

Abstract
Development of biased ligands targeting G protein-coupled receptors (GPCRs) is a promising approach for current drug discovery. Although structure-based drug design of biased agonists remains challenging even with an abundance of GPCR crystal structures, we present an approach for translating GPCR structural data into β-arrestin-biased ligands for aminergic GPCRs. We identified specific amino acid-ligand contacts at transmembrane helix 5 (TM5) and extracellular loop 2 (EL2) responsible for Gi/o and β-arrestin signaling, respectively, and targeted those residues to develop biased ligands. For these ligands, we found that bias is conserved at other aminergic GPCRs that retain similar residues at TM5 and EL2. Our approach provides a template for generating arrestin-biased ligands by modifying predicted ligand interactions that block TM5 interactions and promote EL2 interactions. This strategy could facilitate the structure-guided design of arrestin-biased ligands at other GPCRs, including polypharmacological biased ligands.
Graphical Abstract
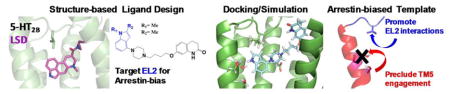
Introduction
G protein-coupled receptors (GPCRs), which form the largest target class in the druggable genome, are crucial for nearly every physiological process1. Aminergic GPCRs, including histamine, adrenergic, dopamine, serotonin, and muscarinic receptors, are of particular importance to drug discovery as they are targeted by one quarter of currently approved drugs2, 3. Functional selectivity4, or signaling bias, is a process whereby GPCR ligands can either activate G proteins or recruit β-arrestins to activate select downstream signaling pathways at a given receptor5–7. In many instances, one signaling pathway is potentially responsible for therapeutic effects, while the other is implicated in side effects8–10. Biased ligands which could yield drugs with optimized on-target effects include agonists for the D2 dopamine receptor (D2R)8, D1 dopamine receptor (D1R)11, angiotensin II type 1 receptor (AT1R)10, δ-opioid receptor (DOR)12, and the μ-opioid receptor (MOR)9. G protein biased MOR agonists are potentially analgesic with fewer side-effects (e.g. respiratory depression and constipation13).
The development of biased ligands remains challenging, even when using high-throughput screening and extensive interrogation of the signaling properties of existing ligands10, 14–17. Recently, our understanding of GPCR ligand recognition and receptor activation dynamics as it pertains to biased signaling has been catalyzed by a ‘golden era’ of GPCR structural biology, with several key aminergic receptor structures being published in the last decade18–23. Despite this wealth of information, no logical process exists for efficiently incorporating insights gleaned from GPCR structures into a design strategy for biased ligand development.
The D2R remains an essential target for antipsychotic drug discovery24, 25 with the newest atypical antipsychotic drugs (e.g. aripiprazole, cariprazine) being partial agonists at D2R and other receptors 26. We previously conducted extensive medicinal chemistry exploration of aripiprazole and although aripiprazole is a partial agonist at multiple GPCRs26, it shows similar potency and efficacy in Gi/o signaling and β-arrestin recruitment at D2R8, 27. Those studies culminated in the discovery of the first D2R β-arrestin-biased ligands8, which show therapeutic potential in animal models of schizophrenia28. Our results suggested that D2R β-arrestin signaling contributes to the antipsychotic efficacy of these drugs, whereas G protein-signaling may contribute to extra-pyramidal side effects8.
In this study, we used D2R as a model system to identify GPCR-ligand contacts that mediate biased signaling and used this information to develop an approach for the structure-based drug design (SBDD) of β-arrestin-biased ligands for other aminergic GPCRs.
Results
Structure-Inspired Design of Indole-Aripiprazole Hybrid Ligands
We analyzed prior aminergic GPCR structural and mechanistic data to identify residues implicated in G protein versus β-arrestin signaling. We focused on the orthosteric site, as this is both the most common and well conserved binding site for class A GPCRs. In the binding pockets of the β1 and β2 adrenergic receptors (β1AR and β2AR, respectively), transmembrane helix 5 (TM5) transduces ligand-induced G protein activation via conserved serine residues (5.42, 5.43, and 5.46)-findings supported by structural21, mutagenesis29, and NMR30 studies. For the nanobody-stabilized β2AR crystallized in complex with epinephrine21, the catechol of epinephrine, which is also present on dopamine, forms an extensive hydrogen bond network with these conserved TM5 serines (Fig. 1a), which have been previously posited to form the structural basis of agonist and partial agonist action31 at β1AR and β2AR. D2R also contains TM5 serine residues (Supplementary Fig. 1a), where mutagenesis studies support that they both contribute to ligand efficacy and overall G protein activation 32, 33, and are also essential for aripiprazole recognition34.
Figure 1. Structure-Inspired Design of Indole-Aripiprazole Hybrid Ligands.

D2 ligand design based on comparison of three aminergic crystal structures. a) β2 adrenergic receptor nanobody-stabilized with epinephrine bound (4LDO) indicates the catechol of epinephrine is involved in an extensive hydrogen bond network with transmembrane (TM) 5 serines. b) Structure of the 5-HT2B receptor with LSD bound (5TVN) indicates that EL2.52 Leu209 forms hydrophobic cap over ligand, preventing ligand egress. c) Thermostabolized β1 adrenergic receptor with 4-indole piperazine bound (3ZPQ) shows that the indole N-H interacts with Ser5.42 in a hydrogen bond d) Design of indole-aripiprazole hybrid compounds by addition of 4-indole (blue) replacing the dichlorophenyl (green) of aripiprazole resulting in compound 1. e) Docking of 1 in D2 homology model places the unsubstituted 4-indole moiety of the indole-aripiprazole hybrid 1 in the D2 orthosteric binding pocket making contact with TM5 Ser5.42.
Structural clues for binding pocket residues that mediate arrestin recruitment are illuminated by the 5-HT2B receptor structures in complex with ergotamine22 and lysergic acid diethylamide35 (LSD; Fig. 1b). In the 5-HT2B-LSD structure study, mutation of the conserved hydrophobic extracellular loop 2 (EL2) residue Leu209 selectively reduced LSD arrestin recruitment by increasing ligand on- and off-rates at the receptor. EL2 as a structural motif was proposed to function as a “lid” over the binding pocket thereby enhancing ligand residence time and also functioning as a major determinant of arrestin recruitment efficacy35. Given that hydrophobic residues located in EL2 are relatively well-conserved for aminergic GPCRs (Supplementary Fig. 1a), we posited that targeting the homologous D2 EL2 hydrophobic residue isoleucine184 (I184EL2) may enhance β-arrestin recruitment at this receptor, thus leading to novel β-arrestin-biased ligands.
First, we required a ligand scaffold to test our hypotheses for the differential involvement of TM5 and EL2 for biased signaling. We recently disclosed β-arrestin-biased ligands that are close structural analogs of aripiprazole8, 36, and chose these as starting points. We also required a small fragment that is predicted to form defined interactions with conserved TM5 serines located in the orthosteric site, which could be substituted in such a way as to disrupt the TM5 serine interactions associated with G protein-dependent activation. Crystal structures of the thermostabilized turkey β1AR in complex with indole-piperazine clearly illustrate how the indole group is positioned in the orthosteric site near TM5 and EL2, with the indole N-H forming a hydrogen bond with TM5 residue S5.4237 (Fig. 1c).
Our design strategy, therefore, was to replace the dichlorophenyl-piperazine portion of aripiprazole with the indole-piperazine fragment found in the β1AR crystal structure leading to an indole-aripiprazole hybrid, compound 1 (Fig. 1d). To generate reliable assumptions regarding the binding pose of 1, we constructed hundreds of D2R homology models based on the crystal structure of the D3 receptor19, and subsequently docked compound 1. In the docked D2 structure, the indole-piperazine portion of 1 occupies the orthosteric site, and the indole N-H group forms a hydrogen bond with S1935.42 (Fig. 1e), which is consistent with D2 docking of aripiprazole38, 39, and with β1AR crystal structure pose of the indole-piperazine37. Additionally, we confirmed 1’s docking pose at TM5 serine mutants, where compound 1’s affinity (Supplementary Fig. 1b) and Gi/o-mediated potency (Supplementary Fig. 1c) were selectively decreased at the TM5 S193A5.42 mutant.
