

Abstract
Objective
A rare cause of familial frontotemporal dementia (FTD) is a mutation in the CHMP2B gene on chromosome 3 (FTD-3), described in a Danish family. Here we examine whether CSF biomarkers change in the preclinical phase of the disease.
Methods
In this cross-sectional explorative study, we analyzed CSF samples from 16 mutation carriers and 14 noncarriers from the Danish FTD-3 family. CSF biomarkers included total tau (t-tau) and neurofilament light chain (NfL) as a marker for neurodegeneration, phosphorylated tau (p-tau) as a marker for tau pathology, β-amyloid (Aβ) 38, 40, and 42 (Aβ38, Aβ40, and Aβ42) to monitor Aβ metabolism, and YKL-40 as a marker of neuroinflammation. Aβ isoform concentrations were measured using a multiplexed immunoassay; t-tau, p-tau, NfL, and YKL-40 concentrations were measured using sandwich ELISAs.
Results
CSF NfL concentration was significantly increased in mutation carriers vs noncarriers. Further, CSF NfL concentration was significantly higher in symptomatic mutation carriers compared to presymptomatic carriers, and also significantly higher in presymptomatic carriers compared to noncarriers. No differences in t-tau and p-tau and YKL-40 concentrations between controls and mutation carriers were observed. CSF concentrations of the Aβ peptides Aβ38 and Aβ40 but not Aβ42 were significantly lower in mutation carriers compared to noncarriers.
Conclusions
Increased NfL levels in presymptomatic individuals and in symptomatic patients with FTD-3 indicate a continuous process of neurodegeneration from the presymptomatic to symptomatic state. Although not specific for FTD-3 pathology, our data suggest that CSF NfL could serve as a valuable biomarker to detect onset of neurodegeneration in FTD-3 mutation carriers.
A truncating mutation in the CHMP2B gene (c.532-1G>C) on chromosome 3 results in early-onset frontotemporal dementia (FTD) (chromosome 3–linked FTD [FTD-3] or CHMP2B-FTD).1 This rare cause of FTD was first described in a large Danish family2 and later a different CHMP2B mutation was identified in an unrelated Belgian patient with familial FTD.3 The disease is mapped through 6 generations from the first known case in 1876 and the family now counts more than 500 individuals. FTD-3 is characterized by progressive cognitive deficits with behavioral changes. FTD-3 brains present primarily with frontal degeneration as well as temporal and dominant parietal lobe dysfunction.2
The histopathologic hallmarks of CHMP2B-FTD include ubiquitin-positive inclusions, autofluorescent aggregates, and p62-positive inclusions, while there is no tau, TAR DNA-binding protein 43, or fused in sarcoma pathology.4
The core CSF biomarkers used for dementia diagnosis are β-amyloid 42 (Aβ42), total tau (t-tau), and phosphorylated tau (p-tau).5 A large subset of FTD cases have pathologic tau changes with robust increases in CSF t-tau and p-tau levels but normal levels of Aβ42.6 CSF biomarkers in familial FTD have been investigated in individuals with mutations in MAPT, GRN, or the C9ORF79 repeat expansion7 but not in CHMP2B mutation carriers.
In this cross-sectional explorative study, we analyzed the CSF biomarkers Aβ42, t-tau, and p-tau181 in 30 individuals from the Danish FTD-3 family. We further included neurofilament light (NfL) as a marker of neuronal injury and YKL-40 as a marker of neuroinflammation in our analysis.
Methods
Study population
The FTD-3 family has been subject to extensive studies over more than 20 years within the Frontotemporal Dementia Research in Jutland Association (FReJA) collaboration, and biological material has been collected during the years for linkage analyses, gene identification, and functional studies.2 The disease has been tracked through 6 generations, and clinical characteristics have been recorded in 45 cases of disease, providing information about natural history, clinical characteristics, and age at onset (AAO).
