

Abstract
Monotherapy clinical trials with mutation-targeted kinase inhibitors, despite some success in other cancers, have yet to impact glioblastoma (GBM). Besides insufficient blood-brain-barrier penetration, combinations are key to overcoming obstacles such as intratumoral heterogeneity, adaptive resistance, and the epistatic nature of tumor genomics that cause mutation-targeted therapies to fail. With now hundreds of potential drugs, exploring the combination space clinically and pre-clinically is daunting. We are building a simulation-based approach that integrates patient-specific data with a mechanistic computational model of pan-cancer driver pathways (receptor tyrosine kinases, RAS/RAF/ERK, PI3K/AKT/mTOR, cell cycle, apoptosis, and DNA damage) to prioritize drug combinations by their simulated effects on tumor cell proliferation and death. Here we illustrate a first step, tailoring the model to 14 GBM patients from The Cancer Genome Atlas defined by an mRNA-seq transcriptome, and then simulating responses to three promiscuous FDA-approved kinase inhibitors (bosutinib, ibrutinib, cabozantinib) with evidence for blood-brain-barrier penetration. The model captures drug binding to primary and off-targets based on published affinity data, and simulates responses of 100 heterogeneous tumor cells within a patient. Single drugs are marginally effective or even counter-productive. Common copy number alterations (PTEN loss, EGFR amplification, NF1 loss) have negligible correlation with single drug or combination efficacy, reinforcing the importance of post-genetic approaches that account for kinase inhibitor promiscuity to match drugs to patients. Drug combinations tend to be either cytostatic or cytotoxic, but seldom both, highlighting the need for considering targeted and non-targeted therapy. Although we focus on GBM, the approach is generally applicable.
Keywords: combination therapy, mechanistic models, cancer precision medicine, stochastic simulation, brain tumors, quantitative systems pharmacology
Graphical Abstract
Introduction
Glioblastoma (GBM—see a list of abbreviations in Supplementary Table 1) is a highly invasive and deadly brain tumor with ~1 year median survival rates and few efficacious treatments1. It is the most common malignant brain tumor with the poorest prognosis2. Aberrant tyrosine kinase signaling is a hallmark of GBM, and is present in 88% of patients3. Over 140 non-synonymous somatic mutations of kinases in GBM have been documented4, nearly 20 kinase genes serve as prognostic biomarkers for tumor recurrence due to their enrichment5, and the four GBM transcriptional subtypes (proneural, classical, mesenchymal, and neural6) are statistically associated with distinctive genetic aberrations in kinases or genes that functionally interact with kinases (PDGFRA mutations, EGFR amplification, PTEN loss, NF1 deletions)7.
In other cancer types, oncogene-targeted small molecule kinase inhibitors, like imatinib for BCR-ABL8–13 in leukemias, have transformed chemotherapy by improving outcomes and providing treatments that are matched to specific mutations. However, such precision medicine approaches are not always efficacious. In some cases, mutation-matched patients do not respond to the drug14–17, or alternatively, resistance develops16,18–21. Moreover, monotherapy can even activate the target pathway, depending on cellular context22,23. In GBM, such small molecule kinase inhibitors have not been efficacious, due to in part to a lack of brain-penetration24, but also perhaps to intra-tumoral, spatial, genomic, and phenotypic heterogeneity of the disease25.
Some kinase inhibitors are selective for the primary target, but many bind to a large proportion of the kinome at therapeutically-relevant concentrations26. Conclusive determination of whether such off-target activity contributes to efficacy is difficult. However, the facts that sorafenib (primary targets of VEGFR, PDGFR and RAF family kinases) and sunitinib (primary targets of VEGFR and PDGFR family kinases) (i) have indications for and clinical efficacy against several cancer types which may or may not involve these primary targets (not counting off-label use) (ii) have been shown, respectively, to bind 21% and 60% of the assayed catalytic kinome with Kd<3 μM26, and (iii) that cell viability IC50’s are often orders of magnitude above primary target Kd’s27,28, support this thought.
Combination therapy is a logical and clinically-proven path forward. For example, in other cancer types, one can target two separate driver/resistance pathways29–33, or even the same pathway (e.g. Raf and MEK)34–36, an irrational genomic-based pharmacological approach. There are now at least 28 FDA-approved small molecule kinase inhibitors37 hitting varied on and off-targets. Rather than developing new kinase inhibitors, we aim to rationalize combination therapy with current drugs and maintain acceptable safety as toxicity can be severe for many kinase inhibitors38,39. There are nearly 400 two-way and over 3,000 three-way combinations, not even considering doses, standard-of-care drugs, and the temporal order of drug administration. Obtaining clinical evidence to support such decision making is scientifically challenging, and may require new cross-company collaboration incentives.
The multiple basic and clinical observations lead us to hypothesize that a kinase network, rather than a single kinase of interest, will be a better therapeutic target in GBM, as well as other cancer types. Determining which inhibitors to administer to patients, in which combinations, and at what concentrations via traditional experimental means is time- and materials-prohibitive due to the hundreds of 2-way combinations that would need to be tested for currently available drugs. The current lack of success with single drug therapies, the unpredictable promiscuity of many kinase inhibitors, and the vast number of combinatorial experiments needed to find promising GBM treatments support the need for simulation approaches that can more rapidly account for all known drug-binding affinities to determine potential downstream effects of inhibitor administration.
In this work, we present a simulation-based approach towards choosing drug combinations in GBM. Namely, we integrate transcriptomic data from TCGA GBM patients with a mechanistic model of pan-cancer driver pathways we recently constructed40. This model predicts how multiple microenviromental signals such as growth factors influence stochastic proliferation and death of individual cells, in the presence of a variety of kinase inhibitor drugs. The combination of these two creates a “virtual patient” which can be used to simulate responses to multiple drugs. We illustrate this approach using three FDA-approved kinase inhibitors with evidence for blood-brain-barrier penetration41.
Results
Mechanistic Systems Pharmacology Model of Pan-Cancer Driver Pathways
We have recently developed a mechanistic model of pan-cancer driver pathways that predicts how varied doses of microenvironmental signals influence the dynamics of stochastic proliferation and death in single cells40. The model represents signaling through multiple receptor tyrosine kinases, proliferation and growth pathways (RAS/RAF/ERK; PI-3K/AKT/mTOR), the cell cycle, DNA damage and apoptosis (Fig. 1). There are a total of 141 genes. Expression comprises epigenetics, transcription and translation for each gene, and captures stochastic gene expression that is important for heterogeneous responses to chemotherapy42,43. Some genes are functionally redundant, so the 141 mRNAs are summed during translation to create 102 “protein conglomerates” that represent functionally unique but genetically redundant proteins (e.g. ERK1 and ERK2 summed to ERK). It is important to note that genetic redundancy is with respect to modeled function, and we do not imply these genes are completely functionally redundant in all contexts44,45. The model is composed of 1197 total species (genes, mRNAs, lipids, proteins, and post-translationally modified proteins/protein complexes). Besides stochastic gene expression, the model is a system of compartmental ordinary differential equations (ODEs).