Indole-Aripiprazole Hybrid D2R SFSR
Next, we evaluated the structure-functional selectivity relationships (SFSR) of indole N-substitutions (e.g., methyl, n-propyl, i-propyl, benzyl) to 1 (Fig. 2a) intended to disrupt interactions with TM5. These substitutions introduce steric repulsion between the ligand and TM5, and are expected to eliminate the S1935.42-ligand hydrogen bond. To assess ligand bias at G protein versus β-arrestin recruitment pathways, ligands were tested by measuring Gi/o-mediated cAMP inhibition and β-arrestin2 recruitment assays40 conducted in parallel. D2R expression was similar in both D2- assay platforms (Supplementary Table 1). D2R-mediated cAMP inhibition, but not D2 β-arrestin2 recruitment, was dependent on pertussis-toxin sensitive Gi/o proteins (Supplementary Fig. 2a). Previously, we confirmed that compound 1 is a D2R partial agonist (~75% of quinpirole, Fig. 2b,c) in both Gi/o signaling and β-arrestin2 recruitment activity, while 1 shows weak preference for arrestin recruitment over Gi/o signaling (bias factor = 2.5) relative to quinpirole (Fig. 2c).
Figure 2. Indole-Aripiprazole Hybrid D2R SFSR.

Structure-Functional Selectivity Relationships (SFSRs) of indole N1-substituted analogs of indole-aripiprazole hybrids, which lead to either D2 arrestin-bias or antagonism dependent on substitution. a) Chemical structures of N1-substituted indole-aripiprazole hybrids b-g) Profiling of indole-aripiprazole hybrids measuring D2 G protein activity (Gαi/o-mediated cAMP inhibition; red) and β-arrestin2 recruitment (Tango; blue), normalized to percent quinpirole activity. Data represent n=5 performed in triplicate and in parallel using the same drug dilutions. h) SFSR summary for indole-aripiprazole hybrids. Unsubstituted indole (1) shows weak preference for arrestin with respect to quinpirole (bias factor = 2.5; D2 Gi/o EC50= 0.98 nM, Emax = 66%, D2 β-arrestin2 EC50= 0.71 nM Emax = 69%) comparing Gi/o and arrestin activity but N-methyl (2, D2 β-arrestin2 EC50= 6.3 nM, Emax = 36%) and N-n-propyl (3, D2 β-arrestin2 EC50= 81 nM, Emax = 32%) show arrestin-bias with no measureable Gi/o activity with respect to quinpirole. Larger substitutions such as N-i-propyl (4) and N-benzyl (5) show no activity and instead act as competitive antagonists i) Orthologous assay for D2 G protein activity utilizing D2 Gi1-γ2 dissociation as measured by BRET, showing partial agonism for 1 (EC50 = 0.49 nM, Emax = 55%) and no activity by 2, compared to quinpirole (EC50 = 1.6 nM). Data represent total BRET as calculated using GFP/Rluc ratio j) Orthologous assays for β-arrestin2 recruitment utilizing BRET measuring Venus-tagged-β-arrestin2 and D2long-tagged Rluc association comparing recruitment by 1 (EC50 = 0.52 nM, Emax = 39%) and 2 (EC50 = 11 nM, Emax = 33%) to quinpirole (EC50 = 13 nM). Data are representative and indicate the change in Net BRET with respect to no Venus-β-arrestin2 expressed.
As predicted, N-alkyl or aryl substitution completely abolished G protein-mediated signaling relative to quinpirole and compound 1 (Fig. 2d–g). However, the N-methyl (2) and N-n-propyl (3) retained arrestin-recruitment efficacy thus exhibiting arrestin-bias relative to quinpirole (Fig. 2d,e). Interestingly, N-isopropyl (4) and N-benzyl (5) substitutions showed no activity in both assays (Fig. 2f,g) but still retained appreciable affinity for D2R (77 and 22 nM, respectively) as measured by radioligand binding (Supplementary Table 2). In fact, both compounds 4 and 5 are potent and competitive antagonists of quinpirole-stimulated D2R cAMP inhibition (compound 4 KB = 11.3 nM; compound 5 KB = 8.1 nM; Supplementary Fig. 2b,c). The added bulk by N-isopropyl or N-benzyl likely avoids hydrogen bonding with TM5 and EL2 engagement, pushing on TM5 preventing activation potentially explaining its antagonist activity. In short, a clear D2R SFSR for the indole-aripiprazole hybrids emerged demonstrating either arrestin preference or antagonism dependent on the indole N-substitution (Fig. 2h).
In addition, because the interpretation of ligand bias can be skewed by system-dependent factors (e.g. receptor reserve, cellular background, assay platforms), we subjected compound 2 to an orthologonal assay of D2R G protein-activity, measuring Gαi1-γ2 dissociation by bioluminescent resonance energy transfer (BRET). In this assay, compound 2 showed no agonist activity whereas compound 1 was a partial agonist with respect to quinpirole (Fig. 2i), recapitulating our findings obtained measuring Gi/o-dependent cAMP inhibition activity. Further confirmation of arrestin-bias employing an orthologonal platform for arrestin recruitment using BRET, revealed 2 to be a potent agonist for arrestin recruitment relative to quinpirole (EC50 = 17 nM, Emax = 33%, Fig. 2j). Although no G protein-mediated agonism could be detected by any method, and therefore no bias factor could be formally calculated, we further tested compound 2 as an antagonist of quinpirole-stimulated Gi/o-mediated cAMP inhibition (KB = 3.6 nM; Supplementary Fig. 2d) to demonstrate 2 indeed acts as a competitive antagonist. Finally, in light of recent findings that the kinetic context can influence bias interpretations35, 41, we also profiled the kinetics of signaling of 2, which revealed no Gi/o-mediated cAMP inhibition up to 90 minutes (Supplementary Fig. 2e) and robust arrestin recruitment peaking between 15–60 minutes (Supplementary Fig. 2f). In summary, compound 2 was extensively profiled and confirmed as an arrestin-biased D2 partial agonist.
D2R MD Simulations Predict EL2 Engagement for Arrestin-bias
To identify binding pocket residues involved in G protein-signaling versus β-arrestin recruitment, we studied compounds 1 and 2 by molecular dynamics (MD) simulations although it was initially clear that the N-methylated compound 2 is incapable of forming a hydrogen bond with S1935.42. Like compound 1, compound 2 will likely position its indole-piperazine portion in the orthosteric site, with the protonated nitrogen of the piperazine ring forming a salt bridge with the conserved D1143.32 in TM3. Less clear, however, is the effect of N-methylation translates to attenuation of G protein-signaling with retention of β-arrestin recruitment. Therefore, we performed MD simulations with the head groups of compounds 1 and 2, i.e. without the dihydroquinolin-2-one and alkyl linker (Supplementary Fig. 3, Supplementary Table 3). Compounds 1 and 2 are identical aside from the head group moiety, but because of the uncertainty in the orientation of their flexible tail region, we chose to use the head groups to investigate potential structural features that lead to biased signaling.
Simulations of both head groups were initiated from the same position in D2R, which was based on the position of 4-(piperazin-1-yl)-1H-indole (equivalent to the head group of compound 1) in the thermostabilized turkey β1AR crystal structure (3ZPQ). These initial poses incorporated an ionic interaction between the cationic ammonium of the ligand and D1143.32. The head group of 1 retained a stable hydrogen bond with S1935.42 throughout each simulation (Fig. 3a), in agreement with the docked pose of the full-length molecule. The N-methyl indole moiety of 2, on the other hand, moved away from TM5 toward the extracellular surface of the D2 orthosteric site, where it associated closely with I184EL2 (Fig. 3b). These results—which were consistent across several sets of simulations (Fig. 3c,d, Supplementary Fig. 4)—indicate that the two head groups, which differ only by a single methyl group, prefer substantially different positions in the orthosteric site. Compound 1’s head group prefers TM5 S1935.42 interaction, whereas compound 2’s head group prefers EL2 interaction with I184. This pose difference potentially confirms compound 2’s preference for β-arrestin recruitment via EL2 interactions.