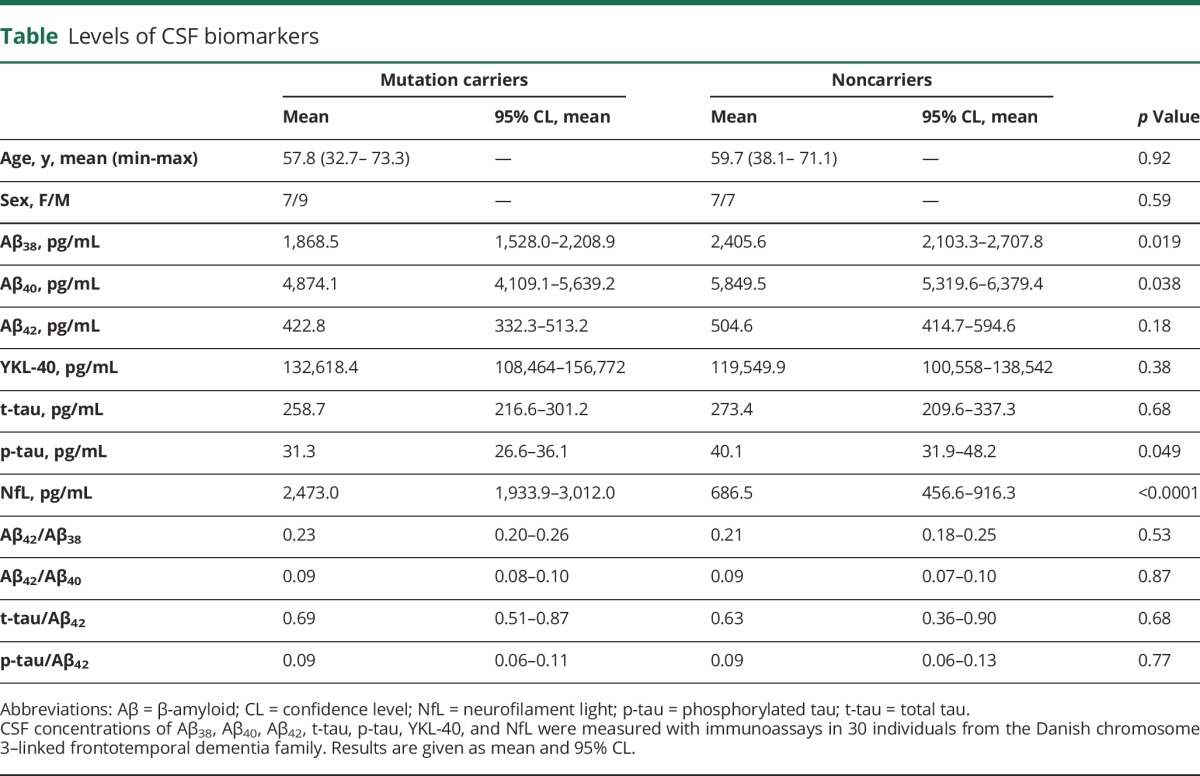
Samples from a total of 30 individuals were included: 10 affected CHMP2B mutation carriers, 6 presymptomatic CHMP2B mutation carriers, and 14 noncarrier family members (table). In the affected FTD-3 patients, the mean AAO was 60.5 years (SD 4.8 years).
Table.
Levels of CSF biomarkers
Standard protocol approvals, registrations, and patient consents
The study was approved by the Ethics Committee of the Capital Region of Denmark (H-1-2012-041), and written informed consent was obtained from each participant before enrollment. Subject age was recorded as the age of the individual on the day of lumbar puncture.
