

Abstract
Diaryl urea derivatives have exhibited a broad spectrum of biochemical effects and pharmaceutical applications. Several diaryl urea derivatives such as sorafenib, regorafenib, linifanib, and tivozanib and lenvatinib are in clinical trial or clinical use. Therefore, development of small molecules within the diaryl urea scaffold with the ability of binding to variety of enzymes and receptors in the biological system are an interesting topic for researchers. Sorafenib as a diaryl urea derivative is a well-known anticancer agent. Corresponding to available information about biological activities of quinoxaline moieties, based on sorafenib scaffold, several structures were designed by replacement of pyridyl carboxamide group of sorafenib with quinoxalindione moiety. A total of 14 novel compounds in 7 synthetic steps were synthesized. Briefly, the amino group of p-aminophenol was first protected followed by O-arylation of 4-acetamidophenol with 5-chloro-2-nitroaniline to provide 5-(4-acetamidophenoxy)-2-nitroaniline. Reduction of the nitro group of 5-(4-acetamidophenoxy)-2-nitroaniline and cyclization of diamine N-(4-(3,4-diaminophenoxy) phenyl) acetamides with oxalic acid afforded compound N-(4-((2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)oxy)phenyl) acetamides which on deacetylation gave compounds 6-(4-aminophenoxy) quinoxaline-2,3 (1H, 4H)-diones. Then resultant compounds, 6-(4-aminophenoxy) quinoxaline-2,3 (1H, 4H)-diones were reacted by appropriate isocyanates/ carbamates to give the target compounds 1-(4-((2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)oxy)phenyl)-3-phenylureas. The structures of compounds confirmed by proton nuclear magnetic resonance (1H NMR), mass spectrum and Fourier transform infrared (FT-IR).
Keywords: Diaryl urea, Quinoxalindione, Sorafenib derivatives
INTRODUCTION
Diaryl urea derivatives have broad spectrum of biochemical effects and pharmaceutical applications. In recent years, there have been intense research focused on the design and development of diaryl urea derivatives as a class of antitumor agents (1,2,3). There are several diaryl urea derivatives that are on clinical trial or have been used clinically such as sorafenib, regorafenib, linifanib, tivozanib and lenvatinib (Fig. 1). Sorafenib (Nexavar®) is the first diaryl urea derivatives that target the RAS-RAF-MEK-ERK signaling pathway in numerous cancer cell lines in vitro. Sorafenib was approved by the U.S. Food and Drug Administration (FDA) for the treatment of advanced renal cell carcinoma in 2005 and unresectable hepatocellular carcinoma in 2007 (4,5). Regorafenib (Stivarga®) approved by FDA for the treatment of colorectal cancer in 2012 and gastrointestinal stromal tumors in 2013.
Fig. 1.
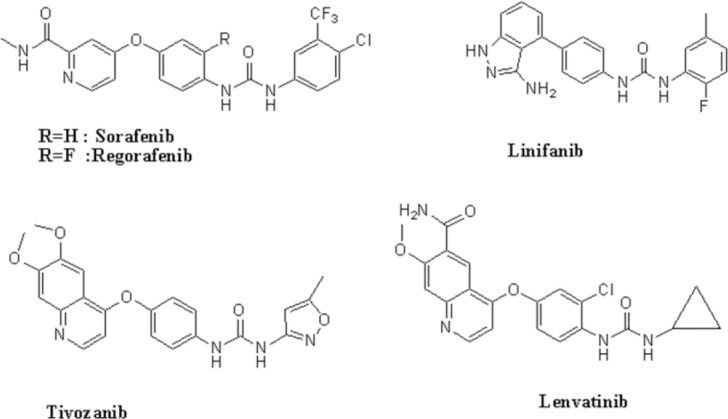
Some diaryl urea derivatives that have been in clinical trial or clinical use.
In 2015, lenvatinib (Lenvima®; Eisai®), an oral, multireceptor tyrosine kinase inhibitor, was approved by FDA to treat patients with locally recurrent or metastatic, progressive, radioactive iodine–refractory differentiated thyroid cancer (6). Tivozanib (AV-951®) an orally bioavailable quinoline-urea derivative inhibitor of vascular endothelial growth factor receptors has demonstrated safety and efficacy for patients with advanced or metastatic renal cell carcinoma (7). Linifanib (ABT-869®) shows significant clinical activity as monotherapy in patients with advanced hepatocellular carcinoma (8). Nowadays development of small molecules within the diaryl urea scaffold, with the binding ability to a variety of enzymes and receptors in the biological systems is a topic of interest for researchers (9,10,11,12,13).
In this study, we focused on modification of pyridyl ring of sorafenib by replacing of this group with quinoxalindione moiety (Fig. 2) according to the structure activity relationship studies (SAR). Nitrogen atoms in the pyridyl carboxylamide moiety of sorafenib are essential for binding to ATP-binding pocket of kinase. Quinoxalindione is an aromatic heterocycle containing two nitrogen atoms in its structure which is comparable with pyridyl carboxamide moiety in terms of size and polarity. Presence of amide in this structure could impart susceptibility to act at target sites of sorafenib. Based on previous SAR of sorafenib analogs, rigidity is beneficial for improvement of antitumor activity of these derivatives; for instance, pyridoimidazolone and pyridopyrazinone analogs of sorafenib showed enhanced inhibitory potency toward B-Raf inhibition and cellular assay (14,15). Our previous quantitative SAR (QSAR) model studies on a series of diaryl urea derivatives revealed that rigidity and aromaticity are two important factors to improve activity for these derivatives. Therefore, quinoxalindione as a bicyclic heterocycle with six membered rigid ring, can afford rigidity in sorafenib framework (16,17,18,19,20,21,22). Quinoxaline and its derivatives showed a wide range of applications in medicine due to their biological activities, which include antimicrobial, antiproliferative, and anticancer activities (23,24). Thus significant biological activities of quinoxalindione derivatives makes it a good fragment in drug design (16,17,18,19,20,21,22). In this work, a total of 14 novel diarylurea derivatives were designed and their successful synthesis were confirmed by proton nuclear magnetic resonance (1H NMR), mass spectroscopy and Fourier transform infrared (FT-IR).
Fig. 2.

The structure of sorafenib and the target compounds 15a-15g and 16a-16g.