Figure 3. D2R MD Simulations Predict EL2 Engagement for Arrestin-bias.

MD simulations of the head groups of compound 1 (a) and compound 2 (b) reveal that β arrestin-biased 2 preferentially interacts with I184 in EL2 over S193 in TM5. By contrast, 1 maintains a stable hydrogen bond with S1935.42 throughout simulation, without interacting substantially with I184EL2. Relative positioning of the head groups to TM5 and EL2 was tracked by the distance from the ligand indole nitrogen to the hydroxyl oxygen of S1935.42 (magenta), and the distance from the center of the indole ring to the β-carbon of I184EL2 (cyan), for compound 1 (c) and 2 (d). The starting pose of the head group simulations, equating to the crystal structure of thermostabilized β1AR (3ZPQ) in complex with indole 4-(piperazin-1-yl)-1H-indole, is shown in light grey, while the green ligand and the protein show a representative snapshot from simulation. In (c) and (d), thin traces are sampled every 100 ps and thick traces are smoothed with a 1 ns moving average.
D2 TM5 and EL2 Mutants Confirm Arrestin-Biased Binding Pose
To investigate changes in bias based on ligand contacts with key TM5 and EL2 residues, we tested 2 at the S1935.42 and I184EL2 mutants and quantified Gi/o-mediated cAMP inhibition and β-arrestin2 recruitment. The design of these mutants reflects our previous observations that S5.42 is required for activation of G protein-signaling at β2AR, and that the conserved hydrophobic EL2 residue corresponding to I184EL2 specifically dampens LSD’s β-arrestin recruitment at the 5-HT2B and 5-HT2A receptors35.
As previously mentioned, the S193A5.42 mutation resulted in a loss of affinity and potency of 1, confirming our prediction that the indole N-H forms a hydrogen bond with S1935.42, as found in the β1AR crystal structure. Furthermore, the affinity of 2 was also tested at TM5 mutants and no substantial affinity changes relative to D2R wild-type were observed for any of the TM5 serine mutations (Supplementary Fig. 5a). By contrast, the G protein-mediated signaling of 2 (Fig. 4b) was selectively recovered by the TM5 S193A5.42 mutation, resulting in balanced signaling between G protein and β-arrestin2 (Fig. 4c) with respect to quinpirole. We reasoned that the D2R S193A5.42 mutant creates a hydrophobic space for the N-methyl group of 2 to fit, allowing it to recapitulate the hydrogen bond between compound 1 and S1935.42 at wild-type D2R leading to G protein-signaling. Docking of 2 to the D2R S193A5.42 model showed that the steric clash between compound 2 and S1935.42 in wild-type D2R is abolished at the D2 S193A5.42 mutant (Supplementary Fig. 5b). In fact, MD simulations of the head group of 2 further support this hypothesis, where at wild-type D2R the head group of 2 moves away from TM5 and interacts with I184EL2. In contrast, the head group of 2 at the S193A5.42 mutant engages TM5 in a pose that is almost identical within the binding pocket to compound 1 head group at wild-type D2R (Fig. 4d).
Figure 4. D2 TM5 and EL2 Mutants Confirm Arrestin-Bias Binding Pose.

a) The pose resulting from MD simulation of the head group of arrestin-biased N-methyl indole-aripiprazole hybrid (2) places the N-methyl indole moiety in contact with I184 on EL2, having moved away from S193 on TM5. b) N-methyl indole-aripiprazole hybrid 2 only shows arrestin recruitment activity at D2 wild-type. Data represent mean and standard error of the mean performed in triplicate (Gi/o GloSensor; red, n=3) and β-arrestin2 recruitment (Tango; blue, n=3, EC50 = 3.7 nM, Emax = 36%) c) S193A5.42 transforms arrestin-bias of 2 into balanced signaling with respect to quinpirole. Data represent Gαi/o-mediated cAMP inhibition (Gi/o GloSensor; red, n=3, EC50 = 2.5 nM, Emax = 67%) and β-arrestin2 recruitment (Tango; blue, n=3, EC50 = 2.6 nM, Emax = 69%). d) Representative pose of compound 2 head group from simulation at wild-type (WT) and S193A D2R constructs, and compound 1 head group from wild-type D2R simulation. At S193A, 2 moves to a pose almost identical to 1 at D2 wild-type. e) Mutation of EL2 I184 (I184A) completely abolishes arrestin recruitment for arrestin-biased ligand 2 (Tango; n=5 in triplicate) f) I184A mutation transforms 2 into a D2R β-arrestin2 recruitment antagonist as measured in Tango (n=2, in triplicate), as seen by comparing D2 wild-type (black, IC50 = 6.3 nM) to EL2 I184A (green, IC50 = 13 nM). g) Compound 2 head group is unstable throughout simulation at I184A D2R, sampling many orientations within the ligand-binding pocket. Ligand poses are shown for three points in time during a single simulation.
Next, we tested compound 2’s arrestin recruitment at the EL2 I184AEL2 mutation and found that arrestin recruitment by 2 was completely abolished at this mutant (Fig. 4e), confirming that EL2 is essential for compound 2’s β-arrestin recruitment. In fact, I184AEL2 resulted in no measureable activity of 2 in either G protein-signaling or arrestin recruitment activity (Fig. 4e). By contrast, β-arrestin recruitment efficacy for the balanced agonists 1 and quinpirole was spared at I184AEL2 (Supplementary Fig. 5c). In addition, 2’s affinity at the I184A mutant was spared (Supplementary Fig. 5d) demonstrating antagonist activity at the D2 I184AEL2 mutant (Fig. 4f, Supplementary Fig. 5e). To confirm that mutations of EL2 may be directly related to 2’s ligand binding kinetics, we measured an increase in on- and off-rate of 2 of 2.2 and 8.7-fold, respectively, at the I184AEL2 mutant compared to D2R wild-type (Supplementary Table 4; Supplementary Fig. 5f), which is consistent with EL2 mutations affecting LSD’s residence time at 5-HT2B and 5-HT2A receptors35. Furthermore, MD simulations confirm that compound 2’s head group is unstable in I184AEL2 D2R simulation and samples many orientations within the binding pocket (Fig. 4g, Supplementary Fig. 6a,6b), which may partially explain the increased off-rate of 2 at the I184AEL2 mutant. Overall, our mutagenesis and computational studies confirm that I184EL2 and S1935.42 are critical contacts for compound 2’s bias profile.
MD-Assisted Rational Design of Arrestin-biased Compounds
Based on the signaling profiles of compounds 1 and 2 at the D2R I184EL2A mutant, and MD observations that β-arrestin biased compound 2 preferentially interacts with EL2 over TM5, we designed compounds 6 and 7 to test whether additional EL2 engagement would lead to superior arrestin recruitment efficacy. Compound 7 is an analog of 2 containing a 2-methyl substitution to the indole ring, which would be expected to engage I184EL2 in a hydrophobic contact (Fig. 5a). Compound 6 is the 2-methyl analog of 1 and was proposed as a control compound that would have similar properties to 7, but predicted to form a hydrogen bond with S1935.42 and demonstrate a balanced signaling profile relative to quinpirole. Both 6 and 7 were synthesized and tested at the D2 receptor for bias (Fig. 5a). Consistent with our prediction, 6 displayed no preference for arrestin recruitment over G protein activation, again demonstrating that predicted engagement with S1935.42 invariably leads to activation of G protein-signaling. Unsurprisingly, simulations reveal that the head groups of 1 and 6 remain closer to TM5 during simulation than those of 2 and 7, due to the presence of a hydrogen bond between S1935.42 and the indole N-H of 1 and 6 (Supplementary Fig. 7).
Figure 5. MD-Assisted Rational Design of Arrestin-biased Compounds.

