

Abstract
Diabetic kidney disease, a leading cause of end-stage renal disease, has become a serious public health problem worldwide and lacks effective therapies. Autophagy is a highly conserved lysosomal degradation pathway that removes protein aggregates and damaged organelles to maintain cellular homeostasis. As important stress-responsive machinery, autophagy is involved in the pathogenesis of various diseases. Emerging evidence has suggested that dysregulated autophagy may contribute to both glomerular and tubulointerstitial pathologies in kidneys under diabetic conditions. This review summarizes the recent findings regarding the role of autophagy in the pathogenesis of diabetic kidney disease and highlights the regulation of autophagy by the nutrient-sensing pathways and intracellular stress signaling in this disease. The advances in our understanding of autophagy in diabetic kidney disease will facilitate the discovery of a new therapeutic target for the prevention and treatment of this life-threatening diabetes complication.
Keywords: mTOR, AMPK, Sirt1, ER stress, Reactive oxygen species, Hypoxia
Diabetic kidney disease
Diabetic kidney disease (DKD) is a serious complication of diabetes mellitus and one of the most significant contributing factors to end-stage renal disease (ESRD) [1]. Around 35–40% of patients with type 1 or 2 diabetes eventually develop DKD, which accounts for a significant increase in mortality in these patients and poses a grave threat to the clinical outcome of diabetic patients [2]. According to projected data from the International Diabetes Federation, the number of diabetic patients worldwide will increase from 382 million in 2013 to 592 million by 2035 [3]. The rapidly increasing prevalence of diabetes and its complications will further escalate an already high cost of therapies and impose a substantial economic burden on patients and society.
Based on clinical observations, initial evidence indicative of DKD generally does not emerge until around 10–20 years after the onset of diabetes [4]. The development of DKD in diabetic patients is a result of multifactorial interactions between metabolic and hemodynamic pathways, which are often disturbed in the settings of diabetes. Hyperglycemia, hypertension, and genetic pre-disposition are the main risk factors besides elevated serum lipids, smoking, overweight or obesity, physical inactivity, and the amount of dietary proteins [2, 5]. The pathogenesis of DKD is extremely complex. It involves hyperglycemia-mediated alterations of intracellular metabolism, including accumulation of advanced glycation end products (AGEs), activation of protein kinase C (PKC), and increased glucose flux through polyol and hexosamine pathways. Intracellular stress including hypoxia, oxidative stress, and endoplasmic reticulum (ER) stress are also thought to be the pathological factors in DKD [6–11]. In addition, hemodynamic changes such as systemic and glomerular hypertension associated with a hyperactive renin–angiotensin system (RAS) have been implicated in the progression of DKD [12–16]. Moreover, the metabolic and hemodynamic abnormalities, interacting with intracellular stress, can also activate the production of a variety of cytokines, chemokines and growth factors, leading to inflammation and kidney fibrosis associated with DKD [2, 5]. Recently, increasing evidence further indicates the contribution of mitochondrial dysfunction to renal damage during DKD [17–19].
Virtually all four renal components, including glomeruli, tubules, interstitium and blood vessels, are affected by diabetes; however, DKD traditionally has been defined as a progressive microvascular complication that affects glomeruli as the first step. The clinical hallmark pathology of DKD is persistent albuminuria or proteinuria followed by decreased glomerular filtration rate (GFR) and subsequent tubular cell damage as well as tubulointerstitial lesions, eventually leading to renal failure [20–23]. As a result, detection of DKD generally depends on the measurement of urinary albumin level [24]. Additional histopathological features of DKD include an accumulation of extracellular matrix (ECM) components, thickening of both glomerular basement membrane (GBM) and tubular basement membrane, mesangial expansion, glomerular sclerosis, podocyte effacement, tubular atrophy, and afferent and efferent arteriolar hyalinosis [19].
The prevention and management of DKD has been multi-targeted, advocating a healthy lifestyle and targeting cellular and molecular factors involved in the pathogenesis of this disease. Intensive interventions such as glycemic control, blood pressure control and inhibition of the RAS have been shown to delay or reduce the risks for the onset and progression of albuminuria, but these treatments are unable to prevent loss of GFR or progression to ESRD as some patients develop treatment-resistant albuminuria [19, 25, 26]. Therefore, there is an urgent need to discover effective therapeutic options to improve the prognosis of diabetes and to prevent its complications including DKD. Recent studies reveal an impaired autophagy and its association with the development of aging- and diabetes-related diseases [27, 28]. Increasing evidence further shows defective autophagy in diabetic kidneys [29]. Given these findings, it is hypothesized that autophagy deficiency in diabetic kidneys may increase the susceptibility of kidney cells to diabetes-associated damage, which in turn leads to treatment-resistant albuminuria and a progressive decrease in renal function. Thus, restoration of autophagy activity may become a new therapeutic option to protect kidneys from DKD [26].
Autophagy
Autophagy, a term derived from Greek, refers to “self-eating”. It is an evolutionarily conserved cellular process in which various intracellular components, such as damaged organelles and protein aggregates, are delivered to lysosomes for degradation, clearance and recycling [30, 31]. Autophagy occurs at a basal level in most cells to maintain cellular homeostasis, whereas stress-induced autophagy primarily attempts to serve as an adaptive and defensive mechanism for cell survival [30, 31]. Autophagy is involved in physiology such as cell differentiation, animal development and nutrient metabolism [32, 33]. More importantly, dysregulation of autophagy contributes to the pathogenesis of an increasing number of diseases including neurodegenerative disease, cancer, aging, infectious and inflammatory disease, metabolic disease, cardiovascular disease, liver disease, pulmonary disease, and kidney disease [31, 34–36].
There are three types of autophagy identified in mammalian cells: macroautophagy, microautophagy and chaperone-mediated autophagy [31]. Macroautophagy, generally referred to as autophagy, is the focus of this review. The process of autophagy is characterized by a cascade of cellular events. It is initiated by the formation and expansion of an isolation membrane or a phagophore around the sequestered cytoplasmic targets, followed by closure of the isolation membrane to form a double-membrane autophagosome. The autophagosome then fuses with a lysosome to form an autolysosome, in which the encapsulated materials are eventually digested by lysosomal acid hydrolases. Alternatively, the autophagosome can fuse with an endosome to generate an amphisome, prior to fusion with the lysosome [31]. This complete dynamic process of autophagy is also termed as autophagic flux. The autophagosome membrane originates from the membrane compartments enriched in phosphatidylinositol 3-phosphate (PtdIns3P) and is likely connected to the ER. An omega-shaped protrusion from the ER is known as the omegasome, which positions at the beginning of isolation membrane formation and serves as a scaffold for autophagosome biogenesis [37–41]. Although autophagy can non-selectively break down bulk cytosol, in many cases autophagy recognizes and degrades specific organelles, proteins, and pathogens [31].
Autophagy is tightly regulated to ensure an optimal balance between synthesis and degradation, use and recycling of cellular components. Autophagosome formation is regulated coordinately at different steps by the core machinery that consists of more than 30 autophagy-related (Atg) genes [42, 43]. The Unc-51-like (ULK) 1/2 complex, ULK1/2–ATG13–FIP200–ATG101, is essential for autophagy initiation. Phagophore nucleation is dependent on the class III phosphatidylinositol 3 kinase (PtdIns3K) complex comprising BECN1, VPS34, VPS15 and ATG14L. The delivery of membrane from other sources to the forming autophagosome is regulated by WIPI1/2, a PtdIns3P scaffold protein, and ATG9L, a transmembrane traffic protein. Two ubiquitin-like conjugation systems, the microtubule-associated protein light chain 3-phosphatidyl ethanolamine (MAPLC3/LC3-PE) and the ATG12–ATG5–ATG16L complex, participate in autophagosome elongation and closure. The conversion of a cytosolic LC3-I to a membrane-bound LC3-II is indicative of autophagy induction and autophagosome formation. The maturation and fusion of autophagosome with lysosome involves UVRAG interaction with PtdIns3K complex and subsequent activation of the GTPase RAB7 [44, 45]. Upstream of the core machinery, autophagy is regulated by a complex signaling network [43, 46, 47]. Multiple signaling pathways that are stimulated by the signals from nutrient, growth factors and energy may integrate to regulate autophagy. Autophagy is also regulated by a variety of cellular stress such as DNA damage, ER stress, hypoxic stress and oxidative stress. Notably, pharmacological agents and genetic methods that target these regulatory pathways to either activate or inhibit autophagy are being discovered for therapeutic applications.
It is noteworthy that autophagy and apoptosis are not mutually exclusive. The two pathways share many of the same regulatory signals and each can regulate and modify the activity of the other, thus influencing differentially the fate of a stressed cell. The functional cross-talk between autophagy and apoptosis is very complex and generally presents as three scenarios [48–50]. First, autophagy antagonizes apoptotic cell death by promoting cell survival through, for example, the removal of damaged organelles that are source of oxidative stress, or catabolizing cellular molecules to provide a source of energy and nutrients, or the degradation of unfolded protein aggregates to limit ER stress. Second, autophagy acts either upstream of apoptosis to enable apoptotic signaling or during the final stage of apoptosis to participate in certain ATP-dependent morphological changes such as phosphatidyl serine exposure, membrane blebbing, and the formation of apoptotic bodies. Third, autophagy and apoptosis can either cooperate in parallel, or autophagy may assist apoptosis to promote cell death.
Autophagy was first detected by transmission electron microscopy (TEM) in the 1950s. Since then, an increasing array of techniques has been developed to monitor autophagy in cells or tissues [51]. Although TEM is a valid and important method both for the qualitative and quantitative analysis of changes in various autophagic structures that sequentially forms the phagophore, autophagosome, amphisome, and autolysosome, it is also one of the most problematic due to misinterpretations mostly deriving from sampling artifacts. To ensure more accurate and objective assessment of autophagy, LC3-based assays have been identified and extensively used in both in vitro and in vivo experimental settings. LC3 precursor (proLC3) is cleaved by a cysteine protease (ATG4) to convert to LC3-I, and is further modified into the PE-conjugated form, LC3-II, through an ubiquitin-like mechanism. As LC3-II is reliably associated with both the inner and outer membranes of forming phagophores as well as completed autophagosomes, the conversion of LC3-I to LC3-II or expression of LC3-II detected by western blotting has become a well-accepted biomarker for autophagy. Detection of endogenous LC3 by immunostaining or green fluorescent protein (GFP)-LC3 by fluorescence microscopy further visualizes the localization and distribution of the two different forms of LC3, with LC3-I showing homogenous staining and punctate LC3-II signal indicative of autophagosomes. Additional assays are also used to monitor the dynamic process of autophagic flux. These include: (1) LC3 turnover assay to compare LC3-II abundance in the presence and absence of lysosome inhibitors such as chloroquine or bafilomycin A1; (2) Analysis of the degradation of autophagic substrates such as p62/Sequestosome 1 (SQSTM1); (3) The use of a tandem monomeric red fluorescent protein (mRFP)/mCherry-GFP-LC3 reporter for simultaneous detection of both autophagosomes (LC3 puncta labelled both red and green) and autolysosomes (red-only LC3 puncta) [51].
Dysregulated autophagy in DKD
Generally speaking, when kidney cells are exposed to stress conditions, including hypoxia, genotoxic damage, oxidative stress, and ER stress, autophagy is activated and plays a critical role for cell survival [36]. In the states of nutrient/energy excess, autophagy is downregulated. Although the downregulation is beneficial in short term, defective autophagy may ultimately contribute to the accumulation of cellular damage, resulting in the development of metabolic or age-related kidney diseases [52]. Although the role of autophagy in DKD remains to be elucidated, emerging evidence has suggested that autophagy is impaired in glomerular and tubular cells under both type 1 and 2 diabetes. Autophagy was suppressed in proximal tubules of streptozotocin (STZ)-induced early diabetic rats, associated with tubular hypertrophy [53]. Inhibition of autophagy was also shown in distal tubules of early diabetic rats, which was reversed by insulin or islet transplantation [54]. In STZ-induced diabetic mice, autophagy was inhibited in podocytes, as indicated by p62/SQSTM1 accumulation [55]. Similar observations were also shown in Wistar fatty rats with type 2 diabetes [56]. Moreover, an accumulation of p62/SQSTM1 was further detected in proximal tubule epithelial cells (PTECs) in kidney biopsy from type 2 diabetic patients, suggesting that human diabetic kidneys are also deficient in autophagy activity [57]. Of note, although the capacity for autophagy is reduced in diabetic kidneys, the need for protective autophagy is significantly increased due to a high exposure to cellular stresses. This contradictory autophagic response may contribute to the development and progression of renal injury in DKD [52].
