

Abstract
One of the most significant challenges in contemporary lipidomics lies in the separation and identification of lipid isomers that differ only in site(s) of unsaturation or geometric configuration of the carbon-carbon double bonds. While analytical separation techniques including ion mobility spectrometry (IMS) and liquid chromatography (LC) can separate isomeric lipids under appropriate conditions, conventional tandem mass spectrometry cannot provide unequivocal identification. To address this challenge, we have implemented ozone-induced dissociation (OzID) in-line with LC, IMS and high resolution mass spectrometry. Modification of an IMS-capable quadrupole time-of-flight mass spectrometer was undertaken to allow the introduction of ozone into the high-pressure trapping ion funnel region preceding the IMS cell. This enabled the novel LC-OzID-IMS-MS configuration where ozonolysis of ionized lipids occurred rapidly (10 ms) without prior mass-selection. LC-elution time alignment combined with accurate mass and arrival time extraction of ozonolysis products facilitated correlation of precursor and product ions without mass-selection (and associated reductions in duty cycle). Unsaturated lipids across 11 classes were examined using this workflow in both positive and negative ion modalities and in all cases the positions of carbon-carbon double bonds were unequivocally assigned based on predictable OzID transitions. Under these conditions geometric isomers exhibited different IMS arrival time distributions and distinct OzID product ion ratios providing a means for discrimination of cis/trans double bonds in complex lipids. The combination of OzID with multidimensional separations shows significant promise for facile profiling of unsaturation patterns within complex lipidomes including human plasma.
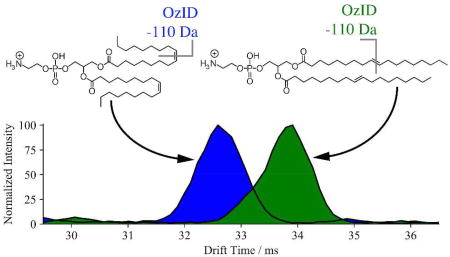
Graphical abstract
Introduction
Advances in the field of lipidomics have been largely underpinned by advances in both mass spectrometry and analytical separations.1–3 In spite of the significant progress made over the past decade, critical limitations still exist in the current workflows used for rapidly identifying and quantifying lipids in biological extracts.4 One of the most significant challenges in lipid analyses lies in separating, uniquely identifying and quantifying the structurally similar lipids within complex mixtures. This challenge is greatest for lipid isomers which, by definition, (i) share an identical mass, (ii) are often very similar in physiochemical properties, and (iii) are usually not separated effectively by conventional approaches.
The prevailing method for lipid separation is high-performance liquid chromatography (HPLC), which is often coupled to mass spectrometry and tandem mass spectrometry (LC-MS and LC-MS/MS) for lipid identification and structural elucidation. While this approach is widely deployed, and can be effective for resolving certain types of lipid isomers, it has limitations in resolving carbon-carbon double bond positions and orientations. For example, conventional reverse phase LC-MS strategies provide separations of constitutional isomers observed in different lipid subclasses5 such as phosphatidylcholine PC 33:2 and phosphatidylethanolamine PE 36:2 which share the same elemental composition, C41H78NO8P, but are effectively separated on a reversed-phase column.6 However, for lipid regioisomers such as those that differ in sites of unsaturation or positions of substitution on a glycerol backbone, long separation times and complex column chemistries are required and even then, chromatographic separation between isomers can be relatively poor.7–9
Ion mobility spectrometry (IMS) presents a fast and scalable toolkit that is orthogonal to LC and can be used to enhance separations. Various implementations of IMS have been demonstrated to separate lipid classes, double bond positional isomers and glycerolipids differing in acyl chain connectivity to the glycerol backbone.6,10–14 Kyle et al. have shown that it is possible to separate isomeric lipids in biological samples using drift tube ion mobility, however in many cases these isomers were not baseline separated, thus challenging lipid identifications based on drift time or collision cross section (CCS) measurements alone.6 For these reasons, IMS separations of complex mixtures are increasingly being coupled with LC-MS to further distinguish each lipid species, even though many isomers still co-elute in LC. Advances in the resolution of IMS separations, for example by increasing the path length using structures for lossless ion manipulation (SLIM), have also enhanced separations of isomeric lipids and enabled baseline separation in many cases. For example, both double bond positional isomers and cis/trans regioisomers of glycerophosphocholine (PC), which are barely separable using commercial IMS devices, have been baseline separated using ~30 m long SLIM devices.15
The rapid developments in IMS and its effective combination with LC-MS indicate significant promise for resolution of lipid regioisomers sharing similar physicochemical properties and highlight the number of lipid isomers present in biological extracts. In the absence of well-characterised standards however, many of these new mobility-based data features remain unidentified. Current IMS-MS and LC-IMS-MS approaches rely on conventional low energy collision-induced dissociation (CID) for structural characterization. Importantly, many regioisomeric lipids have very similar or identical CID spectra, thus hindering the unique structural assignment of an even well-resolved chromatographic or mobility feature.
