

Abstract
Mammalian target of rapamycin (mTOR) is a phosphoinositide 3-kinase-related protein kinase which controls cell growth and is frequently deregulated in many cancers. Therefore, mTOR inhibitors are used as antineoplastic agents for cancer treatment. In this study, 1,2,4-triazine derivatives containing different arylidene-hydrazinyl moieties were designed and synthesized. Cytotoxicity of the compounds was evaluated on HL-60 and MCF-7 cell lines by MTT assay. S1, S2 and S3 exhibited good cytotoxic activity on both cell lines with an IC50 range of 6.42 - 20.20 μM. In general, substitution of a five-membered heterocyclic ring containing NO2, such as 5-nitrofuran-2-yl, resulted in the best potency. Molecular docking analysis was performed to study the possible interactions and binding modes of all the triazine derivatives with mTOR receptor. The most promising compound, S1, was well accommodated within the active site and had the least estimated free energy of binding (even less than the inherent ligand of the protein, PDB ID: 4JT6). It is concluded from both MTT assay and docking studies that the arylidene moiety linked to the hydrazinyl part of the structure had a prominent role in cytotoxicity and mTOR inhibitory activity.
Keywords: Cancer; mTOR inhibitor; 1,2,4-Triazines; Docking; MTT
INTRODUCTION
Cancer is the main cause of death worldwide and it is estimated that the number of annual cancer deaths will increase to 11.4 million in 2030 (1). Despite various therapeutic approaches, treatment of cancer is still a significant obstacle and attempts to find new, effective and less toxic agents are being continued (2,3,4,5).
The PI3K/Akt/mTOR pathway plays an important role in regulating cell proliferation, migration, survival, angiogenesis, and it is deregulated in many human tumors (6,7). This signaling pathway appeared as an attractive target for the design of new and effective anticancer agents. Therefore, finding inhibitors of mammalian target of rapamycin (mTOR) is considered as a potential strategy in molecular targeted therapy for treatment of cancer (8,9).
1,2,4-Triazine derivatives have been reported to have a variety of pharmacological activities such as anti-HIV (10), antiinflammatory (11), antifungal (12), analgesic (11), antimalarial (13), neuroprotective (14) and antimicrobial (15). Moreover, triazine scaffold is of the great interest to medicinal chemists due to its cytotoxic effects (16,17,18). Yurttas, et al. proved that a 5,6-diphenyl- 1,2,4-triazine derivative bearing piperazine amid moiety, (1) (Fig. 1), showed potential antitumor activity against breast cancer cells (19). Karczmarzyk, et al. reported the synthesis and anticancer activity evaluation of a series of sulfur 1,2,4-triazine analogs and introduced 5,5′,6,6′-tetraphenyl-bis-(1,2,4-triazine)-3,3′-disulfide (2) as the most cytotoxic derivative (20)
Fig. 1.

Structures of the reported cytotoxic agents and mTOR inhibitors; the design strategy for compounds S1-S10.
VLX600 (Fig. 1), containing 1,2,4-triazine with aryl hydrazone moiety, showed cytotoxicity against numerous cancer cells with an IC50 range of 1-10 μM (21).
In recent years, a number of studies on pyrimidine-hydrazone and triazine-hydrazone scaffolds, as mTOR inhibitors, have been performed (22,23,24). Menear, et al. introduced compound (3) (Fig. 1) with a triazine-hydrazone structure as a potent antitumor agent with an mTOR IC50 value of 0.27 μM (24). In addition, Zhu, et al. proved that compounds (4) and (5) (Fig. 1) showed a good cytotoxic activity on H460 and PC-3 cell lines. Furthermore, they demonstrated that compounds (4) and (5) inhibited mTOR with IC50 values of 0.92 and 0.16 μM, respectively (23).
In this study, we designed 5,6-diphenyl- 1,2,4-triazine derivatives containing different arylidene-hydrazinyl moieties based on a number of potent cytotoxic agents and mTOR inhibitors that are reported in the literature. After synthesis, the cytotoxic activity of derivatives was evaluated against two human cell lines. Furthermore, docking was performed to get a distinct insight about the binding modes and interactions of these compounds in the active site of mTOR receptor.
MATERIAL AND METHOD
Chemistry
NMR spectra were recorded on Brucker 500 spectrometer (Brucker Corporation, MA, USA) relative to tetramethylsilane (TMS) as an internal standard using dimethyl sulfoxide (DMSO)-d6 as the solvent. The mass spectra were obtained on an Agilent 6410 instrument (Agilent Technologies, USA). The IR spectra were recorded on an FTIR Perkin-Elmer spectrophotometer (KBr disks) (Perkin-Elmer, Waltham, MA, USA). Melting points were determined on a KoFler hot stage apparatus (KoFler, Germany) and uncorrected.
Synthesis
All 3-(2-arylidenehydrazinyl) 5,6-bis (4-methoxyphenyl)-1,2,4-triazine derivatives were prepared according to the described procedure (25). The 2-hydroxy-1,2-bis (4-methoxyphenyl) ethanone and 1,2-bis (4-methoxyphenyl) ethane-1,2- dione were prepared according to the previously described procedure (26).
Synthesis of 5,6-bis(4-methoxyphenyl)-1,2,4- triazine-3-thiol
The 1,2-bis(4-methoxyphenyl)ethane-1,2- dione (10 g, 37 mmol) was added to 60 mL of acetic acid and the mixture was heated to about 100 °C with stirring. Thiosemicarbazide (6.84 g, 75.04 mmol) was added and the mixture was refluxed for about 2 h. The solid product was filtered and washed with cold acetic acid and water.
