

Abstract
Objective
We sought to identify rare variants influencing brain imaging phenotypes in the Framingham Heart Study by performing whole genome sequence association analyses within the Trans-Omics for Precision Medicine Program.
Methods
We performed association analyses of cerebral and hippocampal volumes and white matter hyperintensity (WMH) in up to 2,180 individuals by testing the association of rank-normalized residuals from mixed-effect linear regression models adjusted for sex, age, and total intracranial volume with individual variants while accounting for familial relatedness. We conducted gene-based tests for rare variants using (1) a sliding-window approach, (2) a selection of functional exonic variants, or (3) all variants.
Results
We detected new loci in 1p21 for cerebral volume (minor allele frequency [MAF] 0.005, p = 10−8) and in 16q23 for hippocampal volume (MAF 0.05, p = 2.7 × 10−8). Previously identified associations in 12q24 for hippocampal volume (rs7294919, p = 4.4 × 10−4) and in 17q25 for WMH (rs7214628, p = 2.0 × 10−3) were confirmed. Gene-based tests detected associations (p ≤ 2.3 × 10−6) in new loci for cerebral (5q13, 8p12, 9q31, 13q12-q13, 15q24, 17q12, 19q13) and hippocampal volumes (2p12) and WMH (3q13, 4p15) including Alzheimer disease– (UNC5D) and Parkinson disease–associated genes (GBA). Pathway analyses evidenced enrichment of associated genes in immunity, inflammation, and Alzheimer disease and Parkinson disease pathways.
Conclusions
Whole genome sequence–wide search reveals intriguing new loci associated with brain measures. Replication of novel loci is needed to confirm these findings.
Brain imaging phenotypes (cerebral and hippocampal atrophy or white matter hyperintensity [WMH] burden) are accepted endophenotypes of Alzheimer disease (AD) and vascular brain injury.1 Identifying genetic loci that influence these measures could lead to the discovery of new biological mechanisms underlying these diseases. Recently, large genome-wide association studies (GWAS) have successfully identified and replicated associations between genetic variants and brain imaging phenotypes.2–7
Two regions were consistently reported by association studies (17q25, WMH and 12q24, hippocampal atrophy).2,4,6,7 The 17q25 region was originally discovered by a GWAS of WMH in stroke-free European individuals from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium (n = 9,361) with replication in 2 cohorts (n = 3,024).7 This locus encompasses 6 genes (WBP2, TRIM65, TRIM47, MRPL38, FBF1, and ACOX1).7 The 12q24 region was initially reported by a GWAS of dementia-free individuals from the CHARGE consortium (n = 9,232) with replication in 2 samples (n = 2,318) and external validation in the Enhancing Neuroimaging Genetics through Meta-Analysis (ENIGMA) consortium (n = 7,794). These associations implicate genes related to apoptosis (HRK) and ubiquitination (FBXW8).2
These prior GWAS have identified common genetic variants (minor allele frequency [MAF] ≥5%) with modest effect sizes that resided mostly in noncoding regions. Besides, the causal variant and the functional basis of associations are unclear at many loci. Thus, more detailed scans of low frequency and rare variants (MAF ≤5%) available from whole genome sequence (WGS) are needed, particularly in GWAS loci and coding regions.
GWAS are performed using single nucleotide variants (SNVs) that are available on commercial genotyping chips or in reference panels. They focus on SNVs that are common in a population (MAF ≥5%). WGS can capture other types of genetic variations such as repeats or indels (insertions/deletions) and have a finer resolution compared to GWAS. Indeed, all genetic variations from an individual can be detected, including rare and individual-specific variations. Thus, WGS association analyses can help to detect new signals that could have been missed by GWAS.
In this study, we sought to identify low-frequency and rare variants associated with brain measures in the Framingham Heart Study by performing WGS association analyses within the Trans-Omics for Precision Medicine Program (TOPMed).
Methods
Description of the Framingham Heart Study
We included participants from the 3 generations of the Framingham Heart Study (FHS). Briefly, the FHS is a prospective, population-based study that began in 1948 to study the determinants of cardiovascular disease. The study has followed participants from the town of Framingham, Massachusetts, whose population was almost entirely white at the beginning of the study. The first generation (original cohort/Gen1) has been followed since 1948 and included 5,209 participants; survivors are still receiving examinations biannualy.8 The second generation (offspring cohort/Gen2) has been followed since 1971 and comprises 5,124 offspring and spouses of the offspring, including 3,514 biological offspring; they have received examinations once every 4–8 years.9 The third generation (Gen3) was enrolled in 2002 and included 4,095 children from the largest families of the offspring cohort; they have received 2 examinations 4 years apart and a third examination is currently underway.10 All cohorts are under active surveillance for cardiovascular events.
Attendees of the 26th Gen1 and 7th Gen2 examinations were invited to participate in brain MRI between March 1999 and June 2005; attendees of the 2nd Gen3 examination were similarly invited to undergo brain MRI from 2009. Among 4,772 individuals with brain MRI data, 96 participants were excluded for stroke or TIA, 73 for dementia, and 102 because of other neurologic conditions such as multiple sclerosis, meningiomas, primary or metastatic brain tumors, or significant head trauma. The remaining 4,501 participants constitute our sample for this study.
Standard protocol approvals, registrations, and patient consents
All participants provided written informed consent. This study was approved by the institutional review board of the Boston University Medical Center.
MRI protocol and phenotyping
Brain MRI acquisition measures and image processing methods have been described in detail elsewhere.11 Briefly, participants were imaged on a 1T (1999–2005) or 1.5T (after 2005) Magnetom scanner (Siemens Medical, Erlangen, Germany). We used 3D T1-weighted coronal spoiled gradient-recalled echo acquisition scans for all participants. In addition, we used T2 fluid-attenuated inversion recovery (FLAIR) sequences for scans acquired after 2005. All MRI were transferred to the University of California–Davis Medical Center for centralized reading. We used QUANTA 6.2 (Ultra 5 workstation, Sun Microsystems, Santa Clara, CA) for image analyses and interpretation was performed blinded to participants' demographic and clinical characteristics in random order.