Ozone-induced dissociation (OzID) has proven to be an effective means for identification of site(s) of unsaturation in both mono- and polyunsaturated lipids,16,17 and when combined with CID can also reveal the backbone substitution of glycerolipids.18 OzID relies on the gas phase ion-molecule reaction between the ionized lipid and neutral ozone gas inside the mass spectrometer. Given the geometry of most mass spectrometers, OzID implementations have typically been limited to the delivery of relatively low number densities of ozone (~109 – 1012 molecules cm−3)16,19 and consequently long reaction times (~1 s) have been required to achieve the requisite signal-to-noise ratio. This has, until recently, limited the compatibility of OzID with fast LC and IMS workflows. Recent studies have demonstrated that increasing the number density of ozone in the reactive region of the mass spectrometer, such as the trapping or travelling-wave regions of IMS instruments have dramatically improved the OzID duty cycle.20–22 In these cases the normal operating pressure of the respective regions of the mass spectrometer enables the ozone number density in the reactive region to be ~1015 molecules cm−3. This ozone concentration allows the OzID reaction time to be reduced to under 100 ms, placing OzID on the timescale of the MS cycle and well-matched with rapid separation technologies such as IMS and fast LC.21 As an example, recent work has shown that isomeric phospholipids differing in only a single position of unsaturation were partially separated by a fast LC protocol and could be uniquely identified by OzID with the resulting extracted ion chromatograms facilitating visualisation of the relative contributions of the two isomers to the precursor ion current.21 These recent studies have effectively exploited the IMS region of the mass spectrometer as a high-pressure reactor, but to this point the combination of the IMS and OzID technologies to provide separation and identification of lipid isomers has not been shown. Here we present a novel LC-OzID-IMS-MS technique for lipid analysis. This has been achieved by simple modification of the trapping ion funnel in an Agilent 6560 IMS-enabled quadrupole time-of-flight (IMS-QTOF) mass spectrometer, to facilitate gas phase ozonolysis of ionized lipids prior to IMS analysis. We show that containing the ions in the high-pressure trapping ion funnel in the presence of ozone results in a rapid ozonolysis reaction, yielding diagnostic OzID product ions that allow unambiguous identification of double bond positions. Ultimately, the high number density of ozone in the reactive region results in rapid OzID reactions thatare fast enough to be compatible with both LC and IMS. Furthermore, the multi-dimensional data acquired from the LC-OzID-IMS-MS workflow is shown to provide significant new dimensions for separations and identifications of lipid isomers.
Experimental Methods
Sample Preparation
Synthetic phospholipids; 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC 18:1(n-9, cis)/18:1(n-9, cis)), 1,2-dipetroselenoyl-sn-glycero-3-phosphocholine (PC 18:1(n-12, cis)/18:1(n-12, cis)), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (PE 18:1(n-9, cis)/18:1(n-9, cis)), and 1,2-dielaidoyl-sn-glycero-3-phosphoethanolamine (PE 18:1(n-9, trans)/18:1(n-9, trans)), obtained from Avanti Polar Lipids (Alabaster, AL), and the PC 18:0_18:2(n-7 trans, n-9 cis) and PC 18:0_18:2(n-6 trans, n-8 cis) standards, purchased from Matreya LLC (State College, PA), were diluted to 5 µM in 2:1 methanol:chloroform and infused directly into the mass spectrometer at a flow rate of 50 µL min−1. The SPLASH Lipidomix mixture of deuterated lipid standards was purchased from Avanti Polar Lipids (Alabaster, Al) and diluted in methanol (Fisher Chemical, Fair Lawn, NJ). Finally, lipids were extracted from human plasma using a modified Folch extraction.23,24 Following approval of the institutional review boards at the participating institutions and consent from the participants, plasma was collected from a control patient as detailed previously.25 Lipids were extracted by taking 25 µL of plasma and adding 600 µL of −20 °C 2:1 chloroform/methanol to the sample. This mixture was then vortexed for 30 s and transferred into a shaker at 22 °C for 60 min at 600 rpm. The sample was vortexed again for 30 s and then 125 µL of water was added to induce a phase separation and 350 µL of the bottom nonpolar lipid layer was removed, dried in a speedvac, and stored at −20 °C in 250 µL of 2:1 chloroform/methanol for lipid analyses. Prior to MS analysis, the total lipid extract was dried down and then reconstituted in 5 µL of chloroform first and then 95 µL of methanol so that all lipids would be brought into solution.
Mass Spectrometry
Experiments were conducted on a 6560 IMS QTOF mass spectrometer (Agilent; Santa Clara, CA) modified to allow introduction of ozone into the trapping funnel of the IMS region, as shown in Figure 1. Ozone was generated online from high-purity oxygen (Matheson Tri-Gas, Basking Ridge, NJ) using an ozone generator (HG-1500, Ozone Solutions; Sioux City, IA) and yielding an ultimate concentration of [O3] = 55 g m−3 as measured by a high concentration ozone monitor (106-H, 2B Technologies; Boulder, CO). A split from this flow was directed into the nitrogen supply for the trapping ion funnel, after the nitrogen mass flow controller, using a PEEK restriction (100 mm long × 0.03” I.D.) and a stainless steel needle valve (SS-SS4, Swagelok; Solon, OH). The ions were contained in the trapping ion funnel and allowed to react with the introduced ozone for 1 to 90 ms prior to injection into the IMS region. The unused portion of ozone was catalytically converted to oxygen using an unheated destruct unit (PN:810-0008-03, IN/USA; San Diego, CA) and exhausted from the laboratory. A low concentration ozone monitor (106-L, 2B Technologies; Boulder, CO) was used to monitor the ambient ozone concentration in the laboratory and interlocked to shut down ozone production if the background ozone concentration rose above 75 ppb. To confirm the lipid class identity and assign the fatty acyl composition of the lipids, all-ions CID was performed on alternate scans (scan time 1 s), where the quadrupole was set to transmit all ions which were subsequently fragmented in the collision cell with a collision energy between 20 – 40 eV.