a) Mutagenesis data indicating that 2 requires I184EL2 for β-arrestin recruitment, and MD findings that 2 preferentially interacts with I184 EL2, led to the design of 2-methyl indole derivative 7 to further engage EL2 and enhance β-arrestin recruitment. Compound 6 is the unsubstituted control compound, which can still form a hydrogen bond with S1935.42. Compound 6 shows balanced D2 signaling with respect to quinpirole (bias factor = 1.3, Gi/o EC50 = 0.49 nM, Emax = 86%, β-arrestin2 EC50 = 0.62 nM, Emax = 78%), but compound 7 shows arrestin-bias with respect to quinpirole (bias factor = 20) comparing Gαi/o-mediated cAMP inhibition (GloSensor; red; n=3, EC50 = 23 nM, Emax = 60%) and β-arrestin2 recruitment (Tango; blue; n=3, EC50 = 2.9 nM, Emax = 78%). b) 2-methyl substitution (7, purple, Emax = 78%) shows higher D2 β-arrestin2 recruitment efficacy compared to compound 2 (blue, Emax = 36%) with respect to quinpirole measured by Tango. Data were normalized to quinpirole and represent n=3 in triplicate. c) Interaction with EL2 confirmed with I184AEL2 mutation selectively abolishing β-arrestin2 recruitment (Tango) for biased ligands 2 and 6 and not for balanced 1 (red) and quinpirole (black). Compound 6 (green) shows decreased arrestin recruitment by I184AEL2 mutation but not complete loss of activity (β-arrestin2 EC50 = 2.7 nM, Emax = 40%). Data were normalized to quinpirole and represent n=3 performed in triplicate. d) Structure-function selectivity relationships for indole-aripiprazole hybrid series as outlined using a heat map comparing log log(τ/KA) activities measuring G protein and β-arrestin2 recruitment.
Compound 7, on the other hand, shows a preference for arrestin recruitment with a calculated bias factor of 20 relative to quinpirole (Fig. 5a), demonstrating much increased arrestin recruitment efficacy (Fig. 5b; Emax = 88% of quinpirole) relative to 2. Although compound 7 still showed G protein-mediated signaling, its G protein activity was much weaker in terms of potency compared to its β-arrestin recruitment activity. To explain the recovery in G protein-signaling by 7, simulations with the head groups of compounds 2 and 7 were performed. Although arrestin-biased 2 moves the furthest away from TM5, the additional 2-methyl on 7 hinders this movement and instead shifts the indole ring towards TM5 (Supplementary Fig. 7), enough to engage TM5 and activate G protein-signaling to a degree. Despite this, both 7 and 2 moved closer to I184EL2 compared to 1 and 6, potentially explaining their arrestin preference.
To provide evidence for this differential EL2 engagement by 7, newly synthesized ligands were tested at the I184AEL2 mutant. As for compound 2, the I184AEL2 mutation almost completely abolishes the arrestin recruitment activity for compound 7 (Fig. 5c) and increases 7’s on- and off-rate by a factor of 6.7 and 6.2-fold respectively (Supplementary Table 4), indicating that I184 is a key interaction for 7’s enhanced β-arrestin recruitment efficacy. Although the arrestin recruitment of quinpirole and compound 1 are spared by I184AEL2 mutation, compound 6 showed a partial but not complete loss of arrestin recruitment efficacy, indicating that 2-methyl substitution is sensitive to EL2 mutation, but may retain other ligand-receptor interactions elsewhere in the binding pocket that lead to arrestin recruitment. To provide support for this notion, we tested the previously discovered β-arrestin-biased ligands, UNC 9994 and UNC 99758, and measured no change in arrestin recruitment efficacy at the I184AEL2 mutation (Supplementary Fig. 8), indicating that β-arrestin-bias may arise from other ligand-receptor interactions distinct from EL2. In summary, a route to attaining β-arrestin biased compounds by modification of the head group of aripiprazole-type ligands emerges: removing interactions with TM5, while enhancing interactions with EL2, is a strategy to improve β-arrestin recruitment efficacy to drive arrestin-biased signaling, an SFSR succinctly summarized in a heat map of relative log(τ/KA) activities (Fig. 5d, Supplementary Table 5).
Prediction and Confirmation of Polypharmacologic Arrestin-bias
Although aminergic GPCRs bind distinct classes of endogenous ligands (i.e. catecholamines, tryptamines and histamines), the orthosteric site encompassing TM5 and EL2 residues is relatively well-conserved (Supplementary Fig. 1a). We examined if ligand bias resulting from a lack of interaction with TM5 residues and the retention of hydrophobic engagement with EL2 is conserved for other aminergic GPCRs. Piperazine-containing ligands, such as aripiprazole, have promiscuous activity at aminergic GPCRs, and possess substantial affinity at D3, D4, 5-HT, and α and β-adrenergic receptors29. We hypothesized that the piperazine-containing ligand 2 will bind to the orthosteric site in a similar way for those receptors, and would demonstrate arrestin-bias at receptors with residues similar to those of D2R at EL2.52 (located 2 residues away from conserved disulfide cysteineEL2.50 that are branched aliphatic, e.g., leucine, isoleucine) and TM5 5.42 (polar residues that have hydrogen bond potential, e.g., serine, threonine).
We examined arrestin bias at receptors where 2 has substantial affinity (D3R, D4R, 5-HT7R, 5-HT1AR, 5-HT2Rs, β2AR and β1AR; Supplementary Table 6). The closely related D3R and D4R contain serines at positions 5.42, 5.43, and 5.46 and a branched aliphatic EL2 residue (isoleucine in D3R, leucine in D4R; Fig. 6a,b). Confirming our predictions, 2 demonstrated arrestin-bias at D3R (Fig. 6a) and D4R (Fig. 6b) relative to quinpirole with minimal detected G protein activity below 1 μM. Importantly, the unsubstituted compound 1 demonstrated no preference for either G protein or arrestin recruitment at D3R and D4R (Fig. 6a,b). 5-HT7R also has an isoleucine present in EL2 and a serine at TM5 5.42, therefore we expected to observe arrestin-bias by 2. Consistent with our prediction, 2 demonstrated full agonist arrestin recruitment activity at 5-HT7R relative to 5-HT, but surprisingly exhibited 5-HT7R-Gαs inverse agonist activity (Fig. 6c). Similarly at D3 and D4, 1 showed 5-HT7R agonist activity in both G protein and arrestin recruitment. In addition, we also tested 2 at 5-HT1AR, which also contains an isoleucine at EL2.52 and Ser at 5.42. Compound 2 also showed arrestin-bias at 5-HT1AR with a calculated bias factor of 60 with respect to 5-HT, which exhibits Gi/o preference (Supplementary Fig. 9a). Finally, we tested 2 at the 5-HT2 receptors, which all contain a Gly at 5.42, where 2 showed no Gq-mediated agonist activity at any of these receptors (Supplementary Fig. 9b–d). However, only at 5-HT2B, which contains a Leu at EL2.52, 2 shows weak arrestin recruitment (~ 25% of 5-HT), indicative of weak arrestin-bias relative to 5-HT (Supplementary Fig. 9d).
Figure 6. Prediction and Confirmation of Polypharmacological Arrestin-bias.

Alignments of D2 TM5 and EL2 residues predict that 2 shows arrestin-bias at D3 (a), D4 (b) with respect to quinpirole and 5-HT7 (c) receptors with respect to 5-HT, where TM5 and EL2 residues in orthosteric sites are well-conserved, except at β2 (d) with respect to isoproterenol, where 2 only shows inverse agonist activity but no arrestin recruitment. G protein-signaling was measured by GloSensor and β-arrestin recruitment was measured by Tango performed in parallel (n=3 in triplicate). e) TM5 and EL2 are key contacts in the orthosteric sites of aminergic GPCRs whereby an arrestin-bias template for ligand design can be used to promote EL2 engagement to enhance β-arrestin recruitment and preclude TM5 engagement to avoid G protein-signaling.