Synthesis of 5,6-Bis(4-methoxyphenyl)-3-(methylthio)-1,2,4-triazine
The sodium hydroxide (0.8 g, 20 mmol) was dissolved in 60 mL of ethanol by warming till 50 °C. The basic solution was cooled to room temperature and 5,6-bis(4-methoxyphenyl)-3-(methylthio)- 1,2,4-triazine (6.7 g, 20 mmol) was added and stirred for 15 min. Methyl iodide (6.7 g, 47 mmol) was added dropwise to the reaction mixture and the mixture immediately became a slurry. Ethanol was added to the reaction mixture and stirring was continued for about 4 h at room temperature. Then, 15 mL water was added and the yellow precipitate was filtered and washed with ethanol.
Synthesis of 3-hydrazinyl-5,6-bis(4-methoxy-phenyl)-1,2,4-triazine
A solution of hydrazine hydrate (3 mL) in 30 mL ethanol and 5,6-bis(4-methoxyphenyl)- 3-(methylthio)-1,2,4-triazine (4 g, 11.8 mmol) was stirred and refluxed for 12 h, and the solid product was filtered off, washed with cold ethanol and recrystallized from ethanol to give dark yellow crystalline 3- hydrazinyl-5,6- bis(4-methoxyphenyl)-1,2,4-triazine.
General method for synthesis of 3-(2- arylidenehydrazinyl)-5,6-bis(4-methoxyphenyl)- 1,2,4-triazine (S1-S10)
To a well-stirred warm (70-75 °C) solution of 3-hydrazinyl-5,6-bis(4-methoxyphenyl)-1,2,4- triazine (1 mmol) in 10 mL ethanol equivalent amount of appropriate aldehyde (1 mmol) was added and treated with a few drops of glacial acetic acid. The reaction mixture was stirred for 2 h. The completion of the reaction was monitored by TLC. After completion, the precipitate was filtered off, washed with cold ethanol and recrystallized from suitable solvent to yield the final pure products S1-S10.
5,6-Bis(4-methoxyphenyl)-3-(2-((5-nitrofuran- 2-yl)methylene)hydrazinyl)-1,2,4-triazine (S1).
Compound S1 was prepared by the described procedure using 5-nitrofuran- 2-carbaldehyde (141 mg, 1 mmol). A pale yellow solid was obtained; yield 78%, m.p.: 248–251 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 12.34 (1H, s, NH), 8.74 (1H, s, N=CH), 7.82 (1H, d, J = 3.9 Hz, ArH), 7.49–7.47 (2H, m, ArH), 7.39–7.37 (2H, m, ArH), 7.21 (1H, d, J = 3.9 Hz, ArH), 6.95–6.97 (4H, m, ArH), 3.79 ppm (6H, s, 2×O–CH3). IR (KBr, ν): 3443, 2839, 1608, 1514, 1353, 1254, 1071, 834 cm-1. MS (EI) m/z (%): 446 (M+, 17), 400 (100), 267 (24), 238 (79), 223 (86), 195 (23), 180 (22), 152 (21), 134 (17), 119 (16), 79 (12).
5,6-bis(4- methoxyphenyl)-3 -(2-((1- methyl-5- nitro-1H-imidazol-2-yl)methylene)hydrazinyl)-1,2,4-triazine (S2).
The titled compound S2 was synthesized by the described procedure using 1-methyl-5-nitro-1-imidazole-2-carbaldehyde (155 mg, 1 mmol). Dark yellow solid was obtained; yield 54%, m.p.: 237-239 °C. 1H NMR (DMSO-d6, 500 MHz): δH: 12.36 (1H, s, NH), 8.26 (1H, s, ArH), 8.21 (1H, s, N=CH), 7.45 (2H, d, J = 8.5 Hz, ArH), 7.34 (2H, d, J = 8.6 Hz, ArH), 6.94-6.98 (4H, m, ArH),4.37 (3H, s, N-CH3), 3.77 ppm (6H, s, 2×O-CH3). 13C NMR (DMSO-d6, 125 MHz): δC: 161.20, 160.77, 157.88, 155.48, 152.94, 151.33, 149.89, 145.39, 140.13, 133.78, 131.23, 130.25, 128.80, 128.42, 127.59, 113.88, 113.84, 113.76, 113.63, 55.23, 55.09, 25.09 ppm. IR (KBr, ν): 3284, 2934, 1608, 1589, 1509, 1366, 1254, 838 cm-1. MS (EI) m/z (%): 460 (M+, 22), 432 (16), 238 (100), 223 (67), 195 (17), 180 (13), 166 (15), 152 (16), 134 (10), 119 (10).
5,6- bis(4- methoxyphenyl)- 3-(2-(3,4,5-trimethoxybenzylidene)hydrazinyl)-1,2,4-triazine (S3).
The titled compound S3 was synthesized by the described procedure using 3,4,5- trimethoxybenzaldehyde (196 mg, 1 mmol). A yellow solid was obtained; yield 75%, m.p.: 266–270 °C; 1H NMR (DMSO-d6 500 MHz), δH: 11.81 (1H, s, NH), 8.18 (1H, s, N=CH), 7.49 (2H, d, J = 8.4 Hz, ArH), 7.37 (2H, d, J = 8.0 Hz, ArH), 7.04 (2H, s, ArH), 6.95–6.98 (4H, m, ArH), 3.86 (6H, s, 2×O–CH3), 3.79 (6H, s, 2×O–CH3), 3.70 ppm (3H, s, O–CH3). IR (KBr, ν) = 3444, 3216, 1609, 1508, 836 cm-1. MS (EI) m/z (%): 501 (M+, 25), 334 (18), 308 (39), 238 (100), 223 (46), 195 (14).