Nutrient-sensing pathways in autophagy regulation during DKD
The best known nutrient-sensing pathways include the mechanistic target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), and the sirtuins (SIRT). mTOR is activated by increased levels of glucose, amino acid and growth factors such as insulin under excessive nutrient conditions [58, 59]. In nutrient/energy-depleted conditions, AMPK and SIRT are activated by increases in intracellular AMP and nicotinamide adenine dinucleotide (NAD+) levels, respectively [60, 61]. Dysregulation of these nutrient-sensing pathways and its association with the pathogenesis of DKD have been suggested [52, 62]. It is well-recognized that autophagy is regulated by the nutrient-sensing pathways [43, 46, 47]. Therefore, alteration of these pathways under diabetic conditions may impair autophagic activity, leading to aggravated renal injury in DKD [52, 62].
mTOR and autophagy in DKD
mTOR is widely considered one of the most critical autophagy regulators in diabetic kidneys. Two mTOR complexes, the mTOR complex 1 (mTORC1) and complex 2 (mTORC2), have been identified. The rapamycin-sensitive mTORC1 consists of mTOR, regulatory associated protein of mTOR (RAPTOR), G protein β-subunit-like protein (GβL), proline-rich Akt substrate of 40 kDa (PRAS40) and DEP domain-containing mTOR-interacting protein (DEPTOR) [58, 63–65]. In comparison, mTORC2, which is less sensitive to rapamycin, is composed of mTOR, GβL, rapamycin-insensitive companion of mTOR (RICTOR), protein observed with RICTOR (PROTOR) and stress-activated protein kinase-interacting protein 1 (SIN1) [58, 63–65]. mTORC1 receives nutrient input via small GTPases, such as Ras-related GTP binding protein (Rag) and Ras homolog enriched in brain (Rheb). Increased levels of amino acid and growth factors activate mTORC1 through Rag and Rheb, respectively. Activation of mTORC1 most prominently results in phosphorylation of two downstream targets, p70 ribosomal S6 kinase/S6 ribosomal protein (p70S6K/S6RP) and eukaryotic translation-initiation factor 4E-binding protein 1 (4E-BP1), stimulating ribosome biogenesis and protein synthesis. The activity of mTORC1 is negatively regulated by tuberous sclerosis complex (TSC) 1/2 via inhibiting Rheb [58, 63–65]. Under normal cellular conditions, mTORC1 negatively modulates autophagy by phosphorylating ULK1 and thereby inhibiting its activity [66, 67]. However, in cells exposed to various stress signals, mTORC1 is suppressed, allowing ULK1, and thus autophagy, to be activated by AMPK pathway [68] (Fig. 1). Unlike mTORC1, the regulatory role of mTORC2 in autophagy remains poorly understood and less appreciated.
Fig. 1.
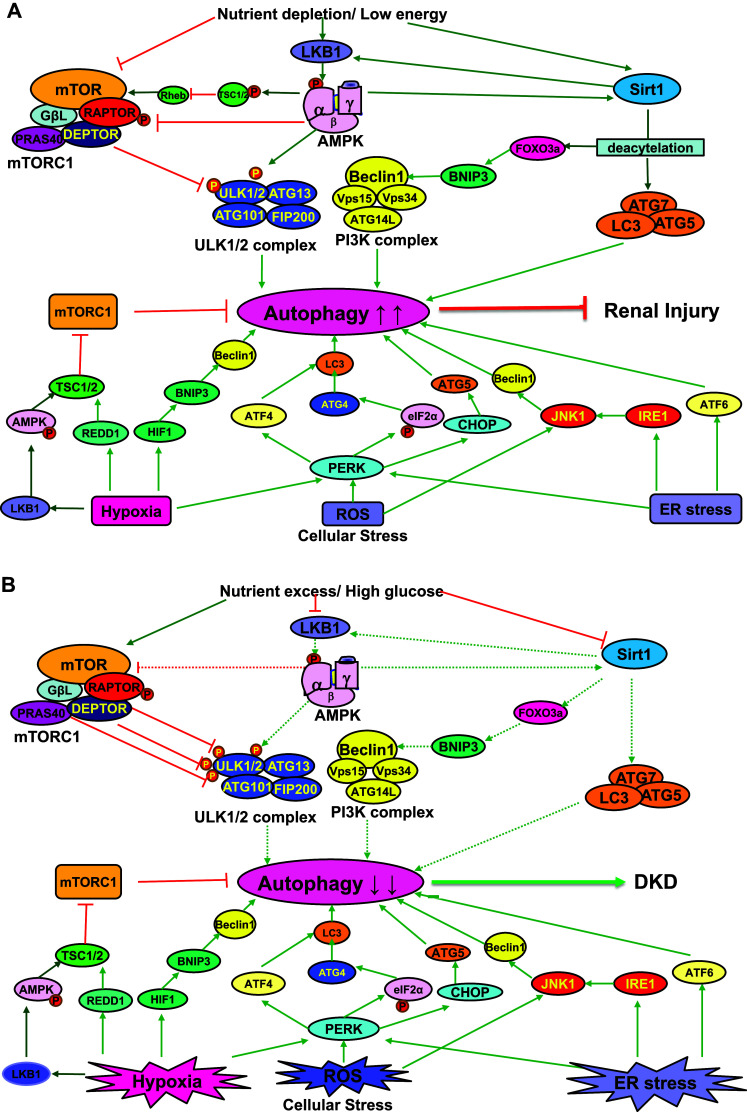
Regulation of autophagy under non-diabetic and diabetic conditions. a Under non-diabetic conditions, the autophagic response in the kidney is normal and competent. Intracellular stresses such as hypoxia, ROS, and ER stress can induce autophagy. Nutrient depletion or low energy may also enhance autophagy by inhibiting mTORC1 and activating AMPK and SIRT1. Autophagy activation under non-diabetic conditions can maintain cellular homeostasis and protect against renal injury. b Under diabetic conditions, hyperglycemia activates mTORC1 and suppresses the activity of AMPK and SIRT1. Hyperactivated mTORC1 can inhibit autophagy by phosphorylating ULK1. The suppression of AMPK and SIRT1 further impairs autophagy under diabetic conditions. In early stage diabetes, hyperglycemia-induced intracellular stresses may activate autophagy as a compensatory response for cell protection. In late stage diabetes, however, sustained disturbance of the nutrient-sensing pathways overwhelmingly suppresses autophagy in the kidney, eventually leading to dysregulated autophagy and the progression of DKD. The solid lines in green with arrowheads represent stimulating effects. The sold lines in red with truncated end indicate inhibiting effects. The dotted lines stand for the regulatory effects that are disturbed and altered under diabetic conditions. AMPK AMP-activated protein kinase, ATF activating transcription factor, ATG autophagy-related, BNIP3 BCL2/adenovirus E1B 19-kDa interacting protein 3, CHOP C/EBP homologous protein, DEPTOR DEP domain-containing mTOR-interacting protein, DKD diabetic kidney disease, eIF2α eukaryotic initiation factor 2α, ER endoplasmic reticulum, FIP200 focal adhesion kinase family interacting protein of 200 kDa, FoxO3a forkhead box O3a, GβL G protein β-subunit-like protein, HIF1 hypoxia-inducible factor 1, IRE1 inositol requiring enzyme 1, JNK1 c-jun N-terminal kinase 1, LKB1 liver kinase B1, MAPLC3/LC3 microtubule-associated protein light chain 3, mTORC1 mechanistic target of rapamycin complex 1, PERK protein kinase RNA-like ER kinase, PRAS40 protein-rich Akt substrate of 40 kDa, PtdIns3K class III phosphatidylinositol 3 kinase, RAPTOR regulatory associated protein of mTOR, REDD1 regulated in development and DNA damage responses 1, Rheb Ras homolog enriched in brain, ROS reactive oxygen species, SIRT1 sirtuin 1, TSC1/2 tuberous sclerosis complex ½, ULK1/2 Unc-51-like ½, VPS vacuolar protein sorting
Hyperactivation of mTORC1 signaling pathway is frequently observed in animals and patients with DKD. p70S6K/S6RP was hyperactivated in glomerular mesangial cells in a murine model of type 2 diabetes [69]. Phosphorylation of both p70S6K/S6RP and 4E-BP1 was also increased in type 1 diabetic mouse kidneys [70]. Similar upregulation of mTORC1 pathway was further shown in the glomeruli of STZ-treated rats, accompanied with various DKD-related renal pathologies [71]. Moreover, podocyte-specific activation of mTORC1 in non-diabetic mice was sufficient to induce DKD-like renal damage including GBM thickening, ECM expansion, podocyte loss, and proteinuria [72]. This causative link between an elevated mTORC1 activity and the progression of DKD was further confirmed in both human and mouse models of DKD [73]. In diabetic proximal tubular cells, hyperactivation of mTORC1 signaling has been shown to induce apoptosis and hypertrophy [74, 75]. By contrast, inhibition of mTORC1 has renoprotective effects against DKD-induced injury. Rapamycin, a pharmacological inhibitor of mTORC1, attenuated the development of kidney injury and suppressed multiple pro-inflammatory and profibrotic cytokines in STZ-induced diabetic rats [76, 77]. Suppression of mTOR by rapamycin also reduced glomerulosclerosis, mesangial expansion, renal hypertrophy, and proteinuria in both STZ-induced diabetic rats and db/db mice [69, 75, 78–80]. In addition to rapamycin, partial suppression of mTORC1 activity in podocytes of heterozygous-Raptor knockout mice further demonstrated a strong connection of hyperactivated mTORC1 signaling with podocyte injury and proteinuria in diabetic kidneys [72, 73]. Autophagy was inhibited in cultured podocytes during prolonged high glucose treatment, which was reversed by rapamycin, suggesting that mTORC1 may participate in podocyte injury under diabetic condition by suppressing autophagy [81]. Consistently, the renoprotective effects of rapamycin via inhibiting mTORC1 for autophagy activation in podocytes were also shown in STZ-induced diabetic mice [82]. Furthermore, inhibition of mTORC1 activity by a very-low-protein diet in diabetic Wistar fatty rats also reversed autophagy impairment in PTECs and thereby ameliorated tubular cell damage, inflammation and interstitial fibrosis [83]. Taken together, these findings suggest that hyperactivation of mTOR signaling pathway, via negatively regulating autophagy, plays a critical role in the pathogenesis of DKD.
It is noteworthy that mTORC1 activity is indispensable to maintain podocyte homeostasis. Thus, mTORC1 inhibition under non-diabetic conditions has serious adverse effects on podocytes [84, 85]. Moreover, complete lack of mTORC1 activity in podocyte-specific Raptor-deficient mice results in severe podocyte injury, glomerulosclerosis and proteinuria, underscoring the crucial importance of maintaining a tightly controlled mTOR signaling pathway to ensure normal renal function [73].
AMPK and autophagy in DKD
AMP-activated protein kinase is an evolutionarily conserved heterotrimeric protein complex that serves as a master regulator integrating a diverse range of energy- and metabolism-related pathways. It monitors the energy status of a cell by sensing the AMP/ATP ratio. Activation of AMPK is mediated by several upstream kinases, and liver kinase B1 (LKB1) is a major kinase phosphorylating AMPK under conditions of energy stress [60, 86]. In contrast to mTORC1, AMPK is a positive regulator of autophagy in response to nutrient/energy depletion. On one hand, AMPK can directly phosphorylate ULK1 to promote autophagy [68, 87]. On the other hand, it can crosstalk with mTORC1 signaling by phosphorylating at least two proteins, TSC1/2 and RAPTOR, to inhibit mTORC1 for autophagy induction [88] (Fig. 1). In addition to the opposite regulation of ULK1 by AMPK and mTORC1, ULK1 has also been shown to phosphorylate and inhibit both of its upstream regulators [88]. The complex crosstalk and feedback of these three interconnected proteins may further fine-tune the autophagic response under metabolic stress conditions.