Figure 1.

Schematic overview for introduction of ozone into the high-pressure trapping ion funnel (TIF) of the Agilent 6560 IMS q-TOF mass spectrometer. Ozone was introduced to the N2 line after the ion funnel mass flow controller (IF MFC).
Chromatography
For the LC analyses, a Waters Aquity UPLC H class system was used. The SPLASH mix was diluted 100-fold in methanol and 10 µL was then injected onto a reversed phase Waters CSH column (3.0 mm × 150 mm × 1.7 µm particle size). The lipids in the mixture were separated over a 34 min gradient (mobile phase A: acetonitrile/water (40:60) containing 10 mM ammonium acetate; mobile phase B: acetonitrile/isopropylalcohol (10:90) containing 10 mM ammonium acetate) at a flow rate of 250 µL min−1. The gradient and column wash are provided in Tables S1 and S2 of the Supporting Information. All m/z only XICs were extracted using the Agilent Mass Hunter Qual program and XICs using both m/z and drift time were extracted with the Agilent IM-MS Browser.
Lipid Nomenclature
The shorthand nomenclature for describing lipids used in this manuscript is based on the recommendations of Liebisch et al.26 The position(s) of unsaturation is indicated to be x-carbons from the methyl end of the acyl chain with the nomenclature (n-x) and the geometry about carbon-carbon double bonds is described as cis (Z) and trans (E) where it is known. For lipids containing stable isotope labelling, the site of labelling is indicated [2Hd], where d is the number of deuterium atoms.
Results and Discussion
Previous work has shown that increasing the number density of ozone in the reactive region of a mass spectrometer improves the duty cycle of OzID by up to 1000-fold. The source interface region of the Agilent 6560 IMS QTOF has several differentially-pumped regions that can potentially be used as high-pressure OzID reaction regions. In these experiments, ozone was introduced into the trapping ion funnel (TIF) of the instrument because this presented a high-pressure region (typical operating pressure of ~4 Torr) where ions can be contained for a user-defined period of time, prior to release into the IMS drift region of the instrument (Figure 1). To test the instrument response to ozone introduction in this region, first PC 18:1(n-9, cis)/18:1(n-9, cis) was infused through the electrospray ionization (ESI) source and analysed without ozone in the trap. The structure of the native lipid and mass spectrum without ozone are shown in Figures 2(a) and 2(b). Prior to ozone introduction, the main features in the mass spectrum are the protonated (m/z 786), sodiated (m/z 808) and potassiated (m/z 824) adducts of PC 36:2. When the needle valve isolating the trapping ion funnel supply gas (N2) from the ozone stream was opened and the ions were trapped for 90 ms, the mass spectrum in Figure 2(c) resulted. New peaks in the mass spectrum were attributed to the oxidative cleavage of the double bond in the n-9 position (i.e., neutral losses of 110 Da and 94 Da for the aldehyde and Criegee ions, respectively) and were readily assigned as neutral losses from both the [M+H]+ and [M+Na]+ precursor ions.27
Figure 2.

(a) Structure of PC 18:1(n-9, cis)/18:1(n-9, cis) showing the oxidative cleavage and neutral losses for one double bond. (b) Positive mode ESI mass spectrum of PC 18:1(n-9, cis)/18:1(n-9, cis) and (c) the same sample acquired in the presence of ozone for 90 ms. (d) Interleaved OzID acquisition showing the Total Ion Chromatogram (solid trace), [M+H]+ precursor (m/z 782; dot-dash trace) and the OzID fragment ions of [M+H]+ n-9 (dotted traces) while alternating ozone delivery to the trapping ion funnel. The externally generated ozone concentration was [O3] = 50 gm−3 for the duration of this experiment.
To understand how quickly the ozone reaction took place, the ion funnel trapping time was varied between 1 to 90 ms. OzID reactions were observed with trapping times as short as 1 ms, however greater product ion yields were noted for trapping times ≥10 ms (Figure S1 in the Supporting Information). Therefore, to optimize efficiency, all experiments used ion funnel trapping times greater than 10 ms since it matched well with the IMS drift times (30 – 90 ms). To investigate the possibility of quickly alternating between “ozone on” and “ozone off” mass spectra, we performed successive opening and closing of the manual needle valve. Figure 2(d) shows the response of the extracted ion chromatograms of the [M+H]+ (m/z 786) precursor and the two OzID product ions. Opening the valve resulted in a prompt response of the ion signal. Notably, the data plotted in Figure 2(d) reveal a slight decrease in the [M+H]+ precursor ion abundance that is accompanied by the appearance of diagnostic OzID fragment ions for the n-9 double bond position derived from both [M+H]+ and [M+Na]+ precursors. Closing the needle valve resulted in a similarly prompt return to the signal intensities prior to ozone introduction. The response time of the ion signal to ozone introduction suggests that in future experiments, delivery of ozone could be toggled on a timescale enabling both conditions to be sampled repeatedly across a chromatographic feature; perhaps using a fast actuating solenoid or pneumatic valve. Even toggling with a frequency of 1 Hz would still allow sampling of both the “ozone on” and “ozone off” conditions several times across an LC peak (typical peak widths for lipids in this workflow were ~10 s, see later). Subtracting the integrated mass spectrum between the 0.60 – 1.05 min “ozone on” and 0.05 – 0.50 min “ozone off” regions in Figure 2d highlights the ions that arise from ozonolysis, with the diagnostic OzID ions appearing as positive signals in the subtraction mass spectrum (Figure S2).