Figure 1. Model Overview.

RTK. proliferation and growth, cell cycle, apoptosis, DNA damage, and gene expression submodels, with genes, compartments and connections indicated.
The mechanism of action of multiple targeted and non-targeted anti-cancer drugs are represented in this model. This gives a direct interface to modeling drug action that allows for systems pharmacology applications to cancer precision medicine. This includes modeling the promiscuity of kinase inhibitors that are thought to be important for both efficacy and toxicity but are as yet very difficult to rationalize26. It is in this sense that such mechanistic descriptions have been labeled as enhanced pharmacodynamics (ePD) models. Such ePD models are of interest to improve our ability to predict patient-specific responses to complex drug combinations and regimens, particularly for diseases such as cancer with multivariate and idiosyncratic etiology46–49. Conveniently, most pharmacokinetic (PK) models are also based on ODEs, so coupling ePD models such as the one used here to existing or new PK models is straightforward. This allows not only in silico prioritization of drug choices, but also optimization of quantitative properties such as dosing and regimen timing that are of utmost importance in pharmacology but are difficult to inform via genetic methods. In this work, we focus on short-term single constant doses and three targeted therapies with promiscuity across multiple modeled kinases, but extensions to these directions are a logical next step that is within close reach (as we have done before50).
While models such as these are often seen as moving in a positive direction for personalized cancer therapy, we must emphasize that such methods are still in very early stages. Much additional work is required to improve the fidelity and predictive capacity of the models across biological contexts and cell types, and even within a single cell type. This includes not only refinement of the already large scope of the current model, but also extension to other biologically important mechanisms and pathways (e.g. metabolism, hypoxia, immune function and heterotypic interactions), and quantification of how uncertainty in both model parameters and structure propagates into uncertainty in model predictions for precision medicine.
Initializing a Virtual Cohort
The model described above was developed in a non-transformed epithelial cell line context, MCF10A. It was trained upon expression data obtained from a serum- and growth factor-starved state, and from a multitude of perturbation response data including biochemical and phenotypic measurements following various doses and combination of growth factors and drugs. Our initialization procedure takes the simulated cell from this starting state to one that best represents an individual patient’s tumor cell behavior, given the available data (Fig. 2). We perform these simulations on a deterministic “average” cell, and introduce stochastic gene expression at a later stage.
Figure 2. Major Steps of the Patient Initialization Procedure.

The details of these steps are described in Methods and in Results. Briefly, the goal here is to take a simulated cell that is non-transformed and in a cell culture environment one step at a time towards a patient’s tumor cell in the tumor environment. This requires a careful and step-wise implementation of superimposing patient data onto the canonical biochemical model of pan-cancer driver pathways. Starting from the non-transformed and serum-starved state allows one to make reasoned expectations about simulated cell behavior along the steps of the initialization procedure. It also allows us to exclude patients for non-sensical or irreconcilable simulation behavior. Because in this study we only investigate kinase inhibitors in cell contexts driven by copy number alteration or loss, the DNA damage and mutation aspects of the initialization are not expanded upon yet. This procedure is applied to a deterministic “average” cell.
The first step is defining the absolute expression levels of mRNA and protein from the transcriptomic data from a patient. To do this, we first convert mRNA-seq data in units of FPKM into molecules/cell40,51. Although there are some transcripts that may have bias from such linearity, in general it has been reported FPKM metrics show a large linear range of detection52. Ideally, unique molecular identifier-based quantification would be used53,54. We define expected protein levels from these mRNA levels given protein-to-mRNA ratios calculated from our MCF10A datasets40. It is often assumed that protein does not correlate well with mRNA, but that is on a genome wide basis. It has been now shown that for a particular gene, the ratio of protein to mRNA is typically constant, although this ratio can vary widely across the genome due primarily to translational regulation51,55,56. Protein abundance in cell lines and tissues is now thought to be largely predictable using gene-specific ratios of protein-to-mRNA levels in lieu of direct protein level measurements51. The ratio of protein-to-mRNA levels for a particular gene but across tissue types is largely conserved, and while transcriptomic data may allow for accurate estimation of unknown protein abundances, technical variation within data may limit accurate predictability56. However, though most genes have this predictability property, not all genes will; there may be important notable exceptions which have yet to be fully elucidated57. Thus, although our modeling approach necessarily predicts protein levels from mRNA levels using gene-specific ratios, a largely reasonable approximation, future work must investigate the degree to which this is suitable for particular genes.
Once expected protein levels are determined, we set these initial conditions in the model and let species equilibrate in a serum-starved context by simulating the model for a long time scale (~1000 hours). Because signaling processes alter protein complexes, post-translational modifications, and protein stability, the initial protein levels do not match the expected protein levels. To reconcile these differences, we adjust the effective translation rate constants and repeat the process until agreement is achieved (Fig. 3A). This step is routinely carried out after most steps in the initialization.
Figure 3. Example Model Behavior During Patient Initialization.

Results are shown from a particular patient (#9) as they progress along the proposed initialization procedure. A. Concordance of protein levels from adjusting translation after model equilibration. B–C. Behavior of the cell cycle (Cyclin A) below (B) and above (C) the critical basal cyclin D synthesis rate. D–E. Behavior of apoptosis as indicated by cleaved PARP (cPARP-red)) vs. PARP (blue), below (D) and above (E) the critical basal Caspase 8 cleavage rate. When cPARP and PARP time courses cross this defines an apoptotic event. F–G. Incorporating ligands expressed based on patient mRNA-seq data increases downstream signaling, as pictured by ligand concentrations (F) and activated AKT (G).
Once the model reflects the patient expression context, there are several empirical parameters we vary to ensure the cell cycle and apoptosis phenotypes are responsive in an appropriate way. We approximate the level of basal cyclin D synthesis that brings the simulated cell to the brink of cycling (Fig. 3B–C), and likewise the basal caspase 8 cleavage rate that brings the cell to the brink of death (Fig. 3D–E). The biological rationale for this is that the average cell at this point has not incorporated any activating mutations, nor are there any growth factors in the microenvironment, so they should on average be primed to enter the cell cycle or to undergo apoptosis with the appropriate stimuli. This assumption is very hard to substantiate with experiments, but is nevertheless necessary to ensure a responsive phenotypic simulation, the results of which without this assumption are largely non-sensical. We noticed that in some patients, apoptosis can proceed gradually as opposed to in a switch-like manner as is commonly seen58; future work will further tune the caspase positive feedback loop responsible for such behavior to ensure qualitative agreement.
