

Key Clinical Message
This case report describes an individual with brain calcifications, cognitive decline, motor dysfunction, and hypocalcaemia. Exome sequencing revealed a previously reported variant in the CASR gene and a variant of uncertain significance in PDGFRB. The clinical phenotype is likely explained by the CASR variant, but we discuss how the PDGFRB variant could also participate in the phenotype.
Keywords: CASR, Fahr's syndrome, autosomal dominant, hypocalcaemia, PDGFRB, primary familial brain calcification, whole‐exome sequencing
Introduction
Calcifications of the basal ganglia were first described in 1850 1, and since that time intracranial calcifications have been linked to multiple disorders and even incidentally discovered at a low frequency within healthy populations 2, 3, 4, 5. Primary familial brain calcification (PFBC, or Fahr's syndrome) is a primarily autosomal dominant disorder characterized by progressive, peri‐capillary bilateral calcifications of the brain, especially in the basal ganglia, but also seen in the cerebellum, thalami, brainstem, and subcortical white matter 3, 6, 7. To identify PFBC, clinicians rely on several core criteria to diagnose patients, including progressive neurological and/or psychological symptoms, the elimination of other underlying etiologies, such as trauma or metabolic disorders, and a family history of basal ganglia calcification 3, 7, 8.
Despite these guidelines, reported phenotypes vary considerably 3, 8, and there is possible confusion with other calcification disorders due to over 35 different descriptive terms being used within the literature 3. With the increased utilization of clinical genetic testing, this diagnostic uncertainty has been reduced, and several genes have been linked to PFBC in recent years 9, 10, 11, 12. Here, we report a novel variant of uncertain significance (VUS) in the platelet‐derived growth factor receptor beta subunit (PDGFRB) gene as well as a likely pathogenic variant of the calcium‐sensing receptor (CASR) gene in the same individual.
Materials and Methods
Ethical compliance
The patient consented to sample collection and subsequent analysis under a protocol approved by the institutional review board of the Mayo Clinic. Written informed consent was obtained from the patient for publication and accompanying images.
Whole‐exome sequencing
Clinical whole‐exome sequencing (WES) was performed by GeneDx (XomeDxPlus). Briefly, genomic DNA was extracted from the proband. As described in the clinical testing methodology by GeneDx, the Agilent Clinical Research Exome capture kit was used for exome enrichment and sequencing was performed on an Illumina HiSeq 2000 that generates 100 bp paired‐end reads. Bidirectional sequences were assembled, aligned to reference gene sequences based on human genome build GRCh37/UCSC hg19, and analyzed for sequence variants using a proprietary analysis tool (Xome Analyzer, GeneDx). Sanger sequencing was used to confirm all disease relevant variants identified in this individual. Variant filtration techniques were performed per standard commercial methods for GeneDx. Sequence alterations were reported according to the Human Genome Variation Society (HGVS) nomenclature guidelines.
Clinical Report
Case description
The patient is a 73‐year‐old man of northern European origin, who presented with a several year history of progressive dysarthria and mild aphasia. The patient reported that he was “…not able to put sentences together.” He further asserted that he had “lost [his] voice,” following each of two previous hip replacement surgeries. Similarly, laryngospasm and tetany were reported. The patient's neurological examination revealed mild to moderate cognitive impairment, characterized by deficits in concentration, executive dysfunction, diminished visual constructional and visuospatial skills, slowed verbal fluency, diminished naming, and inefficient memory retention. These findings were consistent with mild cognitive impairment, secondary to frontal, temporal, and parietal lobe dysfunction. Speech and language assessments revealed a mixed dysarthria with hyperkinetic and hypokinetic features. There was also mild oral apraxia and mild impairment in motor programming.
His most recent imaging examinations (Fig. 1) revealed symmetric, bilateral calcifications of the cerebral deep white matter, basal ganglia, thalami, medial occipital cortices, and deep cerebellar white matter. There was no evidence of atrophy. Prior computed tomography (CT) and magnetic resonance imaging (MRI) examinations also detected extensive calcifications, notably of the basal ganglia, but also across other regions. On MRI, this manifested as increased signal in T1‐weighted images in the corona radiata, centrum semiovale, thalami, dentate nuclei, and cerebellar white matter. In these earlier images, there was evidence of atrophy reported that disproportionately affected the frontal and parietal lobes, and to a milder extent, the cerebellum. The patient had previously undergone MRI in 2009 after suffering a stroke. His records from this time make no mention of brain calcifications, suggesting that this finding was progressive and has persisted for at least 2 years.