5,6- bis(4- methoxyphenyl)-3- (2-(4- methyl-benzylidene)hydrazinyl)-1,2,4-triazine (S4).
The titled compound S4 was synthesized by the described procedure using 4-methylbenzaldehyde (120 mg, 1 mmol). A pale yellow solid was obtained; yield 63%, m.p.: 249–252 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.77 (1H, s, NH), 8.42 (1H, s, N=CH), 7.65-7.74 (4H, m, ArH), 7.37 (2H, d, J = 9.0 Hz, ArH), 7.22 (2H, d, J = 8.0 Hz, ArH), 6.86–6.90 (4H, m, ArH), 3.86 (6H, s, 2×O–CH3), 2.35 ppm (3H, s,CH3). IR (KBr, ν): 3423, 3200, 1613, 1586, 829 cm-1. MS (EI) m/z (%): 425 (M+, 37), 334 (26), 238 (100), 223 (52), 195 (16), 180 (14), 152 (15), 95 (11).
5,6- bis(4- methoxyphenyl)-3-(2- (pyridin-3- ylmethylene)hydrazinyl)-1,2,4-triazine (S5).
The titled compound S5 was synthesized by the described procedure using 3-pyridinecarbaldehyde (107 mg, 1 mmol). Pale yellow solid was obtained; yield: 81%, mp: 255-258 °C. 1H NMR (DMSO-d6, 500 MHz): δH: 12.04 (1H, s, NH), 8.87 (1H, d, J = 1.9, pyridine-H-2), 8.58 (1H, dd, J = 4.9/1.6, pyridine-H-6), 8.30 (1H, s, N=CH), 8.14 (1H, dt, J = 7.9/1.9, pyridine-H-4), 7.50 (2H, d, J = 5.7, ArH), 7.47-7.49 (1H, m, pyridine-H-5), 6.97 (2H, d, J = 8.8 Hz, ArH), 3.79 ppm (6H, s, 2×O-CH3). IR (KBr, ν): 3434, 3146, 1609, 1569, 1507, 1237, 841 cm-1. MS (EI) m/z (%): 412 (M+, 32), 334 (16), 308 (12), 238 (100), 223 (25), 195 (10).
3-(2- (4-Nitrobenzylidene) hydrazinyl) 5,6- bis(4-methoxyphenyl)-1,2,4-triazine (S6).
The titled compound S6 was synthesized by the described procedure using 4-nitrobenzaldehyde (151 mg, 1 mmol). A yellow solid was obtained; yield 88%, m.p.: 265–267 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 12.24 (1H, s, NH), 8.36 (1H, s, N=CH), 8.31 (2H, d, J = 8.6 Hz, ArH), 7.99 (2H, d, J = 8.0 Hz, ArH), 7.52 (2H, d, J = 8.3 Hz, ArH), 7.39 (2H, d, J = 8.6 Hz, ArH), 6.96–6.98 (4H, m, ArH), 3.79 ppm (6H, s, 2×O–CH3). IR (KBr, ν): 3421, 3205, 1607, 1585, 838 cm-1. MS (EI) m/z (%): 456 (M+, 21), 308 (16), 238 (100), 223 (25), 195 (11).
3-(2- Benzylidenehydrazinyl) -5, 6- bis(4- methoxyphenyl)1,2,4-triazine (S7).
The titled compound S7 was synthesized by the described procedure using benzaldehyde (106 mg, 1 mmol). A yellow solid was obtained; yield 83%, m.p: 245–247 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 10.16 (1H, s, NH), 8.24 (1H, s, N=CH), 7.82 (2H, d, J = 7.1 Hz, ArH), 7.66 (2H, d, J = 8.8 Hz, 2H, ArH), 7.50 (2H, d, J = 8.7 Hz, ArH), 7.43–7.38 (3H, m, ArH), 6.93 (2H, d, J = 8.7 Hz, ArH), 6.88 (2H, d, J = 8.7 Hz, ArH), 3.86 ppm (6H, s, 2×O–CH3). IR (KBr, ν): 3440, 3211, 1605, 1571, 831 cm-1. MS (EI) m/z (%): 411.2 (M+, 29), 334 (20), 308 (14), 238 (100), 223 (49), 195 (12), 152 (11).
3-(2-(2,3-Dimethoxybenzylidene)hydrazinyl)- 5,6-bis(4-methoxyphenyl)-1,2,4-triazine (S8).
Compound S8 was synthesized according to the general procedure from 2.3-dimethoxybenzaldehyde (166 mg, 1 mmol). A yellow solid was obtained; yield: 88%, mp: 219-222 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.83 (1H, s, NH), 8.56 (1H, s, N=CH), 7.48-7.53 (2H, m, ArH), 7.37 (2H, d, J = 8.4 Hz, ArH), 7.14-7.09 (3H, m, ArH), 6.97-6.95 (4H, m, ArH), 3.84 (6H, s, 2×O-CH3), 3.78 ppm (6H, s, 2×O-CH3). IR (KBr, ν): 3444, 3211, 1607, 1587, 1255, 831 cm-1. MS (EI) m/z (%): 471 (M+, 16), 334 (10), 308 (24), 238 (100), 223 (46), 195 (14).
3-(2-(3,4-Dimethoxybenzylidene)hydrazinyl)- 5,6-bis(4-methoxyphenyl)-1,2,4-triazine (S9).