The activity of AMPK in the kidney is suppressed in experimental models of both type 1 and 2 diabetes, which, importantly, can be reversed by several AMPK activators, leading to the attenuation of diabetic kidney injury [89–95]. Resveratrol effectively mitigated ECM expansion, inflammation and proteinuria in db/db mice by promoting phosphorylation of AMPK [96]. Consistently, in STZ-induced diabetic kidneys resveratrol also reduced hyperglycemia, inhibited renal hypertrophy, and suppressed inflammation and proteinuria [92, 95]. A similar study by Kitada et al. further revealed a possible involvement of an AMPK-independent pathway in the renoprotective effects of resveratrol against DKD [94]. Metformin, a well-known AMPK stimulator and an antidiabetic drug, has also been shown to protect podocytes from diabetic injuries [97]. Similarly, both metformin and 5-aminoimidazole-4-carboxamide-1β-riboside (AICAR) enhanced AMPK phosphorylation, attenuated mTOR activity, and ameliorated high glucose-induced tubular hypertrophy in vitro as well as STZ-induced diabetic kidney injury in vivo [89, 93]. Activation of AMPK by AICAR has also been shown to improve renal injury and proteinuria in diabetic Akita mice [98]. A recent study further suggested that the renoprotective effects of berberine in diabetic mice were AMPK-dependent [99]. In cultured mouse podocytes Berberine suppressed high glucose-induced cell apoptosis by reactivating AMPK and autophagy [100]. In renal tubular epithelial cells, hydrogen sulfide was also shown to activate AMPK and inhibit high glucose-induced matrix protein synthesis; and interestingly, these effects were abrogated by compound C, a commonly used AMPK inhibitor [101]. Moreover, fenofibrate protected kidneys against diabetic injury by reducing oxidative stress and endothelial dysfunction via enhancing the mRNA expression of LKB1 and AMPK in STZ-induced diabetic rats [102]. Together, these results suggest that inactivation of AMPK may inhibit autophagy and participate in the development of DKD.
In addition to autophagy regulation, AMPK also mediates a number of metabolic processes in the kidney, such as lipid metabolism, glycogen synthesis, and tubular reabsorption of salt and glucose. Decreases in renal AMPK activity under diabetic conditions may compromise these metabolic events along with autophagy inhibition, resulting in tubular and glomerular pathologies of DKD [52].
SIRT1 and autophagy in DKD
Sirtuins (SIRTs) are NAD+-dependent Class III histone deacetylases (HDACs) and play an important role in helping cells cope with redox and metabolic stresses [61, 103, 104]. Seven mammalian SIRTs (SIRT1 to SIRT7) have been identified, and SIRT1 is most studied. SIRT1 functions as an intracellular nutrient sensor by monitoring the NAD+ level and regulates metabolic and redox status. In response to calorie restriction and oxidative stress, SIRT1 is upregulated by elevated intracellular NAD+ levels, whereas lowered NAD+ availability during hypoxia may inactivate SIRT1 [61, 103, 104]. SIRT1 is a positive regulator of autophagy. Once activated, SIRT1 promotes autophagy by deacetylating essential autophagy proteins, such as ATG5, ATG7, and LC3 [105]. Moreover, SIRT1 deacetylates the transcriptional factor forkhead box O3a (FoxO3a), which leads to enhanced expression of proautophagic BCL2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) [106] (Fig. 1). In addition, SIRT1 can crosstalk with AMPK and mTOR pathways, regulating energy metabolism and pro-survival pathways including autophagy [107, 108].
In the kidney, SIRT1 is primarily expressed in the inner medulla and renal interstitium, where it appears to be renoprotective against oxidative stress [109]. Similar to AMPK, the expression and activity of SIRT1 tend to be decreased in renal cells in human and animal models of DKD, and activation of SIRT1 protects the kidney from diabetic injury [110]. A reduced SIRT1 expression was shown in glomeruli of diabetic mice, leading to AGEs accumulation and FoxO4 acetylation, and resveratrol protected podocytes from AGEs-induced apoptosis [111]. Suppression of SIRT1 was also detected in both podocytes and proximal tubules in diabetic mice prior to the development of proteinuria [112]. Importantly, mice with overexpressed SIRT1 specifically in proximal tubules were resistant to diabetes-related progression of podocyte damage and subsequent proteinuria [112]. In STZ-induced diabetic rats, SIRT1 expression was decreased, and intraperitoneal injection of 3,5-Diiodo-l-thyronine restored SIRT1 expression, and as a result, mesangial expansion was significantly attenuated [113]. However, sirtinol, a SIRT1 inhibitor, blocked the beneficial actions of SIRT1 activation [113]. The protective effects of SIRT1 activation in diabetic kidneys were also shown by Li et al. using tetrahydroxystilbene glucoside [114]. Similarly, calorie restriction, known to increase SIRT activity, activated autophagy in a mouse model of type 2 diabetes and ameliorated glomerular and tubular injury of DKD [56]. In STZ-treated rats, resveratrol was shown to limit diabetic kidney injury by suppressing the secretion of ECM and pro-inflammatory cytokines via the upregulation of SIRT1/FoxO1 signaling [115]. Resveratrol in db/db mice also reduced proteinuria, and ameliorated glomerulosclerosis and tubular apoptosis [96]. In high glucose-treated mesangial cells, resveratrol reduced oxidative stress and cell senescence [116, 117]. A recent study further confirmed that the renoprotective effects of resveratrol in type 2 diabetic rats and in hypoxia-treated PTECs was dependent on its reactivation of SIRT1 and subsequent restoration of autophagy [118]. Together, these findings suggest that SIRT1 is protective in the kidney and decreased SIRT1 activity contributes to renal injury associated with DKD via inhibiting autophagy.
In mammalian cells 18 HDAC genes have been identified and classified into four groups depending on sequence identity and domain organization. While class III SIRTs are NAD+-dependent, the other (class I, II and IV) HDACs are Zn+-dependent for enzymatic activity. In sharp contrast to the renoprotective effects of class III SIRTs, recent studies further revealed a pathological role of class I, II and IV HDACs-mediated epigenetic modifications in the development of DKD. Under these conditions HDACs inhibition appeared to be beneficial, suggesting that different HDACs may have very distinct functions in DKD [119–121]. To determine the role of individual HDACs, Wang et al. analyzed the expression patterns of HDACs using different models of DKD [122]. Among all HDACs examined, HDAC2/4/5 were upregulated in the kidney of STZ-induced diabetic rats, db/db mice, and in kidney biopsies from diabetic patients. Of interest, different types of renal cells showed different expression patterns, with HDAC2 in PTECs, HDAC4 in podocytes and HDAC5 in mesangial cells. Increased expression of HDAC4 was also observed in cultured podocytes treated with high glucose, AGEs, and transforming growth factor (TGF)-β, suggesting that HDAC4 may play a central role in regulating podocyte function during DKD. Indeed, the selective upregulation of HDAC4 in podocyte suppressed autophagy by deacetylating signal transducers and activators of transcription factor 1 (STAT1) and led to podocyte injury. Inhibition of HDAC4 prevented autophagy deficiency in podocytes and thereby protected podocytes against DKD-induced glomerulopathy [122]. Together, these results further confirm the connection between defective autophagy and the progression of DKD. Importantly, given the opposite regulation of autophagy by class III SIRTs and class II HDAC4 in diabetic kidneys, targeting HDACs for the treatment of DKD needs to be HDAC-isoform specific.
Obesity-mediated autophagy deficiency in DKD
Obesity is a widespread health issue and associated with the development of various diseases including type 2 diabetes, hypertension and arthrosclerosis. It is also an independent risk factor for renal dysfunction in patients with chronic kidney disease (CKD). Extensive studies have demonstrated dysregulated autophagy caused by high-fat diet (HFD) and obesity in the hypothalamus, liver, heart, pancreatic β-cells and adipocytes. Recently, emerging evidence further suggest that HFD and obesity negatively regulates autophagy in the kidney and promotes the progression of obesity-related CKD [123]. Yamahara et al. examined the effects of diet-induced obesity on autophagy in PTECs in response to proteinuria [57]. Obesity suppressed proteinuria-induced autophagy in proximal tubules and exacerbated tubular cell damage by proteinuria. mTOR was activated in proximal tubules of obese mice and obese patients with proteinuria, and treatment with an mTOR inhibitor reversed obesity-mediated autophagy inhibition, suggesting a negative regulation of autophagy by mTOR under these conditions [57]. Similar observations were also shown in diabetic Wistar fatty rats [56, 83]. Compared with non-diabetic Wistar lean rats, there were increased phosphorylation of p70S6K/S6RP, decreased SIRT1 expression, and accumulation of damaged mitochondria and p62/SQSTM1 in proximal tubules of diabetic Wistar fatty rats. Dietary restriction rescued autophagy deficiency via the suppression of mTORC1 and activation of SIRT1 pathways, and thereby ameliorated tubulointerstitial damage and renal function loss in these obese rats [56, 83]. In HFD-fed mice, restoration of autophagy and its renoprotection against obesity-induced tubular injury was also achieved by AMPK activation [124]. In addition, megalin, an endocytic receptor, was involved in obesity-induced autophagy impairment in proximal tubules of HFD-fed mice. The megalin-mediated endocytosis of glomerular-filtered lipotoxic substances into proximal tubules primarily induced tubular hypertrophy and senescence with autophagy impairment, which further led to glomerular and peritubular capillary damage and tubulointerstitial fibrosis [125]. A recent study by Yamamoto et al. further showed that HFD-induced autophagy insufficiency was associated with lysosomal dysfunction in PTECs. Enhanced phospholipids accumulated in enlarged lysosomes in both HFD-fed mice and obese patients, leading to impaired autophagic flux that could aggravate lipotoxicity and ischemia–reperfusion injury in the kidney [126]. Tagawa et al. also revealed absence of autophagosomes and accumulation of p62/SQSTM1 and huge damaged lysosomes in the podocytes of Otsuka Long–Evans Tokushima Fatty (OLETF) rats with massive proteinuria and HFD-fed podocyte-specific Atg5 knockout mice. Importantly, this impaired autophagy-lysosomal pathway in podocytes was associated with severe podocyte injury and the progression of DKD under obesity and type 2 diabetes [127].
Intracellular stress in autophagy regulation and DKD
As mentioned above, in addition to the nutrient-sensing pathways, autophagy is also regulated by several intracellular stress signals, such as hypoxia, oxidative stress, and ER stress. Under diabetic conditions, hyperactivation of mTORC1 and/or inactivated AMPK and SIRT1 suppress autophagy in the kidney; however, as a compensatory response, autophagy is induced by these stresses signaling to maintain cell integrity. When this adaptive attempt fails, damaged organelles, such as mitochondria and ER, may accumulate in cells and exacerbate DKD.
Oxidative stress, autophagy and DKD
Reactive oxygen species (ROS) are commonly produced by mitochondrial respiration and redox-mediating enzymes in various metabolic pathways [128, 129]. Excessive ROS are often responsible for cell damage, and as a defensive mechanism, cells are equipped with a set of antioxidant enzymes. Oxidative stress occurs as a consequence of the imbalance between ROS generation and antioxidant defenses [128, 129]. Under diabetic conditions, the production of ROS in the kidney is enhanced by hyperglycemia [130, 131]. High glucose-mediated alterations of intracellular metabolism, including auto-oxidation of glucose, advanced glycation, activation of PKC, and increased polyol pathway flux, are the sources of ROS in the diabetic kidney. High levels of free fatty acids (FFA) may also stimulate ROS generation under diabetic conditions [132]. Moreover, accumulation of damaged mitochondrial in diabetic kidneys significantly contributes to ROS generation [19]. Normalizing mitochondrial superoxide production has been shown to block high glucose-induced metabolic changes [133].
It has been suggested that acute exposure to high glucose may activate autophagy via ROS. In immortalized murine podocytes exposed to high glucose, autophagy was induced within 24 h along with ROS generation, which was inhibited by antioxidant N-acetylcysteine [134]. Exposure of podocytes to angiotensin II for 36 h also enhanced ROS generation and induced autophagy, and treatment with antioxidants inhibited autophagy under this condition [135]. ROS-mediated autophagy was further seen in cultured podocytes treated with palmitic acid, a saturated FFA, for 24 h [136]. Together, these results suggest an active autophagy in podocytes in response to ROS in early stage diabetes. ROS induces autophagy through multiple mechanisms. Exogenous hydrogen peroxide can activate protein kinase RNA-like ER kinase (PERK), which in turn phosphorylates eukaryotic initiation factor 2α (eIF2α), activates ATG4 to accelerate the conversion of LC3-I to LC3-II, and inhibits mTORC1 activity [137]. In addition, activation of mitogen-activated protein kinases, including c-jun N-terminal kinase (JNK1), is also involved in ROS regulation of autophagy [138] (Fig. 1). Oxidative stress can induce autophagy to remove damaged mitochondria (mitophagy). This autophagy-mediated mitochondrial quality control and subsequent reduction of ROS should be essential to protect kidney under diabetic conditions [19].