The semiautomated segmentation protocols for quantifying total intracranial volume (TCV), total cerebral brain volume (TCBV), hippocampal volume (HPV), and WMH have been described elsewhere.12 Briefly, TCV was determined by outlining the intracranial vault lying above the tentorium.13 To accurately distinguish CSF from brain matter, we used a semiautomated analysis of MRI pixel distributions for CSF, gray matter, and white matter.14 HPV was computed by a semiautomatic multiatlas hippocampal segmentation algorithm.15 For analyses of WMH, we included the subsample of participants who were imaged on a 1.5T scanner and had FLAIR sequences, and used a semiautomated procedure13 with high inter-rater reliability16 for the segmentation and quantification of WMH on FLAIR. TCBV is expressed relative to TCV to account for differences in head size. No substantial bias was observed when compared participants with and without MRI.11
Whole genome sequencing
WGS was part of the National Heart, Lung and Blood Institute's TOPMed (nhlbiwgs.org) that serves as an initial step for a larger Precision Medicine Initiative. TOPMed phase I consisted of 11 different studies covering a range of heart, lung, blood, and sleep disorders phenotypes and a total of ∼20,000 samples. Phase I began in October 2014 and completion of sequencing production was done in February 2016. The FHS is part of this phase I with 4,148 sequenced individuals (372 Gen1, 2,194 Gen2, and 1,582 Gen3). The samples were sequenced at >×30 depth of coverage at the Broad Institute of the Massachusetts Institute of Technology and Harvard. Individual genetic variations across the genome were identified in a joint calling of all samples performed by the TOPMed Informatics Resource Center (University of Michigan). Centralized read mapping and genotype calling, along with variant quality metrics and filtering of variants and samples that failed to meet these quality metrics, was also completed by the TOPMed Informatics Research Center. Phenotype harmonization, data management, quality control (QC) to ensure correct sample identification, and general study coordination were provided by the TOPMed Data Coordinating Center. Methods for WGS data acquisition and QC are described in a document that is publicly available on the Database of Genotype and Phenotype (dbGaP) website (ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd006969.1). We excluded variants with a read depth less than 10.
Statistical analysis
We used mixed-effect linear regression models to examine associations between WGS SNVs and brain MRI measures. We adjusted models for age, sex, and TCV for WMH, age, sex, age2, and TCV for HPV and age, sex, and age2 for TCBV. Brain volumes and WMH are related to TCV, and vary with age and between men and women. We created the residuals of the original phenotypes adjusted for covariates and inverse normal transformed them. Then, we tested the association between rank-normalized residuals from these models and individual SNVs while taking into account familial relatedness using an empirical kinship matrix based on analysis of actual genotype similarities between participants. We filtered the results including only variants with an allele count of 10 or greater and used a threshold of p = 1.5 × 10−8 to consider an SNV association as genome-wide significant.17
In further analyses, we performed conditional analyses on the previous reported associations in 2 GWAS loci: 12q24 for HPV and 17q25 for WMH.
Within each gene, we also conducted gene-based tests to test the association between rare variants (MAF ≤1%) and MRI measures using 2 different multimarker methods (i.e., sequence kernel association test [SKAT] and burden test [T1]).18 These methods aggregate individual score test statistics of all rare genetic variations in a gene or a region. SKAT tests are robust to the presence of rare variants with either risk-increasing or risk-decreasing effects. In the T1 test, the summed aggregate effect is considered and only rare variants (MAF <1%) are included to assign a score to each gene or region, and hence this test is most powerful when all variants are either increasing or decreasing risk.
In order to improve our power to detect associations, we used different filters to select only SNVs in a gene that were most likely to affect the phenotype as functional exonic SNVs (missense or loss of function) or all rare SNVs. We also used a sliding-window approach19 in which the gene length was divided into discrete regions or “windows” and gene window-based test was performed within each region; this approach improves the power to detect effects if only one region within the gene is affecting the phenotype. We used a window size of 4 kb with an overlap of 2 kb between adjacent windows and we filtered the results according to a burden count of 10 or greater. We used a threshold of p = 2.5 × 10−6 to declare a gene association as genome-wide significant (Bonferroni correction for number of genes tested, 0.05/20,000). For the sliding-window approach, we used a more stringent threshold of p = 1.8 × 10−7 to declare a gene association as genome-wide significant to account for overlapping windows (correction for the number of tests performed in each gene, 0.05/279,713). We performed functional annotations of the SNVs with the publicly available variant function predicting software Annovar.20 All association analyses were carried out using EPACTS 3.2.6 software (University of Michigan) with the EMMAX test. Finally, we used the Meta-Analysis Gene-set Enrichment of Variant Associations (MAGENTA) method to explore pathway-based associations using single-SNV association results. MAGENTA implements a gene set enrichment analysis-based approach, as previously described.21 In this method, a gene score is calculated for each gene based on the SNV with the lower association p value in a 110 kb upstream, 40 kb downstream window around the gene. This score is corrected for confounding factors (gene size, SNV density, or linkage disequilibrium [LD] between SNVs in the gene) and each gene is ranked on its score. In each pathway, an empirical p value is calculated corresponding to the observed number of genes with a rank above a given significance threshold (95th or 75th percentiles of all gene scores) compared to 106 randomly permuted pathways of the same size.
In total, 10,992 pathways from the Gene Ontology, PANTHER, KEGG, Ingenuity, Reactome, and Biocarta databases were tested for enrichment of associations with each brain MRI phenotype.