Collection of CSF
In brief, CSF samples were obtained by lumbar puncture and collected in polypropylene tubes, centrifuged at 2,000 g for 10 minutes, aliquoted into cryo tubes, and stored at −80°C until further use.8
Immunoassays
Aβ triplex
CSF concentrations of Aβ38, Aβ40, and Aβ42 were measured using V-plex Peptide Panel 1 Kits Aβ38, Aβ40, and Aβ42 (Meso Scale Discovery system, Rockville, MD) according to the manufacturer's protocol: 60 μL of CSF was diluted 2-fold in diluent provided with the kit. Calibrator samples and controls were prepared according to manufacturer's protocol. On 96-well MSD plates precoated with capture antibodies, 25 μL of sample, calibrator, or controls were added to each well followed by addition of 25 μL of detection antibody and incubation at room temperature for 2 hours. Finally, the plate was washed in kit washing buffer and read in a Meso Scale Discovery imager at appropriate wavelength.
Total tau
CSF t-tau concentration was measured by the hTAU Ag ELISA assay (INNOTEST, Fujirebio, Japan) according to the manufacturer's protocol: on a 96-well microtiter plate precoated with anti-human tau antibody, 25-μL samples, controls, and standards ranging from 50 to 2,500 pg/mL were added followed by incubation with a biotinylated detection antibody to tau and addition of peroxidase-conjugated streptavidin. The reaction was developed with tetramethyl benzidine (TMB) chromogen solution and subsequently stopped with 0.9 M sulfuric acid and quantified at 450 nm in a microplate reader.
Phosphorylated tau
The CSF concentration of tau phosphorylated at amino acid 181 (p-tau) was measured by the p-tau (181p) ELISA assay (INNOTEST, Fujirebio, Japan) according to the manufacturer's protocol: on a 96-well microtiter plate precoated with anti-human p-tau antibody, 75-μL samples, controls, and standards ranging from 15.6 to 1,000 pg/mL were added followed by incubation with a biotinylated detection antibody to p-tau and addition of peroxidase-conjugated streptavidin. The reaction was developed with TMB chromogen solution and subsequently stopped with 0.9 M sulfuric acid and quantified at 450 nm in a microplate reader.
Neurofilament light chain
CSF NfL concentration was measured by sandwich ELISA with the antibody NfL21 used for coating the plates (monoclonal mouse antibody against NfL [in-house]). The coating concentration was 0.5 μg/mL in carbonate buffer (pH 9.6).
Briefly, 50 μL CSF (1:2 dilution), 100 μL calibrator, and 50 μL PBST (0.05% tween) were added to the coated plates and incubated 1 hour while shaking at room temperature followed by overnight incubation at +4°C. For detection, a biotinylated NfL23 mouse monoclonal antibody against NfL (in-house) was used together with streptavidin–horseradish peroxidase diluted 1:20,000 (100 μL/well).
Following wash, 100 μL TMB substrate (TMB One ready-to-use substrate, Kem En Tec Diagnostic, Taastrup, Denmark) was added to each well and plates were incubated for 20 minutes in the dark at room temperature. The reaction was stopped with 100 μL/well of 0.2 M H2SO4 and absorbance was quantified at 450/650 nm in a microplate reader.
YKL-40
CSF YKL-40 concentration was measured using a solid-phase sandwich ELISA (Human Chitinase 3-like 1 Quantikine ELISA kit, R&D Systems, Oxon, UK) according to the manufacturer's protocol: 50 μL of CSF was diluted 2-fold in diluent provided with the kit. Calibrators and controls were prepared according to protocol.
On a capture-antibody precoated 96-well microtiter plate, 50 μL of CSF sample, calibrator, or controls were added followed by addition of 200 μL of conjugate and incubation at room temperature for 2 hours. Finally, each well was washed in the provided washing buffer, followed by addition of 200 μL substrate solution, and subsequently stopped with 50 μL of the provided stop solution. Quantification was carried out at 540/570 nm in a microplate reader.
All biochemical measurements were performed by board-certified laboratory technicians who were blinded to clinical information. The measurements were performed in one round of experiments using one batch of reagents. Intra-assay coefficients of variation were below 10%.