MATERIALS AND METHODS
All chemicals used for the synthesis of the compounds were purchased from Merck or Sigma (Germany). The purity of the compounds was investigated by thin layer chromatography (TLC) on silica gel plate using appropriate solvents. All reactions were monitored by TLC as described. Melting points were determined with an electro thermal 9200 capillary melting point apparatus (Germany). The IR spectra were recorded with a WQF-510 ratio recording FTIR spectrometer (China) using a KBr disc (γ, cm-1). The 1H NMR spectra were recorded using a Brucker 400 MHz spectrometer (Germany) and (D6) dimethyl sulfoxide (DMSO) or deuterated chloroform (CDCl3) were used as solvents, and chemical shifts (δ) were expressed in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard. Splitting patterns are as follows: s, singlet; d, doublet; m, multiplet; b, broad; dd (doublet of doublet.) Mass spectra were acquired with a Platform II Mass Spectrometer from Micro mass (Shimadzu, Japan). Electron impact ionization was performed at an ionizing energy of 70 ev.
RESULTS
Details of preparation procedures and chemistry of synthesized compounds
Synthetic route for the preparation of target compounds 15a-15g and 16a-16g are illustrated in Scheme 1 where the different starting materials 1 and 2 were utilized respectively and the details of synthesis provided in the following sections.(Scheme 1)
Scheme 1.
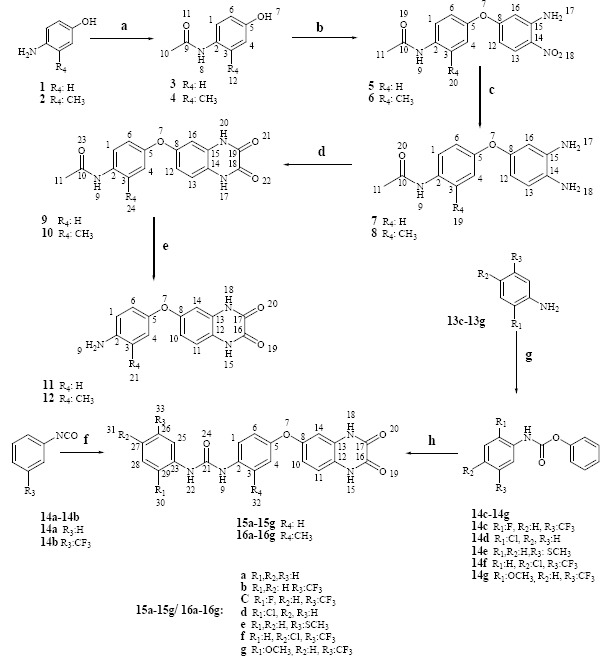
General procedure for the synthesis of the target compounds 15a-15g and 16a- 16g. Reagents and conditions: (a) acetic anhydride, H2O; (b) 5- chloro-2-nitroaniline, NaOH, DMSO; (c) Na2S2O4, EtOH; (d) oxalic acid, HCl; (e) EtOH, HCl; (f) amines 11- 12, DMSO; (g) phenyl chloroformate, tetrahydrofuran; (h) amines 11- 12, DMSO.
Synthesis of N-(4-hydroxyphenyl) acetamide (3) and N- (4-hydroxy-2-methylphenyl) acetamide (4)
A mixture of p-aminophenol (146 mmol, 16 g), H2O (43 mL) and acetic anhydride (18 mL) was heated at 100 °C until solid was dissolved, the mixture was heated for an additional 20 min to complete the reaction, the mixture was then cooled in ice bath. Crystals were collected by filtration and purified by recrystallization in ethanol to afford compound 3 (13 g, 60%) as a white solid, M.p. 170-171°C; 1H NMR (400 MHz, (D6)DMSO): δ 1.97 (s, 3H, Me10), 6.67 (d, J = 8.8 Hz, 2H, H6,4), 7.33 (d, J = 8.8 Hz, 2H, H1,3), 9.1 (s, 1 H, NH), 9.6 (s, 1 H, OH); FT-IR KBr (cm-1): 3320, 3176 (N–H), 1655 (C=O), 1566, 1507 (C=C), 1371 (C-N), 1222 (C-O).
Compound 4. white solid, yield: 70%, M.p. 165-167 °C; 1H NMR (400 MHz, (D6) DMSO): δ 1.97 (s, 3H, Me10), 2.07 (s, 3H, Me12), 6.52 (dd, J = 4 Hz and 8 Hz, 1H, H6), 6.56 (d, J = 4 Hz, 1H, H4), 7.01 (d, J = 8 Hz, 1H, H1), 9.0 (s, 1H, NH), 9.1 (s, 1H, OH)
Synthesis of N-(4-(3-amino-4-nitrophenoxy) phenyl) acetamide (5) and N-(4-(3-amino-4-nitrophenoxy)-2-methylphenyl) acetamide (6)
N-(4-hydroxyphenyl) acetamide 3 (30 mmol, 4.53 g) was added to a solution of NaOH (30 mmol, 1.2 g) in H2O (2 mL) and DMSO (11 mL) and the mixture was stirred for 30 min. Then 5-chloro-2-nitroaniline (50 mmol, 9.06 g) was added and heated at 120 °C overnight. After cooling 500 mL of water was added and the resulting precipitates were collected by filtration and the filtrate was recrystallized with ethanol to give compound 5 (6.75 g, 78%) as yellow solid, M.p. 214-216 °C; 1H NMR (400 MHz, (D6) DMSO): 2.05 (s, 3H, Me11), 6.28 (m, 2H, H12,16), 7.11 (d, J = 8.8 Hz, 2H, H4,6), 7.48 (b, 2H, NH2), 7.66 (d, J = 8.8 Hz, 2H, H1,3), 7.99 (d, J = 9.6 Hz, 1 H, H13), 10.06 (s, 1 H, NH); FT-IR KBr (cm-1): 3458, 3332 (N–H), 3147 (C–H, aromatic), 1664 (C=O), 1572, 1333 (NO2), 1506 (C=C), 1340(C-N), 1234, 1213(C-O).