Results
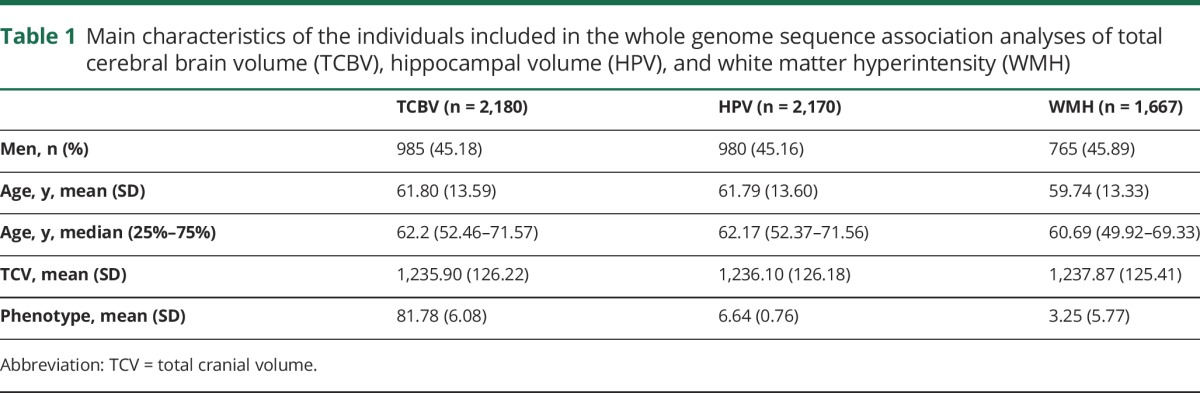
We included a total of 2180, 2,170, and 1,667 individuals from FHS in the WGS association analyses of TCBV, HPV, and WMH, respectively (table 1). Most participants were women (∼55%) and the mean age (SD) of individuals was 61.8 years (13.6) for TCBV and HPV and 59.7 years (13.3) for WMH.
Table 1.
Main characteristics of the individuals included in the whole genome sequence association analyses of total cerebral brain volume (TCBV), hippocampal volume (HPV), and white matter hyperintensity (WMH)
SNV associations
QQ plots and Manhattan plots for the WGS association analyses of TCBV, HPV, and WMH are presented in figures e-1 and e-2 (links.lww.com/WNL/A32). WGS association analyses revealed at the genome-wide level a new locus in 1p21 for TCBV located between the RWDD3 and PTBP2 genes (top SNV rs181221422, MAF = 0.005, p = 10−8, table 2). A second new locus in 16q23 for HPV within LOC102724084 (top SNV rs9921114, MAF = 0.05, p = 2.7 × 10−8, table 2) was close to the genome-wide level. We confirmed previously identified associations in 12q24 for HPV (rs7294919, MAF = 0.11, p = 4.4 × 10−4) and in 17q25 for WMH (rs7214628, MAF = 0.19, p = 2.0 × 10−3) (table e-1, links.lww.com/WNL/A33). Our best 12q24 HPV association was rs7132910 (MAF = 0.15, p = 3.4 × 10−5) in modest LD with the previously reported association (r2 = 0.49, D' = 0.94 with rs7294919). Conditional analysis revealed that the association observed with rs7132910 was distinct from the one observed with rs7294919 (p = 1.2 × 10−5). The SNP rs7132910 lies at 1.4 kb from the 5′ region of HRK gene and it is an expression quantitative trait locus (eQTL) for HRK and FBXW8 genes in blood.22 Strong promoter histone marks are described in brain tissues.22 Our best 17q25 WMH association was rs9889965 (MAF = 0.15, p = 1.2 × 10−6) in modest LD with the previously reported association (r2 = 0.68, D' = 0.88 with rs7214628). Conditional analysis revealed that the association observed with rs9889965 was distinct from the one observed with rs7214628 (p = 3.0 × 10−5). The SNP rs9889965 lies at 113 bp from the 5′ region of TRIM47 gene and it is an eQTL for TRIM65 gene in nerve tibial tissue and TRIM47 in brain, skin, and blood tissues.22 Strong active transcription starting site histone marks are described in brain tissues.22 Single-SNV results in 12q24 and 17q25 regions are presented in table e-1 and regional plots are presented in figure 1.
Table 2.
Single nucleotide variants (SNVs) in main loci associated with total cerebral brain volume (TCBV) and hippocampal volume (HPV) in single-variant analyses at p ≤ 5 × 10−8
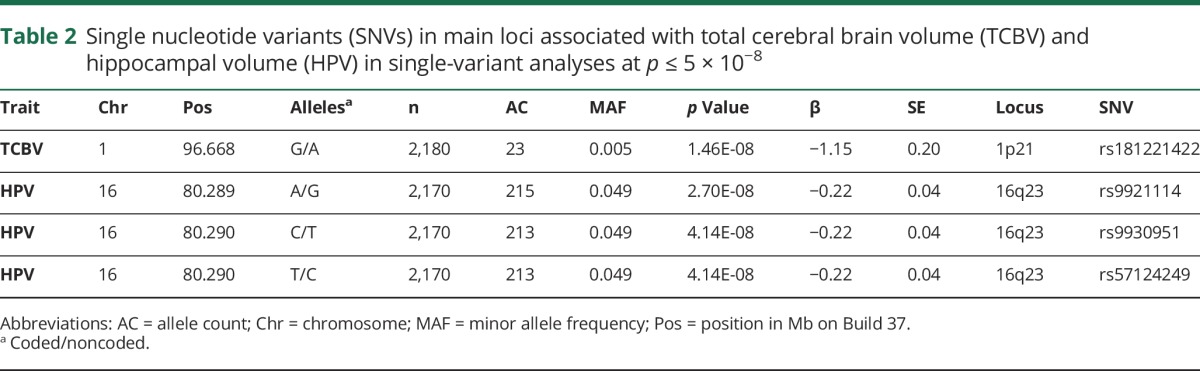
Figure 1. Regional association plots.