ER stress, autophagy and DKD
The ER is involved in protein synthesis and maturation. It contains a quality-control mechanism to handle misfolded proteins, and thus ER stress refers to the physiological or pathological states that result in the accumulation of misfolded proteins [139, 140]. Hyperglycemia and high levels of FFA induced ER stress in podocytes, leading to podocyte apoptosis and subsequent proteinuria in DKD [141, 142]. Interestingly, proteinuria filtered from glomeruli further aggravated the ER stress response in proximal tubules, resulting in tubulointerstitial lesions [143, 144]. In tubular cells exposed to high glucose and albumin, ER stress was also induced, along with tubular cell apoptosis [144, 145]. In addition, the presence of ER stress in aged diabetic mice activated C/EBP homologous protein (CHOP), leading to increased diabetic kidney injury. CHOP-deficient mice were protected from DKD, further suggesting that diabetes-induced ER stress plays an important role in the development of kidney injury [146].
ER stress, through the unfolded protein response (UPR), can also activate autophagy for cell protection. The UPR consists of three main branches that are controlled by the ER membrane proteins: PERK, activating transcription factor 6 (ATF6), inositol requiring enzyme 1 (IRE1) [139, 140]. PERK induces autophagy by mediating the transcriptional activation of LC3 and ATG5 through the ATF4 and CHOP, respectively [147]. ATF6 has also been suggested to induce autophagy. IRE1 plays a dual role in autophagy regulation, with stimulating autophagy by JNK1-mediated BECN1 activation, whereas inhibiting autophagy via the downstream effector X-box binding protein 1(XBP1) [47, 148] (Fig. 1). Autophagy also plays an important role in maintaining the structural and functional integrity of the ER. Therefore, defective autophagy under diabetic conditions may lead to prolonged ER stress and kidney injury. Therapeutic strategies that can activate autophagy for ER degradation (ERphagy) may be required to protect kidney from cytotoxic ER stress. Several recent studies using chemical chaperones have demonstrated that enhanced protein folding can reduce ER stress and suppress diabetic kidney injury. In cultured mouse podocytes, tauroursodeoxycholic acid (TUDCA), a chemical chaperon, was shown to inhibit AGEs-induced apoptosis by reducing ER stress [149]. The renoprotective effects of TUDCA were further observed in diabetic mice. Autophagy was restored in TUDCA-treated mice, accompanied by reduced podocyte injury and proteinuria [81, 150]. Furthermore, in both STZ-induced diabetic rats and db/db mice, prolonged ER stress-induced inflammatory response and kidney injury, which was also inhibited by 4-phenylbutyric acid (4-PBA), a chemical chaperon [151–153]. In addition, both TUDCA and 4-PBA were shown to reactivate autophagy in db/db mice and in high glucose-treated podocytes, thus preventing ER stress-induced podocyte apoptosis under diabetic conditions [153]. Together, these results further confirm the involvement of ER stress in the pathogenesis of DKD. The renoprotective effects of the chemical chaperons by inhibiting ER stress may be facilitated through reactivation of autophagy.
Hypoxic stress, autophagy and DKD
Hypoxia is an important event during DKD progression and generally attributed to chronic ischemia that may result from RAS-induced renal vasoconstriction, hyperglycemic injury of red blood cells, decreased nitric oxide activity, and rarefaction of the glomerular and peritubular capillaries due to renal fibrosis [29, 132]. The best understood response to hypoxia is mediated by the hypoxia-inducible factor (HIF) family of transcription factors including HIF1, HIF2, and HIF3. Both HIF1 and HIF2 are involved in hypoxic signaling with overlapping but partly distinct functions, whereas the role of HIF3 is unclear [154–156].
Hypoxia induces autophagy through both HIF-dependent and -independent mechanisms [157, 158]. HIF1 is activated under hypoxia and induces autophagy by transcriptional upregulation of BNIP3 and BNIP3 like (BNIP3L/NIX) to liberate BECN1 from its inhibitory interaction with BCL2 [159–161] (Fig. 1). Importantly, HIF1-mediated BNIP3 may trigger selective mitophagy under hypoxic conditions to remove damaged mitochondria and reduce excessive ROS generation [160]. Depending on the duration and severity of hypoxic stress, two other oxygen-sensing pathways have also been suggested to regulate autophagy. Severe hypoxia (0.01% oxygen or anoxia) may induce ER stress and activate autophagy through the UPR, for example, PERK pathway. In addition, by activating LKB1–AMPK and REDD1 (regulated in development and DNA damage responses), hypoxia can also stimulate TSC1/2 and thereby inhibit mTORC1 activity for autophagy induction [157, 158] (Fig. 1). Regulation of autophagy coordinated by these signaling pathways in diabetic kidneys would promote hypoxia tolerance and protect against hypoxic renal injury.
Podocyte autophagy in DKD
Podocytes are terminally differentiated glomerular epithelial cells and play a critical role in maintaining the integrity of glomerular filtration barrier. Podocyte loss due to apoptosis and podocyte dysfunction results in massive proteinuria in DKD [162, 163]. Thus, a decrease in podocyte number is a valuable predictive factor for the progression of DKD [164]. Maintaining podocyte homeostasis is regarded as a therapeutic target in DKD.
The autophagy-lysosomal pathway plays an essential role in maintaining podocyte function. Podocytes show high rates of autophagy even under basal conditions, suggesting that autophagy is a fundamental self-repair mechanism for podocyte homeostasis [165–167]. Podocyte-specific Atg5 deletion mice developed glomerulopathy with aging, showing accumulation of oxidized and ubiquitinated proteins and ER stress in podocytes that ultimately led to podocyte loss, proteinuria and glomerulosclerosis [165]. Since autophagy degradation involves lysosomes, the impairment of lysosomal function in podocytes by deletion of mTOR, prorenin, and mVps34 genes also led to severe glomerulosclerosis and proteinuria [168–170]. These results highlight the importance of an intact autophagic flux pathway in maintaining podocyte homeostasis.
The role of autophagy in podocytes under diabetic conditions is being uncovered, and emerging evidence has suggested that dysregulated autophagy may be involved in the diabetic glomerular lesions (Table 1). Immortalized murine podocytes exposed to prolonged high glucose for 48 h had decreased autophagic activity along with reduced expression of BECN1 and ATG12–ATG5 complex. Inhibition of basal autophagy in these cultured podocytes by 3-methyladenine or BECN1 siRNA suppressed podocin expression and impaired the filtration barrier function of podocytes, whereas rapamycin restored autophagy and thereby rescued podocytes from injury [81]. Consistently, in STZ-induced diabetic mice and in renal biopsy samples from diabetic patients, autophagy activity was also decreased in podocytes [81]. These results suggest that sustained hyperglycemia reduces autophagy in podocytes, leading to alterations in podocyte function that may accelerate diabetic injury. Along with autophagy impairment, ER stress was induced in these cultured podocytes by prolonged high glucose treatment for up to 60 h [81]. There was a brief increase and then a decrease of eIF2α phosphorylation in podocytes, accompanied with upregulation of proapoptotic CHOP, indicating a switch from the adaptive UPR for cytoprotection to a cytotoxic ER stress response. This switch appeared to be associated with insufficient autophagic clearance of damaged ER, as inhibition of ER stress by salubrinal or TUDCA restored autophagy and attenuated podocyte injury [81].
Table 1.
Dysregulated autophagy in different types of renal resident cells during DKD
| Cell types | Experimental models | Major findings | References |
|---|---|---|---|
| Podocytes | CIMPs, primary mouse podocyte cultures, STZ-induced diabetic mice | Autophagy induction via ROS in response to short-term treatment of high glucose, angiotensin II, or saturated FFA or in mice at 4-wk post-STZ injection | [134–136, 171] |
| Cultured mouse podocytes, STZ-induced diabetic mice, diabetic patient kidney biopsies | Autophagy impairment following prolonged exposure to high glucose and in diabetic mice and patients, insufficient autophagic clearance of damaged ER | [81] | |
| STZ-induced diabetic mice, podocyte-specific Atg5 knockout mice | Autophagy suppression in mice at 8-week post-STZ injection, autophagy deficiency accelerates podocyte injury, injurious effects of podocyte autophagy loss on both podocytes and nearby mesangial cells | [171] | |
| OLETF rats, HFD-fed mice, type 2 diabetic kidney biopsies, immortalized Atg7-deficient podocytes | Autophagy impairment is associated with DKD progression to massive proteinuria and severe podocyte injury, failed clearance of damaged lysosomes, serum factor-dependent intracellular signaling pathway in regulating autophagy | [127] | |
| CIMPs, STZ-induced diabetic mice | mTORC1-dependent autophagy suppression, renoprotective effects of rapamycin via inhibiting mTORC1 for autophagy reactivation | [81, 82] | |
| CIMPs, STZ-induced diabetic rats, db/db mice, diabetic patient kidney biopsies | Upregulation of HDAC4 in diabetic podocytes suppresses autophagy by deacetylating STAT1 | [122] | |
| CIMPs, STZ-induced diabetic rats, db/db mice, diabetic patient kidney biopsies | β-Arrestins suppress autophagy via interaction with ATG7 and downregulation ATG12-ATG5 conjugation | [172] | |
| CIMPs | A negative regulation of autophagy by miR-21 via the inhibition of PTEN | [173] | |
| CIMPs, STZ-induced diabetic mice | FoxO1 promotes mitophagy via PINK1/Parkin pathway | [174] | |
| PTECs | STZ-induced diabetic rats | Autophagy inhibition in both proximal and distal tubules | [53, 54] |
| STZ-induced diabetic mice | SGLT2-mediated glucose uptake inhibits autophagy | [55] | |
| HK-2 cells, STZ-induced diabetic rats, human diabetic kidney biopsies | Upregulation of TXNIP, via activating mTORC1, inhibits autophagy | [180] | |
| Diabetic Wistar fatty rats, type 2 diabetic mice | Autophagy suppression reversed by calorie restriction via activating SIRT1 | [56] | |
| Diabetic Wistar fatty rats | Autophagy suppression reversed by a very-low-protein diet via inhibiting mTORC1 | [83] | |
| HFD-fed mice, PTEC-specific Atg5 knockout mice, human type 2 diabetic kidney biopsies | mTORC1-mediated autophagy inhibition worsens proteinuria-induced tubular injury, reversed by dietary restriction and rapamycin | [57] | |
| HFD-fed mice | Megalin-mediated endocytosis of glomerular-filtered lipotoxic substances impairs autophagy | [125] | |
| HFD-fed mice, kidney biopsies from obese patients | HFD-induced autophagy insufficiency leads to lysosomal dysfunction, which in turn impairs autophagic flux and aggravates lipotoxicity | [126] | |
| HK-2 cells, human diabetic kidney biopsies | AGEs overload induces lysosomal dysfunction and subsequent inhibition of autophagic flux | [182] | |
| Primary PTECs, STZ-induced diabetic mice, PTEC-specific Atg5 knockout mice | Autophagy deficiency by AGEs overload or in diabetic mice fails to promote lysosomal biogenesis, leading to reduced lysosomal clearance of AGEs and AGEs accumulation | [183] | |
| Mesangial cells | STZ-induced diabetic mice, human diabetic kidney biopsies, TIMP3-deficient mice | Downregulation of TIMP3 under diabetic conditions suppresses autophagy in mesangial cells via FoxO1/STAT1 pathway | [184] |
| Cultured rat mesangial cells | AGEs via ROS induced autophagy and mitophagy to protect mesangial cells against AGEs-induced mitochondrial damage and cell apoptosis | [185] | |
| Cultured rat mesangial cells | High glucose suppresses autophagy in mesangial cells via miR-21/PTEN/Akt/mTOR pathway | [186] | |
| Glomerular endothelial cells | Cultured mouse GECs | Autophagy regulates BAMBI turnover in endothelial cells | [187] |
| STZ-induced diabetic mice, type 2 diabetic mice, human diabetic kidney biopsies | Downregulation of BAMBI under diabetic conditions enhances alternative TGF-β signaling and glomerular dysfunction | [188] | |
| STZ-induced diabetic mice, GEC-specific Atg5 knockout mice | Autophagy in GECs preserves both endothelial integrity and podocyte ultrastructure to maintain GFB homeostasis | [171] |
CIMPs conditionally immortalized mouse podocytes, STZ streptozotocin, OLETF Otsuka Long–Evans Tokushima Fatty, HFD high-fat diet, FFA free fatty acid, ER endoplasmic reticulum, DKD diabetic kidney disease, mTORC1 mammalian target of rapamycin (mTOR) complex 1, HDAC4 histone deacetylase 4, STAT1 signal transducers and activators of transcription factor 1, ATG autophagy-related gene, PTEN phosphatase and tensin homolog, FOXO1 forkhead box O1, PINK1 putative kinase 1, SGLT2 sodium–glucose cotransporter 2, TXNIP thioredoxin-interacting protein, SIRT1 sirtuin 1, AGEs advanced glycation end products, TIMP3 tissue inhibitor of metalloproteinase 3, Akt protein kinase B, BAMBI BMP and activin receptor membrane-bound inhibitor, TGF-β transforming growth factor-β, GFB glomerular filtration barrier, PTECs proximal tubular endothelial cells, GECs glomerular endothelial cells
The effects of high glucose on podocyte autophagy seem to be time-dependent. Immortalized murine podocytes showed increased LC3-II accumulation and autophagosome formation within 24 h of high glucose treatment, which was further enhanced by rapamycin and inhibited by 3-methyladenine [134]. Lenoir et al. also showed that 24-h high glucose promoted autophagy flux in primary podocytes and deletion of Atg5 sensitized the podocytes to high glucose-induced apoptosis. In SVI cells, a stable podocyte cell line, autophagy was induced by high glucose at 48-h, but suppressed at 15-day of treatment [171]. In STZ-induced diabetic mice, increased GFP-LC3 staining and LC3-II turnover were observed in podocytes at 4-week post-STZ injection, when mice were hyperglycemic but had not yet developed glomerular lesions. At 8-week post-STZ injection, when mice showed glomerular lesions, GFP-LC3 staining was reduced, along with p62/SQSTM1 accumulation in glomerular cells. Deletion of Atg5 specifically in podocytes resulted in accelerated podocytopathy with a leaky glomerular filtration barrier in these type 1 diabetic mice. TEM ultrastructure analyses further confirmed the presence of substantial mesangial expansion and glomerulosclerosis in podocyte-specific autophagy-deficient mice. The injurious effects of podocyte autophagy loss on themselves and nearby mesangial cells underscored the communication between these two types of cells under diabetic conditions [171]. Together, these in vitro and in vivo results confirm that high glucose induces autophagy in podocytes for renoprotection in short-term (early stage diabetes) but represses it in long-term (late stage diabetes), leading to aggravated glomerular injury and the progression in DKD.