Compound 6. yellow solid, yield: 72%, M.p. 170-172 °C; 1H NMR (400 MHz, (D6) DMSO): 2.13 (s, 3H, Me11), 2.27 (s, 3H, Me20), 6.35 (dd, J = 2.4 Hz and 9.2 Hz, 1H, H12), 6.40 (d, J = 2.8 Hz, 1H, H16), 7.01 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H6), 7.09 (d, J = 2.4 Hz, 1H, H16), 7.53 (d, J = 8.8 Hz, 1H, H1), 7.57 (s, 2H, NH2), 8.05 (d, J = 9.2 Hz, 1H, H13), 9.49 (s, 1H, NH)
Synthesis of N-(4-(3, 4-diaminophenoxy) phenyl) acetamide (7) and N-(4-(3, 4-diaminophenoxy)-2-methylphenyl) acetamide (8)
Na2S2O4 (70 mmol, 12.4 g) was added to a solution of N-(4-(3-amino-4-nitrophenoxy) phenyl) acetamide 5 (23.5 mmol, 6.75 g) in EtOH (200 mL) and H2O (70 mL) and the mixture was refluxed with vigorous stirring until the yellow color of mixture changed to brown. After cooling to room temperature, the mixture was extracted with water and ethyl acetate (EtOAc). The EtOAc layer was dried over MgSO4, filtered, and evaporated to give compound 7 (4.3 g, 71%) as a brown powder, M.p. 187-190 °C; 1H NMR (400 MHz, (D6) DMSO): 2.06 (s, 3H, Me11), 6.12 (dd, J = 2.8 Hz and 8.4 Hz, 1H, H12), 6.27 (d, J = 2.8 Hz, 1H, H16), 6.54 (d, J = 8.4 Hz, 1H, H13),6.87 (d, J = 8.8 Hz, 2H, H4,6), 7.53 (d, J = 8.8 Hz, 2H, H1,3), 9.89 (s, 1H, H9); FT-IR KBr (cm-1) : 3321, 3151 (N–H), 3045 (C–H, aromatic), 1685, 1637 (C=O), 1504 (C=C), 1265, 1211 (C-O).
Compound 8. Brown powder, yield: 75%, M.p. 187-190 °C; 1H NMR (400 MHz, (D6) DMSO): δ 2.07 (s, 3H Me11), 2.16 (s, 3H, Me19), 6.13 (dd, J = 2.8 Hz and 8.4 Hz, 1H, H12), 6.28 (d, J = 2.8 Hz, 1H, H16), 6.54 (d, J = 8.4 Hz, 1H, H13), 6.70 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H6), 6.76 (d, J = 2.8 Hz, 1H, H4), 7.24 (d, J = 8.8 Hz, 1H, H1), 9.27 (s, 1H, NH)
Synthesis of N-(4-(1, 2, 3, 4-tetrahydro-2,3-dioxoquinoxalin-6-yloxy) phenyl) acetamide (9) and N-(4-(1, 2, 3, 4-tetrahydro-2,3-dioxo-quinoxalin-6-yloxy)-2-methylphenyl) acetamide (10)
A solution of oxalic acid dihydrate (14 mmol, 1764 mg) in HCl (25 mL, 0.5 M) was heated to 100 °C, followed by addition of diamine 7 (14 mmol, 3598 mg) with stirring at 100 °C for 3-4 h. Completion of the reaction was confirmed by TLC. The mixture was cooled by addition of ice. The precipitate formed was washed with water to give compound 9 (3500 mg, 80%) as gray powder, M.p. > 250 °C; 1H NMR (400 MHz, (D6) DMSO): δ 2.03 (s, 3H, Me11), 6.71 (d, J = 2.4 Hz, 1H, H16), 6.74 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H12), 6.97 (d, J = 8.8 Hz, 2H, H4,6), 7.11 (d, J = 8.8 Hz, 1H, H13), 7.59 (dd, J = 1.6 Hz and 8.8 Hz, 2H, H1,3), 9.98 (s, 1H, NH9), 11.80 (s, 1H, NH), 11.90 (s, 1H, NH); FT-IR KBr (cm-1) : 3458, 3332 (N–H), 3147 (C–H, aromatic), 1718 (C=O), 1560, 1493 (C=C), 1340 (C-N), 1230 (C-O).
Compound 10: gray powder, yield: 85%, M.p. > 250 °C, 1H NMR (400 MHz, (D6) DMSO): δ 2.1 (s, 3H, Me11), 2.22 (s, 3H, Me24), 6.8 (d, J = 2.4 Hz, 1H, H16), 6.85 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H6,12), 6.93 (d, J = 2.4 Hz, 1H, H4), 7.18 (d, J = 8.8 Hz, 1H, H13), 7.40 (d, J = 8.8 Hz, 1H, H1), 9.35 (s, 1H, NH9), 11.88 (s, 1H, NH), 11.97 (s, 1H, NH).
Synthesis of 6-(4-aminophenoxy) quinoxaline-2, 3(1H, 4H)-dione (11) and 6-(4-amino-3-methylphenoxy) quinoxaline-2, 3 (1H, 4H)-dione (12)
Solution of N-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxoquinoxalin-6-yloxy) phenyl) acetamide 9 (10 mmol, 311 mg) in HCl/ ethanol (3M, 50 mL) was refluxed for 24 h. After cooling, aq NH3 (30%) was added until mixture reached to basic pH. Then the precipitate formed was collected by filtration and dried in vacuo to give compound 10 (1.6 g, 60%) as brown powder, M.p. > 250 °C; 1H NMR (400 MHz, (D6) DMSO): 5.34 (b, 2H, NH2), 6.75 (m, 3H, H1,3,14), 6.82 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H10), 6.90(d, J = 8.8 Hz, 2H, H4,6), 7.20 (d, J = 8.8 Hz, 1H, H11), 11.88 (s, 1H, NH), 11.98 (s, 1H, NH); FT-IR KBr (cm-1): 3327, 3286 (N–H), 3035 (C–H, aromatic), 1720 (C=O), 1504 (C=C), 1255(C-O).
Compound 11. Brown powder, yield: 55%, M.p. > 250 °C, 1H NMR (400 MHz, (D6) DMSO): 2.1 (s, 3H, Me21), 4.83 (b, 2H, NH2), 6.68 (m, 3H, H1,4,6), 6.74 (m, 2H, H10,14), 7.06 (d, 1H, H11), 11.77 (s, 1H, NH), 11.89 (s, 1H, NH)
General procedure for preparation of substituted aromatic carbamate (14c-14g)
To a solution of aniline 13c-13g (1 mmol) in anhydrous tetrahydrofuran (THF) (9 mL) at 0 °C, pyridine (1.3 mmol, 108 μL) was added, followed by phenyl chloroformate (1.2 mmol, 154 μL). The mixture was kept at 0 °C for 15 min, then allowed to warm at room temperature and stirred for 24 h. The solvent was evaporated and the residue poured in EtOA (15 mL).