Regional association plots of the 12q24 hippocampal volume (HPV) (A) and the 17q25 white matter hyperintensity (WMH) (B) regions using Locuszoom software. Single nucleotide variants (SNVs) are plotted with their −log10 (p values) on the left y-axis as a function of the genomic position on the x-axis. Estimated recombination rates on the right y-axis, taken from 1000 Genomes with European panel, are plotted to reflect the local linkage disequilibrium structure around the top associated SNV in purple and correlated proxies, according to a blue to red scale from r2 = 0 to 1.
Gene-based tests
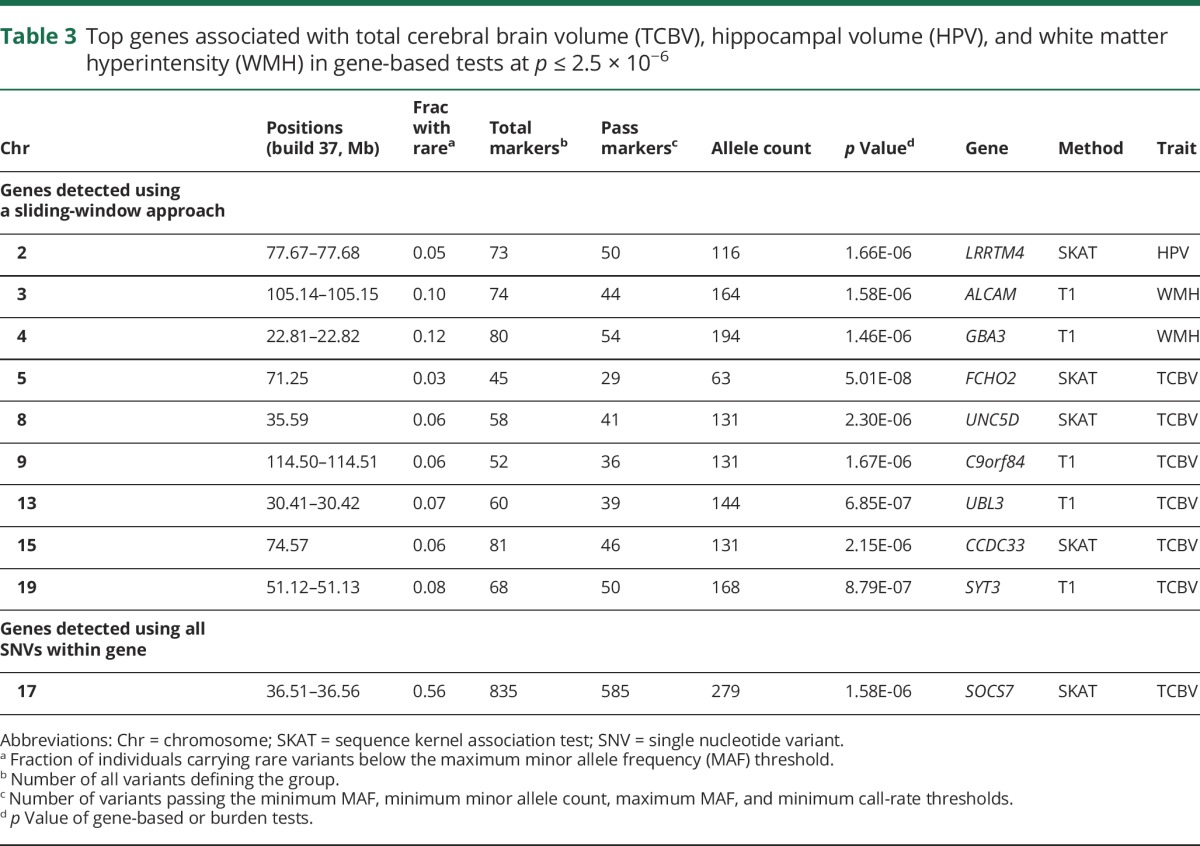
The main gene-based test results are presented in table 3 and regional plots for each gene are provided in figure e-3 (links.lww.com/WNL/A32). Using a sliding-window approach within genes, we detected genome-wide or suggestive associations in new loci for TCBV with the SKAT test (FCHO2-5q13, UNC5D-8p12, CCDC33-15q24) and with the burden test (C9orf84-9q31, UBL3-13q12-q13, SYT3-19q13), for HPV with the SKAT test (LRRTM4-2p12), and for WMH with the burden test (ALCAM-3q13, GBA3-4p15). Using all SNVs within genes, we detected SOCS7 (17q12) significantly associated with TCBV using the SKAT test. No significant results were found when selecting functional exonic SNVs only.
Table 3.
Top genes associated with total cerebral brain volume (TCBV), hippocampal volume (HPV), and white matter hyperintensity (WMH) in gene-based tests at p ≤ 2.5 × 10−6
Pathway analyses
MAGENTA analyses revealed interesting pathways associated with brain MRI phenotypes (table e-2, links.lww.com/WNL/A33). Some of them are linked to immunity or inflammation (B lymphocyte pathway, interleukin-4 and interleukin-6 signaling, antigen presentation pathway) or to AD pathology such as the presenilin pathway. These analyses also confirmed the importance of the ubiquitin proteasome pathway that was found associated with WMH (p = 4 × 10−4, false discovery rate = 0.04 for the 75th percentile enrichment cutoff).
Discussion
We investigated low-frequency or rare SNVs influencing brain MRI phenotypes by performing WGS association analyses in the FHS within the TOPMed.
Using single-variant analyses, we identified rare or low-frequency variants in 1p21 (TCBV) and 16q23 (HPV). In 1p21, SNVs were located at 159 kb from PTBP2 (polypyrimidine tract binding protein 2), a brain-specific homologue of PTBP1. Both genes regulate differentiation of neural precursor cells and promote the proliferation and migration of glioma cell lines.23 In 16q23, the SNVs were located within an ncRNA gene LOC102724084.