Using both type 2 diabetic patient samples and obese rodent models, Tagawa et al. further verified that autophagy insufficiency in podocytes played a key role particularly in the disease progression to massive proteinuria in advanced DKD [127]. Impaired formation of LC3 puncta and accumulation of p62/SQSTM1 were found in the podocytes of type 2 diabetic patients and OLETF rats with massive proteinuria accompanied by podocyte loss, but not in those with absent or minimal proteinuria. Compared with HFD-fed control mice that had minimal proteinuria, autophagy deficiency in podocyte-specific Atg5 knockout mice enhanced HFD-induced injury and these mice developed more severe podocyte damage, exacerbated proteinuria, and increased tubulointerstitial fibrosis. Under these conditions, autophagy impairment led to an accumulation of damaged lysosomes in podocytes, further indicating an important role of autophagy in the clearance of damaged lysosomes and its contribution to the progression of podocyte injury in DKD. Of interest, stimulation of cultured podocytes with sera from patients and OLETF rats with diabetes and massive proteinuria impaired autophagy–lysosomal pathway and induced podocyte apoptosis, suggesting a potential mechanism that may negatively regulate podocyte autophagy through serum factor-dependent intracellular signaling pathways in the progression of DKD [127].
The mechanism underlying diabetes-related impairment of podocyte autophagy is under investigation. In addition to altered nutrient-sensing pathways and intracellular stress signaling described above, a few recent studies have provided additional insights. Liu et al. showed that β-arrestins, negative adaptors of G protein-coupled receptors (GPCRs), were upregulated in the kidney of STZ-induced diabetic mice, db/db mice and kidney biopsies from diabetic patients. Hyperglycemia also induced β-arrestins in cultured podocytes. β-Arrestins interacted with ATG7 to downregulate ATG12-ATG5 conjugation, thereby suppressing autophagy in podocytes [172]. Sun et al. examined microRNA regulation of podocyte autophagy. miR-217 was elevated in podocytes by high glucose, leading to podocyte injury and insulin resistance. Blocking miR-217 expression protected podocytes against high glucose injury by reactivating autophagy. Phosphatase and tensin homolog (PTEN) was a target of miR-217 in podocytes. Downregulation of PTEN restrained autophagy reactivation and attenuated the beneficial role of miR-217 repression in high glucose-treated podocytes. Together, these results suggest a negative regulation of podocyte autophagy by miR-217 via suppressing PTEN and its contribution to high glucose-induced podocyte injury and insulin resistance [173]. Li et al. further demonstrated a role of FoxO1 in regulating mitophagy in podocytes. Aberrant mitochondria accumulated in STZ-induced diabetic mice and in high glucose-treated mouse podocytes. Overexpression of FoxO1 promoted mitophagy via PTEN-induced putative kinase 1 (PINK1)/Parkin pathway, leading to the amelioration of podocyte injury [174].
Based on the findings, we highlighted above autophagy is likely induced in podocytes in early stage DKD via the activation of various intracellular stress signaling such as ROS, ER stress, etc. Induction of autophagy in early stage DKD attempts to eliminate protein aggregates and damaged organelles including mitochondria, ER and lysosomes for renoprotection. However, in late stage DKD (or chronic exposure to high glucose in vitro), sustained disturbances of nutrient-sensing pathways and increasing numbers of newly identified signaling pathways may overwhelmingly impair podocyte autophagy, leading to the accumulation of damaged organelles that may switch the cytoprotective stress signaling to a cytotoxic state with no returning point. Dysregulated autophagy further aggravates proteinuria and podocyte injury, eventually resulting in DKD progression to ESRD.
Proximal tubular epithelial cell (PTEC) autophagy in DKD
The autophagic response in PTECs appears to be different from that induced in podocytes. Renal PTECs display a very low level of basal autophagy under normal conditions [175]. Mice with proximal tubule-specific Atg5 or Atg7 deletion develop premature renal aging, suggesting that a low but sufficient level of basal autophagy is needed to maintain cellular homeostasis in PTECs [175, 176]. Autophagy is remarkably activated in PTECs under various stress conditions including nephrotoxic stress and ischemic/hypoxic stress, and plays a renoprotective role against tubular injury and cell death [36, 177, 178].
In diabetic conditions, hyperglycemia, hyperinsulinemia, and high level of FFA are major metabolic alterations. Among these diabetes-induced metabolic changes, hyperglycemia has been shown to inhibit autophagy in proximal and distal tubular cells in both type 1 and 2 diabetic animals [53, 54, 56]. The uptake and excretion of glucose occurs mainly in proximal tubules via sodium–glucose cotransporter 2 (SGLT2). SGLT2 promotes high-capacity glucose uptake in PTECs, and inhibition of SGLT2 enhances tubular excretion of glucose and reduces blood glucose concentrations [179]. Knockout of Sglt2 attenuated STZ-induced renal accumulation of p62/SQSTM1, suggesting that SGLT2-mediated glucose uptake may contribute to autophagy inhibition in PTECs under diabetic conditions [55]. Huang et al. demonstrated that thioredoxin-interacting protein (TXNIP), a natural inhibitor of thioredoxin, was induced by hyperglycemia and suppressed tubular autophagy in STZ-induced diabetic rats, human diabetic kidney biopsies, and cultured human PTECs exposed to high glucose. The negative regulation of tubular autophagy by TXNIP was mediated by activation of the mTOR pathway [180].
In diabetic patients, the severity of proteinuria-induced tubulointerstitial lesions is correlated strongly with renal outcome [21]. Proteinuria filtered from the glomeruli is a nephrotoxic stress and an increased reabsorption of the protein was shown to activate autophagy in PTECs. Proteinuria-induced tubular cell injury was exacerbated in mice with proximal tubule-specific Atg5 knockout [57]. Obesity was also shown to suppress autophagy in PTECs in a mouse models of type 2 diabetes and in renal biopsy specimens from type 2 diabetic patients, and insufficient autophagy aggravated proteinuria-induced tubulointerstitial injury. Mechanistically, hyperactivated mTORC1 signaling was involved in obesity-induced autophagy inhibition, as diet restriction or rapamycin restored autophagy under this condition [57]. Similarly in diabetic Wistar fatty rats, autophagy was also suppressed in PTECs, as indicated by accumulation of damaged mitochondria and p62/SQSTM1. Again, dietary restriction rescued autophagy deficiency via the suppression of mTORC1 and activation of SIRT1 pathways, which in turn led to the attenuation of diabetic tubular injury and renal dysfunction [56, 83]. Collectively, these results suggest that proteinuria induces autophagy in PTECs primarily serving as a protective mechanism for cell survival. However, diabetic conditions may suppress proteinuria-induced autophagy in PTECs via hyperactivated mTORC1 or decreased SIRT1 pathways, leading to exacerbated tubulointerstitial lesions.
As mentioned above, AGEs are generated in the hyperglycemia state and involved in the progression of DKD. AGEs filtered by glomeruli or delivered from the circulation are endocytosed and degraded in the lysosomes of PTECs [181]. Recent studies demonstrate an interconnection between autophagy impairment and AGEs accumulation in PTECs and its contribution to tubular injury in DKD. In cultured human PTECs (HK-2 cell line) and PTECs from diabetic patients, Liu et al. showed that AGEs overload induced a disrupted autophagy-lysosomal pathway [182]. AGEs, via interaction with AGEs-receptors (RAGEs), evoked oxidative stress and thereby stimulated lysosomal membrane permeabilization and lysosomal dysfunction. As a result, autophagic flux was disrupted in these PTECs, resulting in accumulation of p62/SQSTM1 and ubiquitinated proteins, failed fusion of autophagosomes with lysosomes, and impaired lysosomal turnover of LC3-II [182]. Takahashi et al. further confirmed that AGEs overload gradually suppressed autophagic flux in primary culture of PTECs. Of note, lysosomal-associated membrane protein 1 (LAMP1) expression in response to AGEs overload was upregulated in Atg5-competent primary PTECs, but inhibited in Atg5-deficient cells, suggesting a role for autophagy in promoting lysosomal biogenesis and function under diabetic conditions. Consistently, STZ-treated, PTEC-specific, autophagy-deficient mice failed to upregulate lysosomal biogenesis and exhibited an increased accumulation of AGEs in PTECs, glomeruli and renal vasculature, along with enhanced inflammation and renal fibrosis [183]. Together, these results suggest that autophagy inhibits AGEs accumulation by promoting lysosomal biogenesis and function in PTECs. Impaired autophagy under diabetic conditions fails to remove damaged lysosomes and results in AGEs accumulation. AGEs overload, in turn, further induces lysosomal dysfunction and disrupts lysosomal clearance of AGEs. This vicious cycle ultimately leads to irreversible injury not only in PTECs, but may also largely affect glomeruli and tubulointerstitium, resulting in the progression of DKD.
Mesangial cell autophagy in DKD
Mesangial cells produce ECM components in the mesangium and play an important role in maintaining mesangial matrix homeostasis. In response to injury and progressive CKD, mesangial cells proliferate and produce excessive ECM, leading to glomerulosclerosis and renal fibrosis. Expansion of the cellular and matrix components in the mesangium is a histological characteristic of both type 1 and 2 DKD. Although the function of autophagy in mesangial cells under diabetic conditions is unclear, a few recent studies have just started the journey of exploration (Table 1).
Fiorentino et al. showed that renal expression of tissue inhibitors of metalloproteinase 3 (TIMP3) was decreased in STZ-induced diabetic mice and diabetic patients. Compared with control mice, Timp3-deficient diabetic mice had increased albuminuria, glomerular hypertrophy, GMB thickening, mesangial expansion and interstitial fibrosis. Timp3-deficiency led to a decreased expression of FoxO1 in the kidney and an increased expression of STAT1, a repressor of FoxO1 transcription. As a result, the expression of FoxO1-targeted autophagy-related genes, including Atg5, Becn1 and Lc3, was also suppressed in Timp3-deficient diabetic mice. Consistently, the altered expressions of STAT1, FoxO1 and FoxO1-targeted autophagy-related genes were also shown in kidney biopsies from diabetic patients and in cultured mesangial cells with Timp3 deletion. Re-expression of TIMP3 in these mesangial cells rescued the expression of FoxO1 and its targets, reduced STAT1 expression, and as a result, reversed autophagy inhibition [184]. Together, these findings suggest that under diabetic conditions, downregulation of TIMP3 may suppress autophagy in mesangial cells via FoxO1/STAT1 pathway, resulting in DKD progression.