As previously mentioned, the β2AR binding pocket also contains TM5 serines at positions 5.42, 5.43 and 5.46, but contains Phe at the EL2.52 residue position (Fig. 6d). Compound 2 demonstrated Gαs inverse agonist activity, similar to 5-HT7R, but 2 showed no β-arrestin recruitment at β2AR, consistent with our prediction that smaller aliphatic residues are required for 2 arrestin recruitment efficacy. Although compound 1 showed Gs partial agonism at β2AR, it also showed no arrestin recruitment similar to 2 (Fig. 6d). A similar profile for 2 was also found at β1AR, which also contains a Phe at EL2.52 and Ser at 5.42 (Supplementary Fig. 9e). To test the hypothesis that smaller aliphatic residues present at EL2.52 may be required for compound 2’s arrestin recruitment efficacy, we attempted to rescue compound 2’s arrestin recruitment by mutating β2AR EL2.52 F194 to either alanine, leucine or isoleucine. Although compound 2 showed no recovered arrestin recruitment activity at any of the β2AR EL2 mutants (Supplementary Fig. 10a), β2AR EL2 mutation substantially reduced arrestin recruitment for the full reference agonist isoproterenol (Supplementary Fig. 10b), supporting the notion that EL2 plays a prominent role for arrestin recruitment.
This result confirms our hypothesis that specific interactions by 2 with smaller aliphatic residues present at EL2, even at other distinct aminergic receptors, can predict arrestin-bias. Here, we show that a template can be used to guide biased ligand design at many aminergic receptors, where promoting engagement with aliphatic residues in EL2 and precluding TM5 interaction can induce an arrestin-biased polypharmacological profile (Fig. 6e).
Discussion
Here we illustrate how to design biased ligands by a combined computational, structural, biochemical and molecular dynamics approach. Importantly, our results identify EL2 as a critical conserved region of the receptor that can be targeted to enhance arrestin bias. We anticipate this combined strategy will encourage adoption of MD into SBDD projects.
Our results for the D2 I184AEL2 mutation complement our recent finding that EL2 is important for arrestin-bias and slow binding kinetics35. EL2 appears to play an important role in distinguishing between β2AR active versus inactive states, where the activated state of β2AR involves F193EL2 and TM7 F7.35 coming together to form a lid over the ligand21. Here we provide evidence that EL2 of β2AR is also key for arrestin recruitment (Supplementary Fig. 10), and that further study of β2AR arrestin recruitment as it relates to ligand kinetics is warranted. Apart from aminergic GPCRs, measurements in structural changes in EL2 of rhodopsin reveal this region to be important for the retinal isomerization42, where mutations of rod rhodopsin EL2 Ile189 to proline found in cone rhodopsin directly increased decay rates of the meta II intermediate state of the receptor43. Taken together, EL2 is an important motif which can ‘lock’ the ligand into the binding site, leading to increased ligand residence times. This increased residence time apparently promotes arrestin recruitment and this can be exploited for biased drug design.
Structure-inspired drug design supported the hypothesis that orthosteric site TM5 residues are not only engaged in ligand recognition but also in G protein-signaling and, further, that these interactions can be exploited to modulate biased signaling. Ligand contacts with residues in TM5 have been regarded as a ‘trigger,’ which stabilizes a conformation with a cytoplasmic inward movement of TM544, which in turn moves intracellular loop 2 and TM6 regions that are involved in G protein activation20, 45. Evidence for the involvement of D2R TM5 serines for ligand bias is scant, except for a study suggesting that Ser5.43 may be involved in ligand-dependent arachidonic acid release34. Although we cannot rule out alternative downstream effects stemming from targeting EL2 and avoiding TM5 interaction (e.g. arachidonic acid release, pERK1/2), this study is the first to design ligands predicted to avoid TM5-dependent G protein activity entirely.
Importantly, our design strategy yielded a ligand with bias at multiple related GPCRs. Given that the most clinically effective medications for schizophrenia and depression have a complex polypharmacological profile46 targeting multiple aminergic GPCRs47 (i.e. “magic shotguns’) it is now possible to design promiscuous drugs that manifest arrestin-bias at multiple GPCRs via targeting conserved interactions within the orthosteric site. We thus provide a useful template for the rational design of polypharmacological drugs incorporating ligand bias (i.e. “biased magic shotguns”) and successful design will depend on generating optimal predicted ligand contacts with EL2. One caveat, though, is that this particular strategy may be applicable only to aminergic GPCRs. MOR, for example, was not proposed to trigger G protein-signaling through motion of TM5, and thus is not expected to benefit from this SBDD algorithm48. Conceivably, our template for biased ligand design could also be used to design G protein-biased ligands using the reverse approach (i.e. retain TM5 and exclude EL2 engagement); such compounds would represent extremely desirable tools to dissect the contributions of G protein versus β-arrestin-dependent signaling at various aminergic GPCRs to uncover favorable therapeutic versus side-effect profiles.
The wave of GPCR structures has generated excitement largely because they promise to accelerate the discovery of new and improved drugs49. With knowledge of how ligands can be designed to activate specific signaling pathways, it is apparently possible to leverage GPCR structures to create biased drugs.
Online Methods
General Chemistry Procedures
All reagents were purchased from Sigma-Aldrich or Fisher Scientific. Anhydrous solvents were used unless otherwise noted. Analytical HPLC– Method A: Equipment: Agilent 6110 series with UV detection at 254 nm; Column: Agilent Eclipse Plus 4.6 mm X 50 mm, 1.8 um C18 column. HPLC Solvents: A: 0.1% acetic acid in water; B: 0.1% acetic acid in methanol, with gradient: 10% to 100% B over 5.0 min, followed by 100% B for 2 min, at 1.0 mL/min. Method B: Equipment: Agilent Zorbax 300SC-C18 (5μm) column with UV detection at 254 nm on an Agilent 1200 Series LC/MSD TOF machine. HPLC Solvents: A: 0.1% acetic acid in water; B: 0.1% acetic acid in methanol, with gradient: 1% B for one minute, 1 to 100% B over 3.0 min, followed by 100% B for 4 min, at 1.0 mL/min. LRMS (low resolution mass spectrometry) data were acquired in positive ion mode on an Agilent 6110 single quadrupole mass spectrometer with electrospray ionization (ESI). Nuclear magnetic resonance (NMR) spectra were recorded either on a Varian Mercury spectrometer at 400 MHz for proton (1H NMR) and 100 MHz for carbon (13C NMR), or were recorded on a Bruker DRX spectrometer at 600 MHz for proton (1H NMR) and 150 MHz for carbon (13C NMR). Preparative HPLC (high pressure liquid chromatography) was performed on an Agilent Prep 1200 series with UV detector set to 254 nm, along with a Phenomenex Luna 75 mm X 30 mm, 5 um C18 column with a flow rate of 30 mL/min. High resolution mass spectrometry (HRMS) data was acquired with an Agilent 1200 Series LC/MSD TOF. Medium pressure liquid chromatography (MPLC) was performed on a Combiflash Isco machine. Final compounds had >95% purity as judged by analytical HPLC. Indole synthesis schemes and compound purification details can be found in Supplementary Materials.
Drugs and Reagents
All compounds and aripiprazole were synthesized as described under General Chemistry Procedures. Dopamine hydrochloride, (−)-quinpirole, (+)-butaclamol hydrochloride, 5-hydroxytryptamine creatine sulfate, (−)-isoproterenol bitartrate, and HEPES sodium salt were purchased from Sigma-Aldrich (St. Louis, MO). HBSS (10X) was purchased by Invitrogen and fatty-acid free BSA was purchased from Akron Biotech.
Cloning and Mutagenesis
Mutagenesis was performed according to QuikChange II XL Site-Directed Mutagenesis Kit protocol. Briefly, PCR reactions incorporated wild type D2 long dopamine receptor (pcDNA3.1, cDNA.org) or D2 long-V2-tTA (pcDNA3.1) and primers containing the mutation of interest. Parental wild-type DNA was digested with DpnI (New England Biolabs). PCR products were transformed into supercompetent GC-10 cells and positive clones were selected by ampicillin resistance. Isolated colonies on the plates were picked, cultured and prepped using QIAprep Spin mini prep and Origene maxi prep kits. DNA was then sequenced (Eton Bioscience) using forward (T7) and reverse (BGHreverse and TEV-REV) sequence primers to verify mutant DNA sequence.
