

ABSTRACT
Undefined mesophilic mixed (DL) starter cultures are used in the production of continental cheeses and contain unknown strain mixtures of Lactococcus lactis and leuconostocs. The choice of starter culture affects the taste, aroma, and quality of the final product. To gain insight into the diversity of Lactococcus lactis strains in starter cultures, we whole-genome sequenced 95 isolates from three different starter cultures. Pan-genomic analyses, which included 30 publically available complete genomes, grouped the strains into 21 L. lactis subsp. lactis and 28 L. lactis subsp. cremoris lineages. Only one of the 95 isolates grouped with previously sequenced strains, and the three starter cultures showed no overlap in lineage distributions. The culture diversity was assessed by targeted amplicon sequencing using purR, a core gene, and epsD, present in 93 of the 95 starter culture isolates but absent in most of the reference strains. This enabled an unprecedented discrimination of starter culture Lactococcus lactis and revealed substantial differences between the three starter cultures and compositional shifts during the cultivation of cultures in milk.
IMPORTANCE In contemporary cheese production, standardized frozen seed stock starter cultures are used to ensure production stability, reproducibility, and quality control of the product. The dairy industry experiences significant disruptions of cheese production due to phage attacks, and one commonly used countermeasure to phage attack is to employ a starter rotation strategy, in which two or more starters with minimal overlap in phage sensitivity are used alternately. A culture-independent analysis of the lactococcal diversity in complex undefined starter cultures revealed large differences between the three starter cultures and temporal shifts in lactococcal composition during the production of bulk starters. A better understanding of the lactococcal diversity in starter cultures will enable the development of more robust starter cultures and assist in maintaining the efficiency and stability of the production process by ensuring the presence of key bacteria that are important to the characteristics of the product.
KEYWORDS: amplicon, comparative, dairy, diversity, eps, genomics, lactococcus, Lactococcus lactis, sequencing, starter cultures
INTRODUCTION
Mesophilic mixed starter (DL) cultures used in the production of continental cheeses are composed of undefined mixtures of Lactococcus lactis subsp. lactis, Lactococcus lactis subsp. cremoris, Lactococcus lactis subsp. lactis bv. diacetylactis, and Leuconostoc spp. The latter two provide aroma and texture to the cheese product (1), while L. lactis subsp. lactis and L. lactis subsp. cremoris are the major contributors to the acidification process through the fermentation of lactose. Typically, contemporary starter cultures originate from traditional dairy farm cheese production based on back-slopping starter bacteria from one production to the next. Back-slopping facilitated the coevolution of unknown numbers of strains and their bacteriophages, giving each dairy farm culture its distinct microbial composition, inherently withstanding phage attack (2).
In industrialized cheese production, standardized starter cultures are used to ensure reproducible technical and sensory properties of the product. To preserve their microbial composition, commercial starter cultures are manufactured from frozen seed stock cultures, and care is taken to minimize composition changes during the production process. Even though the starter cultures are standardized, little is known about the microbial diversity and community interactions in the cultures (3). Bacteriophages infecting L. lactis subsp. lactis and L. lactis subsp. cremoris are ubiquitous in dairies and can negatively affect the production process and the quality of the final product (4, 5). Starter cultures originating from traditional cheese farms are considered more robust against phage attack than defined cultures (2), a characteristic gained from their large number of strains with diverse phage sensitivity (6). Because industrial cheese production is dependent on predictable starter culture performance, the use of a frozen batch inoculum is often preferred to back-slopping. This effectively halts the lactococcal evolution, while giving phages the advantage of evolving freely in the dairy environment (5). Thus, the dairy industry experiences significant disruptions of cheese production due to phage attacks.
One countermeasure to phage attack is to employ a starter rotation strategy, in which two or more starters with minimal overlap in phage sensitivity are used alternately. However, the choice of starter culture may affect the taste, aroma, and quality of the final product. Since very little knowledge exists on the genetic diversity of the bacteria or the microbial composition constituting undefined DL starters, it is difficult to decide which starters to use in a rotation strategy (7). Bacteriophages are frequently found in the dairy environment, often in very high titers (4, 8, 9). However, in fermentation failures with DL starter cultures, the diversity of phages rather than their quantity appears to be more important (4).
Knowledge on the microbial diversity of starter cultures is limited, and the complexity and diversity of DL starter cultures beyond the subspecies level are unknown (2). To better predict production performance and advise functional culture rotation strategies, it is of the utmost importance to characterize the strain diversity of DL and other undefined starter cultures. Moreover, the identification of key starter culture strains important to the character of the product will drastically improve the ability to assess the impact of phage attack. With the advances in high-throughput DNA sequencing technology in recent years and the significant increase in lactococcal genomic data available to the scientific community, new opportunities have emerged to achieve this. Here, we present a pangenomic differentiation of lactococci obtained from DL starter cultures and show significant differences in the lactococcal diversity between DL starter cultures using targeted amplicon sequencing.
RESULTS
Isolation and whole-genome sequencing of bacteria.
The microbial diversity of three commercially available DL starter cultures (A, B, and C) was assessed, mainly focusing on culture A. The starter cultures were acquired from three different culture manufacturers. To increase the likelihood of high-diversity representation, two different growth media and phage typing were used (10). Focusing on culture A, 66 isolates were selected from starter culture A and complemented with 15 isolates from culture B and 14 isolates from culture C. The 95 lactococcal isolates were whole-genome sequenced on an Illumina MiSeq platform. Thirty complete Lactococcus lactis genome sequences acquired from the National Center for Biotechnology Information (NCBI) were also included in the study as reference genomes.
Pan-/core-genome analysis.
All the coding sequences (CDS) in the genomes were compared by a blast all-against-all approach to identify orthologous gene groups (OGs) and construct pan- and core matrices. The pan- and core-genome sizes were determined at 8,064 OGs and 551 OGs, respectively (Fig. 1). A pangenomic differentiation of isolates using hierarchal clustering on the pan-matrix clearly separated L. lactis subsp. lactis from L. lactis subsp. cremoris (Fig. 2), as did the core-genome analysis using 551 genes to construct a phylogenetic supertree (Fig. 3). An analysis of the 127 Lactococcus lactis genomes (see Table S1 in the supplemental material) showed that 64 of these belonged to L. lactis subsp. cremoris and 63 to L. lactis subsp. lactis. Interestingly, an analysis of 16S rRNA genes revealed that a number of isolates (CF103, CF117, CF128, CF129, CF207, CF223, and CF229), all identified as L. lactis subsp. cremoris in the pan- and core-genome analyses, contain a novel and unique 16S rRNA gene sequence more closely related to an L. lactis subsp. lactis type than an L. lactis subsp. cremoris type (see Fig. S1). An analysis of the 16S rRNA gene sequences confirmed that all 16S rRNA gene copies in the genomes are of this novel variant. Discrepancies in subspecies identification of lactococci using 16S rRNA genes have also been reported in previous studies (11, 12).
FIG 1.
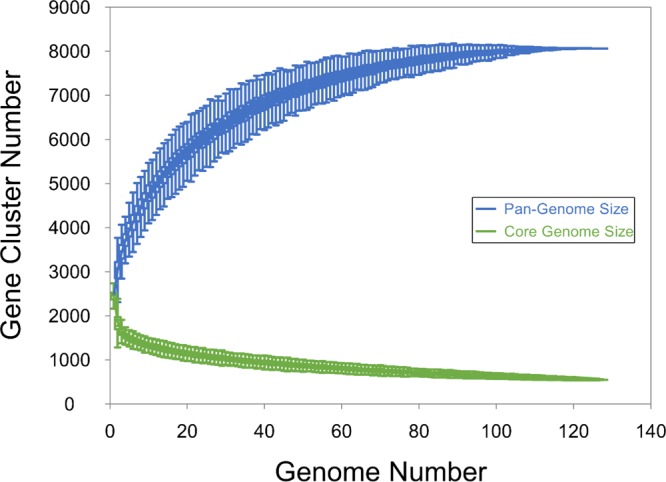
Pan- and core-genome sizes for 127 Lactococcus lactis isolates using PanGP and the traverse all approach were estimated at 8,064 and 551, respectively.
FIG 2.

Heatmap representation of the pangenome contents of 127 Lactococcus lactis isolates. The black regions indicate orthologous groups that are present, while the gray regions indicate orthologous groups that are absent. Using hierarchal single-linkage clustering with a distance cutoff determined using the knee of the curve approach, 63 L. lactis subsp. lactis isolates clustered into 21 genetic lineages, and the remaining 64 L. lactis subsp. cremoris isolates clustered into 28 genetic lineages.
FIG 3.

Differentiation of Lactococcus lactis using 551 core genes.
Differentiation and clustering of genomes.
Robust genotypic discrimination was achieved by an analysis of the pangenome in combination with nucleotide variations in core genes. This provided high-resolution differentiation of isolates beyond the subspecies level (Fig. 2). The 63 L. lactis subsp. lactis isolates clustered into 21 genetic lineages (L1 to L21), while the 64 L. lactis subsp. cremoris isolates clustered into 28 genetic lineages (C1 to C28) (see Table S2). The L. lactis subsp. lactis isolates from our starter cultures fell into 11 of the 21 lineages (see Table S1 in the supplemental material), while the reference genomes occupied the other ten. Notably, the lineages appear culture specific, as no lineage was represented in more than one culture. The reference strains IL1403, 229, and UC77, all isolated from dairy, belong to the same clade as the starter culture isolates, while the other reference L. lactis subsp. lactis strains showed a more distant relationship to the strains in our starter cultures. The L. lactis subsp. cremoris isolates from our starter cultures clustered into 21 of the 28 lineages. With one exception, we also observed a culture-specific lineage distribution for these isolates (see Table S2 in the supplemental material). One isolate from starter culture B clustered with the reference strains 158, UC509.9, and UC109. As shown in Fig. 3, most of the reference strains and all of our starter culture isolates grouped into two clades. Only the reference strains MG1363, NZ9000, and KW2 did not fall into these clades.
Identification of amplicon targets for strain differentiation.
To devise a scheme for the differentiation and quantification of the microbial diversity in each of the starter cultures by amplicon sequencing, core genes and softcore genes were screened for sequence variation reflecting the genomic differentiation. After the curation of targets, the core gene purR, encoding a purine biosynthesis repressor (13), and the softcore gene epsD, part of the eps capsular polysaccharide biosynthesis operon (14, 15), were selected as amplicon targets. Among the core genes, purR was the candidate with the largest number of unique amplicons, with 25 variants (see Fig. S2). The topology of the phylogenetic tree made using the purR amplicon corresponds to the core-genomic supertree, neither of which provides a resolution sufficient to reflect the genetic lineages defined by the pangenome analysis. Importantly, the discrimination between subspecies using the purR amplicon coincided with the subspecies classification made by the pan- and core-genome analyses. An even larger number of variants among our starter isolates was identified in the softcore gene epsD. This gene was present in all except two of our isolates (CF124 and CF223) but in only 9 of the 30 reference strains and presented with 33 variants (see Fig. S3). Altogether, 26 epsD variants were found in the sequenced strain collection from our starter cultures with a sequence distribution corresponding to the pangenomic lineages. No lineage was represented by more than one epsD sequence variant, but a few lineages (L7 and L12; C2 and C7; C5, C9, and C16; and C6 and C23) shared epsD sequences.
Microbial diversity in the starter cultures.
An assessment of the microbial diversity in three starter cultures was performed by targeted amplicon sequencing of three loci, the V1 to V3 region of the 16S rRNA gene, the purR gene (position 324 to 811), and the epsD gene (position 138 to 604). The quantification of microbial diversity was performed on frozen starter cultures and on bulk starters grown at 22°C for 14 h. The results revealed big differences between the starter cultures, as well as shifts in the microbial compositions during bulk starter manufacture. The amplicon data for the 16S rRNA gene showed large differences in the microbial compositions between the starter cultures (Table 1). All cultures were dominated by L. lactis subsp. cremoris, although this was most prominent in culture B with more than 70% L. lactis subsp. cremoris. Small decreases in L. lactis subsp. cremoris were shown in the cultivation of the bulk starters for all three cultures. The contents of leuconostocs varied from <1% in culture B to 24.6% in culture A and 29.4% in culture C. The relative quantification of lactococcal subspecies was performed using the purR amplicon data as well as the commonly used 97% clustering threshold. By comparing the purR and 16S rRNA gene amplicon data, a substantial underestimation of L. lactis subsp. cremoris was identified in the 16S rRNA gene data (Fig. 4). The discrepancy varied from 4.5% in the bulk starter of culture C to 15.5% in the frozen culture of culture B. This demonstrates the impact of strains containing the 16S rRNA gene sequences, which clutter subspecies identification as described earlier. Moreover, this shows that such sequences are not unique to culture A but are present in all three cultures.
TABLE 1.
Microbial diversity and relative abundances in starter cultures A, B, and C given in percentagesa
| Taxon | Relative abundance (% ± SD) |
|||||
|---|---|---|---|---|---|---|
| Culture A |
Culture B |
Culture C |
||||
| Frozen | Bulk | Frozen | Bulk | Frozen | Bulk | |
| L. lactis subsp. cremoris | 58.8 ± 1.6 | 47.6 ± 1.7 | 77.9 ± 0.7 | 73.8 ± 1 | 47.0 ± 1.8 | 33.4 ± 1.0 |
| L. lactis subsp. lactis | 24.7 ± 0.9 | 27.8 ± 1.4 | 21.4 ± 0.1 | 25.7 ± 1.1 | 34.6 ± 0.2 | 37.2 ± 1.1 |
| Leuconostoc spp. | 16.6 ± 0.7 | 24.6 ± 1.8 | 0.8 ± 0.5 | 0.5 ± 0.1 | 18.4 ± 2.0 | 29.4 ± 0.8 |
Analysis was performed by amplicon sequencing of the V1 to V3 region of 16S rRNA gene, clustered at 97% using vsearch.
FIG 4.

Comparison of the quantification of Lactococcus lactis subspecies in starter cultures using the 16S rRNA gene and purR loci. Compared to purR, 16S underreports the relative amount of L. lactis subsp. cremoris compared to L. lactis subsp. lactis. Standard deviations are in parentheses.
Large strain diversity.
To assess the genetic diversity in the three starter cultures, targeted amplicon sequencing of purR and epsD was performed. Using a 99.5% similarity threshold to cluster the amplicon data into operational taxonomic units (OTUs), large differences between the genetic diversity of the starter cultures were revealed. Moreover, a number of OTUs were found to be specific to their culture, showing that a large proportion of the strains did not overlap among the starter cultures.
The purR amplicon sequences clustered into 17 OTUs (Table 2) and enabled relative quantification corresponding to the core-genomic differentiation of strains as shown in Fig. 3. The results show considerable differences in the purR diversity in the three starter cultures and their corresponding bulk starters (Fig. 5). Of the 17 distinct purR OTUs, 10 were found in culture A, 8 in culture B, and 13 in culture C. Two OTUs unique to culture A, one OTU unique to culture B, and two OTUs unique to culture C were identified. The culture-specific OTUs accounted for a substantial proportion in cultures A and C, amounting to 21.7% and 34.3%, respectively, in frozen cultures and declining during bulk starter cultivation to 13.4% and 20.3%, respectively. Cultures A and B were dominated by OTU2, corresponding to several genetic lineages. The same OTU was also abundant in culture C. A noteworthy difference between the cultures was observed for OTU1, an L. lactis subsp. lactis-type OTU reflecting the higher abundance of L. lactis subsp. lactis in culture C compared to those in cultures A and B. The remaining purR OTUs were detected in all three starter cultures, OTU5, OTU6, OTU9, OTU12, and OTU13 in considerable amounts and OTU10, OTU11, OTU14, OTU15, and OTU16 in trace amounts (Table 2). Five of the 17 OTUs were novel variants not found in any of our genomes.
TABLE 2.
Genetic diversity and relative abundances of Lactococcus lactis OTUs in starter cultures A, B, and C using targeted amplicon sequencing of purR
| OTU IDa | % (mean ± SD) of OTUs |
|||||
|---|---|---|---|---|---|---|
| Culture A |
Culture B |
Culture C |
||||
| Frozen | Bulk | Frozen | Bulk | Frozen | Bulk | |
| OTU1b | 3 ± 0.4 | 5.5 ± 0.3 | 2.3 ± 0.6 | 6.2 ± 1.5 | 16.9 ± 2.2 | 30.5 ± 1.2 |
| OTU2c | 52 ± 1.3 | 50.4 ± 1.5 | 81.4 ± 2 | 75 ± 2.6 | 24.6 ± 2 | 13 ± 0.4 |
| OTU3b | 6.1 ± 0.9 | 9.7 ± 1.2 | 0 ± 0.1 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU4c | 15.6 ± 0.7 | 3.7 ± 1.2 | 0 ± 0.3 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU5c | 2.2 ± 0 | 1.8 ± 0.1 | 3.8 ± 0.2 | 5.4 ± 1.4 | 2 ± 0 | 6.1 ± 0.2 |
| OTU6c | 11.8 ± 0.4 | 14.8 ± 0.7 | 3.4 ± 0 | 2.3 ± 0.2 | 3.3 ± 0.1 | 6.4 ± 0.1 |
| OTU7c | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 19.1 ± 0.6 | 17.4 ± 0.1 |
| OTU8c | 0 ± 0 | 0 ± 0 | 0.9 ± 0.2 | 0 ± 0 | 15.2 ± 0.2 | 2.9 ± 0.2 |
| OTU9c | 2.5 ± 0.1 | 6.2 ± 0.8 | 0 ± 0.1 | 1 ± 0.2 | 5.1 ± 0.1 | 4.4 ± 0.3 |
| OTU10b | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 1.3 ± 0.3 | 1.9 ± 0.4 |
| OTU11c | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0.2 | 1 ± 0.4 | 1.6 ± 0.5 |
| OTU12b | 0.9 ± 0.1 | 1.7 ± 0.1 | 0 ± 0.1 | 1.8 ± 0.3 | 5 ± 1 | 8.8 ± 0.5 |
| OTU13c | 1.2 ± 0 | 1.3 ± 0 | 0 ± 0 | 1.2 ± 0.1 | 1.8 ± 0.2 | 2.5 ± 0.4 |
| OTU14b | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 1.2 ± 0.2 | 0 ± 0.1 |
| OTU15c | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0.2 | 1.1 ± 0.4 |
| OTU16c | 1 ± 0.1 | 1 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0.1 |
| OTU17c | 0 ± 0 | 0 ± 0 | 4.3 ± 0 | 2.5 ± 0 | 0 ± 0 | 0 ± 0.2 |
ID, identification. OTUs were generated by clustering purR sequences at a 99.5% similarity threshold.
OTUs identified as L. lactis subsp. lactis.
OTUs identified as L. lactis subsp. cremoris.
FIG 5.

Lactococcus lactis diversity in three commercial starter cultures (A, B, and C) using targeted amplicon sequencing of purR. OTU1, -3, -10, -12, and -14 were identified as L. lactis subsp. lactis, while OTU2, -4, -5, -6, -7, -8, -9, -11, -13, -15, -16, and -17 were identified as L. lactis subsp. cremoris.
The epsD amplicon sequences clustered into 52 OTUs (Table 3), enabling high-resolution quantification of the genetic diversity among eps-positive strains present in the starter cultures. The results show substantial differences in epsD diversity between the three starter cultures and their corresponding bulk starters (Fig. 6). Of these 52 OTUs, 31 were found in culture A, 28 in culture B, and 18 in culture C. Most of these epsD OTUs, 13 in culture A, 9 in culture B, and 11 in culture C, were culture specific. The specific OTUs amounted to a large proportion of the total population. The OTUs unique to culture A (OTU15, OTU20, OTU24, OTU26, OTU31, OTU36, OTU38, OTU40, OTU41, OTU43, OTU44, OTU48, and OTU49) amounted to 18.9% of the population in the frozen starter and 32.6% in the bulk starter. Culture B-specific OTUs (OTU1, OTU8, OTU14, OTU21, OTU25, OTU33, OTU42, OTU50, and OTU52) amounted to 54.0% of the population in the frozen starter and 52.5% of the population in the bulk starter. Lastly, OTUs unique to culture C (OTU6, OTU7, OTU9, OTU12, OTU17, OTU22, OTU32, OTU35, OTU37, OTU39, and OTU47) amounted to 71.9% of the population in the frozen starter and 65.8% of the population in the bulk starter. This showed that a substantial proportion of the genetic diversity did not overlap among the starter cultures. The remaining 19 OTUs were not culture specific but were highly variable with regard to their abundances and degrees of overlap among the starter cultures. Six of the OTUs (OTU2, OTU3, OTU4, OTU5, OTU10, and OTU11) were found in higher abundances in one of the cultures than in the other two. OTU2 was abundant in cultures A and B but not detected at all in culture C. OTU3 was detected in all cultures, although it was more abundant in culture B than in culture A or C. OTU4, OTU5, and OTU11 were detected in all cultures but were more abundant in culture A than in the other two. Lastly, OTU10 was detected in cultures B and C but not A and was more abundant in culture C than in culture B. The remaining 13 OTUs (OTU13, OTU16, OTU18, OTU19, OTU23, OTU27, OTU28, OTU29, OTU30, OTU34, OTU45, OTU46, and OTU51) were more evenly distributed among the starter cultures. However, they all presented with abundances of ∼2% or lower. The epsD OTUs were all assessed using BLAST to identify closely related sequences. Nineteen of the 52 distinct epsD OTUs were a >99.5% match with our isolates from starter cultures, while the remaining 33 epsD OTUs were new variants. Interestingly, these 33 epsD OTUs did not have an identity higher than 99.4% with any sequences included on the NCBI database, showing that they are indeed novel variants.
TABLE 3.
Genetic diversity and relative abundance of Lactococcus lactis OTUs in starter cultures A, B, and C using targeted amplicon sequencing of the epsD gene
| OTU IDa | % (mean ± SD) of OTUs |
|||||
|---|---|---|---|---|---|---|
| Culture A |
Culture B |
Culture C |
||||
| Frozen | Bulk | Frozen | Bulk | Frozen | Bulk | |
| OTU1b | 0 ± 0 | 0 ± 0 | 28.6 ± 3.6 | 23.9 ± 1.2 | 0 ± 0.3 | 0 ± 0 |
| OTU2c | 24 ± 0.4 | 8.6 ± 2.9 | 9.8 ± 2.5 | 1.6 ± 0.2 | 0 ± 0.5 | 0 ± 0 |
| OTU3b | 3.3 ± 0.5 | 5.6 ± 0.9 | 17.7 ± 2.2 | 32 ± 1.9 | 2.1 ± 0.5 | 1.8 ± 0.2 |
| OTU4b | 18.6 ± 1.1 | 8.6 ± 0.8 | 1.3 ± 0.3 | 0.3 ± 0 | 1 ± 0.3 | 3.5 ± 0.2 |
| OTU5c | 13 ± 0.6 | 20.7 ± 2.7 | 0.8 ± 0.3 | 0.5 ± 0 | 1.6 ± 0.6 | 1.3 ± 0.1 |
| OTU6d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 7.3 ± 0.1 | 26.8 ± 1.5 |
| OTU7b | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 1.9 ± 0 | 4.4 ± 0.3 |
| OTU8b | 0 ± 0 | 0 ± 0 | 13.3 ± 2.7 | 18 ± 0.7 | 0 ± 0 | 0 ± 0 |
| OTU9d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 35.6 ± 1.5 | 2 ± 0.2 |
| OTU10d | 0 ± 0 | 0 ± 0 | 0.8 ± 0 | 1 ± 0.1 | 13.3 ± 1.8 | 21.7 ± 0.8 |
| OTU11c | 8.2 ± 1.3 | 3.7 ± 0.8 | 2.2 ± 0.2 | 0.8 ± 0.1 | 3.8 ± 0.3 | 1.4 ± 0 |
| OTU12d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 2.8 ± 0.3 | 0 ± 0 |
| OTU13c | 1 ± 0 | 1.4 ± 0 | 0.4 ± 0 | 0.6 ± 0 | 1 ± 0.2 | 1.1 ± 0 |
| OTU14c | 0 ± 0 | 0 ± 0 | 3.8 ± 4.7 | 3.4 ± 0.2 | 0 ± 0 | 0 ± 0 |
| OTU15b | 4.3 ± 0.2 | 8.3 ± 0.9 | 0 ± 0.1 | 0 ± 0 | 0 ± 0.1 | 0 ± 0 |
| OTU16c | 1.9 ± 0.1 | 3.2 ± 0.3 | 2.4 ± 1.5 | 2.1 ± 0.3 | 1 ± 0.1 | 0 ± 0 |
| OTU17d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 3.9 ± 0.4 | 9.8 ± 0.1 |
| OTU18c | 1.1 ± 0.2 | 2.2 ± 0.1 | 0.5 ± 0 | 1.2 ± 0 | 1.4 ± 0 | 1.6 ± 0.1 |
| OTU19c | 0.5 ± 0.1 | 1.4 ± 0.3 | 3.7 ± 1 | 2.6 ± 0.5 | 0 ± 0 | 0 ± 0 |
| OTU20b | 1.7 ± 0.3 | 4.3 ± 0.9 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU21d | 0 ± 0 | 0 ± 0 | 2.4 ± 2.4 | 1.2 ± 0.1 | 0 ± 0 | 0 ± 0 |
| OTU22b | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 7.4 ± 1 | 14.6 ± 0.5 |
| OTU23c | 1.5 ± 0.3 | 1.1 ± 0.3 | 1.3 ± 0.3 | 0.4 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU24b | 2.5 ± 0.4 | 4.7 ± 0.5 | 0 ± 0 | 0 ± 0 | 0 ± 0.2 | 0 ± 0 |
| OTU25c | 0 ± 0 | 0.5 ± 0 | 2 ± 1.1 | 2.8 ± 0.1 | 0 ± 0 | 0 ± 0 |
| OTU26d | 0.9 ± 0.1 | 2.8 ± 0.3 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU27b | 1.1 ± 0.2 | 1.9 ± 0.3 | 1 ± 0.2 | 1.4 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU28d | 2 ± 0.1 | 2.7 ± 0.2 | 0 ± 0 | 0 ± 0 | 1.8 ± 0 | 0.9 ± 0 |
| OTU29c | 0 ± 0 | 0.5 ± 0.1 | 1.5 ± 1.3 | 0.8 ± 0 | 1 ± 0.1 | 0.9 ± 0 |
| OTU30d | 1.4 ± 0.2 | 2.3 ± 0.4 | 0.3 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU31c | 2.1 ± 0.2 | 0.4 ± 0.1 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU32d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 3.7 ± 0.2 | 1.3 ± 0.1 |
| OTU33d | 0 ± 0 | 0 ± 0 | 1.9 ± 1.6 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU34c | 2.5 ± 0.2 | 1.1 ± 0.2 | 0.5 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU35c | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 2.5 ± 0.4 | 0 ± 0 |
| OTU36c | 1.8 ± 0.1 | 1.8 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU37d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.4 ± 0 | 2.6 ± 0.3 | 4.1 ± 0.3 |
| OTU38c | 1 ± 0 | 1.4 ± 0.2 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU39d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 4.2 ± 0.3 | 1.7 ± 0 |
| OTU40d | 0.4 ± 0 | 1.4 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU41c | 1.3 ± 0.1 | 0.9 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU42d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.3 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU43d | 1.2 ± 0 | 2.3 ± 0.3 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU44d | 0 ± 0 | 0.6 ± 0.1 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU45c | 0.9 ± 0.2 | 1 ± 0.2 | 1 ± 0.2 | 0.4 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU46c | 0 ± 0 | 0.4 ± 0 | 0 ± 0.1 | 0.4 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU47d | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 1.2 ± 0 |
| OTU48d | 0.7 ± 0.1 | 0.6 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU49d | 1.3 ± 0 | 3 ± 0.5 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| OTU50b | 0 ± 0 | 0 ± 0 | 1.1 ± 0 | 1 ± 0.1 | 0 ± 0 | 0 ± 0 |
| OTU51c | 0 ± 0 | 0.7 ± 0.1 | 0.7 ± 0 | 1.2 ± 0.2 | 0 ± 0 | 0 ± 0 |
| OTU52d | 0 ± 0 | 0 ± 0 | 0.8 ± 0.1 | 1.9 ± 0.2 | 0 ± 0 | 0 ± 0 |
ID, identification. OTUs were generated by clustering epsD sequences at 99.5% similarity threshold.
OTUs identified as L. lactis subsp. lactis.
OTUs identified as L. lactis subsp. cremoris.
OTUs that could not be assigned to a subspecies.
FIG 6.

Lactococcus lactis diversity and relative abundance in starter cultures using targeted amplicon sequencing of epsD. The OTUs identified as L. lactis subsp. lactis were OTU1, -3, -4, -7, -8, -15, -20, -22, -24, -27, and -50. The OTUs identified as L. lactis subsp. cremoris were OTU2, -5, -11, -13, -14, -16, -18, -19, -23, -25, -29, -31, -34, -35, -36, -38, -41, -45, -46, and -51. OTU6, -9, -10, -12, -17, -21, -26, -28, -30, -32, -33, -37, -39, -40, -42, -43, -44, -47, -48, -49, and -52 could not be assigned subspecies.
DISCUSSION
Lactococcus lactis is predominantly associated with cheese production and has been subject to extensive research regarding both phenotypic traits and genetic diversity. While suggested to have originated from the plant environment (11), the genetic content of dairy-associated L. lactis is easily distinguished from that of its nondairy counterpart. Evidence of genome decay in the process of adapting to the dairy environment has been accentuated in both L. lactis subspecies, but to a larger extent in L. lactis subsp. cremoris (16). The distinction between L. lactis subsp. lactis and L. lactis subsp. cremoris was initially based on phenotypic features. Since then, detailed studies on the genetic relatedness of the subspecies have shown that phenotypic features alone are inadequate to identify subspecies (17). Moreover, there is a discrepancy between the subspecies identification determined by phenotypic features and the genotypic identification determined using 16S rRNA gene sequences (18). Strains of L. lactis identified as subspecies cremoris by genotype have been reported to show an L. lactis subsp. lactis phenotype, and vice versa, making the accurate identification and differentiation of isolates a difficult task (18, 19). Using a wide range of molecular fingerprinting methods and sequencing schemes, a large genetic diversity of L. lactis has been shown to exist within the dairy environment (16, 20, 21).
Our analyses of 127 L. lactis genomes clearly showed a large genetic diversity among dairy strains. The high resolution of the pangenome analysis enabled a differentiation beyond the subspecies level, distributing the L. lactis subsp. lactis isolates into 21 genetic lineages, and the L. lactis subsp. cremoris isolates into 28 genetic lineages. A phylogenetic analysis of 551 core genes clearly distinguished between dairy and nondairy lactococci and also separated DL starter culture isolates from isolates obtained from other dairy sources. Moreover, most of the lactococci from our DL starter cultures were found to fall into culture-specific genetic lineages, reflecting a spatially separated evolution of strains. Previously, the overlap in sensitivity to bacteriophages among starter cultures A, B, and C was shown to be minimal (10), corroborating this finding.
The lactococcal population of an undefined mesophilic starter culture was previously divided into seven groups (TIFN1 to TIFN7) on the basis of amplified fragment length polymorphisms (AFLPs) (20), which were quantified in a metagenome data set using group-specific gene markers (3). None of our isolates contained the gene markers specific for TIFN1 to TIFN6. However, 19 of our L. lactis subsp. cremoris isolates did contain the gene marker specific to TIFN7. These include isolates from both media and were scattered among several pangenomic lineages comprising 36 isolates. Interestingly, none of the isolates belonging to lineages C1, C3, C5, C9, C27, and C28 contained the TIFN7 gene marker. This shows that the method of Erkus is not applicable to cheese cultures in general but was specific to their culture. Moreover, it highlights the limitations of using unique loci as genetic markers compared to using the sequence variation in conserved genes in culture-independent analyses of complex microbial communities.
During propagation by back-slopping regimes, the microbial communities of complex starter cultures are sustained (2). However, the composition of the culture may change significantly over shorter time periods depending on growth conditions and phage predation (3). The dairy industry depends on reliable and reproducible culture performance and avoids day-to-day variations by using frozen seed stock cultures, effectively resetting the microbial composition every day of production. Our analyses showed that starter cultures are indeed complex, and our cultures showed very little overlap in their diversities. We found substantial differences in the lactococcal compositions of three starter cultures acquired from three different culture manufacturers and showed that they changed during propagation in milk. Moreover, the cultures are significantly different in their content of leuconostocs. In a previous study, we showed large differences in Leuconostoc diversity between the same cultures (22). Our results do not show how culture compositions can vary between production batches of the same culture. Fluctuations in the community during manufacture have an effect on the functionality of the starter, such as in acidification or flavor formation (23). A composition analysis of the microbial community is an important tool in the work for maintaining culture diversity, assessing the effects of phage attack, and monitoring the performance of the culture. More-reproducible starter compositions can be obtained by adjusting the culture parameters.
Using targeted amplicon sequencing, the downstream data analysis clusters the sequences into OTUs. The OTU assignments are dependent on the DNA sequence similarity threshold, which has typically been set at 97% in studies involving 16S rRNA genes (24). Several studies have pointed out that this threshold is excessively low and suggest the use of a higher threshold (25–27). Recently, the use of single nucleotide polymorphism (SNP) distances or so-called zero-radius OTUs (zOTUs) has become common, and computer programs have been developed to accommodate this (26, 27). The advantages of increasing the threshold are a higher-resolution OTU assignment and a significant reduction in the inflation of OTU abundances by false positives (25). In a review of molecular fingerprinting and culture-independent methods, the authors concluded that a sufficient analytical resolution could only be achieved by the identification of a conserved but highly variable locus for strain discrimination (28). The DNA sequences of protein-coding genes have been shown to be more effective than 16S rRNA genes when distinguishing between very closely related bacteria (28, 29). Typically, housekeeping genes are the preferred targets when differentiating between strains. By these criteria, purR was the best candidate and enabled the differentiation of clades beyond the subspecies level, as well as the differentiation of subspecies, superior to that with use of the 16S rRNA gene. In comparison with our purR analyses, a considerable underestimation of L. lactis subsp. cremoris by use of the 16S rRNA gene was demonstrated. This highlights the advantage of species-specific amplicon targets compared to that of the 16S rRNA gene. However, the sequence variation within the purR amplicon was insufficient to differentiate between many of the genetic lineages. Thus, the variance within the amplicons found among our core genes is not high enough to expose the complexity of DL starter cultures.
By expanding the amplicon search to include softcore genes represented in at least 95% of the genomes, the amplicon able to differentiate the genetic lineages from each other was epsD. The pangenome analyses discerned 33 epsD variants, 27 of which were found in our starter culture isolates. Using this amplicon, an unprecedented resolution of the differentiation between genetic lineages was achieved. Interestingly, the phylogenetic analysis of epsD did not separate L. lactis subsp. lactis from L. lactis subsp. cremoris at the root of the tree as did the purR and 16S rRNA genes. Rather, the subspecies separation was made on branches further out on the tree, a strong indication of horizontal gene transfer. The analysis also identified new epsD sequence variants present in low abundances. The results showed a small, but not zero, overlap in epsD variants among the starter cultures. Part of this overlap emerges from culture-specific genetic lineages clearly separated in the pangenome analysis, but which all contain the same epsD variant and cannot be distinguished from each other in the amplicon analysis. Most of the overlapping OTUs were low abundance OTUs, and a large proportion of the culture population is composed of culture-specific OTUs.
The discovery of epsD as a suitable target for strain differentiation was surprising, as the gene was only present in 9 of the 30 reference strains. The eps operon has been found to be located both on plasmids (30, 31) and on chromosomes (14). The epsD gene was highly represented among the starter culture strains, missing in only two of our 95 starter culture isolates. Apart from the missing eps operon, we were unable to distinguish the two isolates CF124 and CF223 from their nearest pan- and core-genomic neighbors. In the laboratory, strains harboring eps plasmids have been cured of their eps-positive phenotype by serial transfers (30), and no evidence exists that suggests a chromosomal locality confers higher stability over multiple transfers (14). The high degree of sequence variation in the eps operon, and more specifically, the sequence variation in the epsD amplicon, represents evolutionary diversification, indicating a history of selection pressure. Typically, lactococcal strains with different phage sensitivities also contain different exopolysaccharides (EPS), and strains that do not produce EPS have been demonstrated to exhibit phage sensitivities different from strains that do produce EPS (30). Moreover, the production of EPS has been shown to confer resistance to phages (31, 32). Regardless of what might be the cause of the high degree of sequence variation in the epsD gene, its applied use in the discrimination and quantification of lactococcal diversity provides culture-independent, robust, and reproducible data. Moreover, it provides the means to monitor temporal shifts in lactococcal diversity, as well as to compare the genetic diversity of Lactococcus lactis between starter cultures and starter culture batches.
The great rate of advancement in next-generation sequencing technologies over the past decade has been accompanied by a rapid development of bioinformatics applications. The reduced cost of sequencing has promoted whole-genome sequencing of bacterial isolates, and the vast improvements to the downstream analysis of genomic data have taken comparative analysis to a completely new level. The pangenomic analysis of several hundred genomes enables the characterization and differentiation of bacteria and facilitates the development of rapid and robust methods such as targeted amplicon sequencing of discriminatory loci. Dairy starter cultures are simple compared to the complexity of other environmental samples, such as soil or the mammalian gut, and could be a good model for the development of groundbreaking methods for differentiating bacteria. Our method of comparative genome analyses of whole-genome-sequenced isolates provides a robust method of discovering intraspecies gene markers for targeted amplicon sequencing and could be applicable to other microbial niches. The use of purR and epsD as gene markers for Lactococcus lactis enables intraspecies differentiation of genetic lineages in O, L, D, and LD starter cultures. The application of the analysis to a completely new starter culture should be prefaced by initial amplicon sequencing of the culture to assess the culture diversity and possibly complemented by whole-genome sequencing of isolates to ensure the validity and continuity of the analysis.
In conclusion, our comparative genomic analysis enabled the discrimination of 127 Lactococcus lactis genomes into 38 genetic lineages. Substantial compositional differences were revealed between starter cultures and temporal shifts in the lactococcal population during cultivation using targeted amplicon sequencing of epsD. The EPS genotype is highly conserved, yet epsD displays high sequence variability, which enables a culture-independent identification and quantification of Lactococcus lactis. Using high-resolution culture-independent methods such as targeted amplicon sequencing of epsD and purR, a better understanding of the microbial composition of starter cultures can be achieved. This will enable the development of more robust starter cultures and assist in maintaining the stability of the culture by ensuring the presence of key bacteria that are important to the characteristics of the product.
MATERIALS AND METHODS
Cultivation and isolation of strains.
All bacterial strains used in this study are listed in Table S1 in the supplemental material. The media used for cultivation were M17 (33) supplemented with 0.5% (wt/vol) lactose (Merck, Kenilworth, New Jersey, USA) or 10% (wt/vol) skimmed milk powder (TINE SA, Oslo, Norway) supplemented with 50 mM β-glycerophosphate (Sigma-Aldrich, Munich, Germany) (GM) as proposed by Hugenholtz (34). Bulk starters were produced by incubating commercial starter cultures in 10% (wt/vol) skim milk at 22°C for 14 h in triplicates. Commercial starter cultures were suspended in GM to an optical density at 600 nm (OD600) of 1.0, serially diluted in 10% (wt/vol) skim milk, and spread plated on M17 and GM agar plates in triplicates. The plates were incubated at 22°C for 5 days before colonies were picked. Isolates were transferred to M17 and GM broth media, respectively, and cultivated at 22°C for two passages before aliquots were made with 15% (wt/vol) glycerol (Sigma-Aldrich), which were stored at −70°C.
Genome sequencing, assembly, and annotation.
Genomic DNA from lactococcal isolates was extracted from 1 ml of an overnight culture using a Qiagen DNeasy blood and tissue kit (Qiagen, Hilden, Germany). The cells were lysed with 40 mg/ml lysozyme (Qiagen, Hilden, Germany) prior to column purification. DNA libraries were constructed using the Nextera XT DNA Sample Prep kit (Illumina, San Diego, California, USA) according to the manufacturer's instructions and sequenced on an Illumina MiSeq (Illumina, San Diego, CA, USA) platform using V3 chemistry. Raw sequences were adapter trimmed, quality filtered (Q > 20), de novo assembled using SPAdes V3.10.1 (35), and annotated using the Prokka v1.12 pipeline (36). Contigs shorter than 1,000 bp or with less than 5× coverage were removed from each assembly prior to gene annotation. In addition, 30 publically available complete L. lactis subsp. genomes were acquired from the NCBI genomes database (Table S1) (16, 37–49). These genomes were reannotated using the Prokka v1.12 pipeline.
Pan-/core-genomic analysis.
The protein coding sequences of all isolates were compared by an all-against-all approach using BLASTP (50) and grouped into orthologous clusters using GET_HOMOLOGUES v2.0.10 (51). Pan- and core-genome sizes were estimated using the pangenomic analysis tool PanGP v1.0.1 (52). Orthologous groups (OGs) were identified via the Markov cluster algorithm (MCL) with an inflation value of 2.5 (53) and intersected using the compare_clusters.pl script provided with GET_HOMOLOGUES. The orthologous clusters were curated to exclude significantly divergent singletons, which are likely to be the result of erroneous assembly or annotation. A pangenomic presence/absence matrix was constructed including each gene cluster and each genome. Hierarchal single-linkage clustering analysis of this matrix was performed in R (http://www.r-project.org/) to construct a pangenome heatmap overview using the heatmap.2 function included in the Gplots package v2.16 (54) supplemented by the dendextend package v0.18.3 (55). Genes were divided into three categories, namely, core genes, which are present in all genomes, softcore genes, which are present in above 95% of genomes, and pan-genes, which are all the genes present in one or more genomes. Core genes were included in a multilocus multiple alignment scheme to determine the phylogenetic distances between genomes and to construct a WPGMA (weighted pair group method with averaging) phylogenetic supertree using the sequence alignment metric functions in the Decipher v2.0 (56) and MASS v7.3-47 (57) packages in R. A distance cutoff for the number of clusters was determined using the knee of the curve approach (58), binning the isolates into genomic lineages.
Relative quantification of the microbial community in starter cultures.
A compositional analysis of starter cultures was performed in triplicates on total DNA extracted from the starter cultures using 1 ml of the starter culture diluted to an OD600 of 1. The samples were treated with 20 mg/ml lysozyme (Sigma-Aldrich) and 3 U/liter mutanolysin (Sigma-Aldrich), were mechanically lysed using FastPrep (MP Biomedicals) with 0.5 g acid-washed glass beads (<106 μm) (Sigma-Aldrich), and were purified using the Qiagen DNeasy blood and tissue kit (Qiagen). A suitable amplicon target was identified by screening the softcore genes for nucleotide sequence variation using the sequence alignment metric functions in the DECIPHER package v1.16.1 (56). Genes without flanking consensus regions within a <500-bp variable region adequate for differentiation or which did not provide sufficient discrimination between lineages were discarded. The loci purR and epsD and the V1 to V3 region of the 16S rRNA gene were amplified by PCR using the Kapa HiFi PCR kit (Kapa Biosystems, Wilmington, MA, USA) with primers purR-324F (5′-YACTCCATCAAATCTTCGTAAAAT-3′), purR-811R (5′-TGTCATTAAATATATTTCCCAATTGAACA-3′), epsD-138F (5′-KCTTATYGCGGCTGCATT-3′), epsD-604R (5′-GATARTARAGTTCTAAATCTGCTCGT-3′), 16S-44F (5′-GCGTGCCTAATACATGCAAGTYGA-3′), and 16S-536R (5′-CTGCTGGCACGTAKTTAGCCGTCC-3′). Forward (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG) and reverse (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG) Illumina adapter overhangs were added to the 5′ ends of the primers to enable Nextera XT DNA indexing of the PCR products. The libraries were sequenced on the Illumina MiSeq platform using V3 (2 × 300 bp) reagents. The resulting data were paired-end merged and quality filtered using PEAR (59) and clustered using VSEARCH v2.4.3 (60) with error minimization from USEARCH v10.0.240 (61). When quantifying at the species and subspecies levels, the 16S rRNA gene and purR amplicon data were clustered using the common identity level threshold of 97% (62, 63). When quantifying at the level of genetic lineages, the purR and epsD data were clustered by a similarity threshold of 99.5%, corresponding to a nucleotide difference of two single-nucleotide polymorphisms. For taxonomic classification, the resulting OTU was matched against a local BLAST database produced using the lactococcal genomes sequenced in this study as well as the lactococcal genomes available on the NCBI database.
Accession number(s).
The whole-genome project has been deposited at DDBJ/ENA/GenBank under BioProject number PRJEB23772. The 16S, purR, and epsD amplicon data have been deposited at DDBJ/ENA/GenBank under BioProject number PRJEB23335.
Supplementary Material
ACKNOWLEDGMENTS
We thank TINE SA for providing culture samples.
This work was funded by the Norwegian Research Council and TINE SA.
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02199-17.
REFERENCES
- 1.Vedamuthu ER. 1994. The dairy Leuconostoc: use in dairy products. J Dairy Sci 77:2725–2737. doi: 10.3168/jds.S0022-0302(94)77215-5. [DOI] [Google Scholar]
- 2.Smid EJ, Erkus O, Spus M, Wolkers-Rooijackers JC, Alexeeva S, Kleerebezem M. 2014. Functional implications of the microbial community structure of undefined mesophilic starter cultures. Microb Cell Fact 13:S2. doi: 10.1186/1475-2859-13-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erkus O, de Jager VC, Spus M, van Alen-Boerrigter IJ, van Rijswijck IM, Hazelwood L, Janssen PW, van Hijum SA, Kleerebezem M, Smid EJ. 2013. Multifactorial diversity sustains microbial community stability. ISME J 7:2126–2136. doi: 10.1038/ismej.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kleppen HP, Bang T, Nes IF, Holo H. 2011. Bacteriophages in milk fermentations: diversity fluctuations of normal and failed fermentations. Int Dairy J 21:592–600. doi: 10.1016/j.idairyj.2011.02.010. [DOI] [Google Scholar]
- 5.Rousseau GM, Moineau S. 2009. Evolution of Lactococcus lactis phages within a cheese factory. Appl Environ Microbiol 75:5336–5344. doi: 10.1128/AEM.00761-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucher I, Moineau S. 2001. Phages of Lactococcus lactis: an ecological and economical equilibrium. Recent Res Dev Virol 3:243–256. [Google Scholar]
- 7.Jany J-L, Barbier G. 2008. Culture-independent methods for identifying microbial communities in cheese. Food Microbiol 25:839–848. doi: 10.1016/j.fm.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Verreault D, Gendron L, Rousseau GM, Veillette M, Massé D, Lindsley WG, Moineau S, Duchaine C. 2011. Detection of airborne lactococcal bacteriophages in cheese manufacturing plants. Appl Environ Microbiol 77:491–497. doi: 10.1128/AEM.01391-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neve H, Berger A, Heller KJ. 1995. A method for detecting and enumerating airborne virulent bacteriophage of dairy starter cultures. Kieler Milchwirtschaftliche Forschungsberichte 47:193–207. [Google Scholar]
- 10.Frantzen C, Kleppen HP, Holo H. 2016. Use of M17 and a milk-based medium enables isolation of two distinct and diverse populations of Lactococcus lactis strains from undefined mesophilic starter cultures. Int Dairy J 53:45–50. doi: 10.1016/j.idairyj.2015.09.005. [DOI] [Google Scholar]
- 11.Cavanagh D, Casey A, Altermann E, Cotter PD, Fitzgerald GF, McAuliffe O. 2015. Evaluation of Lactococcus lactis isolates from nondairy sources with potential dairy applications reveals extensive phenotype-genotype disparity and implications for a revised species. Appl Environ Microbiol 81:3961–3972. doi: 10.1128/AEM.04092-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu H, Sun Z, Liu W, Yu J, Song Y, Lv Q, Zhang J, Shao Y, Menghe B, Zhang H. 2014. Multilocus sequence typing of Lactococcus lactis from naturally fermented milk foods in ethnic minority areas of China. J Dairy Sci 97:2633–2645. doi: 10.3168/jds.2013-7738. [DOI] [PubMed] [Google Scholar]
- 13.Kilstrup M, Martinussen J. 1998. A transcriptional activator, homologous to the Bacillus subtilis PurR repressor, is required for expression of purine biosynthetic genes in Lactococcus lactis. J Bacteriol 180:3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dabour N, LaPointe G. 2005. Identification and molecular characterization of the chromosomal exopolysaccharide biosynthesis gene cluster from Lactococcus lactis subsp. cremoris SMQ-461. Appl Environ Microbiol 71:7414–7425. doi: 10.1128/AEM.71.11.7414-7425.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forde A, Fitzgerald GF. 2003. Molecular organization of exopolysaccharide (EPS) encoding genes on the lactococcal bacteriophage adsorption blocking plasmid, pCI658. Plasmid 49:130–142. doi: 10.1016/S0147-619X(02)00156-7. [DOI] [PubMed] [Google Scholar]
- 16.Kelleher P, Bottacini F, Mahony J, Kilcawley KN, van Sinderen D. 2017. Comparative and functional genomics of the Lactococcus lactis taxon; insights into evolution and niche adaptation. BMC Genomics 18:267. doi: 10.1186/s12864-017-3650-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godon J-J, Delorme C, Ehrlich SD, Renault P. 1992. Divergence of genomic sequences between Lactococcus lactis subsp. lactis and Lactococcus lactis subsp. cremoris. Appl Environ Microbiol 58:4045–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nomura M, Kobayashi M, Narita T, Kimoto-Nira H, Okamoto T. 2006. Phenotypic and molecular characterization of Lactococcus lactis from milk and plants. J Appl Microbiol 101:396–405. doi: 10.1111/j.1365-2672.2006.02949.x. [DOI] [PubMed] [Google Scholar]
- 19.Kelly WJ, Ward LJH, Leahy SC. 2010. Chromosomal diversity in Lactococcus lactis and the origin of dairy starter cultures. Genome Biol Evol 2:729–744. doi: 10.1093/gbe/evq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kütahya OE, Starrenburg MJC, Rademaker JLW, Klaassen CHW, van Hylckama Vlieg JET, Smid EJ, Kleerebezem M. 2011. High-resolution amplified fragment length polymorphism typing of Lactococcus lactis strains enables identification of genetic markers for subspecies-related phenotypes. Appl Environ Microbiol 77:5192–5198. doi: 10.1128/AEM.00518-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Passerini D, Beltramo C, Coddeville M, Quentin Y, Ritzenthaler P, Daveran-Mingot M-L, Le Bourgeois P. 2010. Genes but not genomes reveal bacterial domestication of Lactococcus Lactis. PLoS One 5:e15306. doi: 10.1371/journal.pone.0015306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frantzen CA, Kot W, Pedersen TB, Ardö YM, Broadbent JR, Neve H, Hansen LH, Dal Bello F, Østlie HM, Kleppen HP, Vogensen FK, Holo H. 2017. Genomic characterization of dairy associated Leuconostoc species and diversity of leuconostocs in undefined mixed mesophilic starter cultures. Front Microbiol 8:132. doi: 10.3389/fmicb.2017.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smit G, Smit BA, Engels WJ. 2005. Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. FEMS Microbiol Rev 29:591–610. doi: 10.1016/j.fmrre.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Stackebrandt E, Goebel BM. 1994. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Evol Microbiol 44:846–849. doi: 10.1099/00207713-44-4-846. [DOI] [Google Scholar]
- 25.Edgar RC. 2017. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. bioRxiv 2017:192211. doi: 10.1101/192211. [DOI] [PubMed] [Google Scholar]
- 26.Edgar RC. 2016. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016:081257. doi: 10.1101/081257. [DOI] [Google Scholar]
- 27.Mahé F, Rognes T, Quince C, de Vargas C, Dunthorn M. 2014. Swarm: robust and fast clustering method for amplicon-based studies. PeerJ 2:e593. doi: 10.7717/peerj.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ndoye B, Rasolofo EA, LaPointe G, Roy D. 2011. A review of the molecular approaches to investigate the diversity and activity of cheese microbiota. Dairy Sci Technol 91:495. doi: 10.1007/s13594-011-0031-8. [DOI] [Google Scholar]
- 29.Palys T, Nakamura LK, Cohan FM. 1997. Discovery and classification of ecological diversity in the bacterial world: the role of DNA sequence data. Int J Syst Bacteriol 47:1145–1156. doi: 10.1099/00207713-47-4-1145. [DOI] [PubMed] [Google Scholar]
- 30.Deveau H, Van Calsteren M-R, Moineau S. 2002. Effect of exopolysaccharides on phage-host interactions in Lactococcus lactis. Appl Environ Microbiol 68:4364–4369. doi: 10.1128/AEM.68.9.4364-4369.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forde A, Fitzgerald GF. 1999. Analysis of exopolysaccharide (EPS) production mediated by the bacteriophage adsorption blocking plasmid, pCI658, isolated from Lactococcus lactis subsp. cremoris HO2. Int Dairy J 9:465–472. doi: 10.1016/S0958-6946(99)00115-6. [DOI] [Google Scholar]
- 32.Moineau S, Borkaev M, Holler BJ, Walker SA, Kondo JK, Vedamuthu ER, Vandenbergh PA. 1996. Isolation and characterization of lactococcal bacteriophages from cultured buttermilk plants in the United States. J Dairy Sci 79:2104–2111. doi: 10.3168/jds.S0022-0302(96)76584-0. [DOI] [Google Scholar]
- 33.Terzaghi BE, Sandine WE. 1975. Improved medium for lactic streptococci and their bacteriophages. Appl Microbiol 29:807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hugenholtz J, Splint R, Konings WN, Veldkamp H. 1987. Selection of protease-positive and protease-negative variants of Streptococcus cremoris. Appl Environ Microbiol 53:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 37.Bolotin A, Wincker P, Mauger S, Jaillon O, Malarme K, Weissenbach J, Ehrlich SD, Sorokin A. 2001. The complete genome sequence of the lactic acid bacterium Lactococcus lactis subsp. lactis IL1403. Genome Res 11:731–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wegmann U, O'Connell-Motherway M, Zomer A, Buist G, Shearman C, Canchaya C, Ventura M, Goesmann A, Gasson MJ, Kuipers OP, van Sinderen D, Kok J. 2007. Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp. cremoris MG1363. J Bacteriol 189:3256–3270. doi: 10.1128/JB.01768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato H, Shiwa Y, Oshima K, Machii M, Araya-Kojima T, Zendo T, Shimizu-Kadota M, Hattori M, Sonomoto K, Yoshikawa H. 2012. Complete genome sequence of Lactococcus lactis IO-1, a lactic acid bacterium that utilizes xylose and produces high levels of l-lactic acid. J Bacteriol 194:2102–2103. doi: 10.1128/JB.00074-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, Shakhova V, Grigoriev I, Lou Y, Rohksar D, Lucas S, Huang K, Goodstein DM, Hawkins T, Plengvidhya V, Welker D, Hughes J, Goh Y, Benson A, Baldwin K, Lee JH, Diaz-Muniz I, Dosti B, Smeianov V, Wechter W, Barabote R, Lorca G, Altermann E, Barrangou R, Ganesan B, Xie Y, Rawsthorne H, Tamir D, Parker C, Breidt F, Broadbent J, Hutkins R, O'Sullivan D, Steele J, Unlu G, Saier M, Klaenhammer T, Richardson P, Kozyavkin S, Weimer B, Mills D. 2006. Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci U S A 103:15611–15616. doi: 10.1073/pnas.0607117103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siezen RJ, Bayjanov J, Renckens B, Wels M, van Hijum SA, Molenaar D, van Hylckama Vlieg JE. 2010. Complete genome sequence of Lactococcus lactis subsp. lactis KF147, a plant-associated lactic acid bacterium. J Bacteriol 192:2649–2650. doi: 10.1128/JB.00276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linares DM, Kok J, Poolman B. 2010. Genome sequences of Lactococcus lactis MG1363 (revised) and NZ9000 and comparative physiological studies. J Bacteriol 192:5806–5812. doi: 10.1128/JB.00533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao Y, Lu Y, Teng KL, Chen ML, Zheng HJ, Zhu YQ, Zhong J. 2011. Complete genome sequence of Lactococcus lactis subsp. lactis CV56, a probiotic strain isolated from the vaginas of healthy women. J Bacteriol 193:2886–2887. doi: 10.1128/JB.00358-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolotin A, Quinquis B, Ehrlich SD, Sorokin A. 2012. Complete genome sequence of Lactococcus lactis subsp. cremoris A76. J Bacteriol 194:1241–1242. doi: 10.1128/JB.06629-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ainsworth S, Zomer A, de Jager V, Bottacini F, van Hijum S, Mahony J, van Sinderen D. 2013. Complete genome of Lactococcus lactis subsp. cremoris UC509.9, host for a model lactococcal P335 bacteriophage. Genome Announc 1:e00119-. doi: 10.1128/genomeA.00119-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelly WJ, Altermann E, Lambie SC, Leahy SC. 2013. Interaction between the genomes of Lactococcus lactis and phages of the P335 species. Front Microbiol 4:257. doi: 10.3389/fmicb.2013.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang X, Wang Y, Huo G. 2013. Complete genome sequence of Lactococcus lactis subsp. lactis KLDS4.0325. Genome Announc 1:e00962-. doi: 10.1128/genomeA.00962-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oliveira LC, Saraiva TD, Soares SC, Ramos RT, Sa PH, Carneiro AR, Miranda F, Freire M, Renan W, Junior AF, Santos AR, Pinto AC, Souza BM, Castro CP, Diniz CA, Rocha CS, Mariano DC, de Aguiar EL, Folador EL, Barbosa EG, Aburjaile FF, Goncalves LA, Guimaraes LC, Azevedo M, Agresti PC, Silva RF, Tiwari S, Almeida SS, Hassan SS, Pereira VB, Abreu VA, Pereira UP, Dorella FA, Carvalho AF, Pereira FL, Leal CA, Figueiredo HC, Silva A, Miyoshi A, Azevedo V. 2014. Genome sequence of Lactococcus lactis subsp. lactis NCDO 2118, a GABA-producing strain. Genome Announc 2:e00980-. doi: 10.1128/genomeA.00980-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCulloch JA, de Oliveira VM, de Almeida Pina AV, Pérez-Chaparro PJ, de Almeida LM, de Vasconcelos JM, de Oliveira LF, da Silva DEA, Rogez HLG, Cretenet M, Mamizuka EM, Nunes MRT. 2014. Complete genome sequence of Lactococcus lactis Strain AI06, an endophyte of the Amazonian Açaí palm. Genome Announc 2:e01225-. doi: 10.1128/genomeA.01225-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Contreras-Moreira B, Vinuesa P. 2013. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 79:7696–7701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Y, Jia X, Yang J, Ling Y, Zhang Z, Yu J, Wu J, Xiao J. 2014. PanGP: a tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 30:1297–1299. doi: 10.1093/bioinformatics/btu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Enright AJ, Van Dongen S, Ouzounis CA. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. 2009. gplots: various R programming tools for plotting data. http://cran.r-project.org/web/packages/gplots/index.html.
- 55.Galili T. 2015. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright E. 2016. Using DECIPHER v2.0 to analyze big biological sequence data in R. R J 8:352–359. [Google Scholar]
- 57.Venables WN, Ripley BD. 2002. Modern applied statistics with R, 4th ed Springer, New York, NY. [Google Scholar]
- 58.Salvador S, Chan P. 2004. Determining the number of clusters/segments in hierarchical clustering/segmentation algorithms, p 576–584. In Proceedings of the 16th IEEE International Conference on Tools with Artificial Intelligence. IEEE Computer Society Press, Washington, DC. [Google Scholar]
- 59.Zhang J, Kobert K, Flouri T, Stamatakis A. 2014. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rognes T, Flouri T, Nichols B, Quince C, Mahe F. 2016. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 62.Nguyen N-P, Warnow T, Pop M, White B. 2016. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2:16004. doi: 10.1038/npjbiofilms.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A 102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.