

Abstract
A public workshop entitled “Challenges and strategies to facilitate formulation development of pediatric drug products” focused on current status and gaps as well as recommendations for risk-based strategies to support the development of pediatric age-appropriate drug products. Representatives from industry, academia, and regulatory agencies discussed the issues within plenary, panel, and case-study breakout sessions. By enabling practical and meaningful discussion between scientists representing the diversity of involved disciplines (formulators, nonclinical scientists, clinicians, and regulators) and geographies (eg, US, EU), the Excipients Safety workshop session was successful in providing specific and key recommendations for defining paths forward. Leveraging orthogonal sources of data (eg. food industry, agro science), collaborative data sharing, and increased awareness of the existing sources such as the Safety and Toxicity of Excipients for Paediatrics (STEP) database will be important to address the gap in excipients knowledge needed for risk assessment. The importance of defining risk-based approaches to safety assessments for excipients vital to pediatric formulations was emphasized, as was the need for meaningful stakeholder (eg, patient, caregiver) engagement.
Keywords: Excipients, Safety assessment, Pediatric, Formulation development
1. Introduction
The Safety Qualification of Excipients was one of the sessions organized at a public workshop entitled “Challenges and strategies to facilitate formulation development of pediatric drug products” held June 8–9, 2016 at the College Park Marriott Hotel and Conference Center in Hyattsville, MD. The session was designed to bring together various stakeholders (eg, EU- and US-based formulators, regulators, clinicians, and toxicologists) to discuss approaches to the safety assessment of excipients and to identify gaps and challenges in current paradigms to assess excipient safety and evaluate the potential risk thereof in pediatric formulations. An ultimate goal was to develop recommendations for a risk to benefit – based framework to qualify excipients for pediatric use. This report summarizes the plenary presentations and the working group discussions on the current perspectives on approaches to safety assessments of excipients intended for pediatric use. It includes a number of considerations and recommendations for future best practices put forward by the participants and experts in the workshop.
1.1. Overview of challenges and opportunities in safety assessment of excipients in pediatric drug products
Pediatric drug development has been stimulated by recent legislation (US and EU regulations), resulting in significant efforts to develop age-appropriate formulations acceptable for use in pediatric patients. Dr. Buckley opened the session with an overview of the issues and gaps in current safety assessment paradigms for excipients in pediatric patient populations (Fig. 1). The pediatric population, which spans from neonates to adolescents (0–17 years), is quite heterogeneous with regard to a multitude of factors including difference in swallowing abilities, taste preferences, and dosage requirements depending on the age and the clinical state of the patient. This heterogeneity represents a significant challenge for drug development teams for designing safe and age-appropriate formulations for pediatric products. Considerable cross-functional collaboration involving chemists/formulators, drug disposition scientists, clinicians, and toxicologists is required as the development team considers total doses, (maximum) concentrations and amount of excipients, dosing regimen, duration of treatment, route of administration, as well as the indication and the (minimum) age groups (Schmitt, 2015) during safety assessments of excipients. Further, knowledge of these factors, including the intended patient population, is usually in flux or uncertain over the course of development of a drug.
Fig. 1.
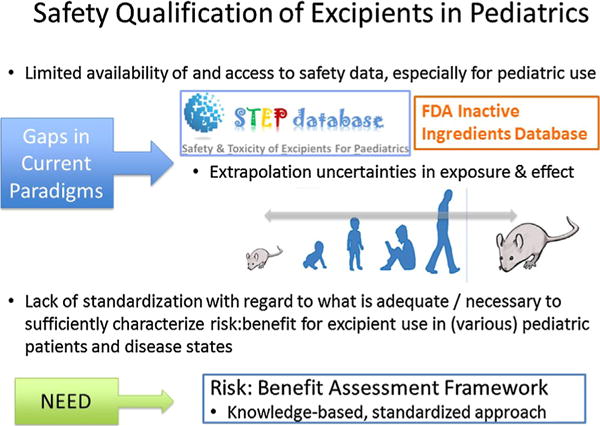
Gaps in current safety assessment paradigms for excipients in pediatric patient populations.