The next step is to move the simulated cell from a serum-starved cell culture situation into a tumor microenvironment. First, we set the cellular and extracellular volumes equal, as opposed to a very large extracellular volume as in cell culture. Next, we use the mRNA-seq data to inform the levels of genetically encoded growth factors. There are little data available to constrain the conversion of transcript levels to extracellular growth factor concentrations. Therefore we must invoke biologically reasonable assumptions. We assume that the transcript levels for a particular ligand, when averaged across the entire virtual patient population, gives an extracellular growth factor concentration equal to the affinity for its cognate receptor (or in the case of multi-targeted ligands, to its highest affinity receptor). Then, a patient with less than average ligand transcript levels would have less ligand concentration than said affinity, and vice versa. This allows the range of ligand expression levels across patients to proportionally affect the downstream signaling in a biologically consistent manner. This does not yet take into account mechanisms such as sequestration mediating by extracellular growth factor binding proteins, but nevertheless is a key step towards capturing growth factor microenvironment. After this step, the simulated cell is now being stimulated with a variety of microenvironment signals, which turns on signaling pathways (Fig. 3F–G).
With signaling pathways turned on, we now revisit the same cell cycle and apoptosis tuning steps to find the critical cyclin D synthesis and basal caspase 8 cleavage rates that bring the simulated cell to the brink of the cell fates. When stochastic simulations are enacted subsequently, this will lead to a fraction of the population cycling and dying, as is ubiquitously seen in tumors.
We applied this procedure to initialize 14 patients with varied transcriptomic aberrations that are likely driver alterations (Table 1). Certainly, point and other mutations are widely important in GBM, but as an illustration of the approach here we focus on transcriptomic tailoring, limiting ourselves to this virtual patient subset that are likely to not be driven by such mutations, but rather the transcriptomic alterations that we are taking into account. We note here that many copy number and other alterations are integrated at the level of the transcriptome. Lastly, we include a step to initialize the levels of basal DNA damage and its repair by enzyme levels given by the mRNA-seq data. However, because in this study we are only considering kinase inhibitors and not DNA damaging agents, it is not immediately relevant.
Table 1. General class of transcriptomic alteration in each patient.
Data across 165 TCGA patients were analyzed for variation across the transcriptome for pathway components, and those showing greater than 1.5 fold standard deviations over the mean were indicative of pathway deregulation from an expression level (mainly related to copy number alteration). We specifically limited ourselves to such patients to avoid those having clear point mutation drivers which are not considered in this work.
| EGFR/ERBB | RAS/NF1 | PTEN/PI3K | PDGFR/PDGF | EIF4E/RSK | |
|---|---|---|---|---|---|
| 1 | |||||
| 2 | |||||
| 3 | |||||
| 4 | |||||
| 5 | |||||
| 6 | |||||
| 7 | |||||
| 8 | |||||
| 9 | |||||
| 10 | |||||
| 11 | |||||
| 12 | |||||
| 13 | |||||
| 14 |
Creating an Intratumorally Heterogeneous Population of Cells for Each Virtual Patient
The initialization procedure above creates a model variant for every considered patient mRNA-seq data set. The model at this point corresponds to an average tumor cell. To model intratumoral heterogeneity arising from stochastic gene expression, we now create 100 separate simulations starting from this initial average cell (Fig. 3). Each simulation proceeds along a different stochastic gene expression trajectory for 72 hours, resulting in 100 simulated tumor cells for each patient, now randomized by stochastic gene expression. This results in heterogeneous cell cycle entry and in some cases apoptosis (top panels in Fig. 4, although this particular virtual patient showed no cell death events), as well as a spectrum of signaling activities (e.g. ERK and AKT activation—bottom panels in Fig. 4). The net result of this step is an emulation of intratumoral heterogeneity within a particular genetic subclone. If assumptions and/or data are available to suggest the presence of multiple subclones within a tumor, then one could simply create a different heterogeneous population for every subclone.
Figure 4. Example Model Behavior During Generation of the Heterogeneous Tumor Cell Population.
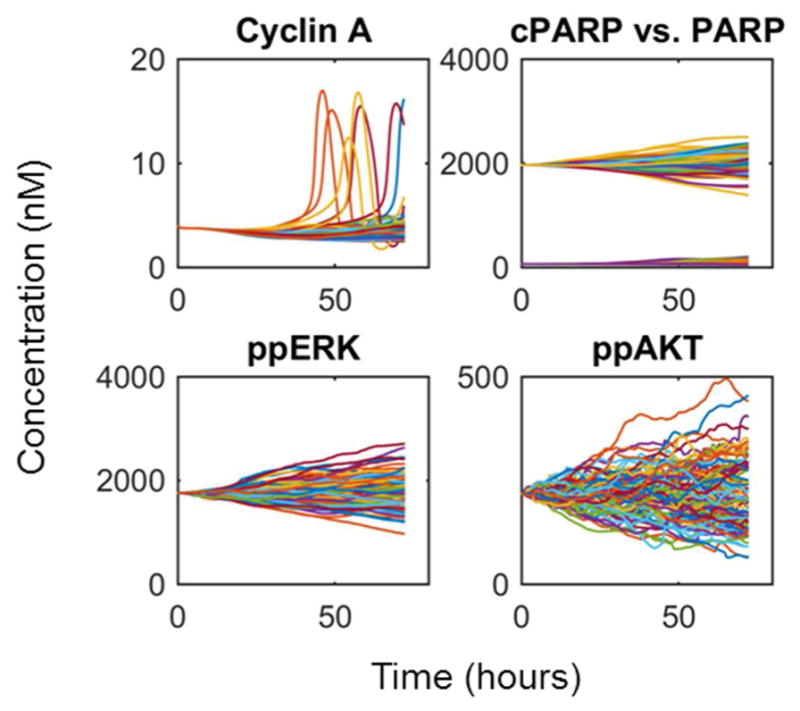
Typical results are shown from a particular patient (#8) for the cell cycle (Cyclin A), apoptosis (cPARP) and two widely important signaling biomarkers (ppERK and ppAkt). Each colored line corresponds to a different stochastic cell simulation (total of 100 per patient). Stochastic gene expression causes the average cell at time point zero to diverge over the 3-day simulation, creating 100 heterogeneous tumor cells ready for virtual drug combination screening.
