

Abstract
Reversed-phase chromatographic separation of glycopeptides tends to be dominated by the peptide composition. In contrast, capillary zone electrophoresis separation of glycopeptides is particularly sensitive to the sialic acid composition of the glycan. In this paper, we combine the two techniques to achieve superior N-glycopeptide analysis. Glycopeptides were first isolated from a tryptic digest using HILIC solid-phase extraction. The glycopeptides were separated using reversed-phase UHPLC to generate four fractions corresponding to different peptide backbones. CZE-ESI-MS/MS was used to analyze the fractions. We applied this method for the analysis of alpha-1-acid glycoprotein (AGP). A total of 268 site-specific N-glycopeptides were detected, representing eight different glycosylation sites from two isomers of AGP. Glycans included tetra-sialic acids with multi N-acetyllactosamine (LacNAc) repeats and unusual pentasialylated terminal sialic acids. Reversed-phase UHPLC coupled with CZE generated ~35% more N-glycopeptides than direct reversed-phase UHPLC-ESI-MS/MS analysis and ~70% more N-glycopeptides than direct CZE-ESI-MS/MS analysis. This approach is a promising tool for global, site-specific glycosylation analysis of highly heterogeneous glycoproteins with mass-limited samples.
Graphical Abstract
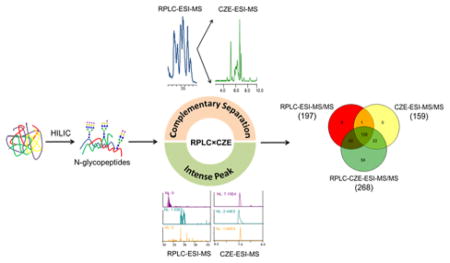
Glycosylation is one of the most common protein post-translational modifications, with at least 50% of the cellular proteome predicted to be glycosylated.1,2 Aberrations in protein glycosylation have been implicated in a variety of diseases, such as immunological disorders,3 neurodegenerative diseases,4 and cancers.5,6 Characterization of these aberrations, especially in the distribution of N-glycoforms at specific sites on proteins, should offer diagnostic and prognostic information, and the characterization of the site-specific microheterogeneity of protein N-glycosylation has drawn recent interest.7–10
Reversed-phase liquid chromatography (RPLC) is commonly coupled with electrospray ionization-tandem mass spectrometry (ESI-MS/MS) for analysis of site-specific heterogeneous structures of glycopeptides. RPLC primarily separates N-glycopeptides based on the peptide’s hydrophobicity, and the glycan has relatively little effect on the separation. As a result, the retention time for a glycopeptide will be similar to the retention time of the deglycosylated peptide, and N-glycopeptides that share the same peptide backbone will elute with similar retention times.11 In this narrow elution window, both the hydrophilicity and the number of terminal sialic acids on glycans contribute to the glycopeptide elution order on RPLC. The retention time is increased with an increase in sialic acid content and decreased by an increase in the number of monosaccharide units.11,12
Capillary zone electrophoresis (CZE) coupled with ESI-MS/MS is emerging as a useful tool in proteomic analysis.13–15 CZE is an orthogonal separation mode to RPLC, producing remarkably efficient separations based on differences in the charge-to-size ratio of peptides. We have shown that CZE coupled with an electrokinetically-pumped nanoelectrospray interface generated much higher peak intensities than reversed-phase UHPLC-ESI-MS for a wide range of peptides.16
CZE has also been shown to be a powerful tool for resolving glycans.17,18 In particular, the electrophoretic migration order of glycans is strongly influenced by the number of sialic acids, each of which carries a negative charge.19 Glycans on the same peptide backbone migrate in the order of increasing numbers of sialic acids.20
In this work, we take advantage of the complementary separations produced by RPLC and CZE for N-glycopeptides. Glycopeptides were first isolated from a tryptic digest using HILIC solid-phase extraction. The extracted glycopeptides were then fractionated with reversed-phase UHPLC, in which glycopeptides with the same peptide backbone were isolated. Each fraction was subsequently subjected to CZE-ESI-MS/MS for the second dimension separation and MS analysis. We also explored this approach for glycan structure characterization with manual precursor mass matching, because reversed-phase separation offers a retention time criterion for confirming peptide sequence, while CZE allows the estimation of the number of sialic acids in the glycan.
CZE produces extremely high sensitivity, favoring a second dimension analysis of fractions collected from single reversed-phase UHPLC run. As an alternative to the conventional two dimension high/low-pH RPLC-ESI-MS/MS method employed for intact N-glycopeptide analysis,12 this system has much lower sample consumption, which is essential for the analysis of mass limited samples.
EXPERIMENTAL SECTION
Materials and Reagents
Bovine pancreas TPCK-treated trypsin (T1426), α1-Acid Glycoprotein (AGP, human plasma), dithiothreitol (DTT), iodoacetamide (IAA), trifluoroacetic acid (TFA), and formic acid (FA) were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (ACN) and hydrofluoric acid (HF) were purchased from Fisher Scientific (Pittsburgh, PA). Methanol and water were purchased from Honeywell Burdick & Jackson (Wicklow, Ireland).