One key difference in this experimental configuration compared to previous OzID implementations is that ozonolysis is occurring without prior mass selection. While utilizing the trapping ion funnel region for OzID presents an advantage in that high ozone number densities can be accessed at no additional pumping cost, additional spectral noise means directly assigning OzID product ions to their respective precursors becomes difficult. This problem has been encountered before in so-called ozone-electrospray ionization mass spectrometry (OzESI-MS), where ozone is delivered in the nebulizing gas of the electrospray ionization source and ozonolysis occurs in the source region, also without mass-selection.28,29 In OzESI-MS of complex extracts, precursor ion scans for specific lipid classes (e.g., m/z 184 for choline-containing lipids) were employed to improve signal-to-noise and, importantly, to aid in the assignment of reaction-derived product ions to the corresponding precursor ion; critical for assignment of an unsaturation site or sites.29 In the present experiment, the additional ion mobility dimension provides an alternative means to reduce noise in the precursor region of the mass spectrum, and consequently aid in assigning OzID products to their associated precursor ions. In Figure 2(c), no IMS selection was used so all ion drift times contribute to the mass spectrum and consequently the precursor ion region containing [M+H]+, [M+Na]+ and [M+K]+ displays additional ions in relatively high abundance compared to the desired precursors. Because these superfluous ions have different arrival time distributions to the ionized lipids, it is possible to use post-acquisition drift time selection to remove them from the mass spectrum. As shown in Figure 3 for PC 18:1(n-12, cis)/18:1(n-12, cis), selecting only ions with drift times falling between 30 and 40 ms significantly reduces the noise in the region above m/z 780 and additional reduction in other areas is possible by further narrowing this range. This, in combination with the high-resolution mass accuracy of the TOF analyzer, allows unambiguous assignment of the OzID product ions to the appropriate precursor ions, confirming the n-12 position of the double bond. This same approach can also be used to assign double bond positions in polyunsaturated species, as shown in the Figure S4 of the supporting information for two polyunsaturated PC 36:2 isomers.
Figure 3.

(a) Structure of PC 18:1(n-12, cis)/18:1(n-12, cis) showing the site of oxidative cleavage and neutral losses for one double bond. (b) Positive ion electrospray mass spectrum of PC 18:1(n-12, cis)/18:1(n-12, cis) showing all drift times and (c) only ions with drift times between 30 and 40 ms. The externally generated ozone concentration was [O3] = 50 gm−3 and ions were stored for 90 ms in the presence of ozone in the trapping ion funnel.
One of the more challenging hurdles for lipid mass spectrometry is the assignment of cis/trans double bond configurations. Previous OzID experiments on mass-selected lipids have shown a difference in reactivity between cis and trans double bond geometries for a range of lipid classes, with trans reacting significantly faster.20,21 As a direct comparison of the efficacy of OzID without mass selection towards cis and trans double bonds, methanolic solutions of PE 18:1(n-9, cis)/18:1(n-9, cis) and PE 18:1(n-9, trans)/18:1(n-9, trans) were directly infused and ionized by ESI in both polarities. The resulting mass spectra, after reacting with ozone in the trapping ion funnel for 30 ms, are shown in Figure 4. The most apparent difference in these spectra is the disparity in OzID product ion abundances between the cis and trans double bond isomers. These data indicate that in both positive and negative ion modes, the trans isomer shows a marked increase in reactivity towards ozone. Taking the combined abundances of the aldehyde and Criegee ions as an indication of reaction efficiency, in positive ion mode the trans isomer reacts ~2.5 times faster than cis, while in negative ion mode the trans isomer reacts ~3.6 times faster than cis. This difference in reaction efficiency has been observed previously for ionized phospholipids19 and has also been described for gas-phase reactions of neutral olefins with ozone.30 It is also interesting to note that for the positive ions, the aldehyde ion abundance is greater than the abundance of the Criegee ion, whereas the relative ion abundance is reversed for the negative ions. Earlier negative ion OzID experiments have also noted this disparity for other glycerophospholipids, where both the charge and basicity of the lipid head group was suggested to influence this Criegee/aldehyde ion branching ratio.16,31 It has been proposed that these differences in reactivity and/or branching ratios could be used to determine double bond geometry in biological samples, although this would require either careful comparison to appropriate standards (which most often are not available) or alternatively, spectral comparisons of at least partially resolved isomeric pairs.19
Figure 4.

Comparative electrospray mass spectra of PE 18:1(n-9, cis)/18:1(n-9, cis) in (a) negative and (b) positive ion mode and PE 18:1(n-9, trans)/18:1(n-9, trans) in (c) negative and (d) positive ion mode. All four spectra were acquired with ozone in the high-pressure trapping ion funnel. The externally generated ozone concentration was [O3] = 50 gm−3. Note that the region between m/z 500 and 600 has been magnified ×10 in panel (a) only.