One of the challenges and key elements of pediatric formulation development is the screening and careful selection of excipients (EMA, 2013), as certain excipients safely used in adult formulations (eg, benzyl alcohol, ethanol, propylene glycol, parabens) are associated with elevated toxicological risks and safety issues when used in children, even at age-adapted lower concentrations (Fabiano et al., 2011). Hence, a comprehensive safety assessment of the excipients in a pediatric formulation is essential. A compilation of all nonclinical and human safety data should be assessed for inclusion of an excipient, and that data placed into context with the risk and the anticipated health benefit of the formulation to the patient. For a well-established excipient commonly used in foods or other therapeutic formulations, known use experience should be leveraged. While additional nonclinical studies may be conducted to support safety of the excipient, such information is typically restricted to regulatory discussions supporting the formulation development of a particular product and is not publicly available. Thus, the safety qualification of excipients for pediatric use is confounded by limited availability of and access to safety data (especially for pediatric use) as well as uncertainties in extrapolations of exposure and effect between adults and children or between nonclinical species and humans. Although regulatory guidance documents provide some direction regarding excipient safety assessment (US FDA, 2005; EMA, 2013), there is a lack of standardization with regard to what is adequate or necessary to sufficiently characterize the risk-benefit profile for excipient use in various pediatric populations and disease states. Throughout the session, participants highlighted a number of challenges and opportunities for realizing the full potential of a better-integrated application of risk assessment framework on safety assessment of excipients and stakeholder decision-making.
1.2. The need for a risk-based assessment framework
In the plenary presentation, Dr Turner presented the current clinical and patient perspectives regarding the use of excipients for neonatal formulations. Assessments of excipient exposure are especially sensitive in the neonatal situation in that exposure always occurs in a clinical context that includes long-term concerns about development as well as treatment of acute illnesses. Dr. Turner emphasized the importance of considering the benefits and potential harms of excipients (in addition to those of the active pharmaceutical ingredient) in the context of the seriousness of the disease state and other conditions not related to the indication (eg, complications of prematurity). While some notable cases of severe excipient toxicity have been obvious against background morbidities, eg, Vitamin E, this is the exception rather than the rule (Arrowsmith et al., 1989).
The clinical example of inotrope use to treat hemodynamic insufficiency during the 72 h after birth before 27 weeks gestational age provides a good example of the need to consider the balance of risk and benefit. The seriousness of the disease state is evident in the associated mortality (30%–50%) and morbidity (brain injury leading to neurodisability in 60%–80% of survivors). In this case, excipients such as sodium metabisulphite are necessarily a part of the inotrope formulation to improve brain perfusion. As some, but not all, adverse events associated with the active ingredient are accepted, potential harms arising from excipients should be similarly accepted if those harms are limited and do not make a material difference to the overall benefit-risk assessment. One must consider how the risk of excipient compares to the risk of the condition. While it is difficult to attribute injury to individual causes (such as drugs) in clinical practice, such understanding may be possible at population level under good circumstances (eg, following a randomized controlled trial).
When determining whether an excipient is safe to use in a pediatric formulation, “Yes” or “No” is not always a helpful answer. There is a need to set limits on excipient exposure that are related to the context-of-use, instead of simply allowing or banning excipient use. Assessing the risks arising from an excipient requires an understanding of relationships between administered dose and exposure (pharmacokinetic, PK) and exposure and response (pharmacodynamic, PD). Thanks to recent technical advances which have enabled quantification of exposure in neonates, dose-exposure relationships can be examined (Mulla, 2015; Pandya et al., 2016). In cases where the exposure-response relationship currently cannot be assessed clinically, PKPD models can be set up using extrapolations from other contexts (animal or laboratory models). In the clinical setting, advice is also needed about what to do if there is a problem that may be attributed to excipients.
1.3. A range of perspectives regarding the safety assessment of excipients
In the subsequent Panel Discussion, perspectives of various Panel members (authors herein) regarding their experiences, expectations, perceived gaps in knowledge and process, as well as possible risk-based approaches were shared. Key observations and suggestions from the panel members follow:
The group noted the importance of leveraging existing data collected at the different stages of safety assessment of excipients and the need to create a mechanism for more seamless exchange of data. The first step in safety assessment of excipients involves comprehensive and critical review of all existing data to acquire a depth of understanding and breadth of knowledge beyond that of a single stakeholder (i.e., excipient maker or formulator alone). Before choosing an excipient, literature and databases such as the STEP database (Salunke et al., 2012, 2013; Salunke and Tuleu, 2015) should be searched to determine 1 if safety has been established and in what context, 2 what gaps exist in the data, and 3 the necessity of any additional studies to assess safety of the excipient. However, the information is currently scattered amongst numerous sources making it difficult for the users to systematically review the existing information. While sharing of nonclinical data and the development of surveillance tools to monitor post marketing safety will certainly help to provide further data to support novel excipient use, safety monitoring is complicated by the other components of the products and therefore, the safety signal is confounded.