The suspension was washed with HCl (15 mL, 1 M), water (15 mL), saturated aqueous NaHCO3 (15 mL) and brine (15 mL). The organic layer was dried over MgSO4 and evaporated. The solid obtained was washed with diethyl ether to afford the products 14c-14g as fallow:
Phenyl 2-jluoro-5-(trifluoromethyl) phenyl-carbamate (14c)
Compound 14c was obtained from 2-fluoro-5-(trifluoromethyl) aniline (8 mmol,1 mL) as a white solid (830 mg, 34%), M.p. 90-91°C. 1H NMR (400 MHz, (D6) DMSO): 7.15 (m, 2H), 7.20 (m, 3H), 7.26 (m, 1H), 7.28 (m, 1H), 7.35 (m, 2H), 8.4 (s, brd, 1H); FT-IR KBr (cm-1): 3251 (N–H), 3049 (C–H, aromatic), 1718 (C=O), 1560, 1493 (C=C), 1340 (C-N), 1230 (C-O).
Phenyl 2-chlorophenylcarbamate (14d)
Compound 14d was obtained from 2-chloroaniline (11.1 mmol, 1 mL) as a white solid (420 mg, 15%), M.p. 53-55 °C. 1H NMR (400 MHz, (CDCL3): 6.97 (m, 1H), 7.20 (m, 4H), 7.34(m, 3H), 8.13 (s, brd, 1H), FT-IR KBr (cm-1): 3418 (N–H), 3064 (C–H, aromatic), 1765 (C=O), 1598, 1490 (C=C), 1299 (C-N), 1186 (C-O).
Phenyl 5-(trifluoromethyl)-2-methoxyphenyl-carbamate (14g)
Compound 14g was obtained from 5-(trifluoromethyl)-2-methoxyaniline (6 mmol, 1.146 g) as a white solid (464 mg, 26%), M.p. 103-105 °C. 1H NMR (400 MHz, (CDCL3): 6.88 (d, J = 8.4 Hz, 2H), 7.13 (m, 2H), 7.19 (m, 2H), 7.28 (m, 1H), 7.33 (m, 1H), 8.39 (s, b, 1H); FT-IR KBr (cm-1) : 3431 (N–H), 3018 (C–H, aromatic), 1749 (C=O), 1541, 1473 (C=C), 1327 (C-N), 1128 (C-O).
Phenyl 4-chloro-3-(trifluoromethyl) phenyl-carbamate (14f)
Compound 14f was obtained from 4-chloro-3-(trifluoromethyl) aniline (800 mg, 4 mmol) as a white solid (445 mg, 40%), M.p.124-125 °C. 1H NMR (400 MHz, (CDCL3): 7.11 (m, 2H), 7.20 (m, 1H), 7.35 (m, 3H),7.54 (m,1H), 7.75 (m,1H); FT-IR KBr (cm-1): 3330 (N–H), 3060 (C–H, aromatic), 1741 (C=O), 1539, 1483 (C=C), 1327 (C-N), 1223 (C-O).
Phenyl 3-(methylthio) phenylcarbamate (14e)
Compound 14e was obtained from 2-chloroaniline (2 mmol, 246 μL) as white solid (207 mg, 40%), M.p. 81-82 °C. 1H NMR (400 MHz, (CDCL3): 6.91 (m, 2H), 7.04 (m, 1H), 7.14 (m, 4H), 7.28 (m, 1H), 7.33 (m, 2H), 7.40 (s, b, 1H); FT-IR KBr (cm-1): 3304 (N–H), 3056 (C–H, aromatic), 1714 (C=O), 1588, 1485 (C=C), 1327 (C-N), 1197 (C-O).
Synthesis and characterization of diaryl urea derivatives (15a-15g) and (16a-16g)
Diary lurea compounds 15a- 15b and 16a- 16b was synthesized from amine 11-12 and appropriate aromatic isocyanate, phenyl isocyanate (14a) or 3-(trifluoromethyl) phenyl isocyanate (14b) and diaryl urea compounds 15c- 15g and 16c-16g from amine 11-12 and appropriate aromatic carbamate 14c-14g that previously was described synthesis procedure’s.
General procedure for synthesis of diaryl urea compounds (15a- 15b) and (16a-16b)
For synthesis of compounds (15a-15b), solution of compound 11 (1 eq) in DMSO (2 mL) and corresponding aromatic isocyanate, 3-(trifluoromethyl) phenyl isocyanate or phenyl isocyanate 14a-14b (1eq) was stirred overnight at room temperature. The reaction mixture quenched by addition of water and the perticipitae was collected and washed with large amount of dichloromethane (DCM) and (EtOAc) to obtain the desired diarylurea compounds 15a- 15b as brown color powder.
Compounds (16a-16b) were synthesized from amine 12 according to procedure described for synthesis of compounds (15a-15b).
General procedure for synthesis of diarylurea compounds (15c- 15g) and (16c-16g)
For synthesis of compounds (15c-15g), solution of compound 11 (1 eq) in DMSO (2 mL) and appropriate aromatic carbamate 14c-14g (1 eq) was heated at 85 °C and stirred overnight. After cooling to room temperature the reaction mixture was quenched by addition of water and the precipitate was collected and washed with large amount of DCM and EtOAC to obtain the desired diary lurea compounds 15c-15g as brown color solids.
Compounds (16c-16g) was synthesized from amine 12 according to the procedure described for synthesis of compounds (15c-15g).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo-quinoxalin-7-yloxy) phenyl)-3-phenylurea (15a)
Brown powder, yield: 20%, M.p. > 300 °C. 1H NMR (400 MHz, (D6) DMSO): 6.88 (d, J = 2.4 Hz, 1H, H14), 6.92 (dd, J = 2.4 Hz and 8.4 Hz, 1H, 1H, H10), 7.20 (m, 3H, H5,6,27), 7.26 (d, J = 8.4 Hz, 1H, H11), 7.42 (t, J = 7.6 Hz, 2H, 2H, H26,28), 7.61(m, 4H, H1,3,25,23), 8.78 (s, 1H, H2), 8.82 (s, 1H, H22), 11.96 (d, J = 8.8 Hz, 2H, H15,18); FT-IR KBr (cm-1): 3280 (b, N–H), 3049 (C–H, aromatic), 1705, 1685 (C=O), 1599, 1500 (C=C), 1209 (C-O); EI-MS: 389.35 ([M + H] +m/z, C21H16N4O4; calc. 389.12).