Statistical methods
Data were analyzed using SAS software (Enterprise Guide 7.1, 2014, SAS Institute Inc, Cary, NC). Concentrations of each biomarker were compared between controls and CHMP2B mutation carriers, and between affected individuals and presymptomatic mutation carriers. As some biomarkers were found to be age-dependent, participant age was included in a subsequent analysis using general linear modeling (GLM). Where it was necessary, logarithmic transformation was performed to obtain normal distribution.
Results
CSF data for all measured markers are shown in the table.
Participant age and sex distribution did not differ significantly between mutation carriers and noncarriers (t[29] = −0.63, p = 0.53 and χ2[1] = 0.28, p = 0.59, respectively).
In our dataset, t-tau, p-tau, and YKL-40 levels were found to be age-dependent (GLM, F1,28 = 6.68, p = 0.0153; F1,28 = 5.14, p = 0.0314; F1,28 = 6.37, p = 0.0176, respectively) while Aβ38, Aβ40, Aβ42, and NfL were not (GLM, F1,28 = 2.45, p = 0.1288; F1,28 = 1.80, p = 0.1906; F1,28 = 0.22, p = 0.6450; F1,27 = 0.38, p = 0.4935, respectively).
When adjusted for age-dependency, statistical analysis showed no difference in t-tau and p-tau levels between controls and mutation carriers (GLM, F3,26 = 3.29, p = 0.5173, and F3,26 = 3.86, p = 0.69, respectively). Levels of Aβ38 and Aβ40 but not Aβ42 were significantly decreased in mutation carriers compared to noncarriers (GLM, F1,28 = 6.23, p = 0.02; F1,28 = 4.75, p = 0.04; and F1,28 = 1.87, p = 0.18, respectively) (figure 1). Aβ42/Aβ38, Aβ42/Aβ40, t-tau/Aβ42, and p-tau/Aβ42 ratios were not found to be significantly different between the 2 groups (GLM, F1,28 = 0.23, p = 0.63; F1,28 = 0.05, p = 0.83; F3,25 = 2.37, p = 0.68; and F3,25 = 1.96, p = 0.73, respectively).
Figure 1. CSF biomarkers in CHMP2B mutation carriers compared to noncarriers.

Box plots show CSF concentrations of β-amyloid 42 (Aβ)38, Aβ40, Aβ42, total tau (t-tau), phosphorylated tau (p-tau), and YKL-40 analyzed by immunoassays. Levels of the Aβ peptides Aβ38 (A) and Aβ40 (C) were significantly decreased in mutation carriers compared to noncarriers (p = 0.02 and p = 0.04, respectively). Levels of Aβ42, t-tau, p-tau, and YKL-40 (B, D–F) were not found to differ significantly between the 2 groups.
YKL-40 CSF levels did not differ significantly between mutation carriers and noncarriers after adjustment for age (GLM, F3,26 = 2.64, p = 0.57) (figure 1).
CSF levels of NfL were significantly higher in mutation carriers compared to noncarriers (GLM, F1,28 = 53.92, p ≤ 0.0001) (figure 2). When adjusting for participant age, this difference persisted (GLM, F3,26 = 29.44, p = 0.021). Symptomatic mutation carriers had significantly higher NfL levels than presymptomatic mutation carriers (GLM, F1,14 = 8.61, p = 0.0109), while presymptomatic carriers had significantly increased NfL levels compared to noncarriers (GLM, F1,17 = 10.97, p = 0.004).
Figure 2. CSF concentrations of neurofilament light (NfL) in symptomatic and presymptomatic CHMP2B mutation carriers (MC) and noncarriers.

Box plots (A, B) and scatterplot (C) show CSF concentrations of NfL. (A) CSF levels of NfL were significantly higher in MCs compared to noncarriers (p ≤ 0.0001), also when adjusted for age correlation (p = 0.021). (B) Symptomatic MCs had significantly higher NfL levels than presymptomatic MCs (p = 0.0109), while presymptomatic carriers had significantly increased NfL levels compared to noncarriers (p = 0.0002). (C) Even the youngest presymptomatic MCs had higher NfL levels than aged noncarriers.