Figure 1.

CT scan in the axial plane showing extensive bilateral calcification of basal ganglia.
Given the link between basal ganglia calcification and metabolic/endocrine dysregulation, the patient was tested for hypoparathyroidism. He displayed low calcium at 8.6 mg/dL (reference range ≥22 years: 8.9–10.1 mg/dL), but creatinine, parathyroid hormone (PTH), and thyroid‐stimulating hormone (TSH) were within normal ranges.
Genetic testing
There were no related clinical findings in the proband's family (see Fig. 2), although his paternal grandfather and son are reported to have either attempted (former) or committed suicide (latter). Notably, depression has been linked with PFBC. The patient also has a son with a history of Graves’ disease with hyperthyroidism. Previous testing by Fulgent for two of the most common PFBC‐associated genes (PDGFB and SLC20A2) did not detect any variants, therefore the patient underwent WES through the Mayo Clinic Department of Clinical Genomics. WES revealed variants in two genes (Table 1) possibly associated with the patient's phenotype: a likely pathogenic variant in the CASR gene (Chr3(GRCh37): g.122003232A>G, NM_000388.2: c.2431A>G, NP_000379.2: p.(Met811Val)) and a novel variant of uncertain significance in PDGFRB (Chr5(GRCh37): g.149512314G>A, NM_002609.3: c.1126C>T, NP_002600.1: p.(Arg376Trp)). The p.(M811V) variant in exon 7 of CASR was not observed in approximately 123,136 exomes and 15,496 genomes in the Genome Aggregation Database (gnomAD). The variant results in a conservative amino acid substitution, which occurs at a position that is conserved across species, down to Danio rerio (zebrafish). In silico analysis predicts this variant is likely damaging to the protein structure and/or function (see Table 2). The p.(R376W) variant in PDGFRB was seen in 3/272342 alleles in gnomAD 13; http://gnomad.broadinstitute.org/). The variant results in a nonconservative amino acid substitution, which is predicted to impact secondary protein structure. The variant falls at the end of exon 7, but is not predicted to impact splicing based on in silico splice prediction tools. This substitution occurs at a position that is moderately conserved across species (including in Gallus gallus). In silico analysis 14 predicts this variant is likely damaging to the protein structure and/or function (see Table 2).
Figure 2.
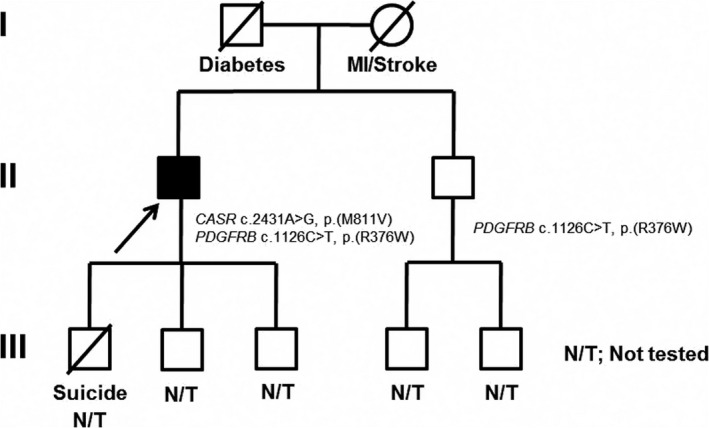
Three‐generation family pedigree. Proband indicated with arrow. Both the proband and his asymptomatic brother share a PDGFRB variant, whilst the proband additionally carries the CASR variant.
Table 1.