Modeling Promiscuous Action of Blood-Brain-Barrier Penetrant Kinase Inhibitors
A recent review highlights targeted kinase inhibitors that have evidence for blood-brain-barrier (BBB) penetration41. While such BBB penetration is often considered a liability for chemotherapeutics due to the sensitivity of neurons, the lack of BBB penetration is thought to be a major limiting factor for effective treatment of GBM (and brain metastases)25. We therefore identified three FDA-approved anti-cancer kinase inhibitors to focus on in this study: bosutinib (BOS), ibrutinib (IBR) and cabozantinib (CAB) (Table 2). The primary targets of these compounds are BCR-ABL (BOS), BTK (IBR), MET/VEGFR (CAB), which show little to no overlap with known genetic driver events in GBM, so using them for GBM is nonsense from a genetic standpoint. However, literature data for the promiscuity of these inhibitors59,60 elucidates why they may have efficacy in GBM, as they bind to and inhibit a variety of kinases within modeled pan-cancer pathways (Table 2). Furthermore, most of these kinases are connected to each other through the pan-cancer network architecture, so it is feasible that varied inhibition of multiple kinases may propagate through the network to achieve desired pharmacodynamic effects, even if direct genetic/primary or off-targets of drugs are not of interest. We modified the original model such that these three drugs, when present, bind to and sequester/inhibit the relevant kinase targets with the experimentally reported affinities (as shown in Table 2).
Table 2. Promiscuity of the Three Considered Kinase Inhibitors.
We assumed rapid binding because these are small molecules, and calculated off rate constants based on published affinity data. We implemented a 10 uM Kd threshold.
| Drug | Gene Targets | Model Targets | k_on (1/s/nM) | k_off (1/s) |
|---|---|---|---|---|
| Bosutinib | MAP2K1/MAP2K2 | MEK | 1 | 288 |
| RPS6KA1/RPS6KA3 | RSK | 1 | 1115 | |
| PRKCA/PRKCG | PKC | 1 | 1567 | |
| CHEK1 | Chk1 | 1 | 1168 | |
| FGFR1 | Fr | 1 | 2206 | |
| IGF1R | Ir | 1 | 2285 | |
| INSR | Isr | 1 | 669 | |
| PDGFRA | Pr | 1 | 3081 | |
|
| ||||
| Ibrutinib | BRAF | Braf | 1 | 1128 |
| EGFR | E1 | 1 | 18 | |
| ERBB3 | E3 | 1 | 1 | |
| FGFR1/FGFR2 | Fr | 1 | 707 | |
| GSK3B | GSK3b | 1 | 2571 | |
| IGF1R | Ir | 1 | 4882 | |
| INSR | Isr | 1 | 1326 | |
| MTOR | mTOR | 1 | 8091 | |
| PDPK1 | PDK1 | 1 | 2448 | |
| PIK3CA/PIK3CB/PIK3CD/PIK3CG | PI3KC1 | 1 | 2039 | |
| RAF1 | Craf | 1 | 2333 | |
| RPS6KA1/RPS6KA3/RPS6KA2 | RSK | 1 | 6447 | |
|
| ||||
| Cabozantinib | BRAF | Braf | 1 | 2961 |
| EGFR | E1 | 1 | 864 | |
| FGFR1/FGFR2 | Fr | 1 | 2153 | |
| IGF1R | Ir | 1 | 8236 | |
| INSR | Isr | 1 | 1880 | |
| MAP2K1 | MEK | 1 | 214 | |
| MET | MET | 1 | 1 | |
| PDGFRA | Pr | 1 | 1 | |
| PIK3CA | PI3KC1 | 1 | 1084 | |
| PIK3R1 | PI3KR1 | 1 | 1084 | |
| RAF1 | Craf | 1 | 1078 | |
Using the Virtual Cohort to Screen Drug Combinations in silico
With the virtual GBM patient cohort, 100 heterogeneous cells for each patient, and an extended model for the promiscuous pharmacodynamics of BOS, IBR and CAB in hand, we now performed simulations to evaluate the potential efficacy of single drugs or two-way drug combinations on the virtual cohort. While as mentioned above, the dynamic and differential equation nature of our model allows interfacing with pharmacokinetics models, we here only investigate responses to an initial long-lasting high dose (10 uM—on the high end of a typical range for kinase inhibitor plasma concentration61) of the drugs or drug combinations, leaving this more complex dynamic optimization of dosing and regimens for future studies.
For every patient and drug, the entire range of biochemistry represented in the model is accessible; we focus on cell division (Cyclin A), apoptosis (cleaved PARP/PARP), and two widely relevant biomarkers for controlling cell division and death, doubly phosphorylated ERK (ppERK) and AKT (ppAKT) (Fig. 5A). From each of these simulated biochemical profiles across the heterogeneous cell population, we count the number of cell divisions (Cyclin A peaks—see Methods) and cell death events (cPARP > PARP) (Fig. 5B). For this particular example of a single virtual patient with PTEN loss, BOS and CAB monotherapy similarly inhibit ppERK, while IBR has little effect on either the ppERK or ppAKT biomarkers. Phenotypically, however, BOS accelerates the cell cycle, while CAB inhibits it. IBR, despite its inability to significantly inhibit either biomarker, does impact the cell cycle, but not cell death. These single agents are largely inferior to the two-way combinations, of which BOS+CAB achieves the highest combined effects on cell cycle and cell death. This effect correlates with the largest ppERK downregulation.
Figure 5. Example Drug Treatment.

A. Results from a particular patient (#12) given a single constant dose (10 uM) for each drug or combination. Results for the cell cycle (Cyclin A), apoptosis (cPARP) and two widely important signaling biomarkers (ppERK and ppAkt) are shown here. Each colored line corresponds to a different stochastic cell simulation (total of 100 per patient). BOS: bosutinib; IBR: ibrutinib; CAB: cabozantinib. B. Results summarized by the number of cell cycles and cell deaths, stratified by drug treatment (different colors).
Across all 14 virtual patients, the effect of these three drugs or drug combinations is typically limited to either cytostatic or cytotoxic, but seldom both (Fig. 6A), as opposed to the highlighted virtual patient above that showed effects on the cell cycle and cell death. There is not a discernable pattern with respect to the type of transcriptomic aberrations (Fig. 6B). While this virtual cohort has a limited number of patients and we have not yet comprehensively addressed mutations, this result is consistent with the generally idiosyncratic response of GBM subtypes to a range of therapies25. Single drugs are typically ineffective both for reducing cycling and inducing death, and can even exacerbate the situation by accelerating the cell cycle or inhibiting death (Fig. 6B–C). However, if single drugs are ineffective, then the combinations also tend to be ineffective for a particular phenotypic modality (proliferation or death). When single drugs show some efficacy, combinations show increased efficacy. IBR+CAB is the predicted combination of choice across most patients that either maximizes cell death or minimizes cell proliferation. However, since none of these kinase inhibitor treatments can strongly induce both cytotoxic and cytostatic effects, additional combinations with more drugs are needed. Moreover, exploration of longer time scales over more complex drug regimen properties considering dose and dynamics, along with pharmacokinetic profiles, will be informative to understand the putative GBM virtual cohort responses to these drugs and their combinations more fully.