Using gene-based tests, we identified 10 potential new genes associated with brain MRI phenotypes. Several of these genes are particularly relevant for brain-related diseases and some are also strongly expressed in the brain.
UNC5D (unc-5 netrin receptor D), related to TCBV, encodes a UNC-5 netrin receptor that plays a role in the regulation of axon guidance and it is strongly expressed in the developing sensory areas of the neocortex in mice24 and in human brain.25 Interestingly, a gene of the same family, UNC5C, was reported associated with AD.26 Moreover, UNC5C genotypes have been found associated with the middle temporal volume and may alter the atrophy of AD regions such as hippocampus and precuneus.27 The expression of C9orf84 (chromosome 9 open reading frame 84), related to TCBV, was found to be upregulated in the hippocampus of individuals with major depression.28 UBL3 (ubiquitin like 3) belongs to the ubiquitin pathway that is implicated in the pathogenesis of neurodegenerative disorders.29,30 UBL3 is strongly expressed in the brain.25 SYT3 (synaptotagmin 3) is highly expressed in cortex, frontal cortex, anterior cingulate cortex, hippocampus, cerebellum, and the cerebral hemispheres. It belongs to a family of brain-specific proteins present on the membrane of synapse vesicles that play a role in the secretion of neurotransmitter.31 LRRTM4 (leucine-rich repeat transmembrane neuronal 4) belongs to the leucine-rich repeat proteins that are important regulators of synapse development and function.32 LRRTM4 was found to regulate excitatory synapse formation in cultured hippocampal neurons.32 LRRTM4 is strongly expressed in the brain.25 ALCAM (activated leukocyte cell adhesion molecule or CD166) is a ligand for CD6 and regulates leukocyte extravasation in the inflamed CNS. ALCAM is expressed on human CNS microvascular endothelium, particularly during neuroinflammatory processes, and plays a key role in the recruitment and migration of leukocytes into the brain.33 GBA3 (glucosylceramidase β3) has been associated with Gaucher disease34 and GBA genes have been associated with late-onset Parkinson and Lewy body dementia.35–37 Furthermore, genetic variants near GBA3 have been found associated with white matter lesion progression.38 SOCS7 (suppressor of cytokine signaling 7) belongs to a family of proteins that play a role in preventing inflammation in the brain. SOCS7 levels were found to be increased in AD human brains.39 SOCS7 is noticeably expressed in murine brain and mice with disrupted SOCS7 gene showed defects in CSF homeostasis.40
The main results were unchanged when including individuals with dementia in the analyses. The significances of ALCAM (WMH), UBL3, and SYRT3 (TCBV) were even improved, whereas LRRTM4 (HPV) was no longer significant, suggesting that this gene may have a role early in the pathogenesis but not in patients with dementia.
The strengths of this study are the population-based sample, the use of quantitative MRI techniques, as well as sequenced data and the fact that we focused on endophenotypes that have substantial variance in our population. This study also has several limitations. First we performed our analyses in a single study. However, the access to datasets with brain MRI phenotypes and WGS data is limited and, to this date, FHS was the only TOPMed phase 1 study with sequenced data and brain MRI measures available. By fine-mapping GWAS regions, we confirmed the association of SNVs that were found associated with the imaging endophenotypes in other studies. A replication or validation in an independent cohort is necessary to confirm the new associations (variants or genes) identified in this study. We were also limited by our sample sizes to discover rare variants (2,180, 2,170, and 1,667 individuals analyzed for TCBV, HPV, and WMH, respectively). Finally, the predominantly European origin of our sample limits the generalization of these results to other ethnic groups.
This FHS WGS-wide search for brain MRI measures reveals rare variants in intriguing new loci associated with brain volumes. Replication of these results in the phase 2 TOPMed studies, whose sequencing is ongoing, is planned to confirm these findings. Further investigation of these loci, such as biological experiments, functional studies, or animal models, will have the potential to validate our findings.
Web resources
TOPMed websites
nhlbi.nih.gov/research/resources/nhlbi-precision-medicine-initiative/topmed
Methods for WGS data acquisition and QC in the FHS are described in a document that is publicly available on dbGaP
ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd006969.1
Acknowledgment
The authors thank the Framingham Heart Study participants, as well as the study team (especially the investigators and staff of the neurology team), for their contributions; whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine Program (TOPMed) was supported by the National Heart, Lung and Blood Institute (NHLBI); WGS for “NHLBI TOPMed: Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study” (phs000974.v1.p1) was performed at the Broad Institute of MIT and Harvard (HHSN268201500014C); centralized read mapping and genotype calling, along with variant quality metrics and filtering, were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1); phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1); the authors thank the studies and participants who provided biological samples and data for TOPMed; Goncalo Abecasis for his work that made the TOPMed data available; Cashell Jaquish for her leadership that has catapulted TOPMed into the largest WGS project and for her support of Framingham within TOPMed; and the contributions of the investigators of the NHLBI TOPMed Consortium (nhlbiwgs.org/topmed-banner-authorship).
