

Key Clinical Message
16p11.2 deletions and duplications are commonly associated with autism spectrum disorder and linked to mirrored phenotypes of physical characteristics and higher penetrance for deletions. A male with a rare 16p11.2 triplication demonstrated a similar phenotypic presentation to deletion carriers with neurocognitive and adaptive skill deficits and above‐average physical growth.
Keywords: 16p11.2 deletion, 16p11.2 duplication, 16p11.2 triplication, ASD risk variant, gene triplication
Introduction
Recurrent deletions and duplications of an approximately 600 kbp region of chromosomal locus 16p11.2 are a frequent genetic etiology of neurodevelopmental disorders such as autism spectrum disorder (ASD), developmental delay, intellectual disability, and schizophrenia 1, 2, 3, 4. 16p11.2 copy number variations (CNVs) occur with a frequency of one of 1000 in the general population 5 and a range of 0.76% to 1.13% in nonsyndromic, idiopathic ASD depending on sample composition 6 making 16p11.2 one of the most prevalent ASD risk variants. Clinical presentations of 16p11.2 CNVs suggest heterogeneous phenotypes including cognitive, motor, and language delays; behavioral problems; congenital anomalies; and dysmorphic features 5, 6, 7, 8, 9, 10, 11. 16p11.2 deletions and duplications are also associated with increased incidence of structural brain abnormalities 1, 4, 12. It has been suggested the 16p11.2 deletion is more penetrant than the duplication, such that deletion carriers show a higher rate of ASD diagnosis 8 and developmental delay 4.
Copy number variation triplications are quite rare, and subsequently, the associated phenotype for 16p11.2 triplication is unknown. Reports of triplication at other loci indicate that triplication cases resemble duplication patients, including behavioral problems associated with 17q21.31 triplication 13 and developmental delay associated with 11q12.3 14. Although instances of triplication in 16p11.2 have been reported 15, this is the first study to report a comprehensive phenotype assessment on the developmental phenotype and trajectory of a child with a 16p11.2 triplication over a period of 4 years.
Methods
The proband was ascertained as part of the longitudinal portion of the Simons Variation in Individuals Project (SVIP) and evaluated during seven study visits between 31 and 72 months old (four clinic visits, three phone calls) with written consent from his parents. This research was approved by the University of Washington Institutional Review Board. Assessments included standardized cognitive, behavioral, adaptive, and ASD evaluations (Appendix S1); neurological examination; and neurologic history and record review by a board‐certified pediatric neurologist (KJS). Comparative data for individuals with 16p11.2 deletion or duplication were downloaded from the SFARI Simons VIP cohort. The comparison sample of Simons VIP participants included individuals with the same recurrent 600 kbp BP4‐BP5 16p11.2 deletion or duplication without other pathogenic CNVs or known genetic diagnoses identified through clinical evaluations 16, 17 and recruited via referral to the Simons VIP Connect Web site (SimonsVIPConnect.org). See the Simons VIP consortium 18 for additional information regarding recruitment and eligibility criteria (http://sfari.org/resources/simons-vip).
Three approaches were used to assay chromosome 16p11.2 copy number in the proband and selected relatives: fluorescence in situ hybridization (FISH), whole‐genome SNP microarray experiments, and molecular inversion probe (MIP) experiments. Specifically, FISH was performed in a clinical cytogenetics laboratory using a bacterial artificial chromosome (BAC) clone (RP11‐74E23) as the probe. Microarray analysis leveraged the Affymetrix Whole Genome‐Human SNP Array 6.0, while MIP experiments utilized probes targeting 54 genetic markers (43 informative with high confidence) distinguishing duplicated sequences on each side of the chromosome 16p11.2 critical region (telomeric/BP4, GRCh37/hg19 chr16:29562241‐29606852; centromeric/BP5, GRCh37/hg19 chr16:30302259‐30346868) from each other and from all other paralogous sequences 19. All MIP experiments incorporated a molecular‐tagging strategy 20 and a previously described analysis pipeline 21.