Cell Culture
HEK 293T cells (ATCC CRL-11268; 59587035; mycoplasma free) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS, Invitrogen) and 0.5% penicillin/streptomycin. HTLA cells expressing β-arrestin-TEV protease and tTA-driven luciferase (provided by Dr. Richard Axel) were cultured similarly as HEK293T cells except media contained selection antibiotics (100 μg/mL hygromycin B and 5 μg/mL puromycin). Cells were maintained at 37° C and 5% CO2.
Radioligand Binding Assays
D2R radioligand binding assays utilized [3H] N-methyl Spiperone (NMSP; Perkin Elmer, Specific Activity = 64.1 Ci/mmol). For competitive binding experiments, assays used [3H] NMSP concentrations ranging from 0.7–1.3 nM, unlabeled ligand competitor at concentrations ranging from 100 μM to 1 pM, and membranes resuspended in binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, 0.01% ascorbic acid, pH 7.4). Binding assays were incubated at 25 °C for 2 hours, and assays were terminated by vacuum filtration using a 96-well Filtermate harvester (Perkin Elmer) onto 0.3% polyethyleneiming pre-soaked 96-well filter mats A (Perkin Elmer). Filters were washed three times using cold wash buffer (50 mM Tris, pH 7.4), and scintillation cocktail (Meltilex) was melted onto dried filters. Radioactivity displacement was measured using a Wallac Trilux Microbeta counter (Perkin Elmer). Counts per minute (CPM) were plotted as a function of unlabeled ligand concentration and the Ki was calculated using the One-site-Fit Ki using 5.0. Data were normalized to the top (100%, no competitor) and bottom (0%, non-specific binding defined as 5 μM (+)-butaclamol) to represent percent displacement. For radioligand binding assays at all other receptors, procedures were similar as described, except for the radioligand used and membrane sources. For a list of these binding assays, refer to procedures at https://pdspdb.unc.edu/pdspWeb/ for the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH PDSP).
For the determination of kon and koff for unlabeled compound 2, membranes of D2 wild-type and I184AEL2 were incubated with at least two concentrations of [3H]-NMSP (range 0.08–0.35 nM) and several concentrations of 2 (range 1 μM to 320 pM). On- and off-rates of [3H]NMSP at D2 wild-type and I184AEL2 were previously determined and used to estimate the kon and koff rates of 2 using “Kinetics of competitive binding” equation in Graphpad Prism 5.0 by Motulsky and Mahan (1984)50.
Gi/o-mediated cAMP inhibition Assay
To measure Gi/o-mediated cAMP inhibition, HEK293T cells were co-transfected in a 1:1 ratio with receptor and a split-luciferase-based cAMP biosensor (GloSensor; Promega) as described. After at least 24 hours, transfected cells were plated in poly-lysine coated 384-well white clear bottom cell culture plates with DMEM containing 1% dialyzed FBS at a density of 15,000 cells per 40 μL per well and incubated overnight. On the day of assay, drug dilutions were prepared in filtered fresh assay buffer (20 mM HEPES, 1X HBSS, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) at 3X and 10 μL per well was added to cells containing 20 μL/well of assay buffer. Drug solutions used for G protein-mediated cAMP assays were exactly the same as used for Tango assays to allow relative within-experiment bias comparisons. After plates were allowed to incubate with drug for 15 minutes, 10 μL per well of 1 μM (final concentration) forskolin and glosensor substrate was added. Luminescence counts per second (LCPS) were quantified after 15 minutes using a TriLux microbeta (Perkin Elmer) luminescence counter. LCPS were plotted as a function of drug concentration and normalized to % quinpirole with 100% as the quinpirole cAMP inhibition Emax and 0% as the forskolin-stimulate cAMP baseline. Data were analyzed using log (agonist) vs. response in GraphPad Prism 5.0 (Graphpad Software Inc., San Diego, CA).
Tango β-arrestin Recruitment Assays
The human D2Long Tango construct was designed and assays were performed as previously described8,40. HTLA cells expressing TEV fused-β-Arrestin2 were transfected with D2 Tango construct. For D3 and D4 Tango constructs, GRK2 was co-transfected in a 1:10 ratio of GRK2:receptor. After at least 24 hours, cells were plated in DMEM supplemented with 1% dialyzed FBS (dFBS) in poly-L-lysine coated 384-well white clear bottom cell culture plates at a density of 15,000 cells/well in total of 40 μL. After at least 6 hours, media was decanted and cells were supplemented with 40 μL of 1% dFBS DMEM and drug solutions (3X) prepared in drug buffer (1× HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) were added (20 μL per well) for overnight incubation. Drug solutions used for Tango assay were exactly the same as used for G protein-mediated cAMP assays to allow relative within experiment bias comparisons. The next day, media and drug solutions were decanted and 20 μL per well of BrightGlo reagents (Promega, 1:20 dilution in drug buffer) was added. The plate was incubated for 20 min at room temperature in the dark before being counted using Wallac TriLux microbeta (Perkin Elmer). LCPS were plotted as a function of drug concentration, normalized to % quinpirole with 100% as the quinpirole Emax and 0% as the baseline, and analyzed using log (agonist) vs. response in GraphPad Prism 5.0 (Graphpad Software Inc., San Diego, CA).
Bioluminescence Resonance Energy Transfer (BRET) Assays
To measure D2-mediated β-Arrestin2 recruitment, HEK293T cells were co-transfected in a 1:1:15 ratio with D2Long containing C-terminal renilla luciferase (RLuc), GRK2, and Venus-tagged N-terminal β-arrestin2. After at least 24 hours, transfected cells were plated in poly-lysine coated 96-well white clear bottom cell culture plates in plating media (DMEM containing 1% dialyzed FBS) at a density of 40–50,000 cells in 200 μL per well and incubated overnight. Next day, media was decanted and cells were washed twice with 60 uL of drug buffer (1X HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4), then 60 uL of drug buffer was added per well. Drug stimulation was performed with addition of 30 uL of drug (3X) per well and incubated for at various time points. At 15 minutes before reading, 10 uL of the RLuc substrate, coelenterazine h (Promega, 5 μM final concentration) was added per well, and plates were read for both luminescence at 485 nm and fluorescent eYFP emission at 530 nm for 1 second per well using a Mithras LB940. Plates were read for multiple time points up to 60 minutes after drug addition. The BRET ratio of eYFP/RLuc was calculated per well and the net BRET ratio was calculated by subtracting the eYFP/RLuc per well from the eYFP/RLuc ratio in wells without Venus-β-Arrestin present. The net BRET ratio was plotted as a function of drug concentration using Graphpad Prism 5 (Graphpad Software Inc., San Diego, CA).
To measure D2R-mediated Gαi1-γ2 dissociation, procedures were exactly the same as for D2-mediated β-Arrestin2 recruitment, except HEK293T cells were co-transfected in a 1:5:5:5 ratio of Gαi1-RLuc, Gβ1, GFP2-Gγ2, and D2long, respectively. Gαi1-RLuc, Gβ1 and GFP2-Gγ2 constructs were generously provided by Dr. Michel Bouvier. Gαi1-γ2 dissociation BRET2 assays utilized 10 uL of the RLuc substrate Coelenterazine 400a (Nanolight, 5 μM final concentration), incubated for 10 minutes, and read for luminescence at 400 nm and fluorescent GFP2 emission at 515 nm for 1 second per well using a Mithras LB940. The ratio of GFP2/RLuc was calculated per well and plotted as a function of drug concentration using Graphpad Prism 5 (Graphpad Software Inc., San Diego, CA).
Bias calculation
Transduction coefficients (log (τ/KA)) were calculated using the Black and Leff operational model in Graphpad Prism 5.0, where τ is agonist efficacy and KA is the equilibrium dissociation constant. Using quinpirole as the full agonist reference, transduction coefficients for Gi/o activity and β-Arrestin2 recruitment were calculated and averaged across experiments. Calculation of bias factors utilized the method by Kenakin et al51, where the Δlog(τ/KA) was calculated relative to the reference and the ΔΔlog(τ/KA) was calculated by subtracting the β-Arrestin2 from the Gi/o transduction coefficient.