Toxicology studies in juvenile animals may be necessary if the use of an excipient in a pediatric medicine cannot be justified based on existing information sources (EMA, 2013a; US FDA, 2005, 2016); however, the standardized conduct of juvenile toxicology studies in a routine “box-ticking” manner was not considered appropriate. Safety assessment should primarily focus on potential effects on growth and development, if not previously characterized (US FDA, 2016). When a cause for concern is identified, the juvenile animal toxicology study can be designed to assess the safety of both the excipient and the active moiety (and, if warranted, potential interactions thereof) (Schmitt, 2015; US FDA, 2005, 2016). Finally, both the rationale for conducting a juvenile animal study and the design of the study should be influenced by its relevance and ability to inform clinical safety data.
In addition to nonclinical safety data, existing human data can often be employed to justify expanded use of an excipient. Therefore, it is important that sponsors provide regulatory agencies with all available data detailing human exposure including, for example, data from foods. When appropriate this prior human exposure information can be used to justify use of an excipient and minimize the need to evaluate a full battery of toxicology studies.
Neonates may be uniquely vulnerable to adverse events related to excipients, due to immature absorption, distribution, metabolism, and elimination pathways, polypharmacy, and serious illness. Off-label use of drugs formulated for older populations remains prevalent (Hsieh et al., 2014), as clinical studies that meet regulatory approval standards are historically scarce.
The case study of propylene glycol presented by Dr. Carleer highlighted the need for special regulatory and safety considerations for different use patterns and pediatric patient populations (EMA, 2005, 2013b). Short- to medium- term treatment of pediatric patients (term neonates to adolescent) with a propylene glycol-containing solution for intravenous infusion have been considered clinically justified when urgent treatment was needed or when other routes of administration were not possible. However, safety concerns related to limited metabolic and renal clearance in neonates and young children have driven the requirement for monitoring the potential toxicity of propylene glycol in pediatric studies, particularly in term and preterm neonatal patients (EMA, 2005). In addition, the importance of potential interactions with the active moiety or other excipients which may compete for the same clearance pathway (eg, alcohol dehydrogenase) was emphasized. (Also see Kaletra (US FDA, 2016), Section 5.2, “Toxicity in Preterm Neonates,” which explains that the ethanol (42.4% v/v) and propylene glycol (15.3% w/v) excipients in the solution may increase risk of cardiac toxicity, lactic acidosis, renal failure, CNS depression, and respiratory complications leading to death. These toxicities were reported with neonatal administration of Kaletra, primarily in preterm neonates.)
Safety assessment should use a risk-benefit approach as opposed to one where safety assessment takes place only in the context of potential risk. This is especially important in light of the fact that most drug products could not be made without the use of excipients. Moreover, although excipients are not intended to exert therapeutic benefits alone, they may be necessary to achieve the full therapeutic benefit of the API (US FDA, 2005). Thus, in situations where the risk cannot be adequately characterized prior to use in pediatric patients and use of the excipient cannot be bypassed, but the therapeutic benefit of the active moiety is sufficient, it may be appropriate to proceed carefully and assess safety in the clinical setting.
During risk assessment it is important to remember who tolerates the risk. The risk is not shared equally among a number of groups: regulators; manufacturers; clinicians; families – parents and children; society. Ideally, groups that will suffer from the consequences of adverse events should inform the risk assessment. Thus, patient and caregiver involvement in these assessments is important.
These perspectives shared by the Panel provided a base from which participants engaged in breakout sessions to discuss key questions regarding risk assessment paradigms for new/novel excipients and those excipients with some use history. Specific questions concerned information needs, resources for potential data/information-sharing platforms, and elements for a common template or approach. The ensuing recommendations for the development of a risk-based, standardized approach to safety assessment of excipients for pediatric use are summarized below.
2. Key considerations and recommendations
2.1. Explicitly justify the need for the excipient(s)
The cross-functional exchanges at the workshop were particularly illuminating of gaps in understanding across stakeholders involved in developing or approving excipients in pediatric formulations. For example, non-formulator stakeholders were sometimes unclear as to the real need for such excipient(s) with regard to addressing specific requisites for acceptable pediatric formulations (palatability, ease of swallowing, ability to dose titrate, alternative dosage forms). While the potential use of alternative excipients should always be considered if there are limited or inadequate safety data for a proposed excipient, innovations in the usage of existing/known excipients or in the development of novel excipients are often needed to enable patients to benefit from these advancements. Formulators need to provide insight as to special considerations regarding the need for and use of specific excipients in pediatric formulations (especially with regard to special needs for younger age groups).