Potentially pathogenic variants identified in genes related to the patient's condition through WES
| Gene | Diseases (MIM #) | Mode of Inheritance | Variant | Coding DNA | Zygosity | Mode of Inheritance | gnomAD frequency | dbSNP | ClinVar Accession number | Classification |
|---|---|---|---|---|---|---|---|---|---|---|
| CASR |
239200; 601198; 145980; 612899 |
Autosomal Dominant/Recessive | p.(M811V) | c.2431A>G | Heterozygous | Unknown | Not reported | Not reported | SCV000521154 | Likely Pathogenic Variant |
| PDGFRB |
615007; 616592; 131440; 228550; 601812 |
Autosomal Dominant | p.(R376W) | c.1126C>T | Heterozygous | Unknown | 3/272342 (0.00001102%) | rs142621427 | None | Variant of Uncertain Significance |
Table 2.
Predicted SNP effects for the identified variants from in silico analysis using PredictSNP2 14
| Tool | PredictSNP2 | CADD | DANN | FATHMM | FunSeq2 | GWAVA | |
|---|---|---|---|---|---|---|---|
| CASR chr3:122003232 A>G | Prediction | Deleterious | Deleterious | Deleterious | Deleterious | Neutral | Unknown |
| Score | 1 | 21.4 | 0.9954 | 0.9901 | 2 | 0.48 | |
| Exp. accuracy | 0.87 | 0.54 | 0.6 | 0.83 | 0.62 | 0.48 | |
| PDGFRB chr5:149512314 G>A | Prediction | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious | Deleterious |
| Score | 1 | 23.1 | 0.9982 | 0.9021 | 3 | 0.26 | |
| Exp. accuracy | 0.87 | 0.55 | 0.7 | 0.56 | 0.61 | 0.51 |
The proband's asymptomatic 72‐year‐old brother was also tested and found to have the PDGFRB variant but not the CASR (see pedigree in Fig. 2) variant.
Discussion
Intracranial mineral deposition is common to a number of disorders, including certain neurodegenerative and metabolic diseases, although it also occurs at low levels in the general population and may be related to the normal aging process 2, 3, 4, 5. In PFBC, calcium is deposited in perivascular areas of the brain, especially the basal ganglia and cerebellar white matter, and is most commonly linked to movement and cognitive dysfunctions 3, 6, 8, 15.
In this case study, multiple MRI and head CT scans uncovered extensive bilateral calcifications in a patient originally presenting with dysarthria and reductions in learning and memory abilities. Consistent with PFBC, the patient's calcifications were found in the deep white matter of the cerebrum and cerebellum, thalami, and basal ganglia. WES uncovered two heterozygous genetic variants in this patient: one novel VUS in PDGFRB and one likely pathogenic variant in CASR.
The first gene, PDGFRB (MIM #173410), produces a homodimeric tyrosine kinase (TK) receptor (PDGF‐Rβ) that can bind multiple ligands, including PDGF‐B (platelet‐derived growth factor subunit B), which, along with the receptor, has been linked to PFBC in a number of recent studies 6, 9, 11, 16, 17, 18, 19. PDGF‐Rβ is thought to be involved in angiogenesis and hematopoiesis during development, and Keller et al. 9 proposed that the accumulation of calcium in PDGF‐Rβ/PDGF‐B linked PFBC cases may be related to a dysfunctional blood–brain barrier (BBB).
Most reported cases of PDGFRB variants have been classified as gain‐of‐function, leading to disorders such as infantile myofibromatosis (MIM #228550), Kosaki overgrowth disorder (MIM #616592) 20, and premature aging disorders (MIM #601812) 21. However, several variants in or near the split‐TK domain of the receptor have been found in patients with basal ganglia calcifications 6, 11, 18, 19. Physically closest to our novel variant (c.1126C>T, p.(R376W)) is a missense variant at codon 371 (p.N371K, c.1113C>G), although the patient in this case was diagnosed with Cornelia de Lange syndrome and had no mention of brain calcifications 22. Lastly, whilst our variant falls close to a splice site, splicing predictions were not in favor of a strong effect on splicing.