Xu et al. further examined autophagy in regulating the survival of mesangial cells treated with AGEs [185]. They found that ROS induced by AGEs stimulated mitochondrial depolarization and apoptosis in cultured mesangial cells. Meanwhile, AGEs via ROS also activated autophagy and mitophagy in mesangial cells, which played an important role in reducing mitochondrial injury and ROS generation. Inhibition of autophagy enhanced AGEs-induced mesangial cell apoptosis, further confirming a protective role of autophagy in AGEs-treated mesangial cells [185].
In cultured rat mesangial cells, autophagy was suppressed in response to high glucose treatment, accompanied by collagen I accumulation and cell hypertrophy and proliferation. High glucose also upregulated miR-21 in mesangial cells, along with downregulated PTEN and increased phosphorylation of phosphoinositide 3 kinase (PI3K), protein kinase B (PKB/Akt) and mTOR. These alterations were reversed by ursolic acid, and as a result, autophagy was reactivated, leading to the attenuation of mesangial injury and collagen I production [186]. These results suggest a negative regulation of autophagy in mesangial cells via miR-21/PTEN/Akt/mTOR pathway under diabetic conditions.
Glomerular endothelial cell autophagy in DKD
DKD is a microvascular complication and injury of glomerular endothelial cells in diabetes is strongly associated with the initiation of this kidney disease. Nevertheless, few studies have examined the role of autophagy in glomerular endothelial cells (Table 1). An earlier study by Xavier et al. showed that bone morphogenetic protein and activin membrane-bound inhibitor (BAMBI), a competitive receptor antagonist for the TGF-β receptor family, was upregulated by TGF-β in cultured mouse glomerular endothelial cells. Serum starvation or rapamycin caused marked degradation of BAMBI, whereas the degradation was totally inhibited by lysosomal inhibitor bafilomycin A1 and partially suppressed by 3-methyladenine, but not by proteasomal inhibitors [187]. These results suggest a role of autophagy in regulating BAMBI turnover in endothelial cells. In diabetic mice, BAMBI was downregulated, which enhanced alternative TGF-β signaling and glomerular dysfunction. The renoprotective role of BAMBI against glomerular injury in DKD was further confirmed using BAMBI-deficient mice [188]. Further studies need to determine whether regulation of BAMBI by autophagy in endothelial cells is relevant to the actions of TGF-β and the development of DKD.
Using endothelial-specific Atg5 knockout mice, Lenoir et al. recently demonstrated direct evidence on the role of glomerular endothelial cell autophagy in DKD [171]. Endothelial-specific Atg5 deletion resulted in mild lesions to the glomerular filtration barrier (GFB) in non-diabetic mice. Following STZ injection, endothelial-specific autophagy-deficient mice developed more severe glomerular endothelial injuries, with enhanced proteinuria, increased dilation of glomerular capillaries and endothelial lesions, as compared with control diabetic mice. Notably, TEM revealed GBM thickening and podocyte effacement in endothelial-specific autophagy-deficient diabetic mice, whereas few ultrastructural defects were found in podocytes from control diabetic mice. Together, these results suggest that autophagy in glomerular endothelial cells preserves both endothelial integrity and podocyte ultrastructure to maintain GFB homeostasis, which may involve a crosstalk between glomerular endothelial cells and surrounding podocytes [171].
Autophagy in renal fibrosis
Renal fibrosis, characterized by the excess deposition of ECM in the glomeruli and tubulointerstitium, is the final common pathway leading to ESRD in all progressive CKD including DKD. The pathogenesis of fibrosis is thought to be a failed renal repair process that occurs after initial injuries, involving an enormously complex and dynamic interaction and coordination of virtually all cell types in the kidney. Major cellular events in renal fibrosis include: infiltration of inflammatory cells; fibroblast activation and proliferation; production and deposition of excessive ECM components; tubular atrophy; glomerulosclerosis; and microvascular rarefaction [189].
The role of autophagy in renal fibrosis remains largely unclear and the findings from recent studies are controversial. In mice subjected to unilateral ureteral obstruction (UUO) autophagy was activated in obstructed tubules along with tubular apoptosis [190–192]. Under this condition, autophagy and apoptosis acted in concert to induce tubular atrophy and nephron loss [190]. Oxidative stress leading to mitochondrial damage seemed to drive autophagy and apoptosis in renal tubules, representing an important mechanism of tubular decomposition in UUO injury [192]. Using a tetracycline-controlled mouse model with TGF-β1 overexpression specifically in renal tubules, Koesters et al. further verified the involvement of autophagy in tubular degeneration and interstitial fibrosis [193]. Sustained expression of TGF-β1 induced autophagy in renal tubules, leading to tubular dedifferentiation and the development of widespread peritubular fibrosis. Notably, the nuclei of such degenerating cells showed a normal chromatin pattern without positive TUNEL staining for apoptosis, suggesting that autophagy may be a main force driving tubular atrophy in TGF-β1-induced renal fibrosis [193]. Using pharmacological and genetic inhibitory approaches, we further demonstrated a profibrotic role of autophagy in a mouse model of UUO and in TGF-β1-treated PTECs [194]. Autophagy was persistently activated in proximal tubules following UUO. Inhibition of autophagy by pharmacological inhibitors or proximal tubule-specific Atg7 knockout suppressed interstitial fibrosis, accompanied with the attenuation of tubular cell apoptosis, interstitial macrophage infiltration and production of fibroblast growth factor 2. In primary culture of PTECs, fibronectin accumulation and cell death induced by TGF-β1 were also dependent on autophagy [194]. A profibrotic role of autophagy was further shown in a mouse model of post-ischemic renal fibrosis. Induction of autophagy following ischemic acute kidney injury (AKI) promoted prosenescent changes in proximal tubules of S3 segments during recovery phase. Selective deletion of Atg5 in these proximal tubules suppressed autophagy and thereby prevented the development of a senescent phenotype and AKI progression to CKD [195]. In contrast to these findings, a few studies have demonstrated an antifibrotic role of autophagy in renal fibrosis. Inhibition of autophagy by 3-methyladenine in a rat model of UUO aggravated tubular cell apoptosis and interstitial fibrosis, suggesting that autophagy may limit fibrosis by reducing tubular apoptosis [196]. In primary culture of mouse kidney mesangial cells, both protein and mRNA levels of collagen I were induced by TGF-β1. Inhibition of autophagy by BECN1 knockdown or lysosomal inhibitors further enhanced collagen I protein accumulation without affecting mRNA expression, suggesting a role for autophagy in regulating ECM deposition in mesangial cells by promoting the degradation of collagen I [197]. Ding et al. also revealed the involvement of autophagy in the degradation of mature TGF-β1 in UUO kidneys and in TGF-β1-treated PTECs (both primary culture and cell line), further suggesting that autophagy can prevent renal fibrosis by negative regulation of TGF-β1 [198]. Understanding the cellular and molecular bases of this multifaceted autophagy regulation of renal fibrosis is crucial for identifying potential therapeutic targets to treat progressive CKD.
Therapeutic strategy targeting autophagy for DKD treatment
Autophagy activity is inhibited under diabetic conditions, thus new therapeutic strategy has been focusing on the restoration of autophagy. Diet or calorie restriction is an important glycemic control therapy in diabetic patients and plays a renoprotective role in several metabolic or age-related kidney diseases [56, 83, 106]. Activation of autophagy is essential for calorie restriction-mediated anti-aging effects [199, 200]. Therefore, a calorie restriction regimen that can activate autophagy should be a potent therapeutic strategy to prevent DKD. Indeed, calorie restriction restores autophagy activity in PTECs and attenuates renal damage in type 2 diabetic Wistar fatty rats [56, 83]. Nonetheless, recent human clinical studies have shown that the quality of diet affects the development of insulin resistance and new onset of diabetes. In addition, under diabetic conditions, a high energy/nutrient intake along with a low-protein diet may cause an insufficiency of diet restriction. Furthermore, the types of carbohydrates, amino acids, and fatty acids in different diet regimens that can lead to a better prognosis also need to be identified [62].
In addition to calorie restriction, drugs that can activate autophagy are being discovered. Agents that can modify the activity of mTORC1, AMPK, and SIRT1 may have therapeutic potency for DKD treatment. Rapamycin, an mTORC1 inhibitor, appears to activate autophagy and ameliorate glomerular and tubular damage in diabetic kidneys. However, the side effects of long-term or complete mTORC1 inhibition have been well-recognized. Whether mTOR inhibition by rapamycin or other mTOR inhibitors is safe and effective for treating diabetic patients is still under debate. Targeting AMPK and SIRT1 with activators such as resveratrol, metformin, and AICAR is also being explored. These agents have been shown to activate autophagy in different animal models of diabetes and protect the kidney from diabetic injury, making them be attractive therapeutic options for DKD. In addition, the use of antioxidants to antagonize oxidative stress or chemical chaperons to reduce ER stress and to restore autophagy activity may also be a therapeutic approach to treat DKD.
Conclusions
Despite the application of multiple intensive therapy programs, the incidence of diabetes is still increasing worldwide. DKD, a leading cause of ESRD, is a serious diabetic complication with high morbidity and mortality. Identifying additional effective therapeutic targets for the prevention and treatment of DKD is in urgent need. In this review, we have provided insights into the role of autophagy in the pathogenesis of DKD and the underlying mechanisms that contribute to autophagy regulation in diabetic kidneys. The advances in our understanding of autophagy in DKD will facilitate the discovery of a new therapeutic target to prevent and to treat DKD.
Acknowledgements
This study was supported by grants from National Natural Science Foundation of China (81528004, 81370791), the National Institutes of Health and Department of Veterans Administration of USA.
Footnotes
Danyi Yang and Man J. Livingston are co-first authors.