Homology modeling and docking
Construction and selection of the D2 dopamine receptor homology model was as described52. Briefly, 400 D2 models were built with MODELLER 9v853, using the crystal structure of the D3 dopamine receptor (PDB ID: 3PBL) as the template19. The sequence alignment between D2 and the D3 template were generated using PROMALS3D. The final D2 model was chosen based on its ability to enrich 85 known, diverse, and high-affinity ligands (taken from the ChEMBL10 database) against a background of property-matched decoy molecules and experimentally tested non-binders from ChEMBL10. The model’s ability to recognize both antagonists and biased agonists were tested prospectively in multiple virtual screening campaigns,and based upon this performance, we decided to use the same model in this study to dock ligands with various functional profiles. Here, we used DOCK3.7 to dock substituted aripiprazoles into the binding site of the D2 model, as in previously published protocols. Aripiprazole and indole-aripiprazole hybrid ligands were protonated using the pKa prediction tool built into Marvin from ChemAxon (Marvin version 5.5.1.0 (ChemAxon, 2011)). The flexible-ligand sampling algorithm in DOCK3.7 uses a graph-matching technique to superimpose atoms of the docked molecule onto binding site matching spheres, which represent favorable positions for individual ligand atoms. Complementarity to the protein of each ligand pose is scored using a physics-based scoring function consisting of receptor-ligand electrostatic and van der Waals interaction energies, using modified versions for DOCK of the AMBER potential and QNIFFT point-charge Poisson-Boltzmann electrostatics models, respectively. Energies were corrected for context-dependent ligand desolvation using a variation of AMSOL desolvation energies. Individual ligands were sampled until a maximum of 20,000 favorable conformations were found and scored. The ability to save any number of top poses of a molecule was used here to examine all possible binding orientations.
System setup for Molecular Dynamic (MD) simulations
MD simulations of the dopamine D2 receptor (D2R) were based on a homology model constructed from the crystal structure of the dopamine D3 receptor complexed to the antagonist eticlopride (PDB ID: 3PBL)19. The resulting model was simulated in four conditions: in complex with the head group of compound 1, 2, 6 and 7, i.e. with the dihydroquinolin-2-one and alkyl linker removed (Supplementary Fig 3). The ammonium nitrogen was methylated in order to maintain the same atom-types as the full-length molecules for simulation. Placement of ligands was guided by the crystal structure of thermostabilized turkey β1-adrenoceptor (3ZPQ)37, which is complexed to 4-(piperazin-1- yl)-1H-indole (equivalent to the head group of compound 1).
Hydrogen atoms were added using Prime (Schrödinger Inc.), and protein chain termini were capped with the neutral groups acetyl and methylamide. Titratable residues were left in their dominant protonation state at pH 7.0. All aspartate residues were deprotonated, as is expected in the inactive state of GPCRs, the tertiary amine of the ligands was protonated.
The prepared protein structures were aligned on the transmembrane helices to the Orientation of Proteins in Membranes (OPM)54 structure of PDB 3PBL, and internal waters added with Dowser55. The structures were then inserted into a pre-equilibrated palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer, and solvated with 0.15 M NaCl in explicitly represented water, then neutralized by removing sodium ions. Final system dimensions were approximately 76 × 72 × 88 Å3, including about 108 lipids, 14 sodium ions, 25 chloride ions, and 9047 water molecules.
MD simulation protocol
We used the CHARMM36 parameter set for protein molecules, lipid molecules, and salt ions, and the CHARMM TIP3P model for water; protein parameters incorporated CMAP terms56. Parameters for ligands were generated using the CHARMM General Force Field (CGenFF)57 with the ParamChem server (paramchem.org), version 1.0.0. Parameters associated with the dihedral term shown in (Supplementary Fig. 4) were refit using Paramfit58 to the results of quantum mechanical calculations performed in Gaussian09. Full parameter sets are available upon request. Simulations were performed on GPUs using the CUDA version of PMEMD (Particle Mesh Ewald Molecular Dynamics) in Amber14.
Prepared systems were minimized, then equilibrated as follows: The system was heated using the Langevin thermostat from 0 to 100 K in the NVT ensemble over 12.5 ps with harmonic restraints of 10.0 kcal·mol−1·Å−2 on the non-hydrogen atoms of lipid, protein and ligand, and initial velocities sampled from the Boltzmann distribution. The system was then heated to 310 K over 125 ps in the NPT ensemble with semi-isotropic pressure coupling and a pressure of one bar. Further equilibration was performed at 310 K with harmonic restraints on the protein and ligand starting at 5.0 kcal·mol−1·Å−2 and reduced by 1.0 kcal·mol−1·Å−2 in a stepwise fashion every 2 ns, for a total of 10 ns of additional restrained equilibration.
We performed five simulations of D2R bound to the head group of 1, and five of D2R bound to the head group of 2. We also performed simulations of the head groups of compound 6 and 7 bound to D2R, and the head group of compound 2 bound to both S193A and I184A D2R mutant models (Supplementary Table 5). These simulations were conducted in the NPT ensemble at 310 K and 1 bar, using a Langevin thermostat and Monte Carlo barostat. In each of these simulations, we performed 5 ns of unrestrained equilibration followed by a production run of 250–350 ns.
Simulations used periodic boundary conditions, and a time step of 2.5 fs. Bond lengths to hydrogen atoms were constrained using SHAKE. Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were computed using the particle mesh Ewald (PME) method with an Ewald coefficient β of approximately 0.31 Å and B-spline interpolation of order 4. The FFT grid size was chosen such that the width of a grid cell was approximately 1 Å.
MD simulation analysis
Trajectory snapshots were saved every 100 ps during production simulations. Trajectory analysis was performed using VMD and CPPTRAJ59, and visualization was performed using VMD. Trajectories were aligned to the D2 inactive state homology model on all transmembrane helix Cα atoms. Two metrics were used to determine the position of the head group of 1 and 2 in relation to TM5 and EL2 during simulation: 1) the distance between the indole nitrogen atom of the head group and the side-chain oxygen atom of S1935.42, 2) the distance from the midpoint of the indole C8-C9 bond and the Cβ atom of I184EL2 (Figure 3, Supplementary Fig. 4). The distance from the ligand cationic nitrogen to D1143.32 was also monitored to ensure this interaction essential to D2 agonists was maintained. To allow comparison of the head groups of 1, 2, 6 and 7, the distance between the nearest ligand heavy atom and the side-chain oxygen of S193, and the distance between the nearest ligand heavy atom and Cβ of I184 were used (Supplementary Fig. 7).
Code availability
The DOCK3.6 program is freely accessible at http://dock.compbio.ucsf.edu/DOCK3.6/ to academic labs.
Data availability
Generated and analyzed data sets that support the findings of this study are available from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgments
We thank Scott Hollingsworth for assistance with simulation analysis. This work was supported by the National Institutes of Health (NIH) grant U19MH082441 (to B.L.R. and J.J.), RO1MH112205 (to B.L.R), R01NS100930 (to J.J), the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH PDSP; to B.L.R), the Michael Hooker Chair for Protein Therapeutics and Translational Proteomics (to B.L.R.), the American Cancer Society postdoctoral fellowship PF-14-021-01-CDD (to K.V.B.), by NIH grant GM59957 (to B.K.S.), by Pfizer, Inc. (R.O.D.), by a Terman Faculty Fellowship (to R.O.D.), and by a National Science Foundation Graduate Research Fellowship (to R.M.B.).