In the age-corrected model, levels of NfL were found to be significantly increased in symptomatic mutation carriers when compared to presymptomatic carriers (GLM, F3,12 = 7.02, p = 0.0064), while the significant difference between presymptomatic mutation carriers and noncarriers disappeared (GLM, F3,15 = 9.94, p = 26.22).
As illustrated (figure 2), NfL levels were high in mutation carriers years prior to the expected clinical onset of 58 years.9 Even when adjusting for participant age, this elevation was significant (GLM, F3,26 = 29.44, p = 0.021).
Discussion
In this explorative study, we analyzed CSF samples from 10 affected CHMP2B mutation carriers, 6 presymptomatic CHMP2B mutation carriers, and 14 noncarriers from the Danish FTD-3 family. Further investigations on the diagnostic performance of the markers are needed to clarify the influence of age on the levels of the investigated biomarkers.
In our FTD-3 cohort, mutation carriers had significantly increased levels of CSF NfL compared to controls. Interestingly, this increase could be detected in presymptomatic individuals several years prior to expected AAO. Even the youngest presymptomatic mutation carrier (participant age 32, which is 26 years prior to expected clinical onset) had higher CSF NfL levels than some of the oldest noncarriers (figure 2). This suggests that the neurodegenerative process is ongoing in mutation carriers several decades before onset of symptoms, as was also reported in individuals with autosomal inherited Alzheimer disease (AD) in the Dominantly Inherited Alzheimer Network (DIAN) cohort.10
In contrast to our findings, a recent publication from the GENFI consortium found that CSF NfL levels were significantly increased in symptomatic carriers of mutations in other FTD-causing risk genes, GRN, MAPT, or C9ORF27, when compared to presymptomatic carriers and that presymptomatic carriers and control participants had similar NfL levels.11 In that study, the symptomatic mutation carriers' NfL levels correlated with disease severity, brain atrophy, and survival.11 In recent years, neurofilaments have been investigated in several neurodegenerative diseases12 and a meta-analysis has concluded that both NfL and neurofilament heavy are increased overall in patients with sporadic AD, FTD, and vascular dementia.13 Other studies have shown that CSF NfL levels were increased in sporadic FTD when compared to other dementia groups or healthy controls.14–19 Our results and the results from the studies of sporadic FTD cases suggest that NfL may be applied as biomarker of neurodegenerative progression and disease severity.
We detected a significant decrease in CSF Aβ38 and Aβ40 levels, but not in Aβ42 levels, in mutation carriers compared to noncarriers. The Aβ42/Aβ38 and Aβ42/Aβ40 ratios, however, did not differ between mutation carriers and controls. These results concur with the hypothesis that FTD-3 is not an amyloid disease, as no amyloid pathology has been identified.2,20 Substantiating this further, amyloid ligand PET scans in 4 affected patients with FTD-3 have shown no accumulation of amyloid (unpublished material). Lower Aβ38 and Aβ40 concentrations in CSF have been noted in PSEN1 mutation carriers and may indicate impaired γ-secretase processing of APP.21 Lower Aβ38 and Aβ40 have also been seen in neuroinflammatory conditions.22 Finally, there are some indications that reduced neuronal/synaptic activity may lead to less Aβ production in general.23,24
In MAPT, GRN, and C9ORF72 mutation carriers, levels of Aβ42 are generally found to be within normal range while levels of t-tau and p-tau are not well-characterized due to lack of consistent CSF profile for these patients.7
We did not observe a difference in CSF p-tau levels in mutation carriers compared to noncarriers in the current study. This finding is not surprising as patients with FTD-3 do not display the hyperphosphorylated tau pathology that is otherwise observed in other FTDs.2,20 Also, we found no significant increase in CSF t-tau levels. This was surprising as t-tau is known to be a general marker of neurodegeneration and it is well-described that patients with FTD-3 have brain atrophy even early in disease.20,25,26
The CSF level of the inflammation marker YLK-40 was found to increase with age in both mutation carriers and noncarriers in our FTD-3 cohort. This finding concurs with another study showing that CSF YKL-40 levels did not differ between patients with FTD and controls.27 YKL-40 is a secreted 40-kDa glycoprotein belonging to the member of the human chitinase activity family.28 YKL-40 is expressed in several tissues including astrocytes and microglia in the brain. It has been shown that YKL-40 plays an important role in astrocyte and microglial response to neuroinflammation.29,30 Our finding suggests that the observed increase is a consequence of age rather than mutation status and therefore YKL-40 may not be a valuable marker of disease in FTD-3. However, in some studies, CSF levels of YKL-40 have been shown to be increased in patients with AD and FTD when compared to cognitively healthy controls.31,32 Other studies have found that CSF YKL-40 levels in patients with FTD are increased compared to patients with AD.33,34 Since FTD is a clinically and pathologically heterogeneous disease, differences in the individual cohorts analyzed could explain why CSF YKL-40 concentrations vary among different studies (for review, see reference 35).