Compound S9 was synthesized according to the general procedure from 3.4-dimethoxybenzaldehyde (166 mg, 1 mmol). A yellow solid was obtained; yield 82%, mp: 250-253 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.70 (1H, s, NH), 8.19 (1H, s, N=CH), 7.49 (2H, d J=8.6 Hz, ArH), 7.37-7.35 (3H, m, ArH), 7.21 (1H, d, J = 8.2 Hz, ArH), 7.03 (1H, d, J = 8.2 Hz, ArH), 6.96-6.94 (4H, m, ArH), 3.83 (3H, s, OCH3), 3.80 (3H, s, OCH3), 3.78 ppm (6H, s, 2×O-CH3); 13C NMR (DMSO-d6, 125 MHz), δC: 160.91, 159.31, 158.19, 155.30, 150.07, 148.95, 143.62, 131.14, 130.15, 128.74, 127.96, 127.59, 120.64, 113.80, 113.71, 111.54, 108.32, 55.50, 55.37, 55.27, 55.10 ppm. IR (KBr, ν): 3444, 3216, 1609, 1508, 836 cm-1. MS (EI) m/z (%): 471 (M+, 16), 308 (28), 238 (100), 223 (35), 195 (13), 152 (10).
3-(2-(3- Fluorobenzylidene) hydrazinyl)-5,6- bis(4-methoxyphenyl)-1,2,4-triazine (S10).
Compound S10 was synthesized according to the general procedure from 3- fluorobenzaldehyde (124 mg, 1 mmol). A yellow solid was obtained; yield: 88 %, m.p.: 219-222 °C; 1H NMR (DMSO-d6, 500 MHz), δH: 11.97 (1H, s, NH), 8.26 (1H, s, N=CH), 7.55-7.49 (5H, m, ArH), 7.37 (2H, d, J = 7.4 Hz, ArH), 7.23-7.21 (1H, m, ArH), 6.97-6.95 (4H, m, ArH), 3.79 ppm (6H, s, 2×O-CH3). IR (KBr, ν): 3444, 3223, 2913, 1607, 1589, 1514, 1256, 836 cm-1. 13C NMR (DMSO-d6, 125 MHz), δC: 163.40, 161.46, 160.99, 159.39, 158.21, 155.45, 150.64, 141.88, 137.48, 137.42, 131.19, 130.86, 130.80, 130.21, 128.63, 127.88, 122.86, 116.03, 115.86, 113.83, 113.77, 112.43, 55.27, 55.11. MS (EI) m/z (%): 429 (M+, 37), 334 (26), 238 (100), 223 (52), 195 (16), 180 (14), 152 (15), 95 (11).
Cell lines
Two cell lines were used in this study including HL-60 (Human promyelocytic leukemia cells) and MCF-7 (human breast adenocarcinoma). HL-60 was grown in suspension, while MCF-7 was grown in monolayer culture. Cell lines were obtained from the National Cell Bank of Iran, Pasteur Institute, Tehran, Iran. The cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) (20% FBS for HL-60 cell line) and 100 Units/mL penicillin-G and 100 μg/mL streptomycin at 37 °C in humidified air containing 5 % CO2. Thiazolyl blue tetrazolium bromide (MTT) was from Sigma-Aldrich, Saint louis, MO. RPMI 1640, Dulbecco’s phosphate buffered saline and penicillin-G/streptomycin were products of Biosera, Ringmer, UK and FBS was from Invitrogen, San Diego, CA, USA. Doxorubicin and cisplatin were obtained from Ebewe Pharma, Unterach, Austria.
MTT assay
Cytotoxicity of the synthesized compounds was tested by MTT reduction assay. The cells were seeded into 96-well plates at density of 1 × 104 cells/well and incubated for 24 h at 37 °C. Then, three different concentrations of synthesized compounds, cisplatin or doxorubicin (as positive controls) were added in triplicate and the plates were incubated at 37 °C for another 72 h. After the incubation period, the media was removed; then, MTT solution in PBS at a concentration of 5 mg/mL was added for MTT assay. After 4 h of incubation, formazan crystals formed. Then, 60 μL DMSO was added to dissolve the crystals and, finally, absorbance was measured at a wavelength of 570 nm with background correction at 650 nm using a microplate reader (model 680, Bio-Rad, Japan). IC50 (concentration that results in 50 % inhibition of cell viability) for each compound was calculated with Curve Expert version 1.34 for windows.
Molecular docking analysis
For docking studies, we got X-ray crystal structure of mTOR in complex with PI-103 (PDB ID: 4JT6) from RCSB Protein Data Bank (http://www.rcsb.org). To prepare protein for docking, the innate ligand and water molecules were removed, hydrogen atoms were added, non-polar hydrogens were merged and Gasteiger charges were added using AutoDock Tools. 3D structures of ligands were sketched and minimized under Molecular Mechanics MM+ and then Semiempirical AM1 methods using HyperChem software. A grid of 52, 52, and 52 points in x, y, and z directions with 0.375 Å grid spacing was built and the center was placed on the binding site of the protein’s natural ligand (PI-103). To define the docking parameter file, we chose rigid macromolecule and Lamarckian Genetic Algorithm (LGA). The number of GA runs was set to 100 and other parameters were left as default. Docking process and visualizing of docking results were performed by AutoDock 4.2. and ViewerLite50, respectively. Validity of the docking procedure was examined using co-crystallized inhibitor as ligand and above-mentioned protocol.