Protein Digestion
AGP (200 μg) dissolved in 50 mM NH4HCO3 (pH 8.0) containing 8 M urea at a concentration of 1 μg/μL was denatured at room temperature for 10 min, reduced by the addition of 2 μL of 500 mM DTT at room temperature for 1 h and alkylated with 5 μL of 500 mM IAA in the dark at room temperature for 30 min. Alkylated proteins were then transferred to a Microcon® −10 centrifugal filter unit (Merck, Darmstadt, Germany) for sample cleanup.21 Excess reagents were first removed by centrifugation for 30 min at 18,000 g. The proteins on the membrane were then washed three times with 200 μL of 50 mM NH4HCO3 (pH 8.0) via centrifugation at 16,000 g for each 15 min wash. Subsequently, the proteins were redissolved in 100 μL of 50 mM NH4HCO3 (pH 8.0) and transferred to a new centrifuge tube. This step was repeated, and the protein solutions were pooled. Finally, trypsin was added into the protein solution at an enzyme/substrate ratio (m/m) of 1:50 and incubated for 16 h at 37 °C. After terminating the digestion with TFA, the protein digest was lyophilized with a vacuum concentrator (Thermo Fisher Scientific, Marietta, OH)
Glycopeptide Enrichment
Glycopeptide enrichment was performed with a locally-constructed hydrophilic interaction chromatography (HILIC) solid phase extraction (SPE) column packed with ZIC® glycocapture resin (ProteoExtract® Glycopeptide Enrichment Kit, EMD Millipore, Billerica, MA). The AGP digest (~200 μg) was redissolved in 200 μL of ZIC® loading buffer and then loaded onto two equilibrated HILIC SPE with 100 μL for each. After centrifugation at 1500×g for 2 min, the SPE column was washed with 300 μL of ZIC® loading buffer four times to remove the non-specifically adsorbed peptides and then eluted twice with 100 μL of ZIC® eluting buffer. The eluted glycopeptide fraction from the two HILIC SPE columns was collected, combined, and lyophilized using a vacuum concentrator. The dried glycopeptide fraction was stored at −20 °C before further analysis.
Reversed-phase UHPLC-ESI-MS/MS analysis
Unfractionated AGP glycopeptides were analyzed by a reversed-phase UHPLC-ESI-MS/MS system consisting of a nanoACQUITY UltraPerformance LC® (UPLC®) M-Class system (Waters, Milford, MA, USA). Chromatography was performed using a commercial C18 reversed phase column (Waters, 100 μm ×100 mm, 1.7 μm particle BEH130C18). Solvents A (0.1% FA in H2O) and B (0.1% FA in ACN) were used to establish a 80 min gradient, comprised of 14 min of 2% B, then 1 min of 2–8% B, 60 min of 8–28%, 1 min of 28–80% B, and finally maintained at 80% B for 4 min, with the flow rate at 0.7 μL/min. The column was equilibrated for 15 min with 2% B at 0.7 μL/min before the run. AGP glycopeptide sample was dissolved in 30 μL of 2% ACN with 0.1% FA. The injection volume is 1 μL (with glycopeptides from ~6.7 μg of initial digest).
A Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) was operated in positive mode with a 1.8 kV applied spray voltage. Full MS scans were acquired in the Orbitrap mass analyzer over m/z 700–2000 with a resolution of 60000. The top-ten HCD scans per full scan were acquired with a normalized collision energy of 28% and an isolation window of ±2.0 Da. The resolution of the MS/MS scan was set at 15000. One microscan was set for each MS and MS/MS scan. The target value was 3.00 × 106 and the maximum injection time was 50 ms. The dynamic exclusion duration was 30s.
Reversed-phase UHPLC-CZE-ESI-MS/MS analysis
Offline reversed-phase UHPLC prefractionation was performed based on the same chromatography condition as the direct reversed-phase UHPLC-ESI-MS/MS analysis mentioned above, including both the commercial C18 reversed phase column and separation gradient. Briefly, 1 μL of AGP glycopeptides (from ~6.7 μg of initial digest) was loaded onto the C18 column at a flow rate of 0.7 μL/min. An 80 min mobile phase gradient was generated using Solvent A and B. Eluates from 6.0 to 15.0 min, 27.0 to 33.0 min, 33.1 to 40.0 min, and 40.1 to 50 min were collected and lyophilized, to generate four fractions for the second dimension CZE-ESI-MS/MS analysis.
CZE separation of AGP glycopeptide fraction was performed using an uncoated fused silica capillary (20 i.d. × 150 μm o.d. × 46 cm length, Polymicro Technologies, Phoenix, AZ). CZE was coupled to ESI-MS/MS via a third generation electrokinetically pumped sheath-flow interface (Figure S-1).22 Briefly, an emitter was prepared from a borosilicate glass tube (0.75 mm i.d. × 1.0 mm o.d. × 10 cm length), which was pulled to a ~20 μm diameter tip using a P-1000 flaming/brown micropipet puller (Sutter Instruments, Novato, CA). One end of the separation capillary was etched to reduce the o.d. to ~50 μm using hydrofluoric acid (Caution: use appropriate safety procedures while handling HF solutions). The electrospray interface was constructed from a plastic cross. The emitter was placed in one arm of the cross. The separation capillary was threaded through the cross into the emitter. The other two arms of the cross were connected to a sheath buffer reservoir with plastic tubing and to a syringe filled with sheath buffer for system flushing. The sheath electrolyte was 10% (v/v) methanol and 0.5% (v/v) FA.
The dried AGP glycopeptide fraction was resuspended in 2 μL of 0.1% NH3.H2O (~pH 10) to adjust the pH to ~8. The injection end of the capillary was fixed in a block that allowed pumping fluids with either pressure or voltage.23 Samples were injected hydrodynamically for 2.5 seconds at 15 psi, producing an injection volume of ~2.2 nL. The background electrolyte for the separation was 5% CH3COOH. The capillary was rinsed at 30 psi for 15 min with 1 M NaOH, water, and separation electrolyte prior to injection.