Footnotes
Author Contributions
J.D.M designed experiments, performed mutagenesis, ligand binding and signaling studies, analyzed the data, and wrote the manuscript. K.V.B. designed and synthesized all ligands and performed analytical chemical analysis, and wrote the manuscript. B.K. performed and analyzed MD simulations, used the results to design ligands, and wrote the manuscript. K.R. assisted with mutagenesis and signaling studies. J.K. built the D2 homology model and performed the docking experiments, and edited the manuscript. R.M.B. determined ligand parameters and performed preliminary MD simulations. B.K.S. supervised the docking experiments and edited the manuscript. R.O.D. supervised the MD simulation studies and assisted with preparing the manuscript. J.J supervised ligand synthesis, designed experiments, and edited the manuscript. B.L.R designed the experiments, was responsible for the overall project strategy and management, and prepared the manuscript.
Competing financial interests
The authors declare no competing financial interests.
Reference List
- 1.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 3.Wacker D, Stevens RC, Roth BL. How Ligands Illuminate GPCR Molecular Pharmacology. Cell. 2017;170:414–427. doi: 10.1016/j.cell.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Urban JD, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 5.Dewire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 6.Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 2011;36:457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Allen JA, et al. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soergel DG, et al. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain. 2014;155:1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 10.Violin JD, et al. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335:572–579. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 11.Urs NM, et al. Targeting beta-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc Natl Acad Sci U S A. 2015;112:E2517–E2526. doi: 10.1073/pnas.1502740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charfi I, Audet N, Bagheri TH, Pineyro G. Identifying ligand-specific signalling within biased responses: focus on delta opioid receptor ligands. Br J Pharmacol. 2015;172:435–448. doi: 10.1111/bph.12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manglik A, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016:1–6. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewire SM, et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 15.Gesty-Palmer D, et al. beta-arrestin-selective G protein-coupled receptor agonists engender unique biological efficacy in vivo. Mol Endocrinol. 2013;27:296–314. doi: 10.1210/me.2012-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masri B, et al. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci U S A. 2008;105:13656–13661. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tchernychev B, et al. Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc Natl Acad Sci U S A. 2010;107:22255–22259. doi: 10.1073/pnas.1009633108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chien EY, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ring AM, et al. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502:575–579. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wacker D, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, et al. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340:610–614. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapur S, Remington G. Atypical antipsychotics: new directions and new challenges in the treatment of schizophrenia. Annu Rev Med. 2001;52:503–517. doi: 10.1146/annurev.med.52.1.503. [DOI] [PubMed] [Google Scholar]
- 25.Wadenberg ML, Soliman A, VanderSpek SC, Kapur S. Dopamine D(2) receptor occupancy is a common mechanism underlying animal models of antipsychotics and their clinical effects. Neuropsychopharmacology. 2001;25:633–641. doi: 10.1016/S0893-133X(01)00261-5. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro DA, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology. 2003;28:1400–1411. doi: 10.1038/sj.npp.1300203. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, et al. Discovery of G Protein-Biased D2 Dopamine Receptor Partial Agonists. J Med Chem. 2016;59:10601–10618. doi: 10.1021/acs.jmedchem.6b01208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park SM, et al. Effects of beta-Arrestin-Biased Dopamine D2 Receptor Ligands on Schizophrenia-Like Behavior in Hypoglutamatergic Mice. Neuropsychopharmacology. 2016;41:704–715. doi: 10.1038/npp.2015.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ambrosio C, Molinari P, Cotecchia S, Costa T. Catechol–binding serines of beta(2)-adrenergic receptors control the equilibrium between active and inactive receptor states. Mol Pharmacol. 2000;57:198–210. [PubMed] [Google Scholar]
- 30.Isogai S, et al. Backbone NMR reveals allosteric signal transduction networks in the beta1-adrenergic receptor. Nature. 2016;530:237–241. doi: 10.1038/nature16577. [DOI] [PubMed] [Google Scholar]
- 31.Warne T, et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neve KA, Wiens BL. Four ways of being an agonist: multiple sequence determinants of efficacy at D2 dopamine receptors. Biochem Soc Trans. 1995;23:112–116. doi: 10.1042/bst0230112. [DOI] [PubMed] [Google Scholar]
- 33.Wiens BL, Nelson CS, Neve KA. Contribution of serine residues to constitutive and agonist-induced signaling via the D2S dopamine receptor: evidence for multiple, agonist-specific active conformations. Mol Pharmacol. 1998;54:435–444. doi: 10.1124/mol.54.2.435. [DOI] [PubMed] [Google Scholar]
- 34.Fowler JC, Bhattacharya S, Urban JD, Vaidehi N, Mailman RB. Receptor conformations involved in dopamine D(2L) receptor functional selectivity induced by selected transmembrane-5 serine mutations. Mol Pharmacol. 2012;81:820–831. doi: 10.1124/mol.111.075457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wacker D, et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell. 2017;168:377–389. doi: 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen X, et al. Structure-functional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. J Med Chem. 2012;55:7141–7153. doi: 10.1021/jm300603y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christopher JA, et al. Biophysical fragment screening of the beta1-adrenergic receptor: identification of high affinity arylpiperazine leads using structure-based drug design. J Med Chem. 2013;56:3446–3455. doi: 10.1021/jm400140q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kling RC, Tschammer N, Lanig H, Clark T, Gmeiner P. Active-state model of a dopamine D2 receptor-Galphai complex stabilized by aripiprazole-type partial agonists. PLoS One. 2014;9:e100069. doi: 10.1371/journal.pone.0100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luedtke RR, et al. Comparison of the binding and functional properties of two structurally different D2 dopamine receptor subtype selective compounds. ACS Chem Neurosci. 2012;3:1050–1062. doi: 10.1021/cn300142q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kroeze WK, et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol. 2015;22:362–369. doi: 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein Herenbrink C, et al. The role of kinetic context in apparent biased agonism at GPCRs. Nature Communications. 2016;7:10842. doi: 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahuja S, et al. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat StructMol Biol. 2009;16:168–175. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuwayama S, Imai H, Hirano T, Terakita A, Shichida Y. Conserved proline residue at position 189 in cone visual pigments as a determinant of molecular properties different from rhodopsins. Biochemistry. 2002;41:15245–15252. doi: 10.1021/bi026444k. [DOI] [PubMed] [Google Scholar]
- 44.Warne T, Tate CG. The importance of interactions with helix 5 in determining the efficacy of beta-adrenoceptor ligands. Biochem Soc Trans. 2013;41:159–165. doi: 10.1042/BST20120228. [DOI] [PubMed] [Google Scholar]
- 45.Deupi X, Standfuss J. Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr Opin Struct Biol. 2011;21:541–551. doi: 10.1016/j.sbi.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 46.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 47.Besnard J, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang W, et al. Structural insights into micro-opioid receptor activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobilka B, Schertler GF. New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol Sci. 2008;29:79–83. doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 50.Motulsky HJ, Mahan LC. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1984 Jan;25(1):1–9. [PubMed] [Google Scholar]
- 51.Kenakin T, et al. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012 Mar 21;3(3):193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss DR, et al. Conformation Guides Molecular Efficacy in Docking Screens of Activated β-2 Adrenergic G Protein Coupled Receptor. ACS Chem Biol. 2013;8:1018–1026. doi: 10.1021/cb400103f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coleman RG, Sterling T, Weiss DR. SAMPL4 & DOCK3.7: lessons for automated docking procedures. J Comput Aided Mol Des. 2014;28:201–209. doi: 10.1007/s10822-014-9722-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 55.Zhang L, Hermans J. Hydrophilicity of cavities in proteins. Proteins Struct Funct Genet. 1996;24:433–438. doi: 10.1002/(SICI)1097-0134(199604)24:4<433::AID-PROT3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 56.Best RB, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vanommeslaeghe K, MacKerell AD. Automation of the CHARMM general force field (CGenFF) I: Bond perception and atom typing. J Chem Inf Model. 2012;52:3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Betz RM, Walker RC. Paramfit: Automated optimization of force field parameters for molecular dynamics simulations. J Comput Chem. 2015;36:79–87. doi: 10.1002/jcc.23775. [DOI] [PubMed] [Google Scholar]
- 59.Roe DR, Cheatham TE., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular synamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Generated and analyzed data sets that support the findings of this study are available from the corresponding authors upon reasonable request.