RESULTS
Synthesis
The synthetic reactions used for the synthesis of 1,2,4-triazine derivatives (S1-S10) are illustrated in Scheme 1. 2-Hydroxy-1,2-bis (4-methoxyphenyl) ethanone (B) was prepared by thiamine hydrochloride-mediated coupling of 4-methoxy benzaldehyde (A). 1,2-bis (4- methoxyphenyl) ethane-1,2-dione (C) was obtained by oxidation of compound B. Cyclization of compound (C) with thiosemicarbazide afforded 5,6-diaryl-1,2,4- triazine-3-thiol (D). Compound (D) was treated with methyl iodide to afford the corresponding methylthio compound (E). In the presence of excess amount of hydrazine hydrate, compound (F) was acquired. The final compounds (S1–S10) were prepared by treatment of (F) with equimolar amounts of the corresponding aldehydes.
Scheme 1.
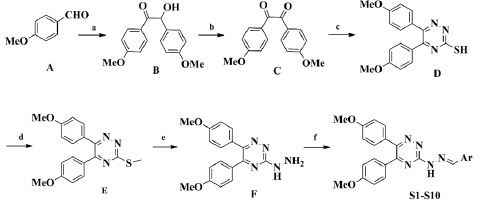
Synthesis pathways for preparation of triazine-hydrazone derivatives. Reagents and conditions: (A) thiamine hydrochloride, NaOH, EtOH, reflux, 3 h; (B) HNO3, 60 °C; (C) thiosemicarbazide, CH3COOH, reflux, 3 h; (D) CH3I, NaOH, EtOH, 2 h, rt; (E) NH2NH2.H2O, EtOH, reflux, 12 h; (F) ArCHO, EtOH, 1–10 h, rt.
Biological evaluation
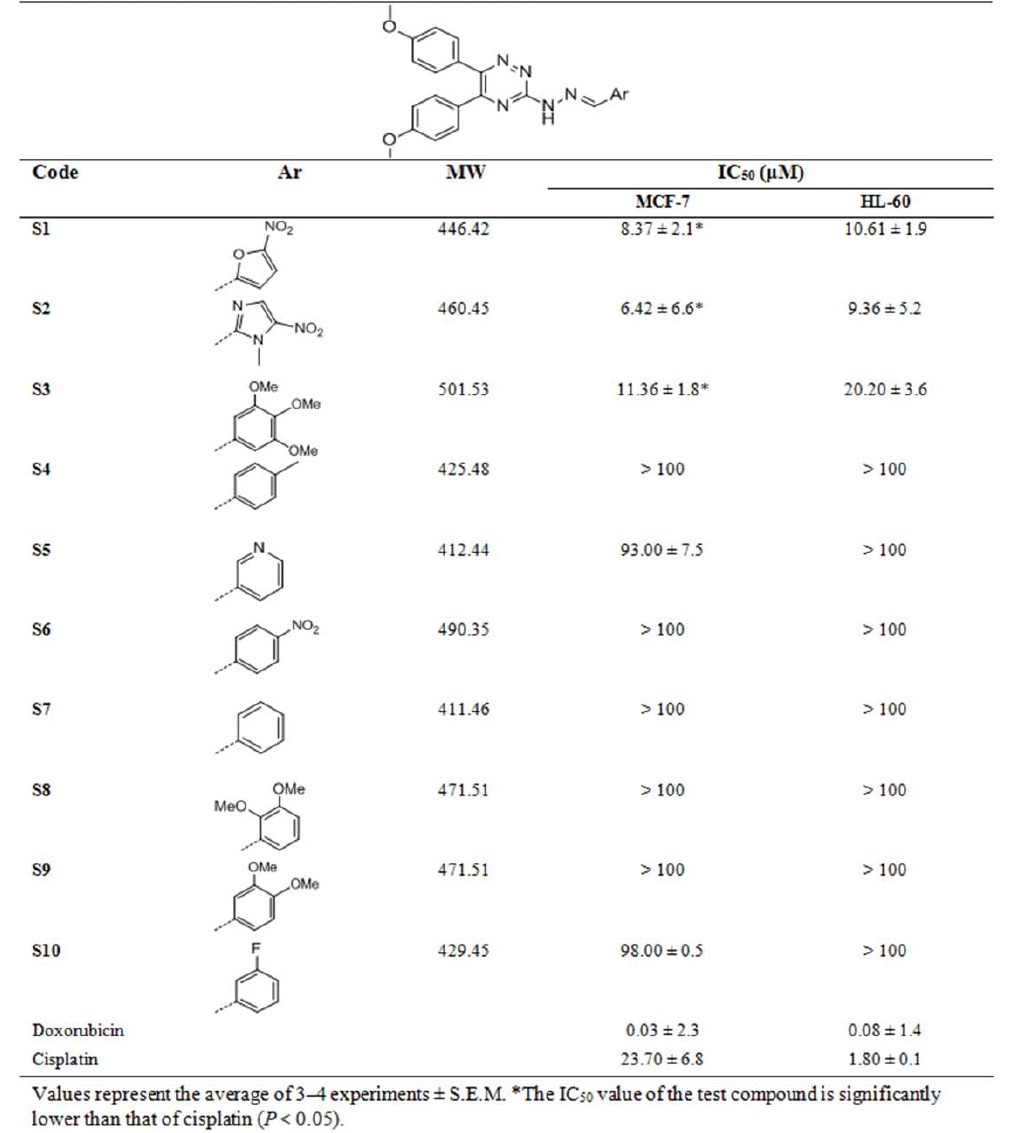
The cytotoxicity of the synthesized compounds was evaluated on HL-60 and MCF-7 cell lines using MTT assay and the results are demonstrated in Table 1. S1 (Ar: 5- nitrofuran-2-yl), S2 (Ar: 1-methyl-5-nitroimidazole-2-yl), and S3 (Ar: 3,4,5-trimethoxyphenyl) are the active compounds with IC50 values of 8.37 ± 2.1, 6.42 ± 6.6 and 11.36 ± 1.8 μM, respectively on MCF-7 cell line. The same results were obtained for HL-60 cell line as S1, S2 and S3 compounds showed good cytotoxicity on this cell line with IC50 values of 10.61 ± 1.9, 9.36 ± 5.2 and 20.2 ± 3.6 μM, respectively.