Glossary
- AD
Alzheimer disease
- CHARGE
Cohorts for Heart and Aging Research in Genomic Epidemiology
- dbGaP
Database of Genotype and Phenotype
- eQTL
expression quantitative trait locus
- FHS
Framingham Heart Study
- FLAIR
fluid-attenuated inversion recovery
- GWAS
genome-wide association studies
- HPV
hippocampal volume
- LD
linkage disequilibrium
- MAF
minor allele frequency
- MAGENTA
Meta-Analysis Gene-set Enrichment of Variant Associations
- QC
quality control
- SKAT
sequence kernel association test
- SNV
single nucleotide variant
- TCBV
total cerebral brain volume
- TCV
total intracranial volume
- TOPMed
Trans-Omics for Precision Medicine Program
- WGS
whole genome sequence
- WMH
white matter hyperintensity
Contributor Information
Collaborators: TOPMed Neurocognitive Working Group, Donna K Arnett, Diane Becker, Joshua Bis, Eric Boerwinkle, Michael Bowers, Jan Bressler, Adolfo Correa, Anita DeStefano, Jingzhong ding Ding, David Fardo, Myriam Fornage, Alex Garcia Lugo, David Glahn, Hector Gonzalez, Kathleen Hayden, Susan Heckbert, Karin Hoth, Timothy Hughes, Cashell Jaquish, Xueqiu Jian, Emma Knowles, Lenore Launer, Honghuang Lin, Will Longstreth, Richard Mayeux, Biswapriya Misra, Thomas Mosley, Paul Nyquist, Michael Olivier, Emmanuel Peprah, Bruce Psaty, Shaun Purcell, Jerome Rotter, Chloé Sarnowski, Claudia Satizabal, Gerard Schellenberg, Sudha Seshadri, Jeannette Simino, Jennifer Smith, Sylvia Smoller, Beverly Snively, Sophie Sokolow, Kate Wehr, Lisa Yanek, Qiong Yang, NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, Abe Namiko, Abecasis Goncalo, Albert Christine, Allred Nicholette (Nichole) Palmer, Almasy Laura, Alonso Alvaro, Ament Seth, Anderson Peter, Anugu Pramod, Applebaum-Bowden Deborah, Arking Dan, Arnett Donna K, Ashley-Koch Allison, Aslibekyan Stella, Assimes Tim, Auer Paul, Avramopoulos Dimitrios, Barnard John, Barnes Kathleen, Barr R. Graham, Barron-Casella Emily, Beaty Terri, Becker Diane, Becker Lewis, Beer Rebecca, Begum Ferdouse, Beitelshees Amber, Benjamin Emelia, Bezerra Marcos, Bielak Larry, Bis Joshua, Blackwell Thomas, Blangero John, Boerwinkle Eric, Borecki Ingrid, Bowler Russell, Brody Jennifer, Broeckel Ulrich, Broome Jai, Bunting Karen, Burchard Esteban, Cardwell Jonathan, Carlson Sara, Carty Cara, Casaburi Richard, Casella James, Chaffin Mark, Chang Christy, Chasman Daniel, Chavan Sameer, Chen Bo-Juen, Chen Wei-Min, Chen Yii-Der Ida, Cho Michael, Choi Seung Hoan, Chuang Lee-Ming, Chung Mina, Cornell Elaine, Correa Adolfo, Crandall Carolyn, Crapo James, Cupples L Adrienne, Curran Joanne, Curtis Jeffrey, Custer Brian, Damcott Coleen, Darbar Dawood, Das Sayantan, David Sean, Davis Colleen, Daya Michelle de Andrade Mariza, DeBaun Michael, Deka Ranjan, DeMeo Dawn, Devine Scott, Do Ron, Duan Qing, Duggirala Ravi, Durda Peter, Dutcher Susan, Eaton Charles, Ekunwe Lynette, Ellinor Patrick, Emery Leslie, Farber Charles, Farnam Leanna, Fingerlin Tasha, Flickinger Matthew, Fornage Myriam, Franceschini Nora, Fu Mao, Fullerton Malia, Fulton Lucinda, Gabriel Stacey, Gan Weiniu, Gao Yan, Gass Margery, Geng Xiaoqi (Priscilla), Germer Soren, Gignoux Chris, Gladwin Mark, Glahn David, Gogarten Stephanie, Gong Da-Wei, Goring Harald, Gu C. Charles, Guan Yue, Guo Xiuqing, Haessler Jeff, Hall Michael, Harris Daniel, Hawley Nicola, He Jiang, Heavner Ben, Heckbert Susan, Hernandez Ryan, Herrington David, Hersh Craig, Hidalgo Bertha, Hixson James, Hokanson John, Hong Elliott, Hoth Karin, Hsiung Chao (Agnes), Huston Haley, Hwu Chii Min, Irvin Marguerite Ryan, Jackson Rebecca, Jain Deepti, Jaquish Cashell, Jhun Min A, Johnsen Jill, Johnson Andrew, Johnson Craig, Johnston Rich, Jones Kimberly, Kang Hyun Min, Kaplan Robert, Kardia Sharon, Kathiresan Sekar, Kaufman Laura, Kelly Shannon, Kenny Eimear, Kessler Michael, Khan Alyna, Kinney Greg, Konkle Barbara, Kooperberg Charles, Kramer Holly, Krauter Stephanie, Lange Christoph, Lange Ethan, Lange Leslie, Laurie Cathy, Laurie Cecelia, LeBoff Meryl, Lee Seunggeun Shawn, Lee Wen-Jane, LeFaive Jonathon, Levine David, Levy Dan, Lewis