Results
Genetic analysis
FISH experiments performed in a clinical laboratory revealed increased copy number at chromosome 16p11.2 in the proband and in his mother. We verified this diagnosis and assessed inheritance by performing SNP microarray (Fig. 1A) and/or MIP genotyping (Fig. 1B), 21 on the proband, his parents, his sister, his maternal aunt (monozygotic twin of his mother), and his maternal grandparents using genomic DNA. These higher‐resolution analyses revealed a typical chromosome 16p11.2 duplication in the mother, occurring on the short arm of chromosome 16 and spanning approximately 597 kbp (GRCh37/hg19 chr16:29488112‐30085308). We identified a triplication of this same region in the proband, likely having originated via nonallelic homologous recombination (NAHR) during meiosis in the mother. No chromosome 16p11.2 rearrangements were observed in the grandparents, and we inferred that the maternal duplication originated in the grandmother 22. Thus, remarkably, two distinct de novo, likely NAHR‐mediated rearrangement events at chromosome 16p11.2, occurred within this family over two generations: an initial de novo duplication followed by the de novo expansion of this duplication to the triplication state (See Fig. 2 Pedigree of 16p11.2 proband).
Figure 1.
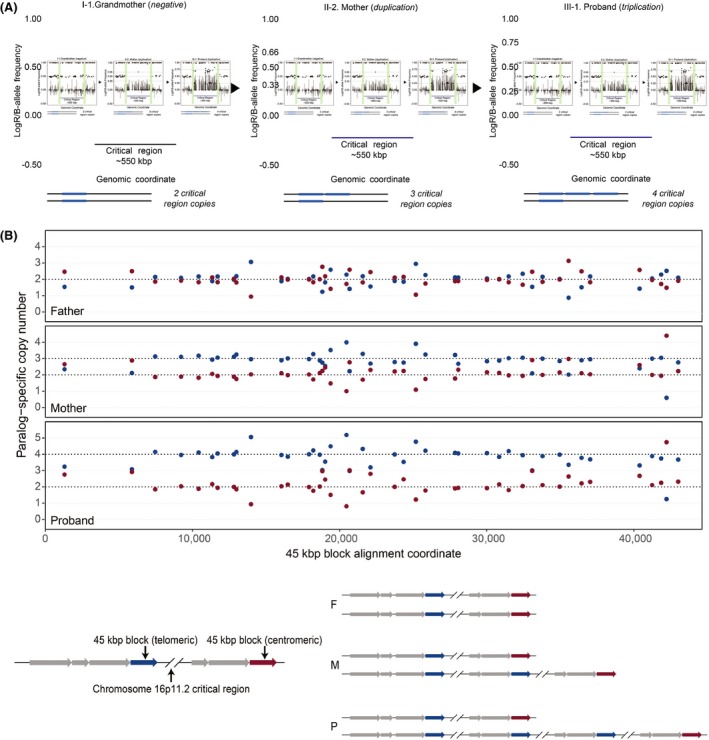
Genetic Overview. (A) Copy number expansion of the 16p11.2 critical region. LogR (lines) and B‐allele frequency (dots) plots show expansion of the 16p11.2 critical region from the maternal grandmother (two copies), to the mother with a duplication (three copies), to the proband with triplication (four copies). The logR values increase from generation to generation. In particular, the ratio of the intensity between proband (III‐3.) and mother (II‐4.) is 1.3 on average, consistent with a ratios of four copies (triplication) to three copies (duplication) (4/3 = 1.3). Green bars indicate the location of segmental duplications associated with breakpoints 4 and 5‐collapsed here for ease of display. (B) MIP data (top) and schematics depicting haplotypes inferred from these data (bottom) for the proband (P) carrying a chromosome 16p11.2 triplication, his mother (M) carrying a chromosome 16p11.2 duplication, and his father (F) showing normal copy number status at chromosome 16p11.2. The plots show paralog‐specific copy number across a 45‐kbp duplication block shared between breakpoint regions flanking the critical region 19. Points indicate paralog‐specific copy number estimates from 43 informative markers targeted in the MIP experiment. Dashed lines signify copy number calls inferred using an automated caller and confirmed by visual inspection.