Figure 6. Simulated Drug and Drug Combination Responses Across Patients.

For every initialized patient’s heterogeneous cell population, all three drugs alone or in combination were given at a constant concentration of 10 uM for 72 hours. The number of cell division events (Cyclin A peaks) or apoptosis events (cleaved PARP crossing) were counted, and then analyzed relative to a no drug control. A–B. Simulation results across all patients (different colors), for all drug treatment conditions. In (B), different marker types correspond to different transcriptomic aberrations, whereas color still is matched for patients as in (A). C–D. Heatmap representations of the number of apoptosis events (B) or cell cycle events (C) across patients and drugs.
Discussion
Aberrant tyrosine kinase signaling in GBM has proven difficult to successfully target in clinical trials with single kinase inhibitors, so an efficient method for determining efficacious, and perhaps even optimal combinations of brain-penetrant inhibitors is needed on a patient-specific basis. We represented the mechanism of action of three FDA approved brain-penetrant anti-cancer kinase inhibitor drugs (Bosutinib, Ibrutinib, and Cabozantinib), accounting for their promiscuity, in a mechanistic computational model of multiple glioma driver pathways. Using this same model then tailored to transcriptomic data from TCGA glioma patients, we simulated patient-specific responses to single doses or combinations of drugs. Our preliminary virtual cohort simulations showed observable differences across patients, indicating that expanding upon these methods and incorporating more drugs could be a viable method for predicting efficacious drug(s) for a specific patient based on the transcriptomic makeup of their tumor.
Combining the details of mechanistic modeling with genomics, and drug pharmacokinetics and pharmacodynamics, we developed an enhanced pharmacodynamic (ePD) model46, a modeling approach that has successfully described antibody-ligand interactions62 and the VEGFR pathway in cancer50,63. ePD models consider drug effects not from the typical empirical Emax sigmoid model, but rather from a first-principles and mechanistic perspective where possible47. Thus, ePD models leverage prior knowledge of basic biological mechanisms of the target cells or systems of interest to make predictions of drug effect. Such a model simulates how all considered species will respond to drug treatments, so while key outputs can be analyzed (such as proliferation or apoptosis), drug effects on every modeled protein can be monitored to explore hypotheses for other downstream or off-target effects. Fittingly, such modeling typically also gives insight into these mechanisms when compared to new data, such as those encountered during pharmacological studies. This comparison highlights when current mechanistic understanding needs to be refined to account for new data.
Physiologically-based pharmacokinetic (PBPK) modeling is another common mechanistic approach to predict drug behavior in humans, but from the drug concentration dynamics standpoint. They incorporate physiologic parameters in the form of different compartments corresponding to different organs in the body connected by blood circulation to guide drug selection and dosing64,65. PBPK models have been utilized in drug discovery for candidate selection, risk simulation, dosing frequency and determining which organ systems will be affected by the drugs.64,65 They are in principle straightforward to couple with ePD models since they are all primarily based on differential equation formalism, although this is beyond the scope of the current work. We envision such coupling to eventually be a powerful predictive tool in both drug development and precision medicine.
We defined 14 virtual patients from TCGA transcriptomic profiles to screen for drug combinations that maximize apoptosis and/or minimize cell proliferation. None of the combinations were antagonistic with respect to apoptosis and proliferation. Efficacious single drugs, when combined, induced equal or greater cell death and equal or lower proliferation. Such efficacy interactions were not quite additive nor synergistic. Within simulation results, we noted that typically if one cell of a heterogenous simulated tumor were sensitive to the first drug, it is likely sensitive to the second. Thus, addition of more drugs seems to increase the likelihood of a new cell becoming sensitive to therapy. With the incorporation of more drugs in future simulations, perhaps greater additive or synergistic effects will become apparent as different subclones within the tumor are targeted. Targeting such subclonal heterogeneity is thought to be a major key to treating not only gliomas, but many cancer types66,67,7.
Two of the virtual patients’ cells (Patients 1 and 2) never underwent apoptosis in the presence or absence of drugs, but also maintained low levels of cell division, even decreasing in the presence of drugs, particularly the combinations. Patients 1 and 2 exhibited PDGFR/PDGF and EIF4E/RSK transcriptomic alterations, respectively. Cabozantinib, with a very high binding affinity for PDGFRA, has the greatest impact on decreasing proliferation in Patient 1 who is characterized by highly elevated PDGFRA levels. Patient 2, exhibiting higher levels of RSK/EIF4E, responded best to Ibrutinib, which had a low binding affinity for RSK, and high affinity for EGFR, which when inhibited has been shown to decrease EIF4E levels, thus resulting in decreased apoptotic resistance68. This is perhaps why simulations indicate increased apoptosis in Patient 2 following greater EGFR inhibition. Five of the virtual patients (3, 6, 8, 9, and 14) showed no changes in apoptosis following drug treatment, but did show differences in cell division events. The two patients from this subset with EGFR amplifications (9, 14) both exhibited the weakest response to the Bosutinib/Ibrutinib combination, and responded best when Cabozantinib was present. The other two EGFR amplification patients (4,11) showed greatest cell death in the presence of Cabozantinib. Cabozantinib and Ibrutinib both bind EGFR, which could explain their success in treating patients with EGFR amplifications. Ibrutinib is not as successful as Cabozantinib in these patients, perhaps because Cabozantinib also strongly binds MET and MAP2K1 (MEK1), the latter of which is downstream of both EGFR and MET, thus shutting down EGFR signaling more completely than Ibrutinib. The three patients with NF1 loss (5, 6, and 13) all responded best to the combination of Ibrutinib and Cabozantinib, with either increased apoptosis (5, 13) or decreased proliferation (6). Both Ibrutinib and Cabozantinib bind to BRAF and CRAF, and Cabozantinib binds to MEK1 (as mentioned above). These are all part of the RAS pathway which is upregulated as a result of NF1 loss, leading to increased cell survival and proliferation69, likely explaining the success of this inhibitor combination in combatting the upregulation of the RAS pathway. In the four patients with decreased PTEN levels (3, 7, 8, and 12), Bosutinib treatment resulted in the worst outcome as measured by both cell proliferation and apoptosis. Bosutinib has the greatest affinity for MEK, but the anti-tumor properties of MEK inhibition are impaired in patients with PTEN loss70, which may explain these simulation results. The mechanistic nature of our modeling approach allows reasoning about such phenomena on a patient-specific basis which is difficult to envision a priori.