Table 1.
Structure and cytotoxicity of synthesized compounds.
Molecular docking study
Validation of molecular docking was done by extracting the structure of the co-crystallized ligand and re-docking it into the receptor (self-docking). The root mean square deviation (RMSD) between the best pose of co-crystallized ligand docked into the binding site of mTOR and the one in the crystal structure was 0.355 Å (Fig. 2). The molecular docking analysis of S1-S10 revealed that among all the docked compounds, compound S1, containing 5-nitrofuryl group, possessed the lowest estimated free energy of binding (-10.59 kcal/mol) and estimated inhibition constant (17.30 nM) (Table. 2). S1 is well accumulated in the binding pocket of mTOR by hydrogen bonds, Pi-H and Pi-Pi interactions. Oxygen atom of NO2 group on furan ring made a strong hydrogen bonding with side chain N-H of Lys2171 and nitrogen atom of hydrazine moiety formed a hydrogen bond interaction with backbone N-H of Cys2243. Triazine ring made a hydrophobic interaction with Trp2239 and phenyl ring involved in Pi-H interaction with Ile2356 (Fig. 3).
Fig. 2.
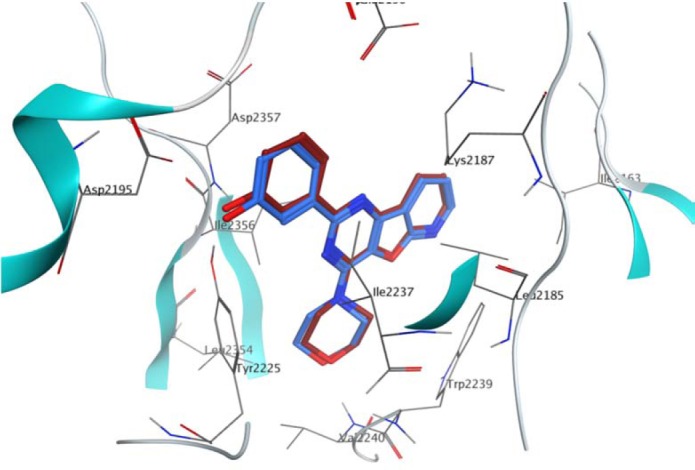
Representation of the co-crystallized inhibitor (blue) docked into the binding site and superimposed on co-crystallized inhibitor (red) in the crystal structure of mTOR (PDB ID: 4JT6).
Table 2.
Interaction data of synthesized derivatives and the innate ligand with mTOR (PDB ID: 4JT6).
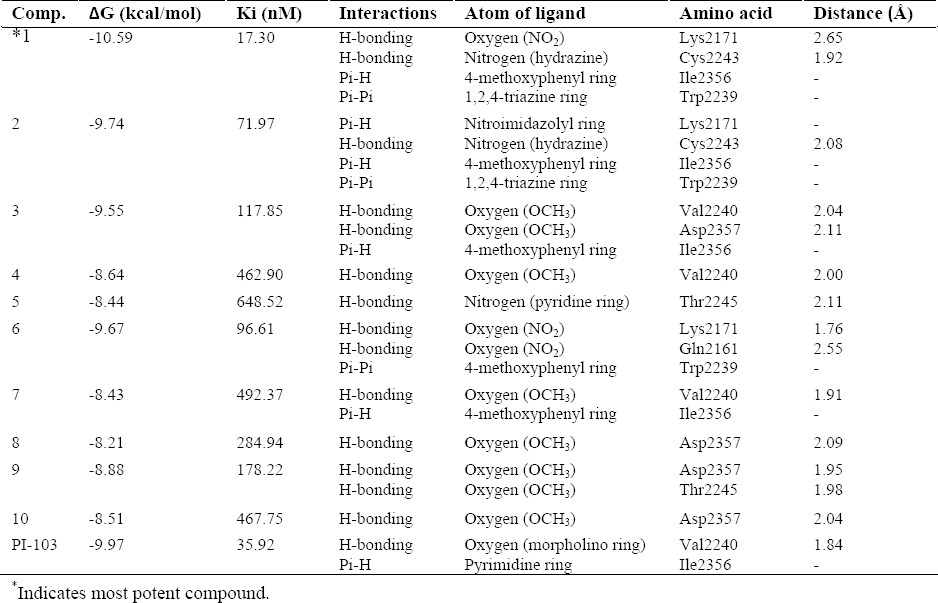
Fig. 3.

(A) The binding orientation of compound S1 within the active site of mTOR. (B) Molecular docking of S1 within the active site of mTOR. The amino acids Lys2171 and Cys2243 make hydrogen bond with compound S1 while Trp2239 and Ile2356 involve in Pi-Pi and H-Pi interactions with compound S1, respectively.