Figure 2.
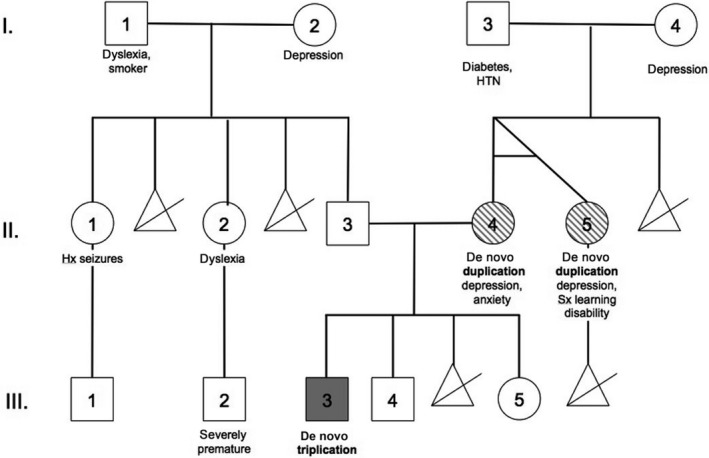
Pedigree of 16p11.2 proband. De novo 16p11.2 copy number variations noted by shading, including the triplication (proband: III.‐3, solid grey) and duplications (mother: II.‐4; maternal aunt: II.‐5, dashed grey). HTN, hypertension; Hx, history of; Sx, suspected.
Medical and developmental history
The proband is a Caucasian male with 16p11.2 triplication identified at 18 months. He was born via spontaneous vaginal delivery at 40 weeks gestation to a 31‐year‐old woman whose pregnancy was complicated only by 3 days of moderate vaginal bleeding of undetermined cause during first trimester. The proband weighed 9lbs 9 oz and measured 21.5 inches long at birth. A medical genetics evaluation at 24 months demonstrated multiple minor dysmorphic features including upslanting palpebral fissures, mild telecanthus, bilateral epicanthal folds, highly arched palate, cupped ears with overfolded helices, a small and broad uvula, a prominent anterior hair whorl, and delays in intramembranous ossification. Echocardiogram identified a likely right‐sided aortic arch and two small, nonhemodynamically significant atrial septal defects. Ophthalmologic evaluation revealed mild temporal pallor of the optic nerves in both eyes and mild myopic astigmatism. Other medical issues include a history of chronic constipation and suspected environmental allergies.
Neuroimaging
An MRI of the brain performed at 14 months demonstrated scattered foci of T2 hyperintensity in bilateral periventricular and parietal subcortical white matter. These findings were interpreted by the reading radiologist as nonspecific and possibly physiologic for age. Follow‐up MRI at 24 months demonstrated unchanged scattered areas of T2 hyperintensity in the parietal subcortical regions and decreased periventricular T2 hyperintensities, which were interpreted as remaining nonspecific in etiology, possibly representing in‐utero hypoxia/ischemia.
Anthropometric and neurological exam findings
Research‐based neurological exams starting at 31 months indicated the proband had a single CAL in his left axilla; mild excessive lordosis; and multiple minor dysmorphic features including a broad forehead, broad nasal root, mild hypertelorism, bilateral epicanthal folds, bilateral prominent ears, wide‐spaced nipples, small though mildly protuberant ears with thickened superior helices, and a double hair whorl (anterior hairline and cranial vertex). He exhibited facial hypotonia, copious drooling, and hypotonia. Notably, the proband's head circumference was similar to 16p11.2 deletion, and his weight and height were above the mean compared to both deletion and duplication cases (Fig. 3A). By 48 months, dysarthria became notable and persisted, along with significant facial hypotonia and excessive drooling, through the 72‐month visit. Lordosis was no longer seen at the 72‐month visit, although truncal hypotonia remained notable. At 48 months, the proband's gait was marked by decreased bilateral arm swing and posturing of his arms in front when walking or running. His gait had normalized by 60 months, but he was unable to jump. By 72 months, he had learned to jump, but was still unable to walk on heels, hop on either foot, or balance on either foot for five‐seconds.