1-(4 - (1, 2, 3, 4-tetrahydro-2,3-dioxo-quinoxalin -7-yloxy)- 2 - methylphenyl) - 3 - phenylurea (16a)
Brown powder, yield: 15%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 2.22(s, 3H, Me33), 6.73 (d, J = 2.8 Hz, 1H, H14), 6.77 (dd, J = 2.4 Hz and 8.4 Hz, 1H, H10), 6.82 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H6), 6.90 (d, J = 2.8 Hz, 1H, H4), 6.95 (t, J = 7.6 Hz, 1H, H27), 7.11 (d, J = 8.8 Hz, 1H, H11), 7.27 (t, J = 7.6 Hz, 2H, H26,28), 7.45 (d, J = 7.6 Hz, 2H, H23,25), 7.75 (d, J = 8.8 Hz, 1H, H1), 7.91 (s, 1H, H9), 8.94 (s, 1H, H22), 11.79 (s, 1H, H15), 11.88 (s, 1H, H18); FT-IR KBr (cm-1): 3288 (b, N–H stretching), 3053 (C–H, aromatic), 2941 (C–H, aliphatic), 1709, 1689 (C=O), 1597, 1495 (C=C), 1211 (C-O); EI-MS: 403.01 ([M + H] +m/z, C22H18N4O4; calc. 403.13).
(1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo-quinoxalin-7-yloxy) phenyl)-3-(3-(trifluoro-methyl) phenyl) urea (15b)
Brown powder, yield: 65%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 6.72 (d, J = 2.8 Hz, 1H, H14), 6.77 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H10), 6.98 (d, J = 9.2 Hz, 2H, H4,6),7.11 (d, J = 8.4 Hz, 1H, H11), 7.30 (d, J = 7.6 Hz, 1H, H29), 7.49 (m, 3H, H1,3,28), 7.57 (d, J = 8 Hz, 1H, H27), 8.01 (s, 1H, H25), 8.83 (s, 1H, H9), 9.04 (s, 1H, H22), 11.80 (s, 1H, H15), 11.89 (s, 1H, H18); FT-IR KBr (cm-1): 3288 (b, N–H), 3060 (C–H, aromatic), 1685 (C=O), 1556, 1502 (C=C), 1338 (C-N), 1120 (C-O).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo-quinoxalin-7-yloxy)-2-methylphenyl)-3-(3-(trifluoromethyl) phenyl) urea (16b)
Brown powder, yield: 72%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 2.22 (s, 3H, Me33), 6.74 (d, J = 2.8 Hz, 1H, H14), 6.78 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H10), 6.83 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H6), 6.90 (d, J = 2.4 Hz, 1H, H4), 7.11 (d, J = 8.8 Hz, 1H, H11), 7.29 (d, J = 7.2 Hz, 1H, H29), 7.52 (m, 2H, H1,28), 7.70 (d, J = 8.8 Hz, 1H, H27),8.02 (d, J = 2.4 Hz, 2H, H26, 9), 9.29 (s, 1H, H22), 11.80 (s, 1H, H15), 11.89 (s, 1H, H18); FT-IR KBr (cm-1): 3288, 3176 (N–H), 3060 (C–H, aromatic), 2947 (C–H, aliphatic), 1689 (C=O), 1560, 1493 (C=C), 1338(C-N), 1207 (C-O);
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo-quinoxalin-7-yloxy) phenyl)-3-(2-fluoro-5- (trifluoromethyl) phenyl) urea (15C)
Brown powder, yield: 73%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 6.72 (d, J = 2.4 Hz, 1H, H14), 6.77 (dd, J = 2.4 Hz and 8.4 Hz, 1H, H10), 6.98 (d, J = 9.2 Hz, 2H, H4,6), 7.11 (d, J = 8.8 Hz, 1H, H14), 7.38 (m, 1H, H25), 7.45 (m, 3H, H1,3,27), 8.6 (dd, J = 2 and 7.6, 1H, H28), 8.86 (s, 1H, H9), 9.2(s, 1H, H22),11.80 (s, 1H, H15), 11.89 (s, 1H, H18); FT-IR KBr (cm-1): 3300, 3168, 3111 (N–H), 3060 (C–H, aromatic), 1691 (C=O), 1560, 1504 (C=C), 1340 (C-N), 1215 (C-O); EI-MS: 473.1([M - H]- m/z, C22H14F4N4O4calc. 473.1).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo- quinoxalin-7-yloxy)-2 - methylphenyl) - 3 - (2- fluoro-5-(trifluoromethyl) phenyl) urea (16C)
Brown powder, yield: 73%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 2.23 (s, 3H, Me33), 6.73 (d, J = 2.4 Hz, 1H, H14), 6.77 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H10), 6.83 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H6), 6.91 (d, J = 2.8 Hz, 1H, H4),7.11 (d, J = 8.8 Hz, 1H, H11), 7.36 (m, 1H, H27), 7.49 (m, 1H, H28), 7.76 (d, J =8.8 Hz,1H, H1), 8.46 (s,1H, H9), 8.64 (dd, J = 2 Hz and 7.2 Hz, 1H, H25), 9.24 (d, J = 2.4, 1H, H22), 11.80 (s,1H, H15), 11.89 (s,1H, H18); FT-IR KBr (cm-1:) 3302 (b, N–H), 3064 (C–H, aromatic), 2951 (C–H, aliphatic), 1691 (C=O), 1558, 1496 (C=C), 1338 (C-N), 1217 (C-O).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo- quinoxalin -7- yloxy) phenyl)-3-(2-chloro- phenyl) urea (15d)
Brown powder, yield: 70%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 6.72 (d, J= 2.4 Hz, 1H, H14), 6.78 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H10), 7.02 (m, 3H, H4,6,11), 7.11 (d, J = 8.8 Hz, 1H, H28), 7.29 (t, J =7.2 Hz and 8.4 Hz, 1H, H26), 7.45 (m, 3H, H1,3,27), 8.16 (d, J = 8.4 Hz, 1H, H25), 8.27 (s, 1H, H9), 9.43 (s, 1H, H22), 11.79 (s, 1H, H15), 11.88 (s, 1H, H18). FT-IR KBr (cm-1) : 3288, 3188 (b, N–H), 3060 (C–H, aromatic), 1689 (C=O), 1593, 1500 (C=C, aromatic), 1338 (C-N), 1209 (C-O).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3- dioxoquinoxalin-7-yloxy)-2-methylphenyl)-3- (2-chlorophenyl) urea (16d)