Separation voltage was provided with a Spellman CZE 1000R high-voltage power supplies; 26 kV was applied at the injection end of capillary and 2.0 kV was applied at the sheath buffer reservoir for electrospray. The power supplies were controlled by LabView software.
The operating parameters for Q Exactive HF mass spectrometer were the same as that used in direct reversed-phase UHPLC-ESI-MS/MS analysis, except the dynamic exclusion duration was set as 5 s.
Data Analysis
Reversed-phase UHPLC-ESI-MS/MS and reversed-phase UHPLC-CZE-ESI-MS/MS data were analyzed with Xcalibur software (Thermo Fisher Scientific). The glycopeptide structures were manually characterized by matching experimental precursors with theoretical ones in MS and experimental fragment ions with theoretical ones in MS/MS. Four abbreviations are used to represent monosaccharide residues: HexNAc, N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc); Hex, galactose or mannose; Fuc, fucose; and Neu5Ac, N-acetylneuraminic acid.12 Glycan compositions are represented by assigning the existing number of each monosaccharide with the order of HexNAc; Hex; Fuc; and NeuAc. For example, G6704 represents a glycan composition of 6 HexNAc, 7 Hex, 0 Fuc, and 4 Neu5Ac.
RESULTS AND DISCUSSION
Our workflow is shown in Figure 1. Glycopeptides were isolated from a tryptic digest using HILIC solid-phase extraction. The enriched fractions were then used for three experiments. First, the peptides were deglycosylated and subjected to reversed-phase UHPLC analysis; this procedure provided the peptide backbone structure. Second, the intact glycopeptides were analyzed by reversed-phase UHPLC-ESI-MS/MS to provide a benchmark for the number of glycoform identifications. Third, the intact glycopeptides were fractionated using reversed-phase UHPLC, and four fractions were analyzed using CZE-ESI-MS/MS.
Figure 1.

Workflow for the site-specific N-glycan characterization of AGP with a reversed-phase UHPLC-CZE-ESI-MS/MS approach.
We evaluated the system by analysis of human AGP, which is a plasma protein with molecular weight of 41–43 kDa. The protein is heterogeneously glycosylated on its five N-glycosylation sites.24,25 The tryptic digest contains a variety of peptide backbone sequences combined with a complex set of N-glycans. The protein backbone has two isotypes, AGP1 and AGP2, which share two glycopeptide sequences after trypsin digestion, Sites II and III.21,24
Reversed-phase UHPLC-ESI-MS/MS analysis of N-glycopeptides from AGP
The deglycosylated peptides were first analyzed by single-shot reversed-phase UHPLC-ESI-MS/MS. Nine peptide sequences covering the eight N-glycosylation sites in AGP1 and AGP2 were confirmed (Table 1, Figure 2A).
Table 1.
List of N-glycosylated sites detected from AGP
| Glycosylation Site | Isotype | Peptide Sequence | Y1 ion m/z (charge) |
|---|---|---|---|
| Site I | AGP1 | LVPVPITN15ATLDQITGK | 992.09 (2+) |
| Site I | AGP2 | LVPVPITN15ATLDR | 806.46 (2+) |
| Site II | AGP1/2* | NEEYN38K | 999.43 (1+) |
| Site III | AGP1/2* | YFTPN54KTEDTIFLR (68-81) | 974.49 (2+) |
| FYFTPN54KTEDTIFLR (67-81) | 1048.02 (2+) | ||
| Site IV | AGP1 | QDQCIYN75TTYLNVQR | 1059.99 (2+) |
| Site IV | AGP2 | QNQCFYN75SSYLNVQR | 1062.49 (2+) |
| Site V | AGP1 | EN85GTISR | 979.47 (1+) |
| Site V | AGP2 | EN85GTVSR | 965.45 (1+) |
AGP1/2 represents glycosylation sites shared in both isotypes
Figure 2.

Base peak chromatograms obtained for A) deglycosylated AGP peptides, B) intact AGP glycopeptides. C) MS spectra of Site III glycopeptides with YFTPN54KTEDTIFLR (68-81) backbone at 35.16 min by reversed-phase UHPLC-ESI-MS/MS analysis. Symbols:
 :N-Acetylglucosamine,
:N-Acetylglucosamine,
 : Mannose,
: Mannose,
 : Galactose,
: Galactose,
 : N-acetylneuraminic acid,
: N-acetylneuraminic acid,
 : Fucose.
: Fucose.
The peptide backbone information facilitated the analysis of intact glycopeptides (Figure 2B). The glycosylated forms of the peptide generated a set of peaks with reversed-phase UHPLC-based separation, and intact glycopeptides with different peptide backbones tended to overlap, e.g. Site I (AGP2) coeluted with Site IV (AGP1 and AGP2) and Site III (67-61, AGP1/2) coeluted with Site I (AGP1). In addition, glycopeptides based on Site II and V from both AGP1 and AGP2 were too hydrophilic to be well resolved during the reversed-phase separation.
The reversed-phase UHPLC-ESI-MS/MS analysis of N-glycopeptides from AGP identified 197 N-glycopeptides (Table S-1 to S-5), which is more than reported in earlier studies of AGP site-specific glycan microheterogeneity.21,24,26 We obtained a more comprehensive N-glycan characterization on Site III of AGP1/2 because of the dramatically increased MS intensity of the YFTPN54KTEDTIFLR (68-81) and FYFTPN54KTEDTIFLR (67-81) species, which were produced by the nonspecific trypsin digestion (commonly seen on Site I of AGP). 21,24,26 The amino acid sequences of both YFTPN54KTEDTIFLR and FYFTPN54KTEDTIFLR were confirmed with MS/MS fragmentation, Figure S-2. Earlier work reported the peptide sequence of Site III as SVQEIQATFFYFTPN54K, and a limited number of N-glycans had been assigned to that sequence.21,24,26 The relative abundance of Site III glycopeptides was reported to be ~3%, the lowest of the glycosylation sites.21 As shown in Figure 2A, deglycosylated YFTPN54KTEDTIFLR (Site III, 68-81) produced ~17% of the total abundance of the deglycosylated peptides. This improved intensity allowed identification of many bifucosylated N-glycans on Site III without lectin purification. Highly complex N-glycans with different numbers of sialic acid were also detected (Table S-3). Nevertheless, N-glycans on Site III with different numbers of N-acetyllactosamine (β-Gal(1,4)GlcNAc, LacNAc) and sialic acids tended to coelute using reversed-phase UHPLC under optimized conditions (Figure 2C).