To our knowledge, all studies of YKL-40 as a CSF biomarker in patients with FTD have reported on sporadic cases and therefore might not reflect the pathology of genetic FTD. In spite of our finding that YKL-40 cannot be used as a marker to differentiate mutation carriers from controls, these results do not necessarily imply that neuroinflammation is not an important aspect of FTD-3 pathology. Recently, microglial activation was studied thoroughly in a FTD-3 mouse model. Early microglial proliferation and a clear proinflammatory phenotype was observed in the CHMP2B mutant mice compared to wild-types, which is similar to the inflammatory profile found in FTD-3 brains.4,36 These data suggest that neuroinflammation could be a pathologic driver of FTD-3 disease and that it might be valuable to measure CSF biomarkers of neuroinflammation in patients with FTD-3. However, in order to differentiate between disease development and normal aging, a more specific CSF marker than YLK-40 should be used.
Taken together, our results suggest that the unchanged levels of tau in CSF corroborate the absence of tau pathology in these patients. The decreased levels of Aβ38 and Aβ40 may be due to a general downregulation of APP processing; however, this needs to be further investigated. Our observations of increase in NfL levels in presymptomatic individuals and even further increase of NfL levels in affected patients with FTD-3 indicate a continuous process of neurodegeneration during the presymptomatic to symptomatic state. Furthermore, our study implies that NfL is a sensitive marker of the widespread neurodegeneration occurring in patients with FTD-3, even decades before symptom onset.
Acknowledgment
FReJA members: Adrian Isaacs, Anders Gade, Elisabet Englund, Elizabeth Fisher, Jeremy Brown, Jette Stockholm, John Collinge, Peter Johannsen, Susanne Gydesen, Suzanne Lindquist, Tove Thusgaard, and Troels Tolstrup Nielsen. The authors thank Dr. Jonathan Henry Wardman for linguistic proofreading and FTD-3 family members for continued dedication and support.
Glossary
- AAO
age at onset
- Aβ42
β-amyloid 42
- AD
Alzheimer disease
- FTD
frontotemporal dementia
- FTD-3
chromosome 3–linked frontotemporal dementia
- GLM
general linear modeling
- NfL
neurofilament light
- p-tau
phosphorylated tau
- t-tau
total tau
- TMB
tetramethyl benzidine
Author contributions
Nina Rostgaard: study concept and design, analysis and interpretation of data, drafting/revising the manuscript for content, and obtaining funding. Peter Roos: study concept and design, statistical analysis, analysis and interpretation of data, drafting/revising the manuscript for content, and obtaining funding. Erik Portelius: study concept and design, acquisition of data, interpretation of data, drafting/revising the manuscript for content, and obtaining funding. Kaj Blennow: study concept and design, acquisition of data, interpretation of data, drafting/revising the manuscript for content, and obtaining funding. Henrik Zetterberg: study concept and design, acquisition of data, interpretation of data, drafting/revising the manuscript for content, and obtaining funding. Anja H. Simonsen: study concept and design, analysis and interpretation of data, and drafting/revising the manuscript for content. Jørgen E. Nielsen: study concept and design, analysis and interpretation of data, drafting/revising the manuscript for content, and obtaining funding.