Figure 3.
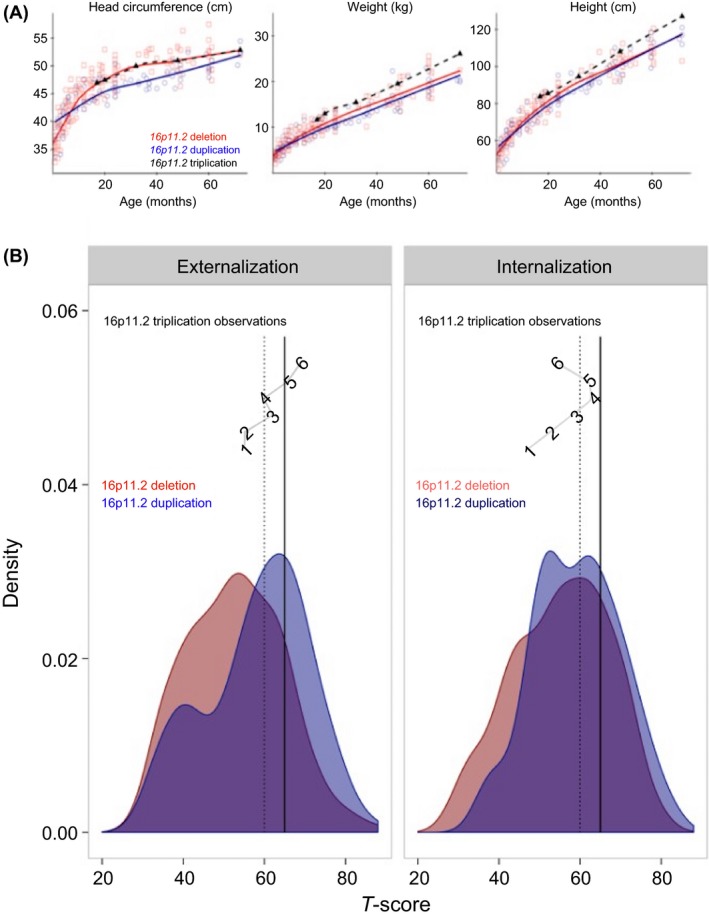
Developmental comparisons to 16p11.2 deletions and duplications. Comparison data was obtained from the publically available SFARI Simons VIP cohort. (A) Physical growth trajectories: head circumference, height, and weight physical growth for 16p11.2 triplication case were compared to 16p11.2 deletions (N = 181) and 16p11.2 duplications (N = 95). (B) Growth trajectories for externalizing and internalizing problems are plotted for the 16p11.2 triplication proband across time with age in months indicated at each time point, corresponding to T‐scores from the CBCL Externalizing (left panel) and Internalizing (right panel) subscales. Vertical lines indicate scores of borderline concern (dashed, T‐scores above 60) and clinical concern (solid, Tscores above 65). The distribution of scores for children with 16p11.2 deletion and duplication are presented for comparison.
Behavioral Phenotyping
Experienced, licensed clinicians derived DSM‐IV‐TR 23 diagnoses at each on‐site visit (Table S1). The proband demonstrated deficits in adaptive functioning and increasing levels of emotional and behavioral difficulties over time particularly in the areas of pervasive developmental problems, attention, affective problems, and externalizing behaviors (Supporting Information and Table S2). To frame behavioral issues in reference to other 16p11.2 CNVs, the proband's individual growth trajectories for the externalizing and internalizing symptoms were compared to the distribution plotted for children with 16p11.2 deletions and duplications (Fig. 3B) suggesting the 16p11.2 triplication proband exhibits increasing problems, similar to the 16p11.2 duplication profile.