CZE-ESI-MS/MS analysis of N-glycopeptides from AGP with reversed-phase UHPLC prefractionation
CZE separations are based on the analyte charge to size ratio, and CZE should be ideally suited for resolution of differentially sialylated glycans that co-elute in reversed-phase UHPLC. Four fractions were generated using reversed-phase UHPLC (Figure 2B). Fraction 1 contained glycopeptides from Site II (AGP1/2) and Site V (AGP1 and AGP2). Fraction 2 contained glycopeptides from Site I (AGP2) and Site IV (AGP1 and AGP2). Fraction 3 contained glycopeptides mostly from Site III (68-61, AGP1/2). Fraction 4 contained glycopeptides from Site III (67-61, AGP1/2) and Site I (AGP1). Each fraction was then subjected to the following CZE-ESI-MS/MS analysis.
CZE separation of fractions
An uncoated fused capillary (20 μm i.d. × 150 μm o.d. × 46 cm) coupled to a Q-Exactive HF mass spectrometer was used for CZE-ESI-MS/MS analysis of the four fractions generated by reversed-phase UHPLC separation of the intact glycopeptides. These separations were completed in 10 min (Figure S-3).
Figure 3 presents the CZE separation of reversed-phase UHPLC Fraction 1. CZE separated differentially sialylated glycopeptides from the same glycosylation site with resolution of greater than 2, Figure 3A. In contrast, glycopeptides containing the same number of sialic acids were separated with much lower resolution; glycan hydrodynamic volume has a much weaker influence on the intact glycopeptide migration compared with the negative charge associated with sialic acid.
Figure 3.

CZE-ESI-MS/MS analysis of AGP glycopeptide fraction collected at 6.0–15.0 min from Fraction 1 from the reversed-phase UHPLC separation. A) Base peak electropherogram. B) Summed MS spectra of monosialylated peptides of Site V migrating at 5.42–5.48 min. C) Summed MS spectra of pentasialylated peptides of Site V migrating at 8.4–8.5 min. Symbols:
:N-Acetylglucosamine (HexNAc),
: Mannose (Hex),
: Galactose (Hex),
: N-acetylneuraminic acid (Neu5Ac),
: Fucose (Fuc). Glycan compositions were represented as G (HexNAc; Hex; Fuc; and NeuAc)
Resolution of glycopeptides based on different peptide backbones could also be observed in CZE, marked as green for Site II and yellow for Site V in Figure 3A. Among all of the factors that play roles in CZE separation of intact glycopeptides, the negative charge on sialic acid contributes most, thus enabling an observation of rearranged glycan-based separation of N-glycopeptides on CZE, which is complementary to conventional RPLC analysis.
We identified 91 N-glycopeptides from Fraction 1 by CZE-ESI-MS/MS analysis, which is nearly double the 55 N-glycopeptides identified with direct reversed-phase UHPLC-ESI-MS/MS analysis (Table 2). The isolation of low intensity CZE peaks that migrated at ~3, ~5.3, and ~8.4 min (Figure 3A) facilitated identification of asialylated Site II, monosialylated Site V (Figure 3B) and pentasialylated Site V (Figure 3C).
Table 2.
Identified AGP N-glycopeptides with Site II and V by reversed-phase UHPLC-ESI-MS/MS and reversed-phase UHPLC-CZE-ESI-MS/MS
| Peptide Sequence | Glycoform | Theoretical m/z | Observed m/z
|
||
|---|---|---|---|---|---|
| Reversed-phase UHPLC-ESI-MS/MS | Reversed-phase UHPLC-CZE-ESI-MS/MS | ||||
| NEEYN38K | Asialylated | G4500 | 806.9800 (3+) | 806.9841 (3+) | 806.9800 (3+) |
| G4510 | 855.6660 (3+) | × | 855.6675 (3+) | ||
| G5600 | 928.6907 (3+) | × | 928.6913 (3+) | ||
| Monosialylated | G3401 | 1172.9476 (2+) | × | 1172.9479 (2+) | |
| G4401 | 849.9942 (3+) | × | 849.9919 (3+) | ||
| G4501 | 904.0118 (3+) | 904.0175 (3+) | 904.0110 (3+) | ||
| G4511 | 952.6978 (3+) | 952.7016 (3+) | 952.6961 (3+) | ||
| G5601 | 1025.7225 (3+) | 1025.7286 (3+) | 1025.7202 (3+) | ||
| G5611 | 1074.4085 (3+) | 1074.4156 (3+) | 1074.4064 (3+) | ||
| G6701 | 1147.4333 (3+) | 1147.4392 (3+) | 1147.4376 (3+) | ||