Study funding
This study was supported by grants from The Jascha Foundation, Desiree & Niels Ydes Foundation, P.A. Messerschmidts and Wife's Foundation, Director Jacob Madsen and Wife Olga Madsens Foundation, the Novo Nordisk Foundation, the Innovation Fund Denmark (“Brainstem” grant no. 4108-00008B), the Lundbeck Foundation (grant no. R167-2013-15940), the Swedish Research Council, the European Research Council, the Torsten Söderberg Foundation, the Knut and Alice Wallenberg Foundation, and Swedish State Support for Clinical Research (ALFGBG).
Disclosure
N. Rostgaard, P. Roos, and E. Portelius report no disclosures relevant to the manuscript. K. Blennow has served as a consultant or on advisory boards for Alzheon, BioArctic, Biogen, Eli Lilly, Fujirebio Europe, IBL International, Pfizer, and Roche Diagnostics, and is co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg. H. Zetterberg has served on advisory boards of Roche Diagnostics, Eli Lilly, and Pharmasum Therapeutics, and is co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg. A. Simonsen and J. Nielsen report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 2005;37:806–808. [DOI] [PubMed] [Google Scholar]
- 2.Gydesen S, Brown JM, Brun A, Chakrabarti L, Gade A, Johannsen P, et al. Chromosome 3 linked frontotemporal dementia (FTD-3). Neurology 2002;59:1585–1594. [DOI] [PubMed] [Google Scholar]
- 3.Isaacs AM, Johannsen P, Holm I, Nielsen JE. Frontotemporal dementia caused by CHMP2B mutations. Curr Alzheimer Res 2011;8:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clayton EL, Mizielinska S, Edgar JR, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol 2015;130:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010;6:131–144. [DOI] [PubMed] [Google Scholar]
- 6.Hales CM, Hu WT. From frontotemporal lobar degeneration pathology to frontotemporal lobar degeneration biomarkers. Int Rev Psychiatry 2013;25:210–220. [DOI] [PubMed] [Google Scholar]
- 7.Rostgaard N, Waldemar G, Nielsen JE, Simonsen AH. Cerebrospinal fluid biomarkers in familial forms of Alzheimer's disease and frontotemporal dementia. Dement Geriatr Cogn Disord 2015;40:54–62. [DOI] [PubMed] [Google Scholar]
- 8.del Campo M, Mollenhauer B, Bertolotto A, et al. Recommendations to standardize preanalytical confounding factors in Alzheimer's and Parkinson's disease cerebrospinal fluid biomarkers: an update. Biomark Med 2012;6:419–430. [DOI] [PubMed] [Google Scholar]
- 9.Isaacs AM, Johannsen P, Holm I, et al. Frontotemporal dementia caused by CHMP2B mutations. Curr Alzheimer Res 2011;8:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 2016;1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perrot R, Berges R, Bocquet A, Eyer J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol 2008;38:27–65. [DOI] [PubMed] [Google Scholar]
- 13.Petzold A, Keir G, Warren J, Fox N, Rossor MN. A systematic review and meta-analysis of CSF neurofilament protein levels as biomarkers in dementia. Neurodegener Dis 2007;4:185–194. [DOI] [PubMed] [Google Scholar]
- 14.Landqvist Waldö M, Frizell Santillo A, Passant U, et al. Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurol 2013;13:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Jong D, Jansen RWMM, Pijnenburg YAL, et al. CSF neurofilament proteins in the differential diagnosis of dementia. J Neurol Neurosurg Psychiatry 2007;78:936–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pijnenburg Y, Janssen JC, Schoonenboom NSM, et al. CSF neurofilaments in frontotemporal dementia compared with early onset Alzheimer's disease and controls. Dement Geriatr Cogn Disord 2007;23:225–230. [DOI] [PubMed] [Google Scholar]
- 17.Sjogren M, Rosengren L, Minthon L, Davidsson P, Blennow K, Wallin A. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology 2000;54:1960–1964. [DOI] [PubMed] [Google Scholar]