Discussion
This is the first in‐depth phenotypic description of an individual with triplication within the 16p11.2 locus. The proband's medical history is significant for congenital heart defects, mild visual impairment, dysmorphisms, chronic constipation, and suspected environmental allergies. Additional phenotypic features included long‐standing articulation difficulties, hypotonia, copious drooling, and abnormal gait and fine motor coordination. Over the course of the evaluations, the proband met DSM‐IV‐TR diagnostic criteria for PDD‐NOS, mild intellectual disability, ADHD, enuresis, and encopresis. He exhibited developmental delays, impaired adaptive and cognitive functioning, and long‐standing pervasive developmental problems in the areas of communication, reciprocal social interaction, repetitive behaviors, and unusual sensory interests and aversions.
CNV triplications are rare chromosomal variants 24, often resulting in a phenotype that is most consistent with duplication of the same locus 25, 26. The proband exhibited increasing externalizing problems, similar to the duplication carriers. However, other features of the 16p11.2 triplication phenotype more closely resemble the deletion carriers, including patterns of physical growth (e.g., head circumference, height, and weight). In other words, while some features convey a more severe phenotype, phenotypic consequences are not uniform.
Recent evidence suggests that 16p11.2 reciprocal CNVs may produce mirrored (i.e., opposing) phenotypes. For instance, deletion carriers tend to exhibit obesity and macrocephaly, while duplication is associated with being underweight and microcephaly 5, 25, 27. These contrasting phenotypes have been proposed to support the presence of dosage sensitive genes within the 16p11.2 locus. This model is also consistent with analyses of gene expression in the context of chromosome 16p11.2 rearrangements, where expression levels of genes within the affected segment have been found to correlate strongly with genomic copy number, with little evidence for dosage compensation 28. Nevertheless, this triplication case conflicts with the notion of a simple graded genetic response between deletion, no‐mutation, duplication, and triplication. This may suggest that the genetic micro‐overexpression of the 16p11.2 locus (i.e., triplication, four copies instead of two) may be deleterious in a manner similar to a 16p11.2 microdeletion. Perhaps, some genes within the affected interval are sensitive to the precise level of increased dosage, such that triplication (but not duplication) may dysregulate them in a manner functionally equivalent to a deletion. It may also be the case that the haploinsufficient dosage within the triplicated segment is mediated in part by a possible complex chromosomal rearrangement that mimics the effect of a deletion of the same region. It is possible that none of these explanations are fully sufficient to describe the triplication phenotype, but rather together clarify how the perturbation of transcription factors may impact the regulation mechanisms of different genes within the 16p11.2 locus. Currently, the mechanistic bases of chromosome 16p11.2 deletion and duplication phenotypes are not well understood, but cellular and mouse modeling projects promise to provide new insights over the next several years. Further studies of human patients, including the identification and phenotypic characterization of additional triplication cases, will undoubtedly complement these efforts and help resolve basic and clinical questions surrounding one of the most common genetic contributors to abnormal neurodevelopment.
Authorship
ASW: performed lead role in writing and revision of the manuscript; involved in manuscript concept and design, clinical assessment, data acquisition, and final approval of manuscript for publication. CMH: involved in review of literature, manuscript concept and design, data acquisition, data analysis and interpretation of the results, drafting/revising the manuscript. KJS: completed neurological exams and review of neurological history and medical records for the case subject; involved in manuscript concept and design, data acquisition, drafting/revising the manuscript. JLP: involved in data acquisition, clinical assessment, writing and revision of the manuscript, final approval of the manuscript for publication. TDD: involved in data acquisition, data preparation and interpretation, revising the article for important intellectual content. MHD: involved in writing and revision of the manuscript; performed genetic analysis and interpretation of genetic testing results. XN: involved in writing and revision of the manuscript; performed genetic analysis and interpretation of genetic testing results. EEE: involved in writing and revision of the manuscript; performed genetic analysis and interpretation of genetic testing results. RAB: involved in design and implementation of the research; manuscript concept and design, data acquisition, revising the article for important intellectual content, final approval of the manuscript for publication.
Conflict of Interest
None declared.
Supporting information
Appendix S1. Methods.
Appendix S2. Clinical report history.
Appendix S3. Clinical assessment results.
Table S1. Longitudinal diagnostic trajectories.