Joshua, Li Yun, Lin Honghuang, Lin Keng Han, Liu Simin, Liu Yongmei, Loos Ruth, Lubitz Steven, Lunetta Kathryn, Luo James, Mahaney Michael, Make Barry, Manichaikul Ani, Manson JoAnn, Margolin Lauren, Martin Lisa, Mathai Susan, Mathias Rasika, McArdle Patrick, McDonald Merry-Lynn, McFarland Sean, McGarvey Stephen, Mei Hao, Meyers Deborah A, Mikulla Julie, Min Nancy, Minear Mollie, Minster Ryan L, Mitchell Braxton, Montasser May E, Musani Solomon, Mwasongwe Stanford, Mychaleckyj Josyf C, Nadkarni Girish, Naik Rakhi, Natarajan Pradeep, Nekhai Sergei, Nickerson Deborah, North Kari, O'Connell Jeff, O'Connor Tim, Ochs-Balcom Heather, Pankow James, Papanicolaou George, Parker Margaret, Parsa Afshin, Pattison Jessica Tangarone, Penchev Sara, Peralta Juan Manuel, Perez Marco, Perry James, Peters Ulrike, Peyser Patricia, Phillips Larry, Phillips Sam, Pollin Toni, Post Wendy, Powers Becker Julia, Preethi Boorgula Meher, Preuss Michael, Prokopenko Dmitry, Psaty Bruce, Qasba Pankaj, Qiao Dandi, Qin Zhaohui, Rafaels Nicholas, Raffield Laura, Ramachandran Vasan, D.C. Rao, Rasmussen-Torvik Laura, Ratan Aakrosh, Redline Susan, Reed Robert, Regan Elizabeth, Reiner Alex, Rice Ken, Rich Stephen, Roden Dan, Roselli Carolina, Rotter Jerome, Ruczinski Ingo, Russell Pamela, Ruuska Sarah, Ryan Kathleen, Sakornsakolpat Phuwanat, Salimi Shabnam, Salzberg Steven, Sandow Kevin, Sankaran Vijay, Scheller Christopher, Schmidt Ellen, Schwander Karen, Schwartz David, Sciurba Frank, Sheehan Vivien, Shetty Amol, Shetty Aniket, Sheu Wayne Hui-Heng, Shoemaker M. Benjamin, Silver Brian, Silverman Edwin, Smith Jennifer, Smith Josh, Smith Nicholas, Smith Tanja, Smoller Sylvia, Snively Beverly, Sofer Tamar, Sotoodehnia Nona, Stilp Adrienne, Streeten Elizabeth, Sung Yun Ju, Sylvia Jody, Szpiro Adam, Sztalryd Carole, Taliun Daniel, Tang Hua, Taub Margaret, Taylor Kent, Taylor Simeon, Telen Marilyn, Thornton Timothy A, Tinker Lesley, Tirschwell David, Tiwari Hemant, Tracy Russell, Tsai Michael, Vaidya Dhananjay, VandeHaar Peter, Vrieze Scott, Walker Tarik, Wallace Robert, Walts Avram, Wan Emily, Wang Fei Fei, Watson Karol, Weeks Daniel E, Weir Bruce, Weiss Scott, Weng Lu-Chen, Willer Cristen, Williams Kayleen, Williams L. Keoki, Wilson Carla, Wilson James, Wong Quenna, Xu Huichun, Yanek Lisa, Yang Ivana, Yang Rongze, Zaghloul Norann, Zhang Yingze, Zhao Snow Xueyan, Zhao Wei, Zheng Xiuwen, Zhi Degui, Zhou Xiang, Zody Michael, and Zoellner Sebastian
Author contributions
Chloé Sarnowski: acquisition and analysis of data, drafting a significant portion of the manuscript or figures. Claudia L. Satizabal: acquisition and analysis of data, drafting a significant portion of the manuscript or figures. Charles DeCarli: acquisition and analysis of data. Achilleas N. Pitsillides: acquisition and analysis of data. L. Adrienne Cupples: acquisition and analysis of data. Ramachandran S. Vasan: acquisition and analysis of data. James G. Wilson: acquisition and analysis of data. Joshua C. Bis: conception and design of the study, acquisition and analysis of data. Myriam Fornage: conception and design of the study, acquisition and analysis of data. Alexa S. Beiser: conception and design of the study, acquisition and analysis of data. Anita L. DeStefano: conception and design of the study, acquisition and analysis of data. Josée Dupuis: conception and design of the study, drafting a significant portion of the manuscript or figures. Sudha Seshadri: conception and design of the study, acquisition and analysis of data, drafting a significant portion of the manuscript or figures. Review of the manuscript: all authors.
Study funding
Supported by the National Heart, Lung and Blood Institute's Framingham Heart Study (contract N01-HC-25195 and HHSN268201500001I); and grants from the National Institute on Aging (R01s AG033193, AG008122, AG054076, AG049505, AG016495, AG049607, NS017950) and the National Heart, Lung and Blood Institute (UH2 NS100605, R01 HL093029, HL096917). Dr. DeCarli is supported by the UCD Alzheimer's Disease Center (P30 AG 010129).