- 18.Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol 2014;75:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skillback T, Farahmand B, Bartlett JW, et al. CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology 2014;83:1945–1953. [DOI] [PubMed] [Google Scholar]
- 20.Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol 2007;66:884–891. [DOI] [PubMed] [Google Scholar]
- 21.Portelius E, Andreasson U, Ringman JM, et al. Distinct cerebrospinal fluid amyloid beta peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer's disease. Mol Neurodegener 2010;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Augutis K, Axelsson M, Portelius E, et al. Cerebrospinal fluid biomarkers of β-amyloid metabolism in multiple sclerosis. Mult Scler J 2013;19:543–552. [DOI] [PubMed] [Google Scholar]
- 23.Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid-?? levels in vivo. Neuron 2005;48:913–922. [DOI] [PubMed] [Google Scholar]
- 24.Pomara N, Bruno D, Sarreal AS, et al. Lower CSF amyloid beta peptides and higher F2-Isoprostanes in cognitively intact elderly individuals with major depressive disorder. Am J Psychiatry 2012;169:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eskildsen SF, Østergaard LR, Rodell AB, et al. Cortical volumes and atrophy rates in FTD-3 CHMP2B mutation carriers and related non-carriers. Neuroimage 2009;45:713–721. [DOI] [PubMed] [Google Scholar]
- 26.Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009;73:1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldacci F, Toschi N, Lista S, et al. Two-level diagnostic classification using cerebrospinal fluid YKL-40 in Alzheimer's disease. Alzheimers Dement 2017;13:1–11. [DOI] [PubMed] [Google Scholar]
- 28.Rejman JJ, Hurley WL. Isolation and characterization of a novel 39 kilodalton whey protein from bovine mammary secretions collected during the nonlactating period. Biochem Biophys Res Commun 1988;150:329–334. [DOI] [PubMed] [Google Scholar]
- 29.Bonneh-Barkay D, Bissel SJ, Wang G, et al. YKL-40, a marker of simian immunodeficiency virus encephalitis, modulates the biological activity of basic fibroblast growth factor. Am J Pathol 2008;173:130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonneh-Barkay D, Wang G, Starkey A, Hamilton RL, Wiley CA. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J Neuroinflammation 2010;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janelidze S, Hertze J, Zetterberg H, et al. Cerebrospinal fluid neurogranin and YKL-40 as biomarkers of Alzheimer's disease. Ann Clin Transl Neurol 2016;3:12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alcolea D, Carmona-Iragui M, Suárez-Calvet M, et al. Relationship between beta-secretase, inflammation and core cerebrospinal fluid biomarkers for Alzheimer's disease. J Alzheimers Dis 2014;42:157–167. [DOI] [PubMed] [Google Scholar]
- 33.Craig-Schapiro R, Perrin RJ, Roe CM, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry 2010;68:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teunissen CE, Elias N, Koel-Simmelink MJA, et al. Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimer's Dement Diagnosis, Assess Dis Monit 2016;2:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldacci F, Lista S, Cavedo E, Bonuccelli U, Hampel H. Diagnostic function of the neuroinflammatory biomarker YKL-40 in Alzheimer's disease and other neurodegenerative diseases. Expert Rev Proteomics 2017;14:285–299. [DOI] [PubMed] [Google Scholar]
- 36.Clayton EL, Mancuso R, Nielsen TT, et al. Early microgliosis precedes neuronal loss and behavioural impairment in mice with a frontotemporal dementia-causing CHMP2B mutation. Hum Mol Genet 2017;26:ddx003. [DOI] [PMC free article] [PubMed] [Google Scholar]