Table S2. Longitudinal neurodevelopmental trajectories.
Acknowledgments
We are sincerely grateful to the proband and his family for their participation in this study and to all the families at the participating Simons Variation in Individuals Project (Simons VIP) sites, as well as the Simons VIP working group (Simons VIP consortium). We appreciate our funding from Simons Foundation (SFARI award 198677 to R.A.B. and SFARI award 303241 to E.E.E.). We are grateful for the Simons VIP working group and access to phenotypic data on SFARI Base. E.E.E. is an investigator of the Howard Hughes Medical Institute, is on the scientific advisory boards (SABs) of DNAnexus, Inc., and was an SAB member of Pacific Biosciences, Inc. (2009–2013) and SynapDx Corp. (2011–2013).
Clinical Case Reports 2018; 6(1): 147–154
References
- 1. Bijlsma, E. K. , Gijsbers C. J., Schuurs‐Hoeijmakers J. H. M., van Haeringen A., Fransen van de Putte D. E., Anderlid B. M., et al. 2009. Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur. J. Med. Genet. 52:77–87. [DOI] [PubMed] [Google Scholar]
- 2. Crespi, B. , Stead P., and Elliot M.. 2010. Comparative genomics of autism and schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 107(Suppl):1736–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCarthy, S. E. , Makarov V., Kirov G., Addington A. M., McClellan J., Yoon S., et al. 2009. Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 41:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shinawi, M. , Liu P., Kang S. L., Shen J., Belmont J. W., Scott D. A., et al. 2010. Recurrent reciprocal 16p.11.2 rearrangements associated with global developmental delay, behavioral problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 47:332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jacquemont, S. , Reymond A., Zufferey F., Harewood L., Walters R. G., Kutalik Z., et al. 2011. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 478:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walsh, K. M. , and Bracken M. B.. 2011. Copy number variation in the dosage‐sensitive 16p11.2 interval accounts for only a small proportion of autism incidence: a systematic review and meta‐analysis. Genet. Med. 13:377–384. [DOI] [PubMed] [Google Scholar]
- 7. Bardakjian, T. M. , Kwok S., Slavotinek A. M., and Schneider A. S.. 2010. Clinical report of microphthalmia and optic nerve coloboma associated with a de novo microdeletion of chromosome 16p11.2. Am. J. Med. Genet. Part A 152A:3120–3123. [DOI] [PubMed] [Google Scholar]
- 8. Fernandez, B. A. , Roberts W., Chung B., Weksberg R., Meyn S., Szatmari P., et al. 2010. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J. Med. Genet. 47:195–203. [DOI] [PubMed] [Google Scholar]
- 9. Hanson, E. , Nasir R. H., Fong A., Lian A., Hundley R., Shen Y., et al. 2010. Cognitive and behavioral characterization of 16p11.2 deletion syndrome. J. Dev. Behav. Pediatr. 31:649–657. [DOI] [PubMed] [Google Scholar]
- 10. Puvabanditsin, S. , Nagar M. S., Joshi M., Lambert G., Garrow E., and Brandsma E.. 2010. Microdeletion of 16p11.2 associated with endocardial fibroelastosis. Am. J. Med. Genet. A 152A:2383–2386. [DOI] [PubMed] [Google Scholar]
- 11. Zufferey, F. , Sherr E. H., Beckmann N. D., Hanson E., Maillard A. M., Hippolyte L., et al. 2012. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J. Med. Genet. 49:660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qureshi, A. Y. , Mueller S., Snyder A. Z., Mukherjee P., Berman J. I., Roberts T. P. L., et al. 2014. Opposing brain differences in 16p11.2 deletion and duplication carriers. J. Neurosci. 34:11199–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gregor, A. , Krumbiegel M., Kraus C., Reis A., and Zweier C.. 2012. De novo triplication of the MAPT gene from the recurrent 17q21.31 microdeletion region in a patient with moderate intellectual disability and various minor anomalies. Am. J. Med. Genet. Part A 158A:1765–1770. [DOI] [PubMed] [Google Scholar]