The configuration of double bond(s) present in an unsaturated molecule can also have a significant effect on the overall shape, and consequently the observed collision cross section. As a result, IMS is well suited to separate such isomeric lipids and has previously been used to resolve cis and trans isomers in standard materials.6,12 To investigate this possibility in the present study, the arrival time distributions for the same pair of PE 18:1(n-9)/18:1(n-9) stereoisomers were extracted in both positive and negative ion modes and are shown in Figure 5. For the [M−H]− ions (Figure 5c), there is not a large difference in arrival time for the precursor ions, with the (cis, cis) isomer detected only slightly earlier than the (trans, trans); however for the [M+H]+ precursors (Figure 5d), separation is significantly better. These results are in good agreement with earlier IMS experiments, which have shown the more compact (cis, cis) isomers arrive earlier than their (trans, trans) counterparts.6 For the OzID product ions, this behavior is maintained in that the n-9 aldehyde ion for the cis isomer arrives first for both [M−H-110]− (Figure 5e) and [M+H-110]+ (Figure 5f), with the arrival time difference between the cis and trans isomers approximately the same as observed for the precursor ions. This result is perhaps unsurprising, since after cleavage of one double bond, one cis or trans double bond remains on the unreacted acyl chain. Cleavage of this remaining double bond results in no structural difference in the product ions, and the arrival time distributions for these second OzID fragments overlap (Figures S5 and S6 of the Supporting Information) indicating that structures of the aldehyde and Criegee ions formed from both cis and trans double bonds are similar or identical. Importantly, the demonstration in Figure 5(c) and 5(e) that both the precursor ions and first generation OzID product ions have distinct IMS arrival time distributions suggests that the combination of these features could be deployed in interrogating biological extracts specifically for trans double bonds and perhaps extracting cis to trans ratios.
Figure 5.

Structures of the phosphatidylethanolamine standards (a) PE 18:1(n-9, cis)/18:1(n-9, cis) and (b) PE 18:1(n-9, trans)/18:1(n-9, trans). Comparison of arrival time distributions for monoisotopic PE 18:1(n-9, cis)/18:1(n-9, cis) (solid lines) and PE 18:1(n-9, trans)/18:1(n-9, trans) (dotted lines): (c) [M−H]−, (d) [M+H]+, (e) [M−H-110]− and (f) [M+H-110]+. The externally generated ozone concentration was [O3] = 50 gm−3 during these experiments.
To determine the efficiency of LC-OzID-IMS-MS across different lipid classes and to establish the possibility of retention time alignment of precursor lipids with OzID product ions, we analysed a commercially available mix of stable isotope-labeled lipid standards in both positive and negative ion mode using a 34 min LC gradient. This mixture is designed as an internal standard for human plasma, and as such contains a cross section of different lipid classes, including the glycerophospholipids LPC [2H7]18:1(n-9, cis), LPE [2H7]18:1(n-9, cis), PC 15:0/[2H7]18:1(n-9, cis), PE 15:0/[2H7]18:1(n-9, cis), PI 15:0/[2H7]18:1(n-9, cis), PG 15:0/[2H7]18:1(n-9, cis), PS 15:0/[2H7]18:1(n-9, cis), PA 15:0/[2H7]18:1(n-9, cis); the sphingomyelin SM [2H9]18:1(n-9, cis); the glycerolipids TG 15:0/[2H7]18:1(n-9, cis)/15:0, DG 15:0_[2H7]18:1(n-9, cis) and MG [2H7]18:1(n-9, cis); and the cholesterol ester CE [2H7]18:1(n-9, cis). The broad lipid class coverage in this standard mix makes it a useful test system for examining how each class will respond to the presence of ozone in the ion funnel region during an LC-OzID-IMS-MS experiment. All sites of unsaturation in this lipid mixture are (n-9, cis) and the sites of deuterium labeling in these standards is at the methyl end of the 18:1 acyl chain, meaning that the OzID neutral loss will include the heavy labels. That is, nominal mass losses for the aldehyde and Criegee ions are expected at 117 and 101 Da for the 2H7-labelled lipids and 119 and 103 Da for the 2H9-labelled SM. Figure 6 illustrates the m/z and drift time extracted XICs of six lipid precursor ions and their OzID products in positive mode (Figure 6a) and six in negative mode (Figure 6b), with the extracted ion chromatograms for PE 15:0/[2H7]18:1(n-9, cis) shown in both polarities. Importantly, OzID product ions were observed from all lipid classes in their preferred ionisation state. XICs of the aldehyde and Criegee ions both show good time alignment with the retention time of the precursor lipids. This retention time alignment, combined with the ability to filter the data by accurate mass and arrival time distribution, allows correlation of precursor and product ions without mass-selection. Further this data-independent approach affords significant improvements in duty cycle over conventional tandem mass spectrometric approaches.
Figure 6.

Drift time and m/z filtered extracted ion chromatograms from (a) positive and (b) negative mode LC-OzID-IMS experiments on the SPLASH mixture of isotopically labelled lipid standards. In all cases, the extracted OzID product ions align with the retention time of the precursor lipid.
Interestingly, the abundance of the two OzID product ions differed significantly between lipid classes and ionisation state. Table 1 reports the relative branching fractions and the OzID conversion efficiency for each of the ionized lipids in the mixture. Across the lipid classes, the Criegee/aldehyde branching ratio changes by a factor of 250, even between similarly ionized lipids (e.g., between deprotonated lipids). Also of note, the relative reaction efficiency (that is the abundance of the OzID product ions relative to the unreacted precursor ion abundance) also changes dramatically across the lipid classes. These data suggest that both the ionisation state and the interaction of the charge with the double bond play an important role in controlling the reactivity of the different lipid classes towards ozone.16,31 The stark variation in relative abundance of the two OzID marker ions provides an additional means for correlation of precursor and product ions as mixture complexity increases with the increased likelihood of co-eluting lipid populations.
Table 1.