The second genetic variant uncovered by WES in the CASR gene (c.2431A>G, p.(M811V)) (MIM #601199) has been reported previously in a family with hypoparathyroidism, hypocalcaemia, and relative hypercalciuria 23. Similarly, the proband described here has hypocalcaemia and borderline low‐normal parathyroid hormone and serum magnesium levels, which is characteristic of autosomal dominant hypocalcaemia caused by an activating CASR variant 24. The variant was shown to segregate with the hypocalcaemic phenotype in three generations of affected individuals in this family, while unaffected family members were negative for the variant, providing strong evidence for its pathogenicity. Activating CASR variants lead to defective sensing and regulation of serum calcium levels, resulting in a reduction in parathyroid hormone and maintained hypocalcaemia. Interestingly, 50% of patients presenting with hypercalciuric hypocalcaemia syndrome type 1 are estimated to have asymptomatic hypocalcaemia, and over 35% have ectopic and/or basal ganglia calcifications 24.
As PFBC is partially diagnosed based on the exclusion of underlying metabolic etiology 3, 7, 8, the presence of hypocalcaemia combined with two potentially contributing genetic variants makes diagnosis difficult. It is possible that both variants contribute to this patient's complex phenotype. For example, in one reported case, a patient rapidly developed extensive unilateral calcifications in both the white and gray matter several weeks after an ischemic stroke in the same brain region 25. The rapid, ectopic accumulation of calcium was thought to occur due to the pre‐existing imbalanced ratio of phosphate to calcium and the weakened vasculature at the site of the stroke 25. This report by Wityk, combined with findings of disrupted BBBs in both mice with PDGF‐B variants 9 and postmortem PFBC human tissue 26, strengthens the hypothesis that an impaired BBB/vascular system could lead to increased susceptibility to mineral deposition in the brain, especially when combined with metabolic imbalances. However, metabolic dysfunction has been reported to cause ectopic mineral deposition independently of any reported increase in BBB or vascular vulnerability, as seen by Kurozumi et al. 27 and Schouten et al. 23. Additionally, a mouse model experiment performed by Vanlandewijck et al. 28 found that PGDFBret/ret mice actually had healthier BBBs in brain regions prone to calcification as compared to regions not prone to calcification 28. The association between BBB impairment and increased mineralization must then merit further investigation.
In this report, the clinical presentation of the patient seems to more closely fit that of PFBC compared to that of activating CASR variants, although his hypocalcaemia is certainly attributable to the latter. In PFBC, the most commonly reported manifestation is movement disorder, closely followed by cognitive and cerebellar impairments, and speech disorder 3, 6, 8, 9. In this case, the patient originally sought medical attention due to cognitive decline and speech impairment, although some tremulous movements in his upper limbs, muscular fasciculations, and dysphagia were noted during examination. Other less commonly reported symptoms of PFBC include stroke 6, 15 and vertigo 29, both of which were identified in this patient prior to or during treatment. In this case, the vertigo was successfully ameliorated using Epley chair repositioning. Additionally, Mufaddel & Al‐Hassani 8 reported that in 40% of patients, psychiatric features are often the first presenting symptom of PFBC and the most common of these is depression. Interestingly, although the patient asserts that he has no current or previous episodes of depression, there is a documented family history of this condition and the patient notes that he has had increased emotional reactivity in recent years.
Follow‐up genetic testing of the patient's asymptomatic 72‐year‐old brother revealed the same heterozygous variant in PDGFRB (c.1126C>T, p.(R376W)), but no changes in CASR were detected. However, this information is the extent of available family history, as no other testing, including CT imaging, was completed for the brother, and other family members were unavailable for testing. Consequentially, we were unable to use the brother's information to determine if the isolated PDGFRB variant is associated with any specific features of the patient's phenotype. The lack of familial confirmation is the largest limitation of the current report as it is impossible to determine if PFBC or hypocalcaemia are present in untested members of the family, the mode of inheritance, and whether either of the variants segregate with the ectopic calcifications, cognitive symptoms, and/or hypocalcaemia as reported in the proband. The limited information provided by the brother's genetic testing and his apparent lack of symptoms may provide clues as to the causative variant in the proband; however, incomplete penetrance and a variable age of onset have commonly been reported for PFBC 3, 6, 15, 30. Of note, PFBC and hypocalcaemia caused by CASR changes are thought to be inherited mainly via an autosomal dominant pattern 9, 19, 23, 26, 27.
Conclusions
In conclusion, we report a 73‐year‐old man with bilateral basal ganglia calcifications and two variants in CASR and PDGFRB with a possible blended phenotype. We were unable to gather substantial information on familial phenotypes to further investigate the effects of the PDGFRB VUS. Despite this, the existence of past cases of PDGFRB variants causing basal ganglia calcifications suggests that the VUS could be implicated in the mineralization present in our proband.