- 14. Yamamoto, T. , Matsuo M., Shimada S., Sangu N., Shimojima K., Aso S., et al. 2013. De novo triplication of 11q12.3 in a patient with developmental delay and distinctive facial features. Mol. Cytogenet 6:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ballif, B. C. , Hornor S. A., Jenkins E., Madan‐Khetarpal S., Surti U., Jackson K. E., et al. 2007. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2‐p12.2. Nat. Genet. 39:1071–1073. [DOI] [PubMed] [Google Scholar]
- 16. Bernier, R. , Hudac C. M., Chen Q., Zeng C., Wallace A. S., Gerdts J., et al. 2017. Developmental trajectories for young children with 16p11.2 copy number variation. Am. J. Med. Genet. Part B 174:367–380. [DOI] [PubMed] [Google Scholar]
- 17. Hanson, E. , Bernier R., Porche K., Jackson F. I., Goin‐Kochel R. P., Snyder L. G., et al. 2015. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol. Psychiatry 77:785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Simons VIP Consortium . 2012. Simons Variation in Individuals Project (Simons VIP): a genetics‐first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron. 73:1063–1067. [DOI] [PubMed] [Google Scholar]
- 19. Nuttle, X. , Giannuzzi G., Duyzend M. H., Schraiber J. G., Narvaiza I., Sudmant P. H., et al. 2016. Emergence of a Homo sapiens‐specific gene family and chromosome 16p11. 2 CNV susceptibility. Nature 536:205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hiatt, J. B. , Pritchard C. C., Salipante S. J., O'Roak B. J., and Shendure J.. 2013. Single molecule molecular inversion probes for targeted, high‐accuracy detection of low‐frequency variation. Genome Res. 23:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nuttle, X. , Huddleston J., O'Roak B. J., Antonacci F., Fichera M., Romano C., et al. 2013. Rapid and accurate large‐scale genotyping of duplicated genes and discovery of interlocus gene conversions. Nat. Methods 10:903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duyzend, M. H. , Nuttle X., Coe B. P., Baker C., Nickerson D. A., Bernier R., and Eichler E. E.. 2016. Maternal modifiers and parent‐of‐origin bias of the autism‐associated 16p11. 2 CNV. Am J Hum Genet. 98:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. American Psychiatric Association . 2000. Diagnostic and statistical manual of mental disorders: DSM‐IV‐TR. American Psychiatric Association, Washington, DC. [Google Scholar]
- 24. Reddy, K. S. , and Logan J. J.. 2000. Intrachromosomal triplications: molecular cytogenetic and clinical studies. Clin. Genet. 58:134–141. [DOI] [PubMed] [Google Scholar]
- 25. Beunders, G. , van de Kamp J. M., Veenhoven R. H., van Hagen J. M., Nieuwint A. W. M., and Sistermans E.. 2010. A triplication of the Williams‐Beuren syndrome region in a patient with mental retardation, a severe expressive language delay, behavioural problems and dysmorphisms. J. Med. Genet. 47:271–275. [DOI] [PubMed] [Google Scholar]
- 26. Ounap, K. , Ilus T., and Bartsch O.. 2005. A girl with inverted triplication of chromosome 3q25.3 –> q29 and multiple congenital anomalies consistent with 3q duplication syndrome. Am. J. Med. Genet. A 134:434–438. [DOI] [PubMed] [Google Scholar]
- 27. Golzio, C. , Willer J., Talkowski M. E., Oh E. C., Taniguchi Y., Jacquemont S., et al. 2012. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485:363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blumenthal, I. , Ragavendran A., Erdin S., Klei L., Sugathan A., Guide J. R., et al. 2014. Transcriptional consequences of 16p11. 2 deletion and duplication in mouse cortex and multiplex autism families. Am. J. Hum. Genet. 94:870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Methods.
Appendix S2. Clinical report history.
Appendix S3. Clinical assessment results.
Table S1. Longitudinal diagnostic trajectories.
Table S2. Longitudinal neurodevelopmental trajectories.