| G6711 | 1196.1192 (3+) | × | 1196.1154 (3+) | ||
| Bisialylated | G4502 | 1001.0436 (3+) | 1001.0496 (3+) | 1001.0468(3+) | |
| G4512 | 1049.7296 (3+) | 1049.7356 (3+) | 1049.7297(3+) | ||
| G5502 | 1068.7367 (3+) | × | 1068.7313(3+) | ||
| G5602 | 1122.7543 (3+) | 1122.7606 (3+) | 1122.7540 (3+) | ||
| G5612 | 1171.4403 (3+) | 1171.4470 (3+) | 1171.4415 (3+) | ||
| G6702 | 1244.465 (3+) | 1244.4731 (3+) | 1244.4539 (3+) | ||
| Trisialylated | G5603 | 1219.7861 (3+) | 1219.7919 (3+) | 1219.7837 (3+) | |
| G5613 | 1268.4721 (3+) | 1268.4785 (3+) | 1268.4686 (3+) | ||
| G5623 | 1317.1581 (3+) | 1317.1625 (3+) | 1317.1570 (3+) | ||
| G6703 | 1341.4969 (3+) | × | 1341.4994 (3+) | ||
| G6713 | 1390.1828 (3+) | × | 1390.1760 (3+) | ||
| EN85GTISR | Asialylated | G4500 | 1199.9873 (2+) | × | 1199.9850 (2+) |
| Monosialylated | G4501 | 897.3593 (3+) | 897.3635 (3+) | 897.3583 (3+) | |
| G5601 | 1019.0700 (3+) | × | 1019.0710 (3+) | ||
| G5611 | 1067.7560 (3+) | × | 1067.7578 (3+) | ||
| G6701 | 1140.7808 (3+) | 1140.7877 (3+) | 1140.7837 (3+) | ||
| G6711 | 1189.4667 (3+) | × | 1189.4662 (3+) | ||
| G6721 | 1238.1527 (3+) | × | 1238.1527 (3+) | ||
| G7801 | 1262.4915 (3+) | × | 1262.4904 (3+) | ||
| G7811 | 1311.1775 (3+) | × | 1311.1776 (3+) | ||
| Bisialylated | G4502 | 994.3911 (3+) | 994.3967 (3+) | 994.3925 (3+) | |
| G5602 | 1116.1018 (3+) | 1116.1080 (3+) | 1116.0985 (3+) | ||
| G5612 | 1164.7878 (3+) | 1164.7932 (3+) | 1164.7898 (3+) | ||
| G6702 | 1237.8234 (3+) | 1237.8234 (3+) | 1237.8124 (3+) | ||
| G6712 | 1286.5132 (3+) | 1286.5132 (3+) | 1286.5021 (3+) | ||
| Trisialylated | G5603 | 1213.1336 (3+) | 1213.1404 (3+) | 1213.1299 (3+) | |
| G5613 | 1261.8196 (3+) | 1261.8257 (3+) | 1261.8143 (3+) | ||
| G5623 | 1310.5056 (3+) | 1310.5090 (3+) | 1310.5037 (3+) | ||
| G6703 | 1334.8444 (3+) | 1334.8511 (3+) | 1334.8402 (3+) | ||
| G6713 | 1383.5303 (3+) | 1383.5378 (3+) | 1383.5253 (3+) | ||
| G6723 | 1432.2163 (3+) | × | 1432.2021 (3+) | ||
| G6733 | 1480.9023 (3+) | × | 1480.8983 (3+) | ||
| G7803 | 1456.5551 (3+) | 1456.5598 (3+) | 1456.5485 (3+) | ||
| G7813 | 1505.2411 (3+) | × | 1505.2319 (3+) | ||
| Tetrasialylated | G6704 | 1431.8762 (3+) | 1431.8826 (3+) | 1431.8717 (3+) | |
| G6714 | 1480.5621 (3+) | 1480.5699 (3+) | 1480.5583 (3+) | ||
| G6724 | 1529.2481 (3+) | 1529.2537 (3+) | 1529.2448 (3+) | ||
| G6734 | 1577.9341 (3+) | 1577.9352 (3+) | 1577.9226 (3+) | ||
| G7804 | 1553.5869 (3+) | 1553.5935 (3+) | 1553.5856 (3+) | ||
| G7814 | 1201.9566 (4+) | 1201.9644 (4+) | 1201.9531 (4+) | ||
| G7824 | 1238.4711 (4+) | × | 1238.4658 (4+) | ||
| G7834 | 1274.9856 (4+) | × | 1274.9803 (4+) | ||
| G8904 | 1256.7261 (4+) | 1256.7307 (4+) | 1256.7178 (4+) | ||
| G8914 | 1293.2397 (4+) | 1293.2472 (4+) | 1293.2349 (4+) | ||
| G8924 | 1329.7542 (4+) | × | 1329.7559 (4+) | ||
| G9(10)04 | 1348.0083 (4+) | × | 1348.0022 (4+) | ||
| G9(10)14 | 1384.5227 (4+) | × | 1384.5321 (4+) | ||
| Pentasialylated | G8905 | 1329.4990 (4+) | × | 1329.5043 (4+) | |
| G8915 | 1366.0135 (4+) | × | 1366.0149 (4+) | ||
| G8925 | 1402.5280 (4+) | × | 1402.5204 (4+) | ||
| G9(10)05 | 1420.7821 (4+) | × | 1420.7756 (4+) | ||
| C9(10)15 | 1457.2966 (4+) | × | 1457.2939 (4+) | ||
| EN85GTVSR | Monosialylated | G4501 | 892.6874 (3+) | × | 892.6888 (3+) |
| G5601 | 1014.3982 (3+) | 1014.4027 (3+) | 1014.3954 (3+) | ||
| G5611 | 1063.0841 (3+) | 1063.0928 (3+) | 1063.0844 (3+) | ||
| G6701 | 1136.1089 (3+) | 1136.1135 (3+) | 1136.1073 (3+) | ||
| G6711 | 1184.7949 (3+) | 1184.7996 (3+) | 1184.7943 (3+) | ||
| Bisialylated | G4502 | 989.7192 (3+) | 989.7225 (3+) | 989.7205 (3+) | |
| G5602 | 1111.4230 (3+) | 1111.4332 (3+) | 1111.4310 (3+) | ||
| G5612 | 1160.1159 (3+) | 1160.1206 (3+) | 1160.1124 (3+) | ||
| G6702 | 1233.1407 (3+) | 1233.1456 (3+) | 1233.1334 (3+) | ||
| G6712 | 1281.8267 (3+) | 1281.8317 (3+) | × | ||
| Trisialylated | G5603 | 1208.4618 (3+) | 1208.4672 (3+) | 1208.4576 (3+) | |