Brown powder, yield: 90%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 2.24 (s, 3H, Me33), 6.74 (d, J = 2.8 Hz, 1H, H14), 6.78 (dd, J = 2.4 Hz and 8.4 Hz, 1H, H10), 6.82 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H6), 6.90 (d, J = 2.8 Hz, 1H, H4), 7.01 (t.d, J =2 Hz and 8Hz, 1H, H27), 7.11 (d, J = 8.8Hz, 1H, H11), 7.28 (t.d, J =2.4 Hz and 8.4 Hz, 1H, H26), 7.45 (dd, J = 2.4 Hz and 8 Hz, 1H, H28), 7.69 (d, J = 8.8 Hz, 1H,H1), 8.13 (dd, J = 1.6 Hz and 8.4 Hz, 1H, H25), 8.59 (s, 1H, H9), 8.62 (s, 1H, H22), 11.80 (s, 1H, H15), 11.88 (s, 1H, H18). FT-IR KBr (cm-1):3300, 3172 (b, N–H), 3060 (C–H, aromatic), 2951 (C–H, aliphatic), 1691 (C=O), 1591, 1495 (C=C), 1338 (C-N), 1207 (C-O).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3- dioxoquinoxalin-7-yloxy) phenyl)-3-(3-(methylthio) phenyl) urea (15e)
Brown powder, yield: 64%, M.p. 247-249 °C. 1H NMR (400 MHz, (D6) DMSO): 2.45 (s, 3H, Me31), 6.71 (d, J = 2.4, 1H, H14), 6.82 (dd, J = 2.4 Hz and 6 Hz, 1H, H10), 6.85 (d, J = 8.4 Hz, 1H, H27), 6.97 (d, J = 9.2 Hz, 2H, H6,4), 7.10 (m, 2H, H11,25), 7.20 (m, 1H, H28), 7.46 (m, 3H,H1,3,29), 8.69 (d, J = 4 Hz, 2H, H9,22), 11.79 (s, 1H, H15), 11.88 (s, 1H, H18); FT-IR KBr (cm-1): 3332 (b, N–H), 3070 (C–H, aromatic), 2949 (C–H, aliphatic), 1695 (C=O), 1545, 1500 (C=C), 1394 (C-N), 1209 (C-O); EI-MS: 433.10 ([M - H]- m/z, C22H18N4O4S calc. 434.1).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo-quinoxalin - 7 - yloxy)- 2 - methylphenyl)-3-(3-(methylthio) phenyl) urea (16e)
Brown powder, yield: 46%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 2.21 (s, 3HMe33), 2.45 (s, 3H, Me31), 6.73 (d, J = 2.4 Hz, 1H, H14), 6.77 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H10), 6.83 (m, 2H, H6, H11), 6.89 (d, J = 2.8 Hz, 1H, H4), 7.12 (m, 2H, H25,27), 7.21 (t, J =8 Hz, 1H, H28), 7.47 (m, 1H, H29), 7.73 (d, J = 8.8 Hz, 1H, H1), 7.93 (s, 1H, H9), 8.99 (s, 1H, H22), 11.79 (s, 1H, H15), 11.88 (s, 1H, H18); FT-IR KBr (cm-1): 3288, 3176 (b, N–H), 3060 (C–H, aromatic), 2920 (C–H, aliphatic), 1689 (C=O), 1589, 1496 (C=C), 1338 (C-N), 1205 (C-O); EI-MS: 447.1 ([M - H]- m/z, C23H20N4O4S calc. 447.12).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo- quinoxalin- 7-yloxy) phenyl)-3-(4-chloro-3- (trifluoromethyl) phenyl) urea (15f)
Brown powder, yield: 52%, M.p. 246-248 °C. 1H NMR (400 MHz, (D6) DMSO): 6.72 (d, J = 2.8 Hz, 1H, H14), 6.77 (dd, J = 2.4 Hz and 8.8 Hz, 1H, H10), 6.98(d, J = 9.2 Hz, 2H, H4,6), 7.11 (d, J = 8.8 Hz, 1H, H11), 7.47 (d, J = 8.8 Hz, 2H, H2,6), 7.61 (m, 2H, H28,29), 8.10 (d, J = 2 Hz, 1H, H25), 8.87 (s, 1H, H9), 9.16 (s, 1H, H22), 11.80 (s, 1H, H15), 11.88 (s, 1H, H18); FT-IR KBr (cm-1): 3354 (b, N–H), 3064 (C–H, aromatic), 1693 (C=O), 1549, 1502 (C=C), 1329 (C-N), 1211 (C-O); 489.05 ([M - H]- m/z, C22H14 ClF3N4O4 calc. 489.07).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo- quinoxalin-7-yloxy)- 2- methylphenyl)- 3 - (4- chloro-3-(trijluoromethyl) phenyl) urea (16f)
Brown powder, yield: 64%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 2.21 (s, 3H, Me33), 6.74 (d, J = 2.4 Hz, 1H, H14), 6.77 (dd, J = 2.4 Hz and 8.4 Hz, 1H, H10), 6.83 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H6), 6.90 (d, J = 2.8 Hz, 1H, H4), 7.11 (d, J = 8.8 Hz, 1H, H11), 7.63 (m, 3H, H1,29,30), 8.07 (s, 1H, H9), 8.10 (d, J = 2 Hz, 1H, H25), 9.40 (s, 1H, H22), 11.80 (s, 1H, H15), 11.88 (s, 1H, H18); FT-IR KBr (cm-1):3300, 3184 (b, N–H), 3064 (C–H, aromatic), 2951 (C–H, aliphatic) 1689 (C=O), 1549, 1485 (C=C), 1329 (C-N), 1205 (C-O); 503.10 ([M - H]- m/z, C23H16 ClF3N4O4 calc. 503.08).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo- quinoxalin-7-yloxy) phenyl)-3-(5-(trifluoro- methyl)-2-methoxyphenyl) urea (15g)
Brown powder, yield: 90%, M.p. >300 °C. 1H NMR (400 MHz, (D6) DMSO): 3.97 (s, 3H, Me30), 6.72 (d, J = 2.4, 1H, H14), 6.77 (dd, J = 2.8 and 8.8, 1H,H10),6.98 (d, J = 9.2 Hz, 2H, H4,6), 7.11 (d, J = 8.8 Hz, 1H, H14), 7.20 (d, J = 8.4 Hz, 1H, H28), 7.31 (dd, J = 0.8 Hz and 8.4 Hz, 1H,H27), 7.47 (d, J = 8 Hz, 2H, H1,3), 8.47 (s, 1H, H9), 8.54 (d, J = 2.4 Hz, 1H, H25), 9.44 (s, 1H, H22), 11.79 (s, 1H, H15), 11.88 (s, 1H, H18); FT-IR KBr (cm-1): 3346, 3199 (b, N–H), 3050 (C–H aromatic), 2949 (C–H, aliphatic), 1697 (C=O), 1549, 1502 (C=C), 1334 (C-N), 1209 (C-O); 485.10 ([M - H]- m/z, C23H17F3N4O5 calc. 485.12).