Components of SPLASH mixture analyzed using LC-OzID-IMS-MS, preferred ionization state, relative OzID product ion abundances and relative reaction efficiencies for the data presented in Figure 6.
| Ionisation State |
Criegee:Aldehyde Ratio |
OzID products /precursor |
|
|---|---|---|---|
| LPE [2H7]18:1(n-9, cis) | [M−H]− | 0.03 | 0.52 |
| PI 15:0/[2H7]18:1(n-9, cis) | [M−H]− | 0.31 | 0.17 |
| PG 15:0/[2H7]18:1(n-9, cis) | [M−H]− | 1.72 | 1.33 |
| PS 15:0/[2H7]18:1(n-9, cis) | [M−H]− | 1.40 | 0.08 |
| PA 15:0/[2H7]18:1(n-9, cis) | [M−H]− | 8.22 | 0.95 |
| PE 15:0/[2H7]18:1(n-9, cis) | [M−H]− | 2.45 | 0.17 |
|
| |||
| PC 15:0/[2H7]18:1(n-9, cis) | [M+H]+ | 0.26 | 0.73 |
| LPC [2H7]18:1(n-9, cis) | [M+H]+ | 0.06 | 1.67 |
| Chol Ester [2H7]18:1(n-9, cis) | [M+NH4]+ | 1.21 | 0.08 |
| TG 15:0/[2H7]18:1(n-9, cis)/15:0 | [M+NH4]+ | 8.65 | 0.10 |
| SM [2H9]18:1(n-9, cis) | [M+H]+ | 0.22 | 0.59 |
| PE 15:0/[2H7]18:1(n-9, cis) | [M+H]+ | 0.11 | 1.98 |
Finally, to examine the efficacy of the LC-OzID-IMS-MS workflow for complex extracts, a human plasma sample was analysed. This lipid extract was evaluated twice, first in the presence of ozone and then in its absence, to determine the different double bond locations in human plasma lipids and verify the reproducibility of the chromatography. To confirm the lipid class identity and assign the fatty acyl composition of the lipids, all-ions CID was performed in the QTOF collision cell for every alternate scan (scan time 1 s). During the 34 min LC gradient, there were many times when lipids co-eluted but due to the different precursor and OzID product ion masses, the lipids and their fragments could be readily distinguished. One example is shown in Figure 7(a), where an examination of the space defined by the precursor ion m/z and drift time reveals the chromatographic coelution of a phosphatidylcholine, PC 36:1, and two sphingomyelins, SM d40:2 and SM d38:1. All-ions CID at this retention time confirms the acyl chain composition of the glycerophospholipid as PC 18:0_18:1, and the observation of two pairs of OzID product ions reveals a mixture of the double bond isomers PC 18:0_18:1(n-7) and PC 18:0_18:1(n-9). Prior investigations have identified the presence of the sphingomyelin isomers SM d18:2/22:0, SM d18:1/22:1 and SM d16:1/24:1 that as [M+H]+ precursor ions could all potentially contribute to the m/z 785.6 ion population in Figure 7(a).32 Importantly however, when the spectra were reacquired in the presence of ozone, signals characteristic of an n-4 site of unsaturation were clearly visible. This double bond position is characteristic of the d18:2 sphingadiene backbone33 and has also recently be observed in deoxyceramides derived from HEK cell lines.34 These OzID signals can thus be used to assign the structure of the sphingomyelin eluting at this time to SM d18:2(n-4)/22:0, while the other two contributors, SM d18:1/22:1 and SM d16:1/24:1, can be excluded from consideration (in this retention time slice) as no losses characteristic of the expected n-7 or n-9 sites of unsaturation in the associated side chains were observed. Interestingly, oxidative cleavage of the n-14 double bond in the sphingadiene SM d18:2/22:0 was not observed, which is consistent with previous reports of the sluggish reaction rate of the allylic alcohol motif in sphingolipids.22,34
Figure 7.

Extracted ions from LC-OzID-IMS of human plasma showing (a) co-eluting PC 18:0_18:1, SM d18:2/22:0 and SM d18:1/20:0, and (b) polyunsaturated PC 14:0_18:2 co-eluting with SM d18:2/16:0. OzID product ions are annotated as (n-xa) or (n-xc) where x is the site of unsaturation with respect to the methyl terminus and a and c correspond to the aldehyde and Criegee ions, respectively.