Authorship
NND and JDB: wrote the manuscript. PRB, JMG, JR and HKA: involved in scientific input and revision. JAvG: collected the clinical data and was involved in scientific input and revision. PSA: is the senior author and was involved in scientific input and revision of the final manuscript.
Conflict of Interests
The authors have no conflict of interests or disclosures.
Acknowledgments
The authors would like to thank the Mayo Clinic Center for Individualized Medicine for supporting this research.
Clinical Case Reports 2018; 6(1): 8–13
References
- 1. Delacour, A. 1850. Ossification des capillairies du cerveau. Ann. Med. Psychol. 2:458–461. [Google Scholar]
- 2. Förstl, H. , Krumm B., Eden S., and Kohlmeyer K.. 1992. Neurological disorders in 166 patients with basal ganglia calcification: a statistical evaluation. J. Neurol. 239:36–38. [DOI] [PubMed] [Google Scholar]
- 3. Manyam, B. V. 2005. What is and what is not ‘Fahr's disease’. Parkinsonism Relat. Disord. 11:73–80. [DOI] [PubMed] [Google Scholar]
- 4. Valdés Hernández, M. C. , Maconick L. C., Tan E. M. J., and Wardlaw J. M.. 2012. Identificaiton of mineral depositions in the brain on radiological images: a systematic review. Eur. Radiol. 22:2371–2381. [DOI] [PubMed] [Google Scholar]
- 5. Yamada, M. , Asano T., Okamoto K., Hayashi Y., Kanematsu M., Hoshi H., et al. 2013. High frequency of calcification in basal ganglia on brain computed tomography images in Japanese older adults. Geriatr. Gerontol. Int. 13:706–710. [DOI] [PubMed] [Google Scholar]
- 6. Nicolas, G. , Pottier C., Charbonnier C., Guyant‐Mare′chal L., Le Ber I., Pariente J., et al. 2013. Phenotypic spectrum of probable and genetically‐confirmed idiopathic basal ganglia calcification. Brain 136:3395–3407. [DOI] [PubMed] [Google Scholar]
- 7. Sobrido, M. J. , Coppola G., Oliveira J., Hopfer S., Geschwind D. H.. 2014. Primary familial brain calcification Pagon R. A., Adam M. P., Ardinger H. H., Wallace S. E., Amemiya A., Bean L. J. H., et al., eds. GeneReviews [Internet]. University of Washington, Seattle, Seattle, Washington. [Google Scholar]
- 8. Mufaddel, A. A. , and Al‐Hassani G. A.. 2014. Familial idiopathic basal ganglia calcification (Fahr's disease). Neurosciences (Riyadh) 19:171–177. [PMC free article] [PubMed] [Google Scholar]
- 9. Keller, A. , Westenberger A., Sobrido M. J., García‐Murias M., Domingo A., Sears R. L., et al. 2013. Mutations in the gene encoding PDGF‐B cause brain calcifications in humans and mice. Nat. Genet. 45:1077–1082. [DOI] [PubMed] [Google Scholar]
- 10. Legati, A. , Giovannini D., Nicolas G., López‐Sánchez U., Quintáns B., Oliveira J., et al. 2015. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet. 47:579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nicolas, G. , Pottier C., Maltête D., Coutant S., Rovelet‐Lecrux A., Legallic S., et al. 2013. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80:181–187. [DOI] [PubMed] [Google Scholar]
- 12. Wang, C. , Li Y., Shi L., Ren J., Patti M., Wang T., et al. 2012. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nature 44:254–256. [DOI] [PubMed] [Google Scholar]
- 13. Lek, M. , Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., et al. 2016. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bendl, J. , Musil M., Štourač J., Zendulka J., Damborský J., and Brezovský J.. 2016. PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput. Biol. 12:e1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lazăr, M. , Ion D. A., Streinu‐Cercel A., and Bādārāu A. I.. 2009. Fahr's syndrome: diagnosis issues in patients with unknown family history of disease. Rom. J. Morphol. Embryol. 50:425–428. [PubMed] [Google Scholar]