2.2. Start planning for pediatric formulation development early
Formulation development starts well in advance of the initiation of pediatric studies in order to assure the formulation has the desired dose flexibility, biopharmaceutics, stability (both chemical and physical), and manufacturing robustness. Formulations are developed for a defined target population(s), and delivery options are, by definition, age appropriate. For example, a solution or suspension formulation may be suitable for a neonate or child under 2 years of age but not ideal for an older child. Likewise, due to developmental changes in metabolism and physiology, safety considerations for excipients are also age appropriate.
Formulating all APIs for possible use in neonates could be quite restrictive and highly impractical. Likewise, assuming all pediatric populations can utilize the adult dosage form ignores the complexity of physiological development and enzymatic ontogeny. Constructing the Paediatric Investigative Plan (PIP) and the Pediatric Safety Plan (PSP) to target age groups most likely to benefit from study is key to developing the most relevant formulation. Shifts in the target population later in development, either desired by the sponsor or requested by regulators, could have serious results for patients, as studies are conducted in sick children and ethical considerations constrain lengthy development timelines to include multiple formulation changes.
Any need for additional nonclinical studies with the excipient could add significantly to the development timelines. Nonetheless, if there are information gaps, these studies are conducted to inform risks of excipients in advance of utilization in pediatric populations.
2.3. Leverage and share existing data
Discussions during the breakout session helped participants to assess the importance of developing a scientifically risk-based framework in which input from in silico, high-throughput screening and conventional toxicity data can form the basis of an integrated evaluation strategy for excipients risk assessment. The use of existing resources and barriers to sharing relevant information within the industry was recognized. The need to integrate the existing safety and toxicity information of excipients from disparate resources under one umbrella was recognized by European Paediatric Formulations Initiative (EuPFI) and drove the collaborative development of the Safety and Toxicity of Excipients for Paediatrics (STEP) database with the USPFI. The database is a publicly accessible evidence base of safety and toxicity information of excipients for the pharmaceutical industry, academics, pharmacists, clinicians and regulators to help make informed decisions and thus expedite pediatric drug development (Salunke et al., 2012, 2013; Salunke and Tuleu, 2015). Participants emphasized that, in addition to referring to existing information sources (such as the STEP database), data collected as part of nonclinical toxicity reports from the pharmaceutical, food, and cosmetics industries could hopefully identify a broader set of established excipients qualified by extensive safety data in animals and humans. However, such data acquired during drug development is not routinely published or shared in public databases owing to the confidential nature of the research that generates the data. Additionally, diverse methods of identifying excipients, differing reliability and review of databases were seen as some of barriers in sharing the information. Several participants expressed their view on the need to promote productive sharing of the safety and toxicity information of excipients by encouraging industry to publish the non-proprietary information on safety and tolerability information available on excipients. This would maximize the use of vast quantities of data and knowledge generated throughout the research development and regulatory review and avoid redundancies in effort, speed the drug development process, and lead to new science and knowledge on excipients.
Meeting the challenge of data sharing will require an extensive and ongoing dialog between the toxicology, risk assessment and other related scientific communities. Several suggestions were put forward by the participants to effectively deal with this issue. In the big picture, the STEP database may benefit users by increasing the flow of information. However, further effort is required by the sponsors to share, either voluntarily or upon request, non-confidential in-house data in the STEP database, to allow the users access to a much larger data pool and prevent the repetition of a number of toxicity studies. The need for expansion of the FDA Inactive Ingredients Database (IID), originally intended as a starting point only from which to establish prior use of an excipient, with regard to content and access was highlighted. It may be necessary, therefore, to expand the IID beyond its current scope to incorporate additional information on the context of use of individual excipients (although expansion of the IID will likely be constrained by issues surrounding the proprietary nature of excipient use). The International Pharmaceutical Excipients Council (IPEC)-America is working with FDA to improve FDA IID database;Pharmtech (2015), however the inclusion of pediatric formulation information may take several years and entail more resources than are currently available. Efforts may be needed to identify suitable way to support the improvement of the FDA IID database. Other regulators may be able to share information subject to compliance with any legal requirements.