1-(4-(1, 2, 3, 4-tetrahydro-2, 3-dioxo-quinoxalin-7-yloxy)-2-methylphenyl)-3-(5-(trijluoromethyl)-2-methoxyphenyl) urea (16g)
Brown powder, yield: 85%, M.p. > 300 °C. 1H NMR (400 MHz, (D6) DMSO): 2.23 (s, 3H, Me33), 3.98 (s, 3H, Me30), 6.73 (d, J = 2.4 Hz, 1H, H14), 6.77 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H10), 6.82 (dd, J = 2.8 Hz and 8.8 Hz, 1H, H6), 6.89 (d, J = 2.8 Hz, 1H, H4), 7.11 (d, J = 8.4 Hz, 1H, H11),7.20 (d, J = 8.4 Hz, 1H, H28), 7.30 (dd, J = 0.6 Hz and 8.4 Hz, 1H, H27),7.71 (d, J = 8.8 Hz, 1H, H1), 8.55 (d, J = 2.4 Hz, 1H, H25), 8.64 (s, 1H, H9), 8.83 (s, 1H, H22), 11.79 (s, 1H, H15), 11.88 (s, 1H, H18); FT-IR KBr (cm-1) :3354, 3186 (N–H), 3064, (C–H, aromatic), 2949 (C–H, aliphatic), 1697 (C=O), 1545, 1496 (C=C), 1334 (C-N), 1205(C-O).
DISCUSSION
NMR characterization
In step 1 protection of amino group of p-aminophenol was confirmed by a singlet peak at 1.97 ppm due to the protons of acetamide methyl group. Moreover, NH of acetamide and phenolic OH groups demonstrated two separate singlet peaks around 9 ppm. In step 2, disappearance of phenolic OH peak at 9 ppm and appearance of 3 aromatic hydrogens peak at 7-8 ppm confirmed O-arylation reaction. In step 3, reduction of nitro group was confirmed by shift of aromatic hydrogen at ortho position of nitro group from 8 ppm to 6 ppm in reduced compound. In step 4, cyclization of diamine and formation of key quinoxalindione moiety intermediate was confirmed by shift of three aromatic protons of this group from high field around 6 ppm to lower field around 7 ppm due to electron withdrawing effect of carbonyl group of quinoxalindione. Furthermore, 2 singlet peaks around 12 ppm represent 2 NH groups of quinoxalindione substructure. In step 5, due to deacetylation methyl peak at 2 ppm is diminished and a broad peak around 5 ppm represent NH2 group. In final step conjugation of isocyanate or N-phenylcarbamate compound with intermediate is confirmed by two significant singlet bonds around 8-9.5 ppm and appearance of several aromatic hydrogens at 7-8 ppm.
FT-IR characterization
The structures of synthesized compounds were confirmed by means of FT-IR spectroscopy. The main IR vibrational bands of the synthesized compounds were found in their expected regions. Protection of amine group of p-aminophenol was confirmed by appearance of acetamide carbonyl group band at 1655 cm-1. O-arylation was confirmed by two significant bands at 1572 and 1333 cm-1 which were attributed to nitro group. In step 3, disappearance of nitro group peaks at 1572 and 1333 cm-1 and appearance of several peaks at 3200-3369 cm-1 region confirmed nitro reduction to amine. In step 4, cyclization of diamine and formation of quinoxalindione was confirmed by appearance of new bond at 1685 cm-1 assigned to carbonyl groups of quinoxalindione moiety. In step 5, after deacetylation, carbonyl groups of quinoxalindione moiety bands were shifted to1720 cm-1 from 1685 cm-1 and the carbonyl of acetamide group band disappeared from 1655 cm-1. In step 6, formation of diaryl urea derivatives was confirmed by strong broad band at 1680-1695 cm-1 assigned to carbonyl stretching of quinoxalindione moiety and carbonyl of urea group.
CONCLUSION
In the current work we report the synthesis of sorafenib derivatives 15a-15g and 16a-16g which differ from sorafenib by replacing pyridyl carboxamide ring of sorafenib with quinoxalindione moiety. A 7-step synthetic pathway includes preparation of N-(4-hydroxyphenyl) acetamides 3-4, N-(4-(3 - amino - 4 - nitrophenoxy) phenyl) acetamides 5-6, N-(4-(3,4- diaminophenoxy) phenyl) acetamides 7-8, N-(4-((2,3- dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)oxy)phenyl) acetamides 9-10, 6- (4- aminophenoxy) quinoxaline-2,3 (1H, 4H)-diones 11-12, phenyl phenylcarbamates 14c-14g and the target compounds 1-(4-((2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)oxy)phenyl) - 3-phenylureas 15a-15g and 16a-16g. The final compounds as well as intermediates were chemically characterized. These compounds will be subjected to biological evaluations to investigate their possible cytotoxic and antimicrobial activities.
ACKNOWLEDGEMENTS
The content of this paper is extracted from the Ph.D thesis (No. 394159) submitted by Sedighe Sadeghian-Rizi which was financially supported by the Isfahan University of Medical Sciences, Isfahan, Iran.