| G5613 | 1257.1477 (3+) | 1257.1548 (3+) | 1257.1482 (3+) | ||
| G6703 | 1330.1725 (3+) | 1330.1768 (3+) | 1330.1670 (3+) | ||
| G6713 | 1378.8585 (3+) | 1378.8638 (3+) | 1378.8590 (3+) | ||
| G6723 | 1427.5444 (3+) | × | 1427.5387 (3+) | ||
| Tetrasialylated | G6704 | 1427.2043 (3+) | 1427.2097 (3+) | 1427.1981 (3+) | |
| G6714 | 1475.8903 (3+) | 1475.8972 (3+) | 1475.8844 (3+) | ||
| G6724 | 1524.5762 (3+) | 1524.5873 (3+) | 1524.5752 (3+) | ||
| G7804 | 1548.9150 (3+) | 1549.2635 (3+) | 1548.9236 (3+) | ||
| G7814 | 1198.4527 (4+) | 1198.4531 (4+) | 1198.4491 (4+) | ||
| G7824 | 1234.9672 (4+) | × | 1234.9587 (4+) | ||
| G8904 | 1253.2222 (4+) | 1253.2255 (4+) | 1253.2178 (4+) | ||
| G8914 | 1289.7358 (4+) | 1269.7380 (4+) | 1289.7334 (4+) | ||
| G9(10)04 | 1344.5044 (4+) | × | 1344.7554 (4+) | ||
| G9(10)14 | 1381.0188(4+) | × | 1381.2648 (4+) | ||
| Pentasialylated | G8905 | 1325.9951 (4+) | × | 1326.0023 (4+) | |
| G8915 | 1362.5096 (4+) | × | 1362.5182 (4+) | ||
| G8925 | 1399.0241 (4+) | × | 1339.0211 (4+) | ||
| G9(10)05 | 1417.2782 (4+) | × | 1417.2744 (4+) | ||
Pentasialylated glycopeptides have rarely been reported.27 Their low mobility results in excellent resolution from other components. Based on the Y1 ions detected in tandem MS analysis, pentasialylated glycans were only assigned on Site V. A dramatically increased relative intensity of sialic acid oxonium ion in MS/MS was observed for glycopeptides migrating at ~8 min compared to those at ~5 min, which further supports our assignment (Figure S-4). We identified 69, 49, and 59 glycopeptides from Fractions 2, 3, and 4, respectively.
Intense peak
In addition to providing a complementary separation to reversed-phase UHPLC, CZE also generated more intense peaks. The fractions for CZE-ESI-MS/MS analysis were collected from single reversed-phase UHPLC separation of AGP glycopeptides (enriched from 0.16 nmol of initial digest). Roughly 0.1% of each fraction was injected onto the CZE column, corresponding to an initial digest amount of 0.18 fmol. Despite this small injection amount, the observed base peak intensity is comparable with that obtained by reversed-phase UHPLC-ESI-MS/MS analysis of much larger sample amounts (Figure 2B and Figure S-3).
We observed a series of novel glycoforms on Site III by direct reversed-phase UHPLC-ESI-MS/MS. These novel glycoforms were relatively easy to detect because Site III glycopeptides (68-81) produced high peak intensity and were only slightly overlapped with other glycopeptides. In contrast, the second dimensional CZE-ESI-MS/MS facilitated identification of additional glycoforms on Site III, including monosialylated and highly complex tetrasialylated species (Table S-3).
As shown in Figure 4A, there was a systematic decrease in peak intensity as the numbers of fucose and LacNAc repeat increased on the tetrasialylated glycans of Site III, for both reversed-phase UHPLC-ESI-MS/MS and CZE-ESI-MS/MS analysis. CZE-ESI-MS/MS generated half of the peak intensity as reversed-phase UHPLC-ESI-MS/MS for the highly abundant G6704 glycopeptide. However, the differences in peak intensities between these two approaches narrowed as the glycopeptides abundance decreased. CZE-ESI-MS/MS produced more intense peaks for the low abundant G7814, G7824, G8904 and G9(10)04 glycopeptides, which allowed the detection of highly complex, multi-fucosylated tetrasialylated glycans on AGP Site III (Table S-3, Figure S-5).
Figure 4.

A) Extracted ion separations of tetrasialylated Site III glycopeptides with the YFTPN54KTEDTIFLR (68-81) backbone by reversed-phase UHPLC-ESI-MS/MS and CZE-ESI-MS/MS analysis. B) Venn diagrams of identified N-glycopeptides by direct reversed-phase UHPLC-ESI-MS/MS, CZE-ESI-MS/MS, and two-dimensional reversed-phase UHPLC-CZE-ESI-MS/MS analysis of the glycopeptides enriched from the AGP digest.