The value in creation of a monograph around the commonly used excipients in pediatric products was discussed. Such monographs could describe and integrate all available data for an excipient – chemical properties, exposure, excipient ADME, systemic effects in animals and humans – and outline recommendations/advice for practitioners, pharmacists, and regulators within various context-of-use scenarios. The panel members suggested creation of vignettes that can illustrate the principles of choice and values to work to elucidate these choices. However, the challenge associated with this are the efforts and resources needed to evaluate the existing data and developing the monographs. The European Study of Neonatal Excipient Exposure (ESNEE) consortium has made some progress in this direction.
2.4. Address challenges associated with novel excipients
Pharmaceutical companies typically avoid the use of new excipients due to additional safety data required to introduce a novel excipient to a pharmaceutical product. The resources, time, and increased development risk associated with this requirement makes formulation scientists hesitant to try new excipients, thus hindering innovation. Academic researchers may not have the GLP facilities to support extensive safety studies. Participants recognized the value of leveraging data available from the food industry, however, that information does not always address specific context-of-use scenarios for pediatric patients. Efforts from both pharmaceutical and excipients manufactures are needed to bridge the gaps and identify the best practices and types of data that are needed for safety assessment of novel excipients. For instance, the Novel Excipients Working Group (members from IPEC-Americas and the IQ Consortium) and a similar group formed within IPEC-Americas are currently exploring the development of joint best practices for nonclinical safety (testing and specification requirements) and creating a process for designing a well-defined nonclinical data package for novel excipients (Challener, 2014). It is the intention that such proposals will be discussed with regulators, as well.
2.5. Develop context-specific risk:benefit analyses for excipients
Dr. Turner proposed a systematic, risk-based, proportionate, prospective approach for selected safety assessments. A systematic, prospective approach would supplement surveillance for adverse events using standard methods to detect potential reactions to excipients, such as spontaneous reports. The proposed risk-based approach is intended to be used proportionately, that is, only when an excipient is likely to be crucial to the success of a formulation in a high impact clinical situation. The proposed approach is summarized in the Figure. The steps are described using the example of a prospectively planned assessment of parabens in products intended for enteral administration to babies born between 24 and 32 weeks of gestation.
Step 1: define the scope of the assessment. This step is critical to identify the content of the assessment and inform decisions about the deployment of resources. A multidisciplinary team should decide which variables are relevant including (age/developmental stage, disease state) and the pharmaceutical program (route of administration, dosing paradigm). An example of a scope could be “the enteral administration of parabens to babies born between 24 and 32 weeks of gestation.”
Step 2: define the goals of the safety assessment in light of the scope. Use the scope to identify which exposures and adverse events are relevant. In the parabens example, “which doses of parabens will avoid long-and short-term adverse events known from animal studies and suggested in human studies (Anderson, 2008).”
Step 3: synthesize the existing knowledge starting with systematic search methods. Synthesis may be narrative or quantitative (for example, by building population PK models for excipients when relevant data is available). Recent EMA documents about excipient labelling are good examples of evidence synthesis (EMA, 2017).
- Step 4: use the knowledge synthesis, scope and goals of the risk assessment to identify knowledge gaps. Then apply reasoned and proportionate judgment to prioritize key knowledge gaps. Examples of knowledge gaps in the parabens example would be:
- How much paraben is included in existing products/how widely are existing products used?
- How does dosage relate to exposure (taking account of patient characteristics)?
- How does exposure relate to effects?
- Step 5: develop a judicious plan to reduce key knowledge gaps. The plan may include nonclinical studies (eg, to provide animal PKPD data that can be bridged with clinical dose-exposure data) and/or clinical work (eg PK studies or structured safety assessment). In the paraben example, studies could include:
- Survey manufacturers of marketed products
- Conduct study of excipient kinetics in babies administered existing paraben-containing products
- Conduct PD studies in suitable models
-
Step 6: design and conduct the appropriate studies. In the paraben example, some selected studies were done:
- Nellis et al. (2015) conducted a survey of excipient use in Europe but found that manufacturers were reluctant to share excipient content of many products
- Mulla et al. (2015) conducted an excipient kinetic study in the target population for methyl and propyl parabens
NB: not all the planned studies were feasible, so that any final assessment must make the best use of imperfect information.