VEGF is an important mediator of angiogenesis in GBM71, a highly vascularized tumor72. VEGF binding to VEGFR transduces signals to the PI3K/Akt pathway73, an important cancer-promoting pathway controlling aspects of cell survival, cycling, and growth that is frequently altered in tumors. Our pan-cancer model in its current state does not include VEGFR because MCF10A cells, the non-tumorigenic breast epithelial cells the model was originally built upon, do not express detectable levels of VEGFR74. VEGFR is predominantly expressed by vascular endothelial cells, but has also been seen in monocytes, macrophages, smooth muscle cells, trophoblasts, osteoblasts, and microglia75. It is the VEGFR expressing macrophages that have been shown to increase glioma angiogenesis, while the glioma tumor cells typically overexpress VEGF75. One of Cabozantinib’s primary targets is VEGFR, and while we model eleven additional high affinity targets, including MET and PDGFR (its two other primary targets), the model in its current state does not yet simulate Cabozantinib’s anti-tumor effects through angiogenesis. This is something which may require more complex models involving multiple cell types within the tumor in cases where VEGFR expression is not driving tumorigenesis from within tumor cells themselves.
The presented model is large and complex, but is certainly not complete, considering only pathways within the pan-cancer scope, and even then, it does not capture all known important aspects of cancer cell biology, some of which related to VEGFR is discussed above. As we expand upon the model and tailor it to cancer-specific cell lines, there are more molecular species it will need to include, but for the purposes of this preliminary study we aim to highlight the feasibility of our method, in part shown by the differences of relevant biological outputs between virtual patients and drug treatments, and general lack of efficacy of single kinase inhibitors. GBM-relevant kinases, as defined by targets of the brain-penetrant kinase inhibitors41, will be added to the model to allow for more accurate predictions of drug effects. While additional cellular pathways, such as those involved in development or immunology/inflammation could add breadth to the model, for the purposes of cancer-specific kinase inhibition, the currently included pathways cover much of the genetically-determined important pathways. The model as yet does not account for mutations, as non-transformed and genetically-stable MCF10A cells have few significant mutations. Modeling effects of some well-studied mutations will be straightforward (e.g. K-Ras G12V), but most mutations are not yet functionally well-understood. Accounting for effects of such ill-understood mutations is a major problem for the entire cancer field that will require collaboration between multiple types of experimentalists and modelers, such as those focused on protein structure and evolutionary biology76,77. Perhaps most importantly, future work will integrate experimental models where both transcriptomic (and other) input data can be acquired in conjunction with follow up drug treatment studies to evaluate the fidelity of simulations.
Future simulations will include more brain-penetrant kinase inhibitors, and at varying doses, to examine efficacy as a function of dosing regimen. A critical piece of this will be using ePD models with with PK and PBPK models. This also allows simulating effects of drug timing and/or sequence for each patient. This concept can also be applied to different types of drugs beyond kinase inhibitors such as traditional chemotherapeutics and DNA damaging agents; in fact, etoposide and temozolomide are currently represented in the presented model, and novel BCL-2 inhibitors can also be modeled. Thus, we have only scratched the surface of what such mechanistic, ePD modeling approaches can begin to address in not only glioma but general precision medicine approaches to cancer. Much work is needed to bring such modeling approaches towards the confidence needed for clinical relevance, but nevertheless, there is significant potential for dealing with these complex issues that do not have current solutions.
Methods
Data Acquisition from TCGA
We downloaded 165 mRNA-seq profiles from TCGA (Supplementary Table 2). For each patient, we converted to molecules per cell as described previously40, which uses a proportionality of a particular gene product from all gene products, and an estimate of total transcript count as 400,000 per cell. The 14 patients used in the simulation studies here are indicated. The genes taken from this larger dataset for tailoring to the model are given in Supplementary Table 3.
Model Simulation
The pan-cancer model was obtained from a biorxiv pre-print40 and simulated using MATLAB and the sundials suite of solvers as described within that publication. We extended this model to include drug binding to kinase targets listed using mass action kinetics and parameters in Table 2.
Computational Specifications
The computational machine used was an MSI GE62 Apache Pro Laptop with 16GB DDR4 RAM and an Intel Core i7 6700HQ 2.6 GHz Processor. Simulations were run on Matlab version 2014b running the signal processing toolbox for the function findpeaks.
Counting Cell Cycle and Apoptosis Events
Simulated Cyclin A dynamic profiles were used to count cell cycle events using the MATLAB function findpeaks with MinPeakProminence of four. Apoptosis events were defined by if cleaved PARP and PARP levels crossed.
Supplementary Material
Model Code
Model Tutorial. A guide that helps users navigate the provided model code for applying the proposed methods and procedures to their own modeling tasks
Supplementary Table 1. List of Abbreviations used in this manuscript
Supplementary Table 2. mRNA-seq data from TCGA in FPKM.
Supplementary Table 3. mRNA-seq data from TCGA for the selected virtual patients and model genes in molecules/cell.
Acknowledgments
MRB acknowledges funding from Mount Sinai, the NIH Grants P50GM071558 (Systems Biology Center New York), R01GM104184 and U54HG008098 (LINCS Center), and an IBM faculty award. MB was supported by a NIGMS-funded Integrated Pharmacological Sciences Training Program grant (T32GM062754). KINOMEscan data were made publicly available by the Harvard Medical School LINCS Center, which is funded by NIH grants U54 HG006097 and U54 HL127365.
Footnotes
Author Contributions
MRB conceived of and supervised the work. MB created and helped alter the model. AMB gathered TCGA patient data, altered the model to include drug interactions, tailored the model to patient-specific contexts, and ran all simulations. AMB and MRB wrote the paper.