- 16. Nicolas, G. , Jacquin A., Thauvin‐Robinet C., Rovelet‐Lecrux A., Rouaud O., Pottier C., et al. 2014. A de novo nonsense PDGFB mutation causing idiopathic basal ganglia calcification with laryngeal dystonia. Eur. J. Hum. Genet. 22:1236–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nicolas, G. , Rovelet‐Lecrux A., Pottier C., Martinaud O., Wallon D., Vernier L., et al. 2014. PDGFB partial deletion: a new, rare mechanism causing brain calcification with leukoencephalopathy. J. Mol. Neurosci. 53:171–175. [DOI] [PubMed] [Google Scholar]
- 18. Sanchez‐Contreras, M. , Baker M. C., Finch N. A., Nicholson A., Wojtas A., Wszolek Z. K., et al. 2014. Genetic screening and functional characterization of PDGFRB mutations associated with basal ganglia calcification of unknown etiology. Hum. Mutat. 35:964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wong, W. C. , Lam C. W., Tong S. F., and Tong C. T.. 2011. Persistent hypocalcaemia in a Chinese girl due to a novel de‐novo activating mutation of the calcium‐sensing receptor gene. Hong Kong Med. J. 17:157–160. [PubMed] [Google Scholar]
- 20. Takenouchi, T. , Yamaguchi Y., Tanikawa A., Kosaki R., Okano H., and Kosaki K.. 2015. Novel overgrowth syndrome phenotype due to recurrent de novo PDGFRB mutation. J. Pediatr. 166:483–486. [DOI] [PubMed] [Google Scholar]
- 21. Johnston, J. J. , Sanchez‐Contreras M. Y., Keppler‐Noreuil K. M., Sapp J., Crenshaw M., Finch N. A., et al. 2015. A point mutation in PDGFRB causes autosomal‐dominant Penttinen syndrome. Am. J. Hum. Genet. 97:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yavarna, T. , Al‐Dewik N., Al‐Mureikhi M., Ali R., Al‐Mesaifri F., Mahmoud L., et al. 2015. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum. Genet. 134:967–980. [DOI] [PubMed] [Google Scholar]
- 23. Schouten, B. J. , Raizis A. M., Soule S. G., Cole D. R., Frengley P. A., George P. M., et al. 2011. Four cases of autosomal dominant hypocalcaemia with hypercalciuria including two with novel mutations in the calcium‐sensing receptor gene. Ann. Clin. Biochem. 48:286–290. [DOI] [PubMed] [Google Scholar]
- 24. Vahe, C. , Benomar K., Espiard S., Coppin L., Jannin A., Odou M. F., et al. 2017. Diseases associated with calcium‐sensing receptor. Orphanet. J. Rare Dis. 12:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wityk, R. J. , Lapeyrolerie D., and Stein B. D.. 1993. Rapid brain calcification after ischemic stroke. Ann. Intern. Med. 119:490–491. [DOI] [PubMed] [Google Scholar]
- 26. Miklossy, J. , Mackenzie I. R., Dorovini‐Zis K., Calne D. B., Wszolek Z. K., Klegeris A., et al. 2005. Severe vascular disturbance in a case of familial brain calcinosis. Acta Neuropathol. 109:643–653. [DOI] [PubMed] [Google Scholar]
- 27. Kurozumi, A. , Okada Y., Arao T., Endou I., Matsumoto T., and Tanaka Y.. 2013. Extrapyramidal symptoms and advanced calcification of the basal ganglia in a patient with autosomal dominant hypocalcemia. Intern. Med. 52:2077–2081. [DOI] [PubMed] [Google Scholar]
- 28. Vanlandewijck, M. , Lebouvier T., et al. 2015. Functional characterization of germline mutations in PDGFB and PDGFRB in primary familial brain calcification. PLoS ONE 10:e0143407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. König, P. 1989. Psychopathological alterations in cases of symmetrical basal ganglia sclerosis. Biol. Psychiatry 25:459–468. [DOI] [PubMed] [Google Scholar]
- 30. Geschwind, D. H. , Loginov M., and Stern J. M.. 1999. Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification (Fahr Disease). Am. J. Hum. Genet. 65:764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]