Step 7: synthesize the consequent new body of knowledge, for example, update PKPD or PoP PK models, conduct a meta-analysis, inform bridging from animals to humans. In the parabens example data from Mulla et al. was referenced in the EMA reflection paper on parabens (2015). In high-impact situations, a bridging exercise could be done between PKPD studies in animals and the available PK data in humans (compare work done with Vancomycin in neonates, Ramos-Martín et al., 2016)
Step 8: perform risk assessment in the light of the new body of knowledge, eg, provide balanced recommendations considering internal vs external validity of the model or data; consider clinical implications; consider risk mitigation strategies. The parabens example includes the dosages for parabens included in the EMA reflection paper (2015). Clinical benefit-risk assessment suggests that it is appropriate to continue using existing products containing methyl and propyl parabens in this population Fig. 2.
Fig. 2.
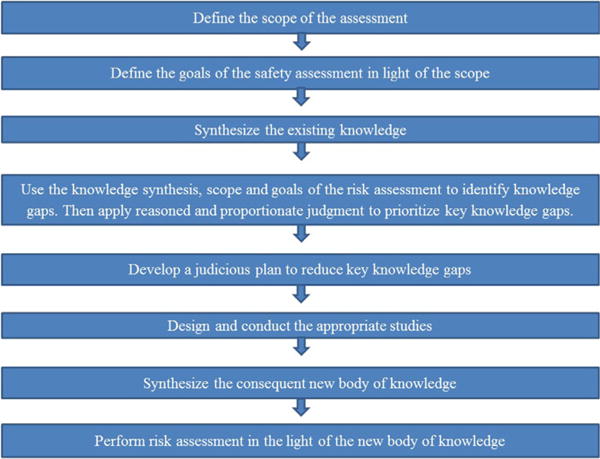
A systematic, risk-based approach to the prospective safety assessment of excipients to be used in pediatric medicines: each step should be adapted to the product and target population.
Interpretation of all the information could result in possible actions for excipients of concern, including termination (when the risk of adversity is too high); the conduct of additional studies to further understand potential risks; or the implementation of clinical monitoring of exposure or biomarkers of safety.
2.5.1. Engage all stakeholders
Decisions about benefit and risk tolerance should be determined by clinicians, regulators, patients, and caregivers. High-impact safety assessments may be built around meaningful stakeholder engagement. Parents and children can present a distinct perspective, especially for serious diseases with few available options. The context-dependence of family tolerance of risk can be observed in trials of novel therapies. For example, among children and their families, the risk tolerance for gene therapy can be much higher if the condition is life-threatening and otherwise untreatable than for other conditions. Less-dramatically, a family’s tolerance for potential harms arising from an excipient in an inotrope may be greater than for an excipient in vitamin drops.
Engaging patient advocate groups to help develop “archetypes” for various risk:benefit scenarios could be very useful for drug developers and regulators by providing the ranges of risk that can be tolerated by a stakeholder group.
2.6. Engage regulators
For development programs for specific drugs, sponsors should consider early interaction in the pre-IND stage (US) or before PIP submission (EU) to gain regulatory insight and alignment regarding for example, the appropriate age range of the target population and the use of excipients to optimize drug product performance and efficacy. Sponsors should provide regulators with all relevant data from prior studies, medical and toxicology literature, and available databases (i.e. STEP, IID, GRAS, etc.). A collaborative discussion can then ensue to determine if additional data are needed for assessment of excipient safety.
In addition to the regular, drug-specific meetings, opportunities to engage in general (non-product-specific) scientific discussions should be leveraged. Similarly, multi-stakeholder collaborative efforts (FDA, (EMA), PMDA, Health Canada and Australia) directed at jointly addressing or discussing a formulation issue in a non-application-specific way could help to improve industry and regulatory confidence around use of excipients in pediatrics. Monthly discussion among regulators normally entails reviews of drug applications but without any formulation discussions. International regulatory groups such as the Pediatric Cluster include regulatory scientists from FDA, EMA, Health Canada, PMDA. Such groups may provide a forum for rich discussion regarding use of excipients in pediatrics and may potentially encourage progress towards harmonized requirements. Finally, various consortia (public-private partnerships) interested in safety topics in drug development may be leveraged for a more broadly informed, collaborative analysis of excipient safety.