REFERENCES
- 1.Kim HJ, Cho HJ, Kim H, El-Gamal MI, Oh CH, Lee SH, et al. New diarylureas and diarylamides possessing acet(benz)amidophenyl scaffold: design, synthesis, and antiproliferative activity against melanoma cell line. Bioorg Med Chem Lett. 2012;22(9):3269–3273. doi: 10.1016/j.bmcl.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 2.Zambon A, Ménard D, Suijkerbuijk BMJM, Niculescu-Duvaz I, Whittaker S, et al. Novel Hinge Binder Improves Activity and Pharmacokinetic Properties of BRAF Inhibitors. J Med Chem. 2010;53(15):5639–5655. doi: 10.1021/jm100383b. [DOI] [PubMed] [Google Scholar]
- 3.Zhao CR, Wang RQ, Li G, Xue XX, Sun CJ, Qu XJ, et al. Synthesis of indazole based diarylurea derivatives and their antiproliferative activity against tumor cell lines. Bioorg Med Chem Lett. 2013;23(7):1989–1992. doi: 10.1016/j.bmcl.2013.02.034. [DOI] [PubMed] [Google Scholar]
- 4.Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res. 2006;12(24):7271–7278. doi: 10.1158/1078-0432.CCR-06-1249. [DOI] [PubMed] [Google Scholar]
- 5.Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs. 2009;69(2):223–2240. doi: 10.2165/00003495-200969020-00006. [DOI] [PubMed] [Google Scholar]
- 6.Fala L. Lenvima (Lenvatinib), a multireceptor tyrosine kinase inhibitor, approved by the fda for the treatment of patients with differentiated thyroid cancer. American health and drug benefits. 2015;8(Spec Feature):176. [PMC free article] [PubMed] [Google Scholar]
- 7.Wong HH, Eisen T. Tivozanib for the treatment of metastatic renal cancer. Expert Rev Anticancer Ther. 2013;13(6):649–660. doi: 10.1586/era.13.40. [DOI] [PubMed] [Google Scholar]
- 8.Marlow M, Al-Ameedee M, Smith T, Wheeler S, Stocks MJ. Linifanib–a multi-targeted receptor tyrosine kinase inhibitor and a low molecular weight gelator. Chem Commun. 2015;51(29):6384–6387. doi: 10.1039/c5cc00454c. [DOI] [PubMed] [Google Scholar]
- 9.Niculescu-Duvaz I, Roman E, Whittaker SR, Friedlos F, Kirk R, Scanlon IJ, et al. Novel inhibitors of B-RAF based on a disubstituted pyrazine scaffold. Generation of a nanomolar lead. J Med Chem. 2006;49(1):407–416. doi: 10.1021/jm050983g. [DOI] [PubMed] [Google Scholar]
- 10.Ramurthy S, Subramanian S, Aikawa M, Amiri P, Costales A, Dove J, et al. Design and synthesis of orally bioavailable benzimidazoles as Raf kinase inhibitors. J Med Chem. 2008;51(22):7049–7052. doi: 10.1021/jm801050k. [DOI] [PubMed] [Google Scholar]
- 11.Wang K, Li Y, Zhang L-J, Chen X-G, Feng Z-Q. Synthesis and in vitro cytotoxic activities of sorafenib derivatives. Chin Chem Lett. 2014;25(5):702–704. [PubMed] [Google Scholar]
- 12.Wu C, Wang M, Tang Q, Luo R, Chen L, Zheng P, et al. Design, synthesis, activity and docking study of sorafenib analogs bearing sulfonylurea unit. Molecules. 2015;20(10):19361–19371. doi: 10.3390/molecules201019361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao J, Chen J, He Z, Sun W, Xu W. Design, synthesis and biological activities of thiourea containing sorafenib analogs as antitumor agents. Bioorg Med Chem. 2012;20(9):2923–2929. doi: 10.1016/j.bmc.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Niculescu-Duvaz D, Gaulon C, Dijkstra HP, Niculescu-Duvaz I, Zambon A, Ménard D, et al. Pyridoimidazolones as novel potent inhibitors of v-Raf murine sarcoma viral oncogene homologue B1 (BRAF) J med chem. 2009;52(8):2255–2264. doi: 10.1021/jm801509w. [DOI] [PubMed] [Google Scholar]
- 15.Zambon A, Ménard D, Suijkerbuijk BM, Niculescu-Duvaz I, Whittaker S, Niculescu-Duvaz D, et al. Novel hinge binder improves activity and pharmacokinetic properties of BRAF inhibitors. J Med Chem. 2010;53(15):5639–5655. doi: 10.1021/jm100383b. [DOI] [PubMed] [Google Scholar]
- 16.Corona P, Carta A, Loriga M, Vitale G, Paglietti G. Synthesis and in vitro antitumor activity of new quinoxaline derivatives. Eur J Med Chem. 2009;44(4):1579–1591. doi: 10.1016/j.ejmech.2008.07.025. [DOI] [PubMed] [Google Scholar]
- 17.Grande F, Aiello F, Grazia OD, Brizzi A, Garofalo A, Neamati N. Synthesis and antitumor activities of a series of novel quinoxalinhydrazides. Bioorg Med Chem. 2007;15(1):288–294. doi: 10.1016/j.bmc.2006.09.073. [DOI] [PubMed] [Google Scholar]
- 18.Tanimori S, Nishimura T, Kirihata M. Synthesis of novel quinoxaline derivatives and its cytotoxic activities. Bioorg Med Chem Lett. 2009;19(15):4119–4121. doi: 10.1016/j.bmcl.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Zarranz B, Jaso A, Aldana I, Monge A. Synthesis and anticancer activity evaluation of new 2-alkylcarbonyl and 2-benzoyl-3-trifluoromethyl-quinoxaline 1,4-di-N-oxide derivatives. Bioorg Med Chem. 2004;12(13):3711–3721. doi: 10.1016/j.bmc.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Kotra VP, Karakavalasa; Vasanthi, R Synthesis, characterization and pharmacological evaluation of some novel quinoxaline derived chalcones. Der Pharma Chemica. 2013;5(4):301–307. [Google Scholar]
- 21.Noorulla S, Sreenivasulu N, Khan A, Sayeed A. Antibacterial activity of novel substituted quinoxaline heterocycles. Pharmanest. 2011;2(2-3):229–238. [Google Scholar]
- 22.Sadeghian-Rizi S, Sakhteman A, Hassanzadeh F. A quantitative structure-activity relationship (QSAR) study of some diaryl urea derivatives of B-RAF inhibitors. Res Pharm Sci. 2016;11(6):445–453. doi: 10.4103/1735-5362.194869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sastry C, Marwah P, Marwah A, Rao G. Synthesis and Biological Activity of Some New N-Arylcarbamoyl and N-Arylthiocarbamoyl Hydrazinoquinoxalin-2-ones. ChemInform. 1990;21(6):885–887. [Google Scholar]
- 24.Yoo HW, Lee YS, Eun Suh M, Kim DJ, Park SW. Cytotoxic effects of quinoxaline derivatives on human cancer cell lines. Archiv Der Pharmazie. 1998;331(10):331–333. doi: 10.1002/(sici)1521-4184(199810)331:10<331::aid-ardp331>3.3.co;2-9. [DOI] [PubMed] [Google Scholar]