References
- 1.USRDS USRDS: the United States Renal Data System. Am J Kidney Dis. 2003;42(6 Suppl 5):1–230. [PubMed] [Google Scholar]
- 2.Cao Z, Cooper ME. Pathogenesis of diabetic nephropathy. J Diabetes Investig. 2011;2(4):243–247. doi: 10.1111/j.2040-1124.2011.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi Y, Hu FB. The global implications of diabetes and cancer. Lancet. 2014;383(9933):1947–1948. doi: 10.1016/S0140-6736(14)60886-2. [DOI] [PubMed] [Google Scholar]
- 4.Rossing P, Hougaard P, Parving HH. Progression of microalbuminuria in type 1 diabetes: ten-year prospective observational study. Kidney Int. 2005;68(4):1446–1450. doi: 10.1111/j.1523-1755.2005.00556.x. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad J. Management of diabetic nephropathy: recent progress and future perspective. Diabetes Metab Syndr. 2015;9(4):343–358. doi: 10.1016/j.dsx.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 7.Noh H, King GL. The role of protein kinase C activation in diabetic nephropathy. Kidney Int Suppl. 2007;106:S49–S53. doi: 10.1038/sj.ki.5002386. [DOI] [PubMed] [Google Scholar]
- 8.Calcutt NA, et al. Therapies for hyperglycaemia-induced diabetic complications: from animal models to clinical trials. Nat Rev Drug Discov. 2009;8(5):417–429. doi: 10.1038/nrd2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitada M, et al. Molecular mechanisms of diabetic vascular complications. J Diabetes Investig. 2010;1(3):77–89. doi: 10.1111/j.2040-1124.2010.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93(1):137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- 12.Brenner BM, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 13.Lewis EJ, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345(12):851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 14.Forbes JM, Fukami K, Cooper ME. Diabetic nephropathy: where hemodynamics meets metabolism. Exp Clin Endocrinol Diabetes. 2007;115(2):69–84. doi: 10.1055/s-2007-949721. [DOI] [PubMed] [Google Scholar]
- 15.Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol. 2010;6(6):319–330. doi: 10.1038/nrneph.2010.58. [DOI] [PubMed] [Google Scholar]
- 16.Har R, et al. The effect of renal hyperfiltration on urinary inflammatory cytokines/chemokines in patients with uncomplicated type 1 diabetes mellitus. Diabetologia. 2013;56(5):1166–1173. doi: 10.1007/s00125-013-2857-5. [DOI] [PubMed] [Google Scholar]
- 17.Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12(4):537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma K, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol. 2013;24(11):1901–1912. doi: 10.1681/ASN.2013020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol. 2014;171(8):1917–1942. doi: 10.1111/bph.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parving HH, et al. A prospective study of glomerular filtration rate and arterial blood pressure in insulin-dependent diabetics with diabetic nephropathy. Diabetologia. 1981;20(4):457–461. doi: 10.1007/BF00253407. [DOI] [PubMed] [Google Scholar]
- 21.Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis. 1992;20(1):1–17. doi: 10.1016/S0272-6386(12)80312-X. [DOI] [PubMed] [Google Scholar]
- 22.Burton C, Harris KP. The role of proteinuria in the progression of chronic renal failure. Am J Kidney Dis. 1996;27(6):765–775. doi: 10.1016/S0272-6386(96)90512-0. [DOI] [PubMed] [Google Scholar]
- 23.Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol. 2006;17(11):2974–2984. doi: 10.1681/ASN.2006040377. [DOI] [PubMed] [Google Scholar]
- 24.American Diabetes A. Standards of medical care in diabetes–2009. Diabetes Care. 2009;32(Suppl 1):S13–S61. doi: 10.2337/dc09-S013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamahara K, et al. The role of autophagy in the pathogenesis of diabetic nephropathy. J Diabetes Res. 2013;2013:193757. doi: 10.1155/2013/193757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kume S, et al. Autophagy: emerging therapeutic target for diabetic nephropathy. Semin Nephrol. 2014;34(1):9–16. doi: 10.1016/j.semnephrol.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez CD, et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7(1):2–11. doi: 10.4161/auto.7.1.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol. 2015;224(1):R15–R30. doi: 10.1530/JOE-14-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 32.Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 2010;21(7):683–690. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 36.Huber TB, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8(7):1009–1031. doi: 10.4161/auto.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Axe EL, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayashi-Nishino M, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11(12):1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 39.Yla-Anttila P, et al. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5(8):1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- 40.Hamasaki M, et al. Autophagosomes form at ER–mitochondria contact sites. Nature. 2013;495(7441):389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 41.Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- 42.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 43.Mehrpour M, et al. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20(7):748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 44.Liang C, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jager S, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(Pt 20):4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 46.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maiuri MC, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 49.Scarlatti F, et al. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009;16(1):12–20. doi: 10.1038/cdd.2008.101. [DOI] [PubMed] [Google Scholar]
- 50.Rubinstein AD, Kimchi A. Life in the balance—a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci. 2012;125(Pt 22):5259–5268. doi: 10.1242/jcs.115865. [DOI] [PubMed] [Google Scholar]
- 51.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12(1):1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kume S, Thomas MC, Koya D. Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes. 2012;61(1):23–29. doi: 10.2337/db11-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barbosa Junior Ade A, et al. Inhibition of cellular autophagy in proximal tubular cells of the kidney in streptozotocin-diabetic and uninephrectomized rats. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;61(6):359–366. doi: 10.1007/BF02890439. [DOI] [PubMed] [Google Scholar]
- 54.Han K, Zhou H, Pfeifer U. Inhibition and restimulation by insulin of cellular autophagy in distal tubular cells of the kidney in early diabetic rats. Kidney Blood Pressure Res. 1997;20(4):258–263. doi: 10.1159/000174155. [DOI] [PubMed] [Google Scholar]
- 55.Vallon V, et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol. 2013;304(2):F156–F167. doi: 10.1152/ajprenal.00409.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kitada M, et al. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res. 2011;2011:908185. doi: 10.1155/2011/908185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamahara K, et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol. 2013;24(11):1769–1781. doi: 10.1681/ASN.2012111080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40(2):323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89(3):1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 61.Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. 2010;31(5):212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kume S, et al. Role of nutrient-sensing signals in the pathogenesis of diabetic nephropathy. Biomed Res Int. 2014;2014:315494. doi: 10.1155/2014/315494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 64.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Inoki K. mTOR signaling in autophagy regulation in the kidney. Semin Nephrol. 2014;34(1):2–8. doi: 10.1016/j.semnephrol.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim J, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mori H, et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun. 2009;384(4):471–475. doi: 10.1016/j.bbrc.2009.04.136. [DOI] [PubMed] [Google Scholar]
- 70.Zhang MZ, et al. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 2014;63(6):2063–2072. doi: 10.2337/db13-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nagai K, et al. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int. 2005;68(2):552–561. doi: 10.1111/j.1523-1755.2005.00433.x. [DOI] [PubMed] [Google Scholar]
- 72.Inoki K, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Investig. 2011;121(6):2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Godel M, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Investig. 2011;121(6):2197–2209. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Velagapudi C, et al. The tuberin/mTOR pathway promotes apoptosis of tubular epithelial cells in diabetes. J Am Soc Nephrol. 2011;22(2):262–273. doi: 10.1681/ASN.2010040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sakaguchi M, et al. Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem Biophys Res Commun. 2006;340(1):296–301. doi: 10.1016/j.bbrc.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 76.Yang Y, et al. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am J Nephrol. 2007;27(5):495–502. doi: 10.1159/000106782. [DOI] [PubMed] [Google Scholar]
- 77.Wittmann S, et al. Long-term treatment of sirolimus but not cyclosporine ameliorates diabetic nephropathy in the rat. Transplantation. 2009;87(9):1290–1299. doi: 10.1097/TP.0b013e3181a192bd. [DOI] [PubMed] [Google Scholar]
- 78.Lloberas N, et al. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol. 2006;17(5):1395–1404. doi: 10.1681/ASN.2005050549. [DOI] [PubMed] [Google Scholar]
- 79.Stridh S, et al. Inhibition of mTOR activity in diabetes mellitus reduces proteinuria but not renal accumulation of hyaluronan. Upsala J Med Sci. 2015;120(4):233–240. doi: 10.3109/03009734.2015.1062442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sataranatarajan K, et al. Regulation of elongation phase of mRNA translation in diabetic nephropathy: amelioration by rapamycin. Am J Pathol. 2007;171(6):1733–1742. doi: 10.2353/ajpath.2007.070412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fang L, et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS One. 2013;8(4):e60546. doi: 10.1371/journal.pone.0060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiao T, et al. Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol Cell Biochem. 2014;394(1–2):145–154. doi: 10.1007/s11010-014-2090-7. [DOI] [PubMed] [Google Scholar]
- 83.Kitada M, et al. A very-low-protein diet ameliorates advanced diabetic nephropathy through autophagy induction by suppression of the mTORC1 pathway in Wistar fatty rats, an animal model of type 2 diabetes and obesity. Diabetologia. 2016;59(6):1307–1317. doi: 10.1007/s00125-016-3925-4. [DOI] [PubMed] [Google Scholar]
- 84.Huber TB, Walz G, Kuehn EW. mTOR and rapamycin in the kidney: signaling and therapeutic implications beyond immunosuppression. Kidney Int. 2011;79(5):502–511. doi: 10.1038/ki.2010.457. [DOI] [PubMed] [Google Scholar]
- 85.Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20(12):2493–2502. doi: 10.1681/ASN.2008111186. [DOI] [PubMed] [Google Scholar]
- 86.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee JW, et al. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5(11):e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alers S, et al. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee MJ, et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am J Physiol Renal Physiol. 2007;292(2):F617–F627. doi: 10.1152/ajprenal.00278.2006. [DOI] [PubMed] [Google Scholar]
- 90.Cammisotto PG, et al. Control of glycogen synthase through ADIPOR1-AMPK pathway in renal distal tubules of normal and diabetic rats. Am J Physiol Renal Physiol. 2008;294(4):F881–F889. doi: 10.1152/ajprenal.00373.2007. [DOI] [PubMed] [Google Scholar]
- 91.Yamazaki T, et al. Combination effects of enalapril and losartan on lipid peroxidation in the kidneys of KK-Ay/Ta mice. Nephron Exp Nephrol. 2009;113(2):e66–e76. doi: 10.1159/000228714. [DOI] [PubMed] [Google Scholar]
- 92.Ding DF, et al. Resveratrol attenuates renal hypertrophy in early-stage diabetes by activating AMPK. Am J Nephrol. 2010;31(4):363–374. doi: 10.1159/000300388. [DOI] [PubMed] [Google Scholar]
- 93.Sokolovska J, et al. Influence of metformin on GLUT1 gene and protein expression in rat streptozotocin diabetes mellitus model. Arch Physiol Biochem. 2010;116(3):137–145. doi: 10.3109/13813455.2010.494672. [DOI] [PubMed] [Google Scholar]
- 94.Kitada M, et al. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes. 2011;60(2):634–643. doi: 10.2337/db10-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chang CC, et al. Resveratrol retards progression of diabetic nephropathy through modulations of oxidative stress, proinflammatory cytokines, and AMP-activated protein kinase. J Biomed Sci. 2011;18(1):47. doi: 10.1186/1423-0127-18-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim MY, et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1alpha axis in db/db mice. Diabetologia. 2013;56(1):204–217. doi: 10.1007/s00125-012-2747-2. [DOI] [PubMed] [Google Scholar]
- 97.Kim J, et al. Renal podocyte injury in a rat model of type 2 diabetes is prevented by metformin. Exp Diabetes Res. 2012;2012:210821. doi: 10.1155/2012/210821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dugan LL, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Investig. 2013;123(11):4888–4899. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao L, et al. Berberine improves kidney function in diabetic mice via AMPK activation. PLoS One. 2014;9(11):e113398. doi: 10.1371/journal.pone.0113398. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 100.Jin Y, et al. Berberine enhances the AMPK activation and autophagy and mitigates high glucose-induced apoptosis of mouse podocytes. Eur J Pharmacol. 2017;794:106–114. doi: 10.1016/j.ejphar.2016.11.037. [DOI] [PubMed] [Google Scholar]
- 101.Lee HJ, et al. Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J Biol Chem. 2012;287(7):4451–4461. doi: 10.1074/jbc.M111.278325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Al-Rasheed NM, et al. Renoprotective effects of fenofibrate via modulation of LKB1/AMPK mRNA expression and endothelial dysfunction in a rat model of diabetic nephropathy. Pharmacology. 2015;95(5–6):229–239. doi: 10.1159/000381190. [DOI] [PubMed] [Google Scholar]
- 103.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13(4):225–238. doi: 10.1038/nrn3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014;25(3):138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee IH, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105(9):3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kume S, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Investig. 2010;120(4):1043–1055. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Canto C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One. 2010;5(2):e9199. doi: 10.1371/journal.pone.0009199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.He W, et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Investig. 2010;120(4):1056–1068. doi: 10.1172/JCI41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yacoub R, Lee K, He JC. The role of SIRT1 in diabetic kidney disease. Front Endocrinol (Lausanne) 2014;5:166. doi: 10.3389/fendo.2014.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chuang PY, et al. Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS One. 2011;6(8):e23566. doi: 10.1371/journal.pone.0023566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hasegawa K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19(11):1496–1504. doi: 10.1038/nm.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shang G, et al. 3,5-Diiodo-l-thyronine ameliorates diabetic nephropathy in streptozotocin-induced diabetic rats. Biochim Biophys Acta. 2013;1832(5):674–684. doi: 10.1016/j.bbadis.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 114.Li C, et al. Tetrahydroxystilbene glucoside ameliorates diabetic nephropathy in rats: involvement of SIRT1 and TGF-beta1 pathway. Eur J Pharmacol. 2010;649(1–3):382–389. doi: 10.1016/j.ejphar.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 115.Wu L, et al. The effect of resveratrol on FoxO1 expression in kidneys of diabetic nephropathy rats. Mol Biol Rep. 2012;39(9):9085–9093. doi: 10.1007/s11033-012-1780-z. [DOI] [PubMed] [Google Scholar]