3. Conclusion
The development of effective and safe formulation of drugs to treat pediatric patients is rife with complexities related to the diversity of the populations (neonate to adolescent), the need for novel technologies and approaches by Formulators to optimize efficacy of active ingredients in ways amenable to those patient populations, and the lack of mechanisms to effectively share data and experience in the scientific community. Industry drug development teams need to developing target product profiles as early as possible, understanding that the dynamics of drug development, including unanticipated (and potentially, disparate) regulatory requests for consideration of additional age groups, will require some flexibility in approach. Development of archetypes to frame constraints for various patient populations could be helpful, keeping in mind that it is the younger patient groups that set the risk posture around excipients in pediatric formations. A collaborative partnership with different disciplinary groups in regulatory agencies (eg, Chemists/Formulators, Pharmtox and Medical Reviewers, and Pediatric experts) at early pre-IND stages (US) or prior to PIP submission (EU) should be considered. However, the lack of harmonization in approach (for example, on the most appropriate age group(s) for a given therapeutic) between regulatory agencies presents further challenges. By enabling practical and meaningful discussion between scientists representing the diversity of involved disciplines (formulators, nonclinical scientists, clinicians, and regulators) and geographies (eg, US, EU), the Excipients Safety workshop session was successful in providing specific and key recommendations for defining paths forward. Leveraging orthogonal sources of data (eg. food industry, agro science), collaborative data sharing, and increased awareness of the existing sources such as the STEP database will be important to address the gap in excipients knowledge needed for risk assessment. The importance of defining risk-based approaches to safety assessments for excipients vital to pediatric formulations was emphasized, as was the need for meaningful stakeholder (eg, patient, caregiver) engagement. As development of pediatric dosage forms progresses, additional experiences with excipients will further an evolving and shared landscape for future pediatric development.
Acknowledgments
Drs. Brian Aylward (Irish Health Products Regulatory Authority) and Jacqueline Carleer (Belgian Federal Agency for Medicines and Health Products) served as Panel members and discussants during the case studies.
We thank Drs. Anthony Nunn and Trupti Dixit for their expert reviews of the manuscript.
This workshop was co-sponsored by the University of Maryland and the US FDA in collaboration with the IQ Consortium and EuPFI.
References
- Anderson J. Final amended report on the safety assessment of methylparaben, ethylparaben, propylparaben, isopropylparaben, butylparaben, isobutylparaben, and benzylparaben as used in cosmetic products. Int J Toxicol. 2008;27(Suppl. 4):1–82. doi: 10.1080/10915810802548359. [DOI] [PubMed] [Google Scholar]
- Arrowsmith JB, Faich GA, Tomita DK, Kuritsky JN, Rosa FW. Morbidity and mortality among low birth weight infants exposed to an intravenous vitamin E product, E-Ferol. Pediatrics. 1989;83(2):244–249. [PubMed] [Google Scholar]
- Challener Exploring a new approval process for continued excipient innovation. Pharm Technol. 2014 ( http://www.pharmtech.com/exploring-new-approval-process-continued-excipient-innovation, Last Accessed on 20 March 2017)
- EMA. Reflection Paper: Formulations of Choice for the Paediatric Population. 2005 (EMEA/CHMP/PEG/194810/2005) [Google Scholar]
- EMA. EU Guideline on Pharmaceutical Development of Medicines for Paediatric Use. 2013a http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/07/WC500147002.pdf.
- EMA. Draft Questions and Answers on Propylene Glycol and Esters in the Context of the Revision of the Guideline on ‘Excipients in the Label and Package Leaflet of Medicinal Products for Human Use’. 2013b (EMA/CHMP/704195/2013) http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/12/WC500177945.pdf.
- EMA. Reflection Paper on the Use of Methyl- and Propylparaben as Excipients in Human Medicinal Products for Oral Use. 2015 http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/11/WC500196733.pdf.
- EMA. Reflection Paper on Extrapolation of Efficacy and Safety in Paediatric Medicine Development. 2016 (01 Apr 2016 DRAFT:) http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2016/04/WC500204187.pdf.
- EMA. Excipients Labeling. 2017 ( http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001683.jsp&mid=WC0b01ac05808c01f6, Last Accessed 22 February 2017)
- European Commission. Guidelines: medicinal products for human use, safety, environment and information. Excipients in the Label and Package Leaflet of Medicinal Products for Human Use. 2003 http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003412.pdf.