In a second example from human plasma extract, the elution profile of a phosphatidylcholine of sum composition PC 32:2 is shown in Figure 7(b). All-ions CID across the corresponding retention time window yielded the spectrum shown in Figure S7 (see supporting information) revealing characteristic neutral losses corresponding to the acyl chain composition, PC 14:0_18:2. The time-aligned OzID spectra showed distinct Criegee/aldehyde ion pairs (Figure 7b), corresponding to n-6 neutral losses and n-9 (polyunsaturated) neutral losses,27 consistent with PC 14:0_18:2(n-6, n-9). The ability of the LC-OzID-IMS-MS workflow to assign site(s) of unsaturation in polyunsaturated lipids could thus reveal changes in the plasma lipidome reflecting diet and environment. For example, the 18:2(n-6, n-9) identified here is most likely originating from plant-based foods in the patient’s diet as humans lack the biosynthetic machinery for the de novo synthesis of linoleic acid.35
Conclusions
In this manuscript, we illustrated that OzID is compatible with high throughput IMS workflows when using the ion funnel trap for reaction. The presence of ozone in the trapping ion funnel is shown to have no impact on the normal experiment duty cycle, and with further optimization, on-demand ozone delivery to this region may present a means to interrogate chromatographic features with both “ozone on” and “ozone off” during a single analysis. From the OzID-IMS-MS data it was possible to use the arrival time distribution of the ozonolysis product ions to assign reaction-derived products to their associated precursors and when the LC dimension was added, the use of LC elution time, IMS drift time and m/z values allowed specific characteristics of each lipid to be acquired. Our results show that it is possible to couple OzID to fast IMS based separations without any compromise to throughput, both in direct infusion and HPLC analyses. The combination of LC-OzID-IMS-MS has been shown to generate a rich and highly informative data set that carries information on the site(s) of unsaturation across all major lipid classes in their preferred ionization state. Importantly, mass and mobility characteristics of OzID product ions can be time-aligned with precursor ions to enable structural assignment of unsaturated lipids without the reduction in duty cycle that arises from precursor ion selection. Indeed, this can be considered as a data-independent “ozonolysis-of-everything” workflow whereby consecutive LC-OzID-IMS-MS analyses of a sample in positive and negative ion mode yield a comprehensive profile of lipid unsaturation (cf. Figure 6). The remarkable stability and reproducibility of IMS data on ionized metabolites has recently been demonstrated,36 so based on these findings it is reasonable to suggest that libraries of accurate mass and collisional cross section data for OzID product ions could be used to interrogate LC-OzID-IMS-MS datasets of complex lipid extracts. Indeed, a cursory analysis of data acquired from human plasma extracts using our approach reveals an underlying complexity that may not be adequately captured by other approaches. Given that a recent analysis of the NIST Standard Reference Material for human plasma (SRM1950) identified and quantified ~70 PC lipids,37 many of which were unsaturated, it is reasonable to assume that including double bond regioisomers could expand this by a factor of two or more. Consistent with other data-independent strategies, the datasets could be re-interrogated to test evolving hypotheses of the lipidome without having to re-analyze the samples. Finally, the marked differences in both OzID reactivity and IMS arrival time distributions between cis and trans double bond geometries indicate that combination of these two technologies could provide a means to reveal the presence of geometrical isomers in biological samples.
Supplementary Material
Acknowledgments
This work was supported through funding from the Australian Research Council Discovery Program (DP150101715), the National Institute of Environmental Health Sciences of the NIH (R01ES022190) and the Laboratory Directed Research and Development Program at Pacific Northwest National Laboratory. This work was performed in the W. R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a DOE national scientific user facility at the Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle for the DOE under contract DE-AC05-76RL0 1830.
Footnotes
Supporting Information Available Two tables summarising the UPLC gradient and additional presentations of mass spectral data are provided as Supporting Information (PDF file).
The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Wenk MR. Cell. 2010;143:888–895. doi: 10.1016/j.cell.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell TW, Brown SH, Blanksby SJ. In: Lipidomics Technologies and Applications. Ekroos K, editor. John Wiley and Sons; 2012. [Google Scholar]
- 3.Blanksby SJ, Mitchell TW. Annu. Rev. Anal. Chem. 2010;3:433–465. doi: 10.1146/annurev.anchem.111808.073705. [DOI] [PubMed] [Google Scholar]
- 4.Hancock SE, Poad BLJ, Batarseh A, Abbott SK, Mitchell TW. Anal. Biochem. 2017;524:45–55. doi: 10.1016/j.ab.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Cajka T, Fiehn O. TrAC - Trend. Anal. Chem. 2014;61:192–206. doi: 10.1016/j.trac.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyle JE, Zhang X, Weitz KK, Monroe ME, Ibrahim YM, Moore RJ, Cha J, Sun X, Lovelace ES, Wagoner J, Polyak SJ, Metz TO, Dey SK, Smith RD, Burnum-Johnson KE, Baker ES. Analyst. 2016;141:1649–1659. doi: 10.1039/c5an02062j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakanishi H, Iida Y, Shimizu T, Taguchi R. J. Biochem. 2010;147:245–256. doi: 10.1093/jb/mvp171. [DOI] [PubMed] [Google Scholar]