AGP glycosylation characterization
Reversed-phased UHPLC-CZE-ESI-MS/MS analysis identified 268 N-glycopeptides from AGP, which is a 35% increase over our direct reversed-phase UHPLC-ESI-MS/MS analysis (Figure 4B), which was an improvement over previous reports.21,24,26 A number of newly characterized N-glycans were primarily located on Site III (AGP1/2) and Site V (AGP1 and AGP2), which appear to have a more complicated glycan distribution compared with other sites of AGP. Besides the monosialylated and pentasialylated Site V, highly branched Site III with various numbers of sialic acid were found (Table 2 and Table S-3).
Six known glycopeptides were not detected on Site IV (AGP1 and AGP2) with our approach (Table S-4). The failure to observe these peptides is likely due to the very similar charge to size ratio of the peptide backbone for Site I (AGP2) and Site IV. Glycopeptides with the same number of sialic acids from these two sites in Fraction 2 co-migrated during CZE separation, causing some low abundant Site IV glycopeptides to be suppressed by glycopeptides from Site I (AGP2). However, since Site IV and Site I (AGP2) did not share tetrasialylated glycans, CZE produced an efficient separation and detection of multiple tetrasialylated glycopeptides based on Site IV (Table S-4), demonstrating the complementary identification of reversed-phase UHPLC-ESI-MS/MS and CZE-ESI-MS/MS for complex glycopeptide mixtures.
Comparison with single-shot CZE-ESI-MS/MS
Unfractionated AGP glycopeptides were also analyzed with single-shot CZE-ESI-MS/MS. An uncoated fused capillary (20 μm i.d. × 150 μm o.d. × 80 cm) was coupled to a Q-Exactive HF mass spectrometer for this experiment (see Supporting Information). A total of 159 site-specific N-glycopeptides representing eight different glycosylation sites from AGP1 and AGP1 were identified (Table S-1 to S-5). The electrophoretic migration order of N-glycopeptides was strongly influenced by the negative charges associated with sialic acids (Figure S-6), facilitating the detection of pentasialylated Site V and tetrasialylated Site IV, which is complementary to the reversed-phased UHPLC-ESI-MS/MS results (Figure 4B). The separation of different peptide backbones could also be observed, but with lower resolution than reversed-phase UHPLC. We attributed the identification of a modest number of AGP N-glycopeptides by single-shot CZE-ESI-MS/MS to its relatively narrow separation window for peptide backbone differentiation, especially when the sample was complex in both peptide backbone and glycan components. Finally, the reversed-phased UHPLC-CZE-ESI-MS/MS approach generated ~70% more N-glycopeptides from AGP over direct CZE-ESI-MS/MS analysis (Figure 4B), demonstrating the value of applying two dimensional separations of N-glycopeptides from highly glycosylated proteins for comprehensive site-specific glycopeptide characterization.
Migration time-assisted N-glycopeptide characterization
The use of CZE for analysis of reversed-phase UHPLC glycopeptide fraction facilitated glycan structural elucidation. The first dimension reversed-phase UHPLC separation of deglycosylated peptides enabled the identification of peptide backbones. Glycopeptides with known peptide sequences were generated by reversed-phase UHPLC fractionation of the intact glycosylated peptides. The second dimension separation by CZE of the glycopeptides produced high-resolution based on the number of sialic acids, which produced a migration time-based prediction of the number of sialic acids. The precursor mass matching of successively eluted precursors observed in a particular cluster become straightforward. Assignment could be processed following the N-glycan growing on the lowest-mass glycopeptide. With 10 ppm mass tolerance window and migration time correction, site-specific glycan structures were characterized with high confidence, even when MS/MS spectra had poor quality. Additionally, the glycopeptide MS spectra could be assigned using commercial software such as Byonic and others. Automated spectral interpretation would simplify analysis of a large number of reversed-phase UHPLC fractions to achieve a more comprehensive characterization of complex samples.
Supplementary Material
Acknowledgments
We thank Dr. William Boggess in the Notre Dame Mass Spectrometry and Proteomics Facility for his help with this project. This work was funded by the National Institutes of Health (Grant R01GM096767).
Footnotes
Single-shot CZE-ESI-MS/MS method for unfractionated AGP N-glycopeptide analysis, schematic diagram of the electrokinetically pumped sheath flow interface (Figure S-1), MS/MS spectra of AGP deglycosylated site III (Figure S-2), base peak electropherograms of AGP fractions by CZE-ESI-MS/MS (Figure S-3), MS/MS spectra of AGP intact N-glycopeptides (Figure S-4 and 5), base peak chromatogram of unfractioned AGP glycopeptides by single-shot CZE-ESI-MS/MS (Figure S-6), and observed glycoforms on AGP N-glycosyltaion sites by reversed-phase UHPLC-ESI-MS/MS, CZE-ESI-MS/MS, and reversed-phase UHPLC-CZE-ESI-MS/MS (Table S-1 to Table S-5).