- Fabiano V, Mameli C, Zuccottii GV. Paediatric pharmacology: remember the excipients. Pharm Res. 2011;63(5):362–365. doi: 10.1016/j.phrs.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Hsieh EM, Hornik CP, Clark RH, Laughon MM, Benjamin DK, Jr, Smith PB. Medication use in the neonatal intensive care unit. Am J Perinatol. 2014;31(9):811–821. doi: 10.1055/s-0033-1361933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IPEC. Position Paper on EU Risk Assessment Guidelines for Excipients (2015/C95/02) 2015 ( http://ipec.org/sites/default/files/IPEC%20Federation_RA_position_statement__Final_0.pdf Last Accessed on 24 February 2017)
- Mulla H, Yakkundi S, McElnay J, Lutsar I, Metsvaht T, Varendi H, Nellis G, Nunn A, Duncan J, Pandya H, Turner M. An observational study of blood concentrations and kinetics of methyl- and propyl-parabens in neonates. Pharm Res. 2015;32(3):1084–1093. doi: 10.1007/s11095-014-1520-2. doi: http://dx.doi.org/10.1007/s11095-014-1520-2. [DOI] [PubMed] [Google Scholar]
- Nellis G, Metsvaht T, Varendi H, Toompere K, Lass J, Mesek I, Anthony J, Nunn AJ, Turner MA. Lutsar I on behalf of the ESNEE consortium. Potentially harmful excipients in neonatal medicines: a pan-European observational study. Arch Dis Child. 2015;100:694–699. doi: 10.1136/archdischild-2014-307793. doi: http://dx.doi.org/10.1136/archdischild-2014-307793. [DOI] [PubMed] [Google Scholar]
- Pandya HC, Mulla H, Hubbard M, Cordell RL, Monks PS, Yakkundi S, McElnay JC, Nunn AJ, Turner MA. ESNEE consortium. Essential medicines containing ethanol elevate blood acetaldehyde concentrations in neonates. Eur J Pediatr. 2016;175(6):841–847. doi: 10.1007/s00431-016-2714-x. doi: http://dx.doi.org/10.1007/s00431-016-2714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PharmTech. IPEC-Americas suggest improvements to FDA’s inactive ingredients database. PTSM: Pharm Technol Sourc Manage. 2015;11:11. http://www.pharmtech.com/ipec-america-suggests-improvements-fda-s-inactive-ingredients-database. [Google Scholar]
- Ramos-Martín V, Johnson A, Livermore J, McEntee L, Goodwin J, Whalley S, Docobo-Pérez F, Felton TW, Zhao W, Jacqz-Aigrain E, Sharland M, Turner MA, Hope WW. Pharmacodynamics of vancomycin for CoNS infection: experimental basis for optimal use of vancomycin in neonates. J Antimicrob Chemother. 2016 Apr;71(4):992–1002. doi: 10.1093/jac/dkv451. doi: http://dx.doi.org/10.1093/jac/dkv451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salunke S, Tuleu C. The STEP database through the end-users eyes Usability study. Int J Pharm. 2015;492(1-2):316–331. doi: 10.1016/j.ijpharm.2015.06.016. http://www.sciencedirect.com/science/article/pii/S0378517315005360. [DOI] [PubMed] [Google Scholar]
- Salunke S, Giacoia G, Tuleu C. The STEP (Safety and Toxicity of Excipients in Paediatrics) database: part 1 a need assessment study. Int J Pharm. 2012;435(2):101–111. doi: 10.1016/j.ijpharm.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Salunke S, Brandys B, Giacoia G, Tuleu C. The STEP (Safety and Toxicity of Excipients for Paediatrics) database: part 2-the pilot version. Int J Pharm. 2013;457(1):310–322. doi: 10.1016/j.ijpharm.2013.09.013. http://www.sciencedirect.com/science/article/pii/S037851731300848X. [DOI] [PubMed] [Google Scholar]
- Schmitt G. Safety of excipients in pediatric formulations a call for toxicity studies in juvenile animals? Children. 2015;2:191–197. doi: 10.3390/children2020191. doi: http://dx.doi.org/10.3390/children2020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MA. Risk assessment of neonatal excipient exposure: lessons from food safety and other areas. Adv Drug Deliv Rev. 2014;73:89–101. doi: 10.1016/j.addr.2013.11.003. doi: http://dx.doi.org/10.1016/j.addr.2013.11.003. [DOI] [PubMed] [Google Scholar]
- US FDA. Guidance for Industry: Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients. 2005 ( https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079250.pdf, Last Accessed 24 February 2017)
- US FDA. Label for Kaletra, Oral Solution. 2016 http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021251s052_021906s046lbl.pdf.