Disclosure
C. Sarnowski, C. Satizabal, C. DeCarli, A. Pitsillides, L. Cupples, R. Vasan, J. Wilson, J. Bis, M. Fornage, A. Beiser, and A. DeStefano report no disclosures relevant to the manuscript. J. Dupuis reports grants from NIH during the conduct of the study. S. Seshadri reports no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Braskie MN, Ringman JM, Thompson PM. Neuroimaging measures as endophenotypes in Alzheimer's disease. Int J Alzheimers Dis 2011;2011:490140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bis JC, DeCarli C, Smith AV, et al. Common variants at 12q14 and 12q24 are associated with hippocampal volume. Nat Genet 2012;44:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hibar DP, Stein JL, Renteria ME, et al. Common genetic variants influence human subcortical brain structures. Nature 2015;520:224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein JL, Medland SE, Vasquez AA, et al. Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet 2012;44:552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traylor M, Zhang CR, Adib-Samii P, et al. Genome-wide meta-analysis of cerebral white matter hyperintensities in patients with stroke. Neurology 2016;86:146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verhaaren BF, Debette S, Bis JC, et al. Multiethnic genome-wide association study of cerebral white matter hyperintensities on MRI. Circ Cardiovasc Genet 2015;8:398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fornage M, Debette S, Bis JC, et al. Genome-wide association studies of cerebral white matter lesion burden: the CHARGE consortium. Ann Neurol 2011;69:928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dawber TR, Kannel WB. The Framingham Study: an epidemiological approach to coronary heart disease. Circulation 1966;34:553–555. [DOI] [PubMed] [Google Scholar]
- 9.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study: design and preliminary data. Prev Med 1975;4:518–525. [DOI] [PubMed] [Google Scholar]
- 10.Splansky GL, Corey D, Yang Q, et al. The third generation cohort of the National Heart, Lung, and Blood Institute's Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol 2007;165:1328–1335. [DOI] [PubMed] [Google Scholar]
- 11.DeCarli C, Massaro J, Harvey D, et al. Measures of brain morphology and infarction in the Framingham Heart Study: establishing what is normal. Neurobiol Aging 2005;26:491–510. [DOI] [PubMed] [Google Scholar]
- 12.Kaur B, Himali JJ, Seshadri S, et al. Association between neuropathology and brain volume in the Framingham Heart Study. Alzheimer Dis Assoc Disord 2014;28:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeCarli C, Fletcher E, Ramey V, Harvey D, Jagust WJ. Anatomical mapping of white matter hyperintensities (WMH): exploring the relationships between periventricular WMH, deep WMH, and total WMH burden. Stroke 2005;36:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeCarli C, Miller BL, Swan GE, et al. Predictors of brain morphology for the men of the NHLBI twin study. Stroke 1999;30:529–536. [DOI] [PubMed] [Google Scholar]
- 15.Aljabar P, Heckemann RA, Hammers A, Hajnal JV, Rueckert D. Multi-atlas based segmentation of brain images: atlas selection and its effect on accuracy. Neuroimage 2009;46:726–738. [DOI] [PubMed] [Google Scholar]
- 16.Carmichael O, Mungas D, Beckett L, et al. MRI predictors of cognitive change in a diverse and carefully characterized elderly population. Neurobiol Aging 2012;33:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu C, Tachmazidou I, Walter K, Ciampi A, Zeggini E, Greenwood CM; UK10K Consortium. Estimating genome-wide significance for whole-genome sequencing studies. Genet Epidemiol 2014;38:281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin WY, Lou XY, Gao G, Liu N. Rare variant association testing by adaptive combination of P-values. PLoS One 2014;9:e85728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang R, Feng T, Sha Q, Zhang S. A variable-sized sliding-window approach for genetic association studies via principal component analysis. Ann Hum Genet 2009;73:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segre AV; DIAGRAM Consortium, MAGIC Investigators, Groop L, Mootha VK, Daly MJ, Altshuler D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 2010;6:e100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res 2016;44:D877–D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheung HC, Hai T, Zhu W, et al. Splicing factors PTBP1 and PTBP2 promote proliferation and migration of glioma cell lines. Brain 2009;132:2277–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takemoto M, Hattori Y, Zhao H, et al. Laminar and areal expression of unc5d and its role in cortical cell survival. Cereb Cortex 2011;21:1925–1934. [DOI] [PubMed] [Google Scholar]
- 25.GTEx Consortium. Human genomics: the genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wetzel-Smith MK, Hunkapiller J, Bhangale TR, et al. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat Med 2014;20:1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun JH, Wang HF, Zhu XC, et al. The impact of UNC5C genetic variations on neuroimaging in Alzheimer's disease. Mol Neurobiol 2016;53:6759–6767. [DOI] [PubMed] [Google Scholar]
- 28.Kim S, Hwang Y, Webster MJ, Lee D. Differential activation of immune/inflammatory response-related co-expression modules in the hippocampus across the major psychiatric disorders. Mol Psychiatry 2016;21:376–385. [DOI] [PubMed] [Google Scholar]
- 29.Debette S, Ibrahim Verbaas CA, Bressler J, et al. Genome-wide studies of verbal declarative memory in nondemented older people: the cohorts for heart and aging research in genomic epidemiology consortium. Biol Psychiatry 2015;77:749–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ying Z, Wang H, Wang G. The ubiquitin proteasome system as a potential target for the treatment of neurodegenerative diseases. Curr Pharm Des 2013;19:3305–3314. [DOI] [PubMed] [Google Scholar]
- 31.Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Sudhof TC. Ca(2+)-dependent and -independent activities of neural and non-neural synaptotagmins. Nature 1995;375:594–599. [DOI] [PubMed] [Google Scholar]
- 32.de Wit J, O'Sullivan ML, Savas JN, et al. Unbiased discovery of glypican as a receptor for LRRTM4 in regulating excitatory synapse development. Neuron 2013;79:696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cayrol R, Wosik K, Berard JL, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol 2008;9:137–145. [DOI] [PubMed] [Google Scholar]
- 34.Beutler E, Beutler L, West C. Mutations in the gene encoding cytosolic beta-glucosidase in Gaucher disease. J Lab Clin Med 2004;144:65–68. [DOI] [PubMed] [Google Scholar]
- 35.Gamez-Valero A, Prada-Dacasa P, Santos C, et al. GBA mutations are associated with earlier onset and male sex in dementia with Lewy bodies. Mov Disord 2016;31:1066–1070. [DOI] [PubMed] [Google Scholar]
- 36.Geiger JT, Ding J, Crain B, et al. Next-generation sequencing reveals substantial genetic contribution to dementia with Lewy bodies. Neurobiol Dis 2016;94:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swan M, Doan N, Ortega RA, et al. Neuropsychiatric characteristics of GBA-associated Parkinson disease. J Neurol Sci 2016;370:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofer E, Cavalieri M, Bis JC, et al. White matter lesion progression: genome-wide search for genetic influences. Stroke 2015;46:3048–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walker DG, Whetzel AM, Lue LF. Expression of suppressor of cytokine signaling genes in human elderly and Alzheimer’s disease brains and human microglia. Neuroscience 2015;302:121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krebs DL, Metcalf D, Merson TD, et al. Development of hydrocephalus in mice lacking SOCS7. Proc Natl Acad Sci USA 2004;101:15446–15451. [DOI] [PMC free article] [PubMed] [Google Scholar]