Compound S2, having 1-methyl-5- nitroimidazolyl moiety, demonstrated the second best estimated free energy of binding (9.74 kcal/mol) and estimated inhibition constant (71.97 nM) (Table. 2). As shown in Fig. 4A, S1 and S2 had the same binding orientations and the triazine-hydrazone part of the compounds made the same interactions with the receptor. Compound S6, bearing 4-nitrophenyl, showed the estimated free energy of binding of -9.67 kcal/mol and estimated inhibition constant (Ki) of 96.61 nM. As illustrated in Fig. 4A, the binding orientation of S6 is a little different from that of S1 and S2. Thus, for compound S6, hydrazone moiety did not have any interaction with the receptor, while methoxyphenyl ring involved in a hydrophobic interaction with Trp2239 and NO2 formed two hydrogen bonds with Lys2171 and Gln2161.
Figure 4.

(A) Dock poses of compounds S1 (yellow), S2 (green) and S6 (purple). (B) Dock poses of compounds S3 (purple), S4 (yellow) and S5 (green) in the active site of mTOR. Different binding orientations of compounds with lower estimated free energy of binding (S1, S2 and S6) and some compounds with higher estimated free energy of binding (S3, S4 and S5) are illustrated in this
S3 (bearing 3,4,5-trimethoxyphenyl), S4 (bearing 4-methylphenyl), S5 (bearing pyridinyl), S7 (bearing phenyl), S8 (bearing 2,3-dimethoxyphenyl), S9 (bearing 3,4-dimethoxyphenyl) and S10 (bearing 3-fluorophenyl) with the highest estimated free energies of binding (Table 2) orientated in the mTOR active site with the same binding modes. Binding orientation of some of these compounds is illustrated in Fig. 4B.
DISCUSSION
Compounds containing 5-nitrofuran-2-yl (S1), 1-methyl-5-nitro-imidazole-2-yl (S2) and trimethoxyphenyl (S3) moieties showed good cytotoxicity. Consequently, the cytotoxic activity is dependent on the nature of arylidene-hydrazinyl unite in 5,6-bis (4-methoxyphenyl)-1,2,4-triazine scaffold.
Molecular docking analysis results indicated that arylidene-hydrazinyl moiety played an important role in binding orientations of 5,6-bis(4-methoxyphenyl)-1,2,4-triazines, and the presence of a five-membered heterocyclic ring with nitro substitution (compounds S1 and S2) could lead to more favorable interactions and better orientations than a six-membered ring.
Therefore, it could be suggested that 3-(2-arylidenehydrazinyl) 5,6-bis (4-methoxyphenyl)-1,2,4-triazine analogs be introduced as cytotoxic agents and potential mTOR inhibitors; however, complementary biological evaluations will be the subject of future studies.
CONCLUSION
3- (2- Arylidenehydrazinyl) 5,6- bis (4-methoxyphenyl)-1,2,4-triazine derivatives have been synthesized and evaluated for their cytotoxic activity against two cell lines including HL-60 and MCF-7. Compounds S1, S2 and S3 were found to be active compounds with favorable cytotoxicity against both cell lines. Docking study revealed that these compounds were placed in the active site of mTOR with hydrogen bonding and Pi interactions. Additionally, nitro substitution on arylidene-hydrazinyl moiety played an important role in drug-receptor interactions and compounds with five-membered heterocyclic rings, S1 and S2, showed the lowest estimated free energy of binding and estimated inhibition constant. Therefore, S1 and S2 could be proposed as effective cytotoxic agents and potential mTOR inhibitors. However, further biological evaluations are necessary to confirm our findings.
ACKNOWLEDGEMENTS
This project was financially supported by the vice chancellery of Tehran University of Medical Sciences, Tehran, I.R. Iran.
REFERENCES
- 1.Mathers CD, Loncar D. Updated projections of global mortality and burden of disease, 2002-2030: data sources, methods and results. Geneva: World Health Organization; 2005. [Google Scholar]
- 2.Hosseinzadeh L, Aliabadi A, Kalantari M, Mostafavi A, Khajouei MR. Synthesis and cytotoxicity evaluation of some new 6-nitro derivatives of thiazole-containing 4-(3H)-quinazolinone. Res Pharm Sci. 2016;11:210–218. [PMC free article] [PubMed] [Google Scholar]
- 3.Sarvmeili N, Jafarian-Dehkordi A, Zolfaghari B. Cytotoxic effects of Pinus eldarica essential oil and extracts on HeLa and MCF-7 cell lines. Res Pharm Sci. 2016;11:476–483. doi: 10.4103/1735-5362.194887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abnous K, Manavi H, Mehri S, Alibolandi M, Kamali H, Ghandadi M, et al. In vitro evaluation of dihydropyridine-3-carbonitriles as potential cytotoxic agents through PIM-1 protein kinase inhibition. Res Pharm Sci. 2017;12:196–203. doi: 10.4103/1735-5362.207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vosooghi M, Yahyavi H, Divsalar K, Shamsa H, Kheirollahi A, Safavi M, et al. Synthesis and In vitro cytotoxic activity evaluation of (E)-16-(substituted benzylidene) derivatives of dehydroepiandrosterone. DARU. 2013;21:34. doi: 10.1186/2008-2231-21-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- 7.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci. 2015;36:124–135. doi: 10.1016/j.tips.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 10.Zhan P, Li X, Li Z, Chen X, Tian Y, Chen W, et al. Structure-based bioisosterism design, synthesis and biological evaluation of novel 1,2,4-triazin-6- ylthioacetamides as potent HIV-1 NNRTIs. BioorgMed Chem Lett. 2012;22:7155–7162. doi: 10.1016/j.bmcl.2012.09.062. [DOI] [PubMed] [Google Scholar]