References
- 1.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-sloan JS. CBTRUS Statistical ReportR: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2006–2010. Neuro Oncol. 2013;12:28–36. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tuncbag N, Milani P, Pokorny JL, Johnson H, Sio TT, Dalin S, Iyekegbe DO, White FM, Sarkaria JN, Fraenkel E. Network Modeling Identifies Patient-specific Pathways in Glioblastoma. Sci Rep. 2016;6:28668. doi: 10.1038/srep28668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bleeker FE, Lamba S, Zanon C, Molenaar RJ, Hulsebos TJM, Troost D, van Tilborg AA, Vandertop WP, Leenstra S, van Noorden CJF, Bardelli A. Mutational profiling of kinases in glioblastoma. BMC Cancer. 2014;14:718. doi: 10.1186/1471-2407-14-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varghese RT, Liang Y, Guan T, Franck CT, Kelly DF, Sheng Z. Survival kinase genes present prognostic significance in glioblastoma. Oncotarget. 2016:7. doi: 10.18632/oncotarget.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olar A, Aldape KD. Using the Molecular Classification of Glioblastoma to Inform Personalized Treatment. 2015;232:165–177. doi: 10.1002/path.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchdunger E, Zimmermann J, Mett H, Meyer T, Müller M, Druker BJ, Lydon NB. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–4. [PubMed] [Google Scholar]
- 9.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 10.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 11.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 12.le Coutre P, Mologni L, Cleris L, Marchesi E, Buchdunger E, Giardini R, Formelli F, Gambacorti-Passerini C. In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. J Natl Cancer Inst. 1999;91:163–8. doi: 10.1093/jnci/91.2.163. [DOI] [PubMed] [Google Scholar]
- 13.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 14.Talpaz M, Silver RT, Druker BJ, Goldman JM, Gambacorti-Passerini C, Guilhot F, Schiffer CA, Fischer T, Deininger MWN, Lennard AL, Hochhaus A, Ottmann OG, Gratwohl A, Baccarani M, Stone R, Tura S, Mahon FX, Fernandes-Reese S, Gathmann I, Capdeville R, Kantarjian HM, Sawyers CL. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. 2002;99:1928–37. doi: 10.1182/blood.v99.6.1928. [DOI] [PubMed] [Google Scholar]
- 15.O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T, Lechner K, Nielsen JL, Rousselot P, Reiffers J, Saglio G, Shepherd J, Simonsson B, Gratwohl A, Goldman JM, Kantarjian H, Taylor K, Verhoef G, Bolton AE, Capdeville R, Druker BJ IRIS Investigators. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 16.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2 -Overexpressing Metastatic Breast Cancer. J Clin Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 17.Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–48. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 18.Druker BJ. Circumventing Resistance to Kinase-Inhibitor Therapy. 2006:2594–2596. doi: 10.1056/NEJMe068073. [DOI] [PubMed] [Google Scholar]
- 19.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 23.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: A systematic review of clinical studies. Neuro Oncol. 2010;12:304–316. doi: 10.1093/neuonc/nop068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levin VA, Tonge PJ, Gallo JM, Birtwistle MR, Dar AC, Iavarone A, Paddison PJ, Heffron TP, Elmquist WF, Lachowicz JE, Johnson TW, White FM, Sul J, Smith QR, Shen W, Sarkaria JN, Samala R, Wen PY, Berry DA, Petter RC. CNS Anticancer Drug Discovery and Development Conference White Paper. Neuro Oncol. 2015:17. doi: 10.1093/neuonc/nov169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 27.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vora SR, Juric D, Kim N, Mino-kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, Lehar J, Wiesmann M, Wartmann M, Chen Y, Cao ZA, Pinzon-ortiz M, Kim S, Schlegel R, Huang A, Jeffrey A. CDK 4/6 inhibitors sensitize PIK3CA Mutant Breast Cancer to PI3K inhibitors. 2015;26:136–149. doi: 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, Will M, Yellen P, de Stanchina E, Baselga J, Scher HI, Barry ST, Sawyers CL, Chandarlapaty S, Rosen N. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell. 2015;27:109–22. doi: 10.1016/j.ccell.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, Fowst C, Huang X, Kim ST, Randolph S, Slamon DJ. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 33.Ni J, Ramkissoon SH, Xie S, Goel S, Stover DG, Guo H, Luu V, Marco E, Ramkissoon LA, Kang YJ, Hayashi M, Nguyen QD, Ligon AH, Du R, Claus EB, Alexander BM, Yuan GC, Wang ZC, Iglehart JD, Krop IE, Roberts TM, Winer EP, Lin NU, Ligon KL, Zhao JJ. Combination inhibition of PI3K and mTORC1 yields durable remissions in mice bearing orthotopic patient-derived xenografts of HER2-positive breast cancer brain metastases. Nat Med. 2016;22:723–726. doi: 10.1038/nm.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chapman PB, Solit DB, Rosen N. Combination of RAF and MEK Inhibition for the Treatment of BRAF-Mutated Melanoma: Feedback Is Not Encouraged. Cancer Cell. 2014;26:603–604. doi: 10.1016/j.ccell.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 35.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM, Schadendorf D. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N Engl J Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 36.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JMG, Utikal J, Dreno B, Nyakas M, Middleton MR, Becker JC, Casey M, Sherman LJ, Wu FS, Ouellet D, Martin AM, Patel K, Schadendorf D METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 37.Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–39. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 38.Moslehi JJ. Cardiovascular Toxic Effects of Targeted Cancer Therapies. N Engl J Med. 2016;375:1457–1467. doi: 10.1056/NEJMra1100265. [DOI] [PubMed] [Google Scholar]
- 39.Jabbour E, Deininger M, Hochhaus A. Management of adverse events associated with tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia. Leukemia. 2011;25:201–10. doi: 10.1038/leu.2010.215. [DOI] [PubMed] [Google Scholar]
- 40.Bouhaddou M, Barrette AM, Koch RJ, Distefano MS, Riesel EA, Stern AD, Santos LC, Tan A, Mertz A, Birtwistle MR. An Integrated Mechanistic Model of Pan-Cancer Driver Pathways Predicts Stochastic Proliferation and Death 2017 [Google Scholar]
- 41.Heffron TP. Small Molecule Kinase Inhibitors for the Treatment of Brain Cancer. J Med Chem. 2016;59:10030–10066. doi: 10.1021/acs.jmedchem.6b00618. [DOI] [PubMed] [Google Scholar]
- 42.Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, Milo R, Cohen-Saidon C, Liron Y, Kam Z, Cohen L, Danon T, Perzov N, Alon U. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–6. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 43.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Thun A, Birtwistle M, Kalna G, Grindlay J, Strachan D, Kolch W, von Kriegsheim A, Norman JC. ERK2 drives tumour cell migration in threedimensional microenvironments by suppressing expression of Rab17 and liprin-β2. J Cell Sci. 2012:125. doi: 10.1242/jcs.092916. [DOI] [PubMed] [Google Scholar]
- 46.Iyengar R, Zhao S, Chung SW, Mager DE, Gallo JM. Merging Systems Biology with Pharmacodynamics. Sci Transl Med. 2012;4:126ps7–126ps7. doi: 10.1126/scitranslmed.3003563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Birtwistle MR, Mager DE, Gallo JM. Mechanistic vs. Empirical network models of drug action. CPT pharmacometrics Syst Pharmacol. 2013;2:e72. doi: 10.1038/psp.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gallo JM, Birtwistle MR. Network pharmacodynamic models for customized cancer therapy. Wiley Interdiscip Rev Syst Biol Med. 2015:7. doi: 10.1002/wsbm.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klinke DJ, Birtwistle MR. In silico model-based inference: An emerging approach for inverse problems in engineering better medicines. Curr Opin Chem Eng. 2015:10. doi: 10.1016/j.coche.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang XY, Birtwistle MR, Gallo JM. A General Network Pharmacodynamic Model-Based Design Pipeline for Customized Cancer Therapy Applied to the VEGFR Pathway. CPT pharmacometrics Syst Pharmacol. 2014;3:e92. doi: 10.1038/psp.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M, Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 52.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2011;9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 54.Xiong Y, Soumillon M, Wu J, Hansen J, Hu B, van Hasselt JGC, Jayaraman G, Lim R, Bouhaddou M, Ornelas L, Bochicchio J, Lenaeus L, Stocksdale J, Shim J, Gomez E, Sareen D, Svendsen C, Thompson LM, Mahajan M, Iyengar R, Sobie EA, Azeloglu EU, Birtwistle MR. A Comparison of mRNA Sequencing with Random Primed and 3′-Directed Libraries. bioRxiv. 2017 doi: 10.1038/s41598-017-14892-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edfors F, Danielsson F, Hallström BM, Käll L, Lundberg E, Pontén F, Forsström B, Uhlén M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol. 2016:1–10. doi: 10.15252/msb.20167144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, Mathieson T, Lemeer S, Schnatbaum K, Reimer U, Wenschuh H, Mollenhauer M, Slotta-Huspenina J, Boese JH, Bantscheff M, Gerstmair A, Faerber F, Kuster B. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 57.Fortelny N, Overall CM, Pavlidis P, Freue GVC. Can we predict protein from mRNA levels? Nature. 2017;547:E19–E20. doi: 10.1038/nature22293. [DOI] [PubMed] [Google Scholar]
- 58.Modeling a Snap-Action, Variable-Delay Switch Controlling Extrinsic Cell Death.