- 8.Kozlowski RL, Mitchell TW, Blanksby SJ. Eur. J. Mass Spectrom. 2015;21:191–200. doi: 10.1255/ejms.1300. [DOI] [PubMed] [Google Scholar]
- 9.Kozlowski RL, Campbell JL, Mitchell TW, Blanksby SJ. Anal. Bioanal. Chem. 2015;407:5053–5064. doi: 10.1007/s00216-014-8430-3. [DOI] [PubMed] [Google Scholar]
- 10.Shvartsburg AA, Isaac G, Leveque N, Smith RD, Metz TO. J. Am. Soc. Mass Spectrom. 2011;22:1146. doi: 10.1007/s13361-011-0114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lintonen TPI, Baker PRS, Suoniemi M, Ubhi BK, Koistinen KM, Duchoslav E, Campbell JL, Ekroos K. Anal. Chem. 2014;86:9662–9669. doi: 10.1021/ac5021744. [DOI] [PubMed] [Google Scholar]
- 12.Groessl M, Graf S, Knochenmuss R. Analyst. 2015;140:6904–6911. doi: 10.1039/c5an00838g. [DOI] [PubMed] [Google Scholar]
- 13.Castro-Perez J, Roddy TP, Nibbering NM, Shah V, McLaren DG, Previs S, Attygalle AB, Herath K, Chen Z, Wang SP, Mitnaul L, Hubbard BK, Vreeken RJ, Johns DG, Hankemeier T. J. Am. Soc. Mass Spectrom. 2011;22:1552–1567. doi: 10.1007/s13361-011-0172-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowman AP, Abzalimov RR, Shvartsburg AA. J. Am. Soc. Mass Spectrom. 2017;28:1552–1561. doi: 10.1007/s13361-017-1675-2. [DOI] [PubMed] [Google Scholar]
- 15.Wojcik R, Webb KI, Deng L, Garimella VS, Prost AS, Ibrahim MY, Baker SE, Smith DR. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18010183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas MC, Mitchell TW, Harman DG, Deeley JM, Nealon JR, Blanksby SJ. Anal. Chem. 2008;80:303–311. doi: 10.1021/ac7017684. [DOI] [PubMed] [Google Scholar]
- 17.Pham HT, Maccarone AT, Campbell JL, Mitchell TW, Blanksby SJ. J. Am. Soc. Mass Spectrom. 2013;24:286–296. doi: 10.1007/s13361-012-0521-9. [DOI] [PubMed] [Google Scholar]
- 18.Pham HT, Maccarone AT, Thomas MC, Campbell JL, Mitchell TW, Blanksby SJ. Analyst. 2014;139:204–214. [Google Scholar]
- 19.Poad BLJ, Pham HT, Thomas MC, Nealon JR, Campbell JL, Mitchell TW, Blanksby SJ. J. Am. Soc. Mass Spectrom. 2010;21:1989–1999. doi: 10.1016/j.jasms.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Vu N, Brown J, Giles K, Zhang Q. Rapid Commun. Mass Spectrom. 2017;31:1415–1423. doi: 10.1002/rcm.7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poad BLJ, Green MR, Kirk JM, Tomczyk N, Mitchell TW, Blanksby SJ. Anal. Chem. 2017;89:4223–4229. doi: 10.1021/acs.analchem.7b00268. [DOI] [PubMed] [Google Scholar]
- 22.Barrientos RC, Vu N, Zhang Q. J. Am. Soc. Mass Spectrom. 2017 doi: 10.1007/s13361-017-1772-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakayasu ES, Nicora CD, Sims AC, Burnum-Johnson KE, Kim Y-M, Kyle JE, Matzke MM, Shukla AK, Chu RK, Schepmoes AA, Jacobs JM, Baric RS, Webb-Robertson B-J, Smith RD, Metz TO. mSystems. 2016;1:e00043–00016. doi: 10.1128/mSystems.00043-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Folch J, Lees M, Stanley GHS. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 25.Kyle JE, Casey CP, Stratton KG, Zink EM, Kim Y-M, Zheng X, Monroe ME, Weitz KK, Bloodsworth KJ, Orton DJ, Ibrahim YM, Moore RJ, Lee CG, Pedersen C, Orwoll E, Smith RD, Burnum-Johnson KE, Baker ES. Rapid Commun. Mass Spectrom. 2017;31:447–456. doi: 10.1002/rcm.7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liebisch G, Vizcaino JA, Kofeler H, Trotzmuller M, Griffiths WJ, Schmitz G, Spener F, Wakelam MJ. J. Lipid Res. 2013;54:1523–1530. doi: 10.1194/jlr.M033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown SHJ, Mitchell TW, Blanksby SJ. BBA - Mol. Cell Biol. L. 2011;1811:807–817. doi: 10.1016/j.bbalip.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 28.Thomas MC, Mitchell TW, Blanksby SJ. J. Am. Chem. Soc. 2006;128:58–59. doi: 10.1021/ja056797h. [DOI] [PubMed] [Google Scholar]
- 29.Thomas MC, Mitchell TW, Harman DG, Deeley JM, Murphy RC, Blanksby SJ. Anal. Chem. 2007;79:5013–5022. doi: 10.1021/ac0702185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greene CR, Atkinson R. Int. J. Chem. Kin. 1992;24:803–811. [Google Scholar]
- 31.Marshall DL, Saville JT, Maccarone AT, Ailuri R, Kelso MJ, Mitchell TW, Blanksby SJ. Rapid Commun. Mass Spectrom. 2016;30:2351–2359. doi: 10.1002/rcm.7715. [DOI] [PubMed] [Google Scholar]
- 32.Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, Bandyopadhyay S, Jones KN, Kelly S, Shaner RL, Sullards CM, Wang E, Murphy RC, Barkley RM, Leiker TJ, Raetz CR, Guan Z, Laird GM, Six DA, Russell DW, McDonald JG, Subramaniam S, Fahy E, Dennis EA. J. Lipid Res. 2010;51:3299–3305. doi: 10.1194/jlr.M009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merrill AH. Chem. Rev. 2011;111:6387–6422. doi: 10.1021/cr2002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steiner R, Saied EM, Othman A, Arenz C, Maccarone AT, Poad BLJ, Blanksby SJ, von Eckardstein A, Hornemann T. J. Lipid Res. 2016;57:1194–1203. doi: 10.1194/jlr.M067033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guillou H, Zadravec D, Martin PGP, Jacobsson A. Prog. Lipid Res. 2010;49:186–199. doi: 10.1016/j.plipres.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Stow SM, Causon TJ, Zheng X, Kurulugama RT, Mairinger T, May JC, Rennie EE, Baker ES, Smith RD, McLean JA, Hann S, Fjeldsted JC. Anal. Chem. 2017;89:9048–9055. doi: 10.1021/acs.analchem.7b01729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zacek P, Bukowski M, Rosenberger T, Picklo M. J. Lipid Res. 2016 doi: 10.1194/jlr.D070656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.