References
- 1.Steen PVd, Rudd PM, Dwek RA, Opdenakker G. Crit Rev Biochem Mol Biol. 1998;33:151–208. doi: 10.1080/10409239891204198. [DOI] [PubMed] [Google Scholar]
- 2.Ford K, Zeng W, Heazlewood JL, Bacic A. Front Plant Sci. 2015;6:674. doi: 10.3389/fpls.2015.00674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perdivara I, Peddada SD, Miller FW, Tomer KB, Deterding LJ. J Proteome Res. 2011;10:2969–2978. doi: 10.1021/pr200397h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abou-Abbass H, Abou-El-Hassan H, Bahmad H, Zibara K, Zebian A, Youssef R, Ismail J, Zhu R, Zhou S, Dong X, Nasser M, Bahmad M, Darwish H, Mechref Y, Kobeissy F. Electrophoresis. 2016:1–13. doi: 10.1002/elps.201500585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon S-J, Park S-Y, Pang P-C, Gallagher J, Gottesman JE, Dell A, Kim J-H, Hakomori S-I. Int J Oncol. 2010;36:193–203. [PubMed] [Google Scholar]
- 6.Anugraham M, Jacob F, Nixdorf S, Everest-Dass AV, Heinzelmann-Schwarz V, Packer NH. Mol Cell Proteomics. 2014;13:2213–2232. doi: 10.1074/mcp.M113.037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pegg CL, Hoogland C, Gorman JJ. Glycoconjugate J. 2016;34:1–17. doi: 10.1007/s10719-016-9750-7. [DOI] [PubMed] [Google Scholar]
- 8.Chandler KB, Leon DR, Meyer RD, Rahimi N, Costello CE. J Proteome Res. 2017;16:677–688. doi: 10.1021/acs.jproteome.6b00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanda M, Zhang L, Edwards NJ, Goldman R. Anal Bioanal Chem. 2017;409:619–627. doi: 10.1007/s00216-016-0041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen R, Cheng K, Ning Z, Figeys D. Anal Chem. 2016;88:11837–11843. doi: 10.1021/acs.analchem.6b03531. [DOI] [PubMed] [Google Scholar]
- 11.Wang B, Tsybovsky Y, Palczewski K, Chance MR. J Am Soc Mass Spectr. 2014;25:729–741. doi: 10.1007/s13361-013-0823-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Q, Yan X, Liang Y, Stein SE. J Proteome Res. 2016;15:1472–1486. doi: 10.1021/acs.jproteome.5b01046. [DOI] [PubMed] [Google Scholar]
- 13.Sun L, Hebert AS, Yan X, Zhao Y, Westphall MS, Rush MJP, Zhu G, Champion MM, Coon JJ, Dovichi NJ. Angew Chem-Int Edit. 2014;53:13931–13933. doi: 10.1002/anie.201409075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L, Zhu G, Yan X, Zhang Z, Wojcik R, Champion MM, Dovichi NJ. Proteomics. 2016;16:188–196. doi: 10.1002/pmic.201500339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kammeijer GSM, Kohler I, Jansen BC, Hensbergen PJ, Mayboroda OA, Falck D, Wuhrer M. Anal Chem. 2016;88:5849–5856. doi: 10.1021/acs.analchem.6b00479. [DOI] [PubMed] [Google Scholar]
- 16.Yan X, Sun L, Zhu G, Cox OF, Dovichi NJ. Proteomics. 2016;16:2945–2952. doi: 10.1002/pmic.201600262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mechref Y, Novotny MV. Mass Spectrom Rev. 2009;28:207–222. doi: 10.1002/mas.20196. [DOI] [PubMed] [Google Scholar]
- 18.Jayo RG, Thaysen-Andersen M, Lindenburg PW, Haselberg R, Hankemeier T, Ramautar R, Chen DDY. Anal Chem. 2014;86:6479–6486. doi: 10.1021/ac5010212. [DOI] [PubMed] [Google Scholar]
- 19.Mitra I, Snyder CM, Zhou X, Campos MI, Alley WR, Novotny MV, Jacobson SC. Anal Chem. 2016;88:8965–8971. doi: 10.1021/acs.analchem.6b00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khatri K, Klein JA, Haserick JR, Leon DR, Costello CE, McComb ME, Zaia J. Anal Chem. 2017;89:6645–6655. doi: 10.1021/acs.analchem.7b00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JY, Lee HK, Park GW, Hwang H, Jeong HK, Yun KN, Ji ES, Kim KH, Kim JS, Kim JW, Yun SH, Choi C-W, Kim SI, Lim J-S, Jeong S-K, Paik Y-K, Lee S-Y, Park J, Kim SY, Choi Y-J, Kim Y-I, Seo J, Cho J-Y, Oh MJ, Seo N, An HJ, Kim JY, Yoo JS. J Proteome Res. 2016;15:4146–4164. doi: 10.1021/acs.jproteome.5b01159. [DOI] [PubMed] [Google Scholar]
- 22.Sun L, Zhu G, Zhang Z, Mou S, Dovichi NJ. J Proteome Res. 2015;14:2312–2321. doi: 10.1021/acs.jproteome.5b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krylov SN, Starke DA, Arriaga EA, Zhang Z, Chan NWC, Palcic MM, Dovichi NJ. Anal Chem. 2000;72:872–877. doi: 10.1021/ac991096m. [DOI] [PubMed] [Google Scholar]
- 24.Imre T, Schlosser G, Pocsfalvi G, Siciliano R, Molnár-Szöllősi É, Kremmer T, Malorni A, Vékey K. J Mass Spectrom. 2005;40:1472–1483. doi: 10.1002/jms.938. [DOI] [PubMed] [Google Scholar]
- 25.Balmaña M, Giménez E, Puerta A, Llop E, Figueras J, Fort E, Sanz-Nebot V, de Bolós C, Rizzi A, Barrabés S, de Frutos M, Peracaula R. J Proteomics. 2016;132:144–154. doi: 10.1016/j.jprot.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Li X, Guo Z, Zhang X, Liang X. Chem-Eur J. 2009;15:12618–12626. doi: 10.1002/chem.200902370. [DOI] [PubMed] [Google Scholar]
- 27.Bendiak B, Harris-Brandts M, Michnick SW, Carver JP, Cumming DA. Biochemistry. 1989;28:6491–6499. doi: 10.1021/bi00441a050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.