- 11.Ashour HM, Shaaban OG, Rizk OH, El-Ashmawy IM. Synthesis and biological evaluation of thieno [2′, 3′: 4, 5] pyrimido [1, 2-b][1,2,4] triazines and thieno [2, 3-d][1,2,4] triazolo [1, 5-a] pyrimidines as anti-inflammatory and analgesic agents. Eur J Med Chem. 2013;62:341–351. doi: 10.1016/j.ejmech.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Sangshetti JN, Shinde DB. One pot synthesis and SAR of some novel 3-substituted 5, 6-diphenyl-1,2,4-triazines as antifungal agents. Bioorg Med Chem Let. 2010;20:742–745. doi: 10.1016/j.bmcl.2009.11.048. [DOI] [PubMed] [Google Scholar]
- 13.El-Sayed Ali T. Synthesis of some novel pyrazolo [3, 4-b] pyridine and pyrazolo [3, 4-d] pyrimidine derivatives bearing 5, 6-diphenyl-1,2,4-triazine moiety as potential antimicrobial agents. Eur J Med Chem. 2009;44:4385–4392. doi: 10.1016/j.ejmech.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 14.Irannejad H, Amini M, Khodagholi F, Ansari N, Tusi SK, Sharifzadeh M, et al. Synthesis and in vitro evaluation of novel 1,2,4-triazine derivatives as neuroprotective agents. Bioorg Med Chem. 2010;18:4224–4230. doi: 10.1016/j.bmc.2010.04.097. [DOI] [PubMed] [Google Scholar]
- 15.Shaikh BM, Chobe SS, Konda SG, Khandare NT, Chavan SA, Dawane BS. An efficient synthesis and in vitro antimicrobial activity of 1,2,4-triazin-6- (5H)-one derivatives. Der Chemica Sinica. 2010;1:86–91. [Google Scholar]
- 16.Bakharev V, Gidaspov A, Yakunina N, Bulychev YN. Synthesis and cytotoxic activity of trinitromethyl derivatives of 1, 3, 5-triazine. Pharm Chem J. 2008;42:241–244. [Google Scholar]
- 17.Sztanke K, Rzymowska J, Niemczyk M, Dybala I, Koziol AE. Synthesis, crystal structure and anticancer activity of novel derivatives of ethyl 1-(4-oxo-8-aryl-4, 6, 7, 8-tetrahydroimidazo [2, 1-c][1,2,4] triazin-3- yl) formate. J Med Chem. 2006;41:539–547. doi: 10.1016/j.ejmech.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 18.Gucký T, Fryšová I, Slouka J, HaJdύch M, Džubák P. Cyclocondensation reaction of heterocyclic carbonyl compounds, Part XIII: Synthesis and cytotoxic activity of some 3, 7-diaryl-5-(3,4,5- trimethoxyphenyl) pyrazolo [4, 3-e][1,2,4] triazines. Eur J Med Chem. 2009;44:891–900. doi: 10.1016/j.ejmech.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 19.Yurttaş L, Demirayak S, Ilgın S, Atlı Ö. In vitro antitumor activity evaluation of some 1,2,4-triazine derivatives bearing piperazine amide moiety against breast cancer cells. Bioorg Med Chem. 2014;22:6313–6323. doi: 10.1016/j.bmc.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Karczmarzyk Z, Wysocki W, Urbańczyk-Llpkowska Z, Kalicki P, Bielawska A, Bielawski K, et al. Synthetic Approaches for Sulfur Derivatives Containing 1,2,4-Triazine Moiety: Their Activity for in Vitro Screening towards Two Human Cancer Cell Lines. Chem Pharm Bull. 2015;63:531–537. doi: 10.1248/cpb.c15-00153. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Fryknäs M, Hernlund E, Fayad W, De Milito A, Olofsson MH, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun. 2014;5:3295. doi: 10.1038/ncomms4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutherlin DP, Sampath D, Berry M, Castanedo G, Chang Z, Chuckowree I, et al. Discovery of (thienopyrimidin-2-yl) aminopyrimidines as potent, selective, and orally available pan-PI3-kinase and dual pan-PI3-kinase/mTOR inhibitors for the treatment of cancer. J Med Chem. 2010;53:1086–1097. doi: 10.1021/jm901284w. [DOI] [PubMed] [Google Scholar]
- 23.Zhu W, Chen C, Sun C, Xu S, Wu C, Lei F, et al. Design, synthesis and docking studies of novel thienopyrimidine derivatives bearing chromone moiety as mTOR/PI3Ka inhibitors. Eur J Med Chem. 2015;93:64–73. doi: 10.1016/j.ejmech.2015.01.061. [DOI] [PubMed] [Google Scholar]
- 24.Menear KA, Gomez S, Malagu K, Bailey C, Blackburn K, Cockcroft X-L, et al. Identification and optimisation of novel and selective small molecular weight kinase inhibitors of mTOR. Bioorg Med Chem Let. 2009;19:5898–5901. doi: 10.1016/j.bmcl.2009.08.069. [DOI] [PubMed] [Google Scholar]
- 25.Khoshneviszadeh M, Ghahremani MH, Foroumadi A, Miri R, Firuzi O, Madadkar-Sobhani A, et al. Design, synthesis and biological evaluation of novel anti-cytokine 1,2,4-triazine derivatives. Bioorg Med Chem. 2013;21:6708–6017. doi: 10.1016/j.bmc.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Bag S, Vaze VV, Degani MS. Microwave assisted benzoin condensation using thiamine as catalyst. J Chem Res. 2006;2006:267–269. [Google Scholar]