- 59.Kitagawa D, Yokota K, Gouda M, Narumi Y, Ohmoto H, Nishiwaki E, Akita K, Kirii Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells. 2013;18:110–22. doi: 10.1111/gtc.12022. [DOI] [PubMed] [Google Scholar]
- 60.HMS LINCS.
- 61.Pyrimidines F, Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U, Jaehde U. Clinical Pharmacokinetics of Tyrosine Kinase Inhibitors. Clin Pharmacokinet. 2011;50:551–603. doi: 10.2165/11593320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 62.Davda JP, Hansen RJ. Properties of a general PK/PD model of antibody-ligand interactions for therapeutic antibodies that bind to soluble endogenous targets. MAbs. 2010;2:576–588. doi: 10.4161/mabs.2.5.12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clegg LE, Mac Gabhann F. A computational analysis of in vivo VEGFR activation by multiple co-expressed ligands. PLoS Comput Biol. 2017 doi: 10.1371/journal.pcbi.1005445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gerlowski LE, Jain RK. Physiologically based pharmacokinetic modeling: Principles and applications. J Pharm Sci. 1983;72:1103–1127. doi: 10.1002/jps.2600721003. [DOI] [PubMed] [Google Scholar]
- 65.Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. 2016;6:430–440. doi: 10.1016/j.apsb.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dry JR, Yang M, Saez-Rodriguez J. Looking beyond the cancer cell for effective drug combinations. Genome Med. 2016;8:125. doi: 10.1186/s13073-016-0379-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, Zweibel J, Collins J, Doroshow JH. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov. 2010;9:843–856. doi: 10.1038/nrd3216. [DOI] [PubMed] [Google Scholar]
- 68.Liu T, Yacoub R, Sun S, Graham T, Tighiouart M, Yang L, O’Regan R. eIF4E Expression Predicts Apoptosis in Response to Epidermal Growth Factor Receptor Inhibition and Mammalian Target of Rapamycin Inhibition in Triple Negative Breast Cancers. Cancer Res. 2014;69:5078LP-5078. [Google Scholar]
- 69.Gioeli D. Targeted Therapies: Mechanisms of Resistance. Springer Science & Business Media; 2011. [Google Scholar]
- 70.Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CMA, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: Multiple Functions in Human Malignant Tumors. Front Oncol. 2015;5:24. doi: 10.3389/fonc.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.VEGF Manipulation in Glioblastoma.
- 72.Conroy S, Wagemakers M, Walenkamp AME, Kruyt FAE, den Dunnen WFA. Novel insights into vascularization patterns and angiogenic factors in glioblastoma subclasses. J Neurooncol. 2017;131:11–20. doi: 10.1007/s11060-016-2269-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takahashi M, Matsui A, Inao M, Mochida S, Fujiwara K. ERK/MAPK-dependent PI3K/Akt phosphorylation through VEGFR-1 after VEGF stimulation in activated hepatic stellate cells. Hepatol Res. 2003;26:232–236. doi: 10.1016/s1386-6346(03)00112-8. [DOI] [PubMed] [Google Scholar]
- 74.Kaliberova LN, Kusmartsev Sa, Krendelchtchikova V, Stockard CR, Grizzle WE, Buchsbaum DJ, Kaliberov Sa. Experimental cancer therapy using restoration of NAD+-linked 15-hydroxyprostaglandin dehydrogenase expression. Mol Cancer Ther. 2009;8:3130–3139. doi: 10.1158/1535-7163.MCT-09-0270. [DOI] [PubMed] [Google Scholar]
- 75.Kerber M, Reiss Y, Wickersheim A, Jugold M, Kiessling F, Heil M, Tchaikovski V, Waltenberger J, Shibuya M, Plate KH, Machein MR. Flt-1 signaling in macrophages promotes glioma growth in vivo. Cancer Res. 2008;68:7342–7351. doi: 10.1158/0008-5472.CAN-07-6241. [DOI] [PubMed] [Google Scholar]
- 76.Dixit A, Yi L, Gowthaman R, Torkamani A, Schork NJ, Verkhivker GM. Sequence and structure signatures of cancer mutation hotspots in protein kinases. PLoS One. 2009:4. doi: 10.1371/journal.pone.0007485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Friedman R, Boye K, Flatmark K. Molecular modelling and simulations in cancer research. Biochim Biophys Acta - Rev Cancer. 2013;1836:1–14. doi: 10.1016/j.bbcan.2013.02.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Model Code
Model Tutorial. A guide that helps users navigate the provided model code for applying the proposed methods and procedures to their own modeling tasks
Supplementary Table 1. List of Abbreviations used in this manuscript
Supplementary Table 2. mRNA-seq data from TCGA in FPKM.
Supplementary Table 3. mRNA-seq data from TCGA for the selected virtual patients and model genes in molecules/cell.