- 116.Xu Y, et al. Resveratrol protects against hyperglycemia-induced oxidative damage to mitochondria by activating SIRT1 in rat mesangial cells. Toxicol Appl Pharmacol. 2012;259(3):395–401. doi: 10.1016/j.taap.2011.09.028. [DOI] [PubMed] [Google Scholar]
- 117.Zhang S, et al. SIRT1 is required for the effects of rapamycin on high glucose-inducing mesangial cells senescence. Mech Ageing Dev. 2012;133(6):387–400. doi: 10.1016/j.mad.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 118.Ma L, et al. Sirt1 is essential for resveratrol enhancement of hypoxia-induced autophagy in the type 2 diabetic nephropathy rat. Pathol Res Pract. 2016;212(4):310–318. doi: 10.1016/j.prp.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 119.Noh H, et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am J Physiol Renal Physiol. 2009;297(3):F729–F739. doi: 10.1152/ajprenal.00086.2009. [DOI] [PubMed] [Google Scholar]
- 120.Gilbert RE, et al. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011;79(12):1312–1321. doi: 10.1038/ki.2011.39. [DOI] [PubMed] [Google Scholar]
- 121.Advani A, et al. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol. 2011;178(5):2205–2214. doi: 10.1016/j.ajpath.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang X, et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014;86(4):712–725. doi: 10.1038/ki.2014.111. [DOI] [PubMed] [Google Scholar]
- 123.Satriano J, Sharma K. Autophagy and metabolic changes in obesity-related chronic kidney disease. Nephrol Dial Transplant. 2013;28(Suppl 4):iv29–iv36. doi: 10.1093/ndt/gft229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sohn M, et al. Delayed treatment with fenofibrate protects against high-fat diet-induced kidney injury in mice: the possible role of AMPK autophagy. Am J Physiol Renal Physiol. 2017;312(2):F323–F334. doi: 10.1152/ajprenal.00596.2015. [DOI] [PubMed] [Google Scholar]
- 125.Kuwahara S, et al. Megalin-mediated tubuloglomerular alterations in high-fat diet-induced kidney disease. J Am Soc Nephrol. 2016;27(7):1996–2008. doi: 10.1681/ASN.2015020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yamamoto T, et al. High-fat diet-induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J Am Soc Nephrol. 2017;28(5):1534–1551. doi: 10.1681/ASN.2016070731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tagawa A, et al. Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes. 2016;65(3):755–767. doi: 10.2337/db15-0473. [DOI] [PubMed] [Google Scholar]
- 128.Bondeva T, Wolf G. Reactive oxygen species in diabetic nephropathy: friend or foe? Nephrol Dial Transplant. 2014;29(11):1998–2003. doi: 10.1093/ndt/gfu037. [DOI] [PubMed] [Google Scholar]
- 129.Wagener FA, et al. The role of reactive oxygen species in apoptosis of the diabetic kidney. Apoptosis. 2009;14(12):1451–1458. doi: 10.1007/s10495-009-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koya D, et al. Effects of antioxidants in diabetes-induced oxidative stress in the glomeruli of diabetic rats. J Am Soc Nephrol. 2003;14(8 Suppl 3):S250–S253. doi: 10.1097/01.ASN.0000077412.07578.44. [DOI] [PubMed] [Google Scholar]
- 131.Brezniceanu ML, et al. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int. 2007;71(9):912–923. doi: 10.1038/sj.ki.5002188. [DOI] [PubMed] [Google Scholar]
- 132.Tanaka Y, et al. Autophagy as a therapeutic target in diabetic nephropathy. Exp Diabetes Res. 2012;2012:628978. doi: 10.1155/2012/628978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nishikawa T, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 134.Ma T, et al. High glucose induces autophagy in podocytes. Exp Cell Res. 2013;319(6):779–789. doi: 10.1016/j.yexcr.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yadav A, et al. ANG II promotes autophagy in podocytes. Am J Physiol Cell Physiol. 2010;299(2):C488–C496. doi: 10.1152/ajpcell.00424.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jiang XS, et al. Autophagy protects against palmitic acid-induced apoptosis in podocytes in vitro. Sci Rep. 2017;7:42764. doi: 10.1038/srep42764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu L, et al. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem. 2008;283(45):31153–31162. doi: 10.1074/jbc.M805056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sakon S, et al. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22(15):3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cunard R. Endoplasmic reticulum stress in the diabetic kidney, the good, the bad and the ugly. J Clin Med. 2015;4(4):715–740. doi: 10.3390/jcm4040715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Taniguchi M, Yoshida H. Endoplasmic reticulum stress in kidney function and disease. Curr Opin Nephrol Hypertens. 2015;24(4):345–350. doi: 10.1097/MNH.0000000000000141. [DOI] [PubMed] [Google Scholar]
- 141.Sieber J, et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Renal Physiol. 2010;299(4):F821–F829. doi: 10.1152/ajprenal.00196.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cao Y, et al. Role of endoplasmic reticulum stress in apoptosis of differentiated mouse podocytes induced by high glucose. Int J Mol Med. 2014;33(4):809–816. doi: 10.3892/ijmm.2014.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cybulsky AV. Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int. 2010;77(3):187–193. doi: 10.1038/ki.2009.389. [DOI] [PubMed] [Google Scholar]
- 144.Lindenmeyer MT, et al. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol. 2008;19(11):2225–2236. doi: 10.1681/ASN.2007121313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ohse T, et al. Albumin induces endoplasmic reticulum stress and apoptosis in renal proximal tubular cells. Kidney Int. 2006;70(8):1447–1455. doi: 10.1038/sj.ki.5001704. [DOI] [PubMed] [Google Scholar]
- 146.Wu J, et al. Induction of diabetes in aged C57B6 mice results in severe nephropathy: an association with oxidative stress, endoplasmic reticulum stress, and inflammation. Am J Pathol. 2010;176(5):2163–2176. doi: 10.2353/ajpath.2010.090386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rouschop KM, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Investig. 2010;120(1):127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ogata M, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen Y, et al. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am J Nephrol. 2008;28(6):1014–1022. doi: 10.1159/000148209. [DOI] [PubMed] [Google Scholar]
- 150.Cao A, et al. Ursodeoxycholic acid ameliorated diabetic nephropathy by attenuating hyperglycemia-mediated oxidative stress. Biol Pharm Bull. 2016;39(8):1300–1308. doi: 10.1248/bpb.b16-00094. [DOI] [PubMed] [Google Scholar]
- 151.Qi W, et al. Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metab Clin Exp. 2011;60(5):594–603. doi: 10.1016/j.metabol.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 152.Luo ZF, et al. Effects of 4-phenylbutyric acid on the process and development of diabetic nephropathy induced in rats by streptozotocin: regulation of endoplasmic reticulum stress-oxidative activation. Toxicol Appl Pharmacol. 2010;246(1–2):49–57. doi: 10.1016/j.taap.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 153.Cao AL, et al. Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Lab Investig. 2016;96(6):610–622. doi: 10.1038/labinvest.2016.44. [DOI] [PubMed] [Google Scholar]
- 154.Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16(10):1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- 155.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 156.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;291(2):F271–F281. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 158.Rouschop KM, Wouters BG. Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr Mol Med. 2009;9(4):417–424. doi: 10.2174/156652409788167131. [DOI] [PubMed] [Google Scholar]
- 159.Tracy K, et al. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27(17):6229–6242. doi: 10.1128/MCB.02246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhang H, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 161.Bellot G, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pagtalunan ME, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Investig. 1997;99(2):342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54(6):1626–1634. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 164.Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia. 1999;42(11):1341–1344. doi: 10.1007/s001250051447. [DOI] [PubMed] [Google Scholar]
- 165.Hartleben B, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Investig. 2010;120(4):1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mizushima N, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15(3):1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sato S, et al. Two types of autophagy in the podocytes in renal biopsy specimens: ultrastructural study. J Submicrosc Cytol Pathol. 2006;38(2–3):167–174. [PubMed] [Google Scholar]
- 168.Chen J, et al. mVps34 deletion in podocytes causes glomerulosclerosis by disrupting intracellular vesicle trafficking. J Am Soc Nephrol. 2013;24(2):198–207. doi: 10.1681/ASN.2012010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cina DP, et al. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol. 2012;23(3):412–420. doi: 10.1681/ASN.2011070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Oshima Y, et al. Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol. 2011;22(12):2203–2212. doi: 10.1681/ASN.2011020202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lenoir O, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy. 2015;11(7):1130–1145. doi: 10.1080/15548627.2015.1049799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Liu J, et al. beta-Arrestins promote podocyte injury by inhibition of autophagy in diabetic nephropathy. Cell Death Dis. 2016;7:e2183. doi: 10.1038/cddis.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sun J, et al. Repression of miR-217 protects against high glucose-induced podocyte injury and insulin resistance by restoring PTEN-mediated autophagy pathway. Biochem Biophys Res Commun. 2017;483(1):318–324. doi: 10.1016/j.bbrc.2016.12.145. [DOI] [PubMed] [Google Scholar]
- 174.Li W, et al. FoxO1 promotes mitophagy in the podocytes of diabetic male mice via the PINK1/Parkin pathway. Endocrinology. 2017 doi: 10.1210/en.2016-1970. [DOI] [PubMed] [Google Scholar]
- 175.Liu S, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8(5):826–837. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 176.Kimura T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22(5):902–913. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Livingston MJ, Dong Z. Autophagy in acute kidney injury. Semin Nephrol. 2014;34(1):17–26. doi: 10.1016/j.semnephrol.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Havasi A, Dong Z. Autophagy and tubular cell death in the kidney. Semin Nephrol. 2016;36(3):174–188. doi: 10.1016/j.semnephrol.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nair S, Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95(1):34–42. doi: 10.1210/jc.2009-0473. [DOI] [PubMed] [Google Scholar]
- 180.Huang C, et al. Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci Rep. 2016;6:29196. doi: 10.1038/srep29196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Saito A, et al. Significance of proximal tubular metabolism of advanced glycation end products in kidney diseases. Ann N Y Acad Sci. 2005;1043:637–643. doi: 10.1196/annals.1333.072. [DOI] [PubMed] [Google Scholar]
- 182.Liu WJ, et al. Autophagy-lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy. J Biol Chem. 2015;290(33):20499–20510. doi: 10.1074/jbc.M115.666354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Takahashi A, et al. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes. 2017;66(5):1359–1372. doi: 10.2337/db16-0397. [DOI] [PubMed] [Google Scholar]
- 184.Fiorentino L, et al. Loss of TIMP3 underlies diabetic nephropathy via FoxO1/STAT1 interplay. EMBO Mol Med. 2013;5(3):441–455. doi: 10.1002/emmm.201201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Xu L, et al. Inhibition of autophagy increased AGE/ROS-mediated apoptosis in mesangial cells. Cell Death Dis. 2016;7(11):e2445. doi: 10.1038/cddis.2016.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lu X, et al. Ursolic acid attenuates diabetic mesangial cell injury through the up-regulation of autophagy via miRNA-21/PTEN/Akt/mTOR suppression. PLoS One. 2015;10(2):e0117400. doi: 10.1371/journal.pone.0117400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xavier S, et al. BAMBI is expressed in endothelial cells and is regulated by lysosomal/autolysosomal degradation. PLoS One. 2010;5(9):e12995. doi: 10.1371/journal.pone.0012995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fan Y, et al. BAMBI elimination enhances alternative TGF-beta signaling and glomerular dysfunction in diabetic mice. Diabetes. 2015;64(6):2220–2233. doi: 10.2337/db14-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7(12):684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li L, et al. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol. 2010;176(4):1767–1778. doi: 10.2353/ajpath.2010.090345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Forbes MS, Thornhill BA, Chevalier RL. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol. 2011;301(1):F110–F117. doi: 10.1152/ajprenal.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Xu Y, et al. Autophagy and apoptosis in tubular cells following unilateral ureteral obstruction are associated with mitochondrial oxidative stress. Int J Mol Med. 2013;31(3):628–636. doi: 10.3892/ijmm.2013.1232. [DOI] [PubMed] [Google Scholar]
- 193.Koesters R, et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177(2):632–643. doi: 10.2353/ajpath.2010.091012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Livingston MJ, et al. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy. 2016;12(6):976–998. doi: 10.1080/15548627.2016.1166317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Baisantry A, et al. Autophagy induces prosenescent changes in proximal tubular S3 segments. J Am Soc Nephrol. 2016;27(6):1609–1616. doi: 10.1681/ASN.2014111059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kim WY, et al. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology. 2012;17(2):148–159. doi: 10.1111/j.1440-1797.2011.01541.x. [DOI] [PubMed] [Google Scholar]
- 197.Kim SI, et al. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J Biol Chem. 2012;287(15):11677–11688. doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ding Y, et al. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol. 2014;25(12):2835–2846. doi: 10.1681/ASN.2013101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Colman RJ, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fontana L, Partridge L, Longo VD. Extending healthy life span—from yeast to humans. Science. 2010;328(5976):321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]