

Abstract
Whole-genome sequencing data allow detection of copy number variation (CNV) at high resolution. However, estimation based on read coverage along the genome suffers from bias due to GC content and other factors. Here, we develop an algorithm called BIC-seq2 that combines normalization of the data at the nucleotide level and Bayesian information criterion-based segmentation to detect both somatic and germline CNVs accurately. Analysis of simulation data showed that this method outperforms existing methods. We apply this algorithm to low coverage whole-genome sequencing data from peripheral blood of nearly a thousand patients across eleven cancer types in The Cancer Genome Atlas (TCGA) to identify cancer-predisposing CNV regions. We confirm known regions and discover new ones including those covering KMT2C, GOLPH3, ERBB2 and PLAG1. Analysis of colorectal cancer genomes in particular reveals novel recurrent CNVs including deletions at two chromatin-remodeling genes RERE and NPM2. This method will be useful to many researchers interested in profiling CNVs from whole-genome sequencing data.
INTRODUCTION
DNA copy number variation (CNV) is a major class of genome variations in the human genome. Germline CNVs are inherited genetic events that could confer susceptibility to various types of cancer (1–3) as well as other diseases (4,5). Somatic CNVs are de novo genetic events that could result in diseases, e.g. deletion of tumor suppressors or amplification of oncogenes can drive tumorigenesis (6). With whole-genome sequencing (WGS) becoming more common, there are opportunities to characterize CNVs with a greater spatial resolution than ever before, but accurate and efficient algorithms must be applied to detect small-scale CNVs while avoiding false positives.
Among the available detection algorithms for WGS data, paired-end mapping (PEM) methods (7–9) identify CNVs by analyzing the configuration of read pairs. These methods are advantageous in their high resolution and their sensitivity for detecting small events. However, when the configuration of CNVs is complex, PEM-based method can have low sensitivity. Read-depth methods (10–16) instead can readily detect these complex CNVs as long as they are relatively large.
Read-depth methods detect CNVs by identifying regions with abnormal sequencing read coverage along the genome, as the presence of CNVs leads to increased or decreased sequencing coverage in those regions. However, sequencing coverage can be easily affected by various biases including the influence of GC-content in the sequenced fragments, the regional variation in the fraction of short reads that can be uniquely aligned, and different nucleotide composition of short reads (17). For somatic CNVs, one strategy for dealing with these biases is to compare to a control (13,14). This approach is model-independent and can capture even unknown sources of biases; but sequencing a control genome doubles the experimental cost, assumes that the biases in the case and control are the same and cannot be used for germline CNVs. Another bias-correction strategy (10–12,15,16) is to explicitly incorporate a mathematical model for bias correction to normalize data. However, most current algorithms (10–12) simply bin the data into equal-sized bins and perform normalization based on the binned data with GC and/or mappability (proportion of uniquely mappable positions in a bin) as explanatory variables. The choice of the bin size is often a subtle but critical parameter, as it has to balance detection resolution and control of the noise level. The best choice of the bin size depends on the read depth: for high depth data, a small bin size can adequately control the noise level, but this is not the case for low depth data. Although equal-sized bins are simple, they can contain a substantially different number of mappable positions, resulting in high variance of some data points that potentially mask real copy number change signals.
In this paper, we present BIC-seq2 for detecting somatic and germline CNVs based on WGS data. Whereas the first version in 2011 used a control genome for normalization (13) and was thus limited to identification of somatic variants, the new version is also applicable to germline variants and is more robust overall. This algorithm first normalizes the sequencing data by taking into account the GC-content, the nucleotide composition of the short reads and the mappability. It then performs segmentation and detects CNVs based on the normalized data using a Bayesian information criterion. Unlike other algorithms, BIC-seq2 performs normalization at a nucleotide level rather than at a large bin level, resulting in its high sensitivity of detection for small CNVs. If there is a control genome or a sample was sequenced on multiple runs, we perform normalization individually for each data set and perform joint segmentation for CNV detection. To demonstrate the superior performance of BIC-seq2, we compared it with a number of other read-depth methods on a set of simulated sequencing data sets and a data set from the 1000 Genome Project (NA12878). We then applied it to data sets from The Cancer Genome Atlas (TCGA) to identify novel regions of cancer susceptibility.
MATERIALS AND METHODS
Overview of BIC-seq2
BIC-seq2 has two main steps, the normalization step and the segmentation step. In the normalization step, BIC-seq2 models the number of reads mapped to a position in the mappability map (i.e. positions to which a read can be aligned under given criteria) to be dependent on local genomic features such as GC-content. This model is a semi-parametric regression model that can calculate the expected number of mapped reads for every position in the mappability map. This regression model accounts for known sources of bias; thus, the ratio between the observed number and the expected number of mapped reads in a region would reflect just the copy number of the region. A significant difference between the ratios of two regions implies that the copy numbers of the two regions are different. In the segmentation step, BIC-seq2 employs the BIC-seq segmentation algorithm (13). This algorithm is based on a non-parametric model and performs segmentation by iteratively merging similar neighboring bins. In each iteration, the neighboring bin pair whose merging will lead to the largest reduction of the Bayesian information criterion (BIC) are merged. At the last step, BIC-seq2 performs post-processing to calculate copy ratios and assign P-values.
In addition to uniquely aligned reads, BIC-seq2 can also incorporate reads that can be aligned to multiple positions (‘multiply-aligned’ reads) if desired. The mapping position of a multiply-aligned read is randomly chosen as one of its mapped positions. If only uniquely aligned reads are used, the mappability map consists of all uniquely mappable positions in the genome; if multiply-aligned reads are considered, it consists of the non-N regions. The uniquely mappable positions were downