significance determined by ANOVA with Tukey’s multiple comparisons test (p<0.05).
Since tumor cells lacked vimentin staining, the epithelial cell marker, E-cadherin, was used to further probe for evidence of EMT. Tumor cells within patient CIPs were E-cadherin positive (18) (Figure 6C) and pro-SPC positive (Figure 6D), which is also consistent with the data in the KLV models. Immunofluorescence of FSP1 was performed to determine if patient CIPs are surrounded by CAFs similar to the KLV model. These data show an expression pattern consistent with the KLV model in which FSP1+ CAFs surround CIPs of tumor cells (Figure 6E). Taken together, these data suggest that in human lung adenocarcinoma samples tumor cells are not undergoing EMT but instead contain invasive CIPs surrounded by vim+/FSP+ CAFs, indicating the predominant role of vimentin in lung adenocarcinoma lies within CAFs and not tumor cells.
To determine the clinical relevance of CIPs, CIP density was stratified by in human lung adenocarcinoma patient samples (Figure 6F, G). Stage 1 patients exhibit significantly higher CIP density than higher stage patients indicating an early dissemination of CIP and CAF invasion, though there are still CIPs present in late stage patients (p<0.05). Taken together, the formation of CIPs in early stage tumors supports an early dissemination model (38) in which metastatic seeds such as CIPs disseminate early during tumor progression.
Discussion
Vimentin expression has historically correlated with increased metastatic potential across numerous solid tumor types (24, 39); however, a functional role for vimentin in cancer progression has yet to be elucidated. Data from the KLV−/− GEMM show that vimentin is required for metastasis and progression but not primary tumor formation, whereby KLV−/− mice had less mediastinal lymph nodes metastasis, less invasive foci, and lower grade tumors compared to their KLV+/+ counterparts (Figures 1 and 2); therefore, this supports a model that vimentin is more than an EMT biomarker and plays a functional role in lung adenocarcinoma invasion and metastasis. Despite decreased metastatic rates in the KLV−/− model, there is no survival advantage with vimentin depletion (Supplemental Figure 2), which is consistent with previous reports demonstrating increased morbidity in vimentin null mice (46). In the KLV−/− model, primary tumor burden could be exacerbating vimentin-dependent deficiencies in arterial remodeling (47) and wound healing (48).
EMT has been the canonical mechanism (9) for how cancer cells lose their epithelial morphology (4), invade through the basement membrane, and navigate the surrounding microenvironment (1, 40); however, this model is challenged in part by studies showing a more epithelial and collective-based migration by tumor cell clusters or packs (18, 41). The data presented here support the concept that a classical EMT does not occur in the majority of lung adenocarcinoma patients or in the lung adenocarcinoma KLV GEMM, but rather cancer cells undergo epithelial-like collective invasion and are surrounded by vim+/FSP1+ CAFs (Figures 3 and 4). Across 96% of lung adenocarcinoma patient samples tested (n=26), independent of driver mutation, vimentin was expressed in CAFs surrounding epithelial CIPs and only rarely in the tumor cells (Figure 6). Similarly, vimentin was rarely expressed within the tumor cells of the KLV+/+ GEMM, and only in the CAFs surrounding collective invasion packs at the primary tumor site and at secondary metastatic lymph nodes (Figure 3 and 4). This supports the concept that malignant cancer cells are not undergoing EMT but partnering with vim+/FSP1+ CAFs to potentially co-metastasize to a secondary site (42). This finding is consistent with studies demonstrating that circulating tumor cell clusters have metastatic potential up to 50 times greater than single circulating tumor cells (43), suggesting that tumor cell cooperativity can lead to greater metastatic success. Furthermore, these heterotypic CAF-CIP clusters are enriched in early stage lung adenocarcinoma patient samples (Figure 6), indicating potential early dissemination (38, 44) of metastatic seeding.
Studies investigating vimentin within CAFs in homotypic (CAFs only) and heterotypic (CAFs + cancer cells) in vitro cell cultures demonstrate that vimentin drives not only homotypic CAF invasion, but also the formation and maintenance of heterotypic collective invasion chains (Figure 5). We show that heterotypic invasive chains in vimentin-depleted CAFs is impaired compared to their wild-type counterparts. Nevertheless, some collective invasion remains suggesting compensatory or redundant mechanisms within vimentin-null CAFs. Since actin, focal adhesion kinase (FAK), and other components of the motility machinery (45) are not directly targeted, it is possible that some degree of vimentin-independent cell motility is maintained within the vimentin-depleted CAFs. This is also consistent with the in vivo studies showing that while significantly reduced, CIPs still form in the KLV−/− mice.
Taken together, the in vivo and in vitro data here support a model where vimentin is required for collective lung cancer invasion and metastasis by facilitating epithelial tumor cell CIP formation via CAF recruitment to the CIP. When vimentin function is impaired, heterotypic interactions are significantly reduced likely due to the inability of vimentin-depleted CAFs to move efficiently. These data support the claim that there are early stage, epithelial, and collective modes of lung cancer metastasis that drive cellular escape from the primary tumor and require vimentin-positive CAFs. We speculate that vimentin could be a target for anti-metastatic therapies that are given at an early stage and in conjunction with traditional cytotoxics (49, 50).
Supplementary Material
Translational Relevance.
Clinical data has historically linked vimentin expression to poor patient prognosis; however, it is unclear if vimentin is required for metastasis; therefore, a genetically engineered mouse model LSL-KrasG12D/Lkb1fl/fl/Vim−/− model (KLV−/−) was developed that is a whole-body vimentin knockout crossed with the conditional Cre-dependent metastatic lung adenocarcinoma model. In this model, vimentin is not required for primary tumor growth, but is required for tumor invasion and metastasis. In KLV+/+ mice, vimentin is not expressed in tumor cells but in collagen-secreting cancer-associated fibroblasts (CAFs) surrounding heterotypic collective tumor invasion packs (CIPs) adjacent to the primary tumor and in metastatic lymph nodes. Tumor cell CIPs are vimentin-negative and E-cadherin-positive, indicating a lack of cancer EMT. A similar pattern is observed in human lung adenocarcinoma samples during early stage dissemination. Taken together, vimentin is necessary for early stage lung adenocarcinoma dissemination by functioning in heterotypic tumor cell-CAF interactions during lung cancer invasion, suggesting that vimentin can serve as a target for preventing lung metastasis.
Acknowledgments
Alessandra M. Richardson- Ruth L. Kirschstein National Research Service Award through the NCI (5F31CA189772-03). Adam I. Marcus- NCI (1R01CA194027, 1RO1CA201340, 1R01CA142858, and 1U54 CA209992). Melissa Gilbert-Ross (1R01CA194027, 1RO1CA201340)
This work was supported through funds from the National Cancer Institute (1R01CA194027, 1RO1CA201340, and 1R01CA142858) awarded to A.I.M. and M.G-R. and through a Ruth L. Kirschstein National Research Service Award awarded to A.M.R. The research in this publication was supported in part by the Winship Cancer Institute NCI/NIH funding (P30CA138292) and several Winship Core Facilities- the Emory Integrated Cellular Imaging, the Winship Core Pathology Laboratory, the Winship Biostatistics and Bioinformatics Shared Resource and the Winship Cancer Animal Models Core.
Special thanks to Jamie Arnst, Rachel Commander, Jamie King, Junghui Koo, Janna Mouw, Brian Pedro, Katelyn Ponder, Emily Summerbell, Carol Tucker-Burden, and Paula Vertino for helpful discussions and technical advice.
Footnotes
Author Contributions
Conceptualization, A.M.R., L.S.H., A.I.M., and M.G-R; Methodology, M.G-R. and L.S.H.; Resources, W.D.M. and W.G.W IV; Investigation, A.M.R., J.M.K., J.S., and A.E.K.; Validation, A.M.R.; Formal Analysis, A.M.R.; Writing- Original draft, A.M.R. and A.I.M.; Writing- Review and Editing, A.M.R., L.S.H., A.E.K., J.M.K., J.S., H.E.G., G.S., M.G-R., A.I.M.; Funding Acquisition, A.I.M., M.G-R. and A.M.R.
The authors declare no potential conflicts of interest.
References
- 1.Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. Journal of cellular biochemistry. 2007 Jul 1;101(4):816–29. doi: 10.1002/jcb.21215. [DOI] [PubMed] [Google Scholar]
- 2.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014 Mar;15(3):178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Molecular cancer. 2016 Feb 24;15:18. doi: 10.1186/s12943-016-0502-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer research. 2008 May 15;68(10):3645–54. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 5.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Current opinion in cell biology. 2005 Oct;17(5):548–58. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer metastasis reviews. 2016 Dec;35(4):645–54. doi: 10.1007/s10555-016-9648-7. [DOI] [PubMed] [Google Scholar]
- 7.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. The Journal of clinical investigation. 2009 Jun;119(6):1429–37. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao D, Dai C, Peng S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011 Dec;9(12):1608–20. doi: 10.1158/1541-7786.MCR-10-0568. [DOI] [PubMed] [Google Scholar]
- 9.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002 Jun;2(6):442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 10.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015 Nov 26;527(7579):472–6. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015 Nov 26;527(7579):525–30. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016 Jan;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Cespedes M. The role of LKB1 in lung cancer. Familial cancer. 2011 Sep;10(3):447–53. doi: 10.1007/s10689-011-9443-0. [DOI] [PubMed] [Google Scholar]
- 14.Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012 Mar 18;483(7391):613–7. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007 Aug 16;448(7155):807–10. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 16.Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016 Nov 17;539(7629):390–5. doi: 10.1038/nature20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer cell. 2013 Feb 11;23(2):143–58. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert-Ross M, Konen J, Koo J, Shupe J, Robinson BS, Wiles WGt, et al. Targeting adhesion signaling in KRAS, LKB1 mutant lung adenocarcinoma. JCI insight. 2017 Mar 09;2(5):e90487. doi: 10.1172/jci.insight.90487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao Y, Xiao Q, Ma H, Li L, Liu J, Feng Y, et al. LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proceedings of the National Academy of Sciences of the United States of America. 2010 Nov 02;107(44):18892–7. doi: 10.1073/pnas.1004952107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colucci-Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell. 1994 Nov 18;79(4):679–94. doi: 10.1016/0092-8674(94)90553-3. [DOI] [PubMed] [Google Scholar]
- 21.Liu S, Liu L, Ye W, Ye D, Wang T, Guo W, et al. High Vimentin Expression Associated with Lymph Node Metastasis and Predicated a Poor Prognosis in Oral Squamous Cell Carcinoma. Scientific reports. 2016 Dec 14;6:38834. doi: 10.1038/srep38834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas PA, Kirschmann DA, Cerhan JR, Folberg R, Seftor EA, Sellers TA, et al. Association between keratin and vimentin expression, malignant phenotype, and survival in postmenopausal breast cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999 Oct;5(10):2698–703. [PubMed] [Google Scholar]
- 23.Javle MM, Gibbs JF, Iwata KK, Pak Y, Rutledge P, Yu J, et al. Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Annals of surgical oncology. 2007 Dec;14(12):3527–33. doi: 10.1245/s10434-007-9540-3. [DOI] [PubMed] [Google Scholar]
- 24.Dauphin M, Barbe C, Lemaire S, Nawrocki-Raby B, Lagonotte E, Delepine G, et al. Vimentin expression predicts the occurrence of metastases in non small cell lung carcinomas. Lung cancer (Amsterdam, Netherlands) 2013 Jul;81(1):117–22. doi: 10.1016/j.lungcan.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Burch TC, Watson MT, Nyalwidhe JO. Variable metastatic potentials correlate with differential plectin and vimentin expression in syngeneic androgen independent prostate cancer cells. PLoS One. 2013;8(5):e65005. doi: 10.1371/journal.pone.0065005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Domagala W, Lasota J, Dukowicz A, Markiewski M, Striker G, Weber K, et al. Vimentin expression appears to be associated with poor prognosis in node-negative ductal NOS breast carcinomas. The American journal of pathology. 1990 Dec;137(6):1299–304. [PMC free article] [PubMed] [Google Scholar]
- 27.Chung BM, Rotty JD, Coulombe PA. Networking galore: intermediate filaments and cell migration. Current opinion in cell biology. 2013 Oct;25(5):600–12. doi: 10.1016/j.ceb.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helfand BT, Mendez MG, Murthy SN, Shumaker DK, Grin B, Mahammad S, et al. Vimentin organization modulates the formation of lamellipodia. Molecular biology of the cell. 2011 Apr 15;22(8):1274–89. doi: 10.1091/mbc.E10-08-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivaska J, Pallari HM, Nevo J, Eriksson JE. Novel functions of vimentin in cell adhesion, migration, and signaling. Experimental cell research. 2007 Jun 10;313(10):2050–62. doi: 10.1016/j.yexcr.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 30.Havel LS, Kline ER, Salgueiro AM, Marcus AI. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene. 2015 Apr 09;34(15):1979–90. doi: 10.1038/onc.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010 Jun;24(6):1838–51. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu QS, Rosenblatt K, Huang KL, Lahat G, Brobey R, Bolshakov S, et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene. 2011 Jan 27;30(4):457–70. doi: 10.1038/onc.2010.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konen J, Wilkinson S, Lee B, Fu H, Zhou W, Jiang Y, et al. LKB1 kinase-dependent and -independent defects disrupt polarity and adhesion signaling to drive collagen remodeling during invasion. Molecular biology of the cell. 2016 Apr 01;27(7):1069–84. doi: 10.1091/mbc.E15-08-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nature protocols. 2009;4(7):1064–72. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao P, Wu S, Li J, Fu W, He W, Liu X, et al. Human alveolar epithelial type II cells in primary culture. Physiological reports. 2015 Feb 01;3(2) doi: 10.14814/phy2.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;4:62. doi: 10.3389/fonc.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konen J, Summerbell E, Dwivedi B, Galior K, Hou Y, Rusnak L, et al. Image-guided genomics of phenotypically heterogeneous populations reveals vascular signaling during symbiotic collective cancer invasion. Nature Communications. 2017 doi: 10.1038/ncomms15078. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature. 2016 Dec 14; doi: 10.1038/nature20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian W, Wang G, Yang J, Pan Y, Ma Y. Prognostic role of E-cadherin and Vimentin expression in various subtypes of soft tissue leiomyosarcomas. Medical oncology (Northwood, London, England) 2013 Mar;30(1):401. doi: 10.1007/s12032-012-0401-y. [DOI] [PubMed] [Google Scholar]
- 40.van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutation research. 2011 Jul-Oct;728(1–2):23–34. doi: 10.1016/j.mrrev.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013 Dec 19;155(7):1639–51. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proceedings of the National Academy of Sciences of the United States of America. 2010 Dec 14;107(50):21677–82. doi: 10.1073/pnas.1016234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014 Aug 28;158(5):1110–22. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roychowdhury A, Samadder S, Islam MS, Chaudhury K, Roy A, Banerjee D, et al. Identification of Changes in the Human Papilloma Virus 16 (HPV16) Genome During Early Dissemination of Cervical Cancer Cells May Complement Histological Diagnosis of Lymph Node Metastasis. Pathology oncology research : POR. 2017 Jan 19; doi: 10.1007/s12253-017-0189-3. [DOI] [PubMed] [Google Scholar]
- 45.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer – a new therapeutic opportunity. Nat Rev Cancer. 2005 Jul;5(7):505–15. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 46.Terzi F, Henrion D, Colucci-Guyon E, Federici P, Babinet C, Levy BI, et al. Reduction of renal mass is lethal in mice lacking vimentin. Role of endothelin-nitric oxide imbalance. The Journal of clinical investigation. 1997 Sep 15;100(6):1520–8. doi: 10.1172/JCI119675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schiffers PM, Henrion D, Boulanger CM, Colucci-Guyon E, Langa-Vuves F, van Essen H, et al. Altered flow-induced arterial remodeling in vimentin-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2000 Mar;20(3):611–6. doi: 10.1161/01.atv.20.3.611. [DOI] [PubMed] [Google Scholar]
- 48.Eckes B, Colucci-Guyon E, Smola H, Nodder S, Babinet C, Krieg T, et al. Impaired wound healing in embryonic and adult mice lacking vimentin. Journal of cell science. 2000 Jul;113(Pt 13):2455–62. doi: 10.1242/jcs.113.13.2455. [DOI] [PubMed] [Google Scholar]
- 49.Yang Z, Garcia A, Xu S, Powell DR, Vertino PM, Singh S, et al. Withania somnifera root extract inhibits mammary cancer metastasis and epithelial to mesenchymal transition. PLoS One. 2013;8(9):e75069. doi: 10.1371/journal.pone.0075069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee J, Hahm ER, Marcus AI, Singh SV. Withaferin A inhibits experimental epithelial-mesenchymal transition in MCF-10A cells and suppresses vimentin protein level in vivo in breast tumors. Molecular carcinogenesis. 2015 Jun;54(6):417–29. doi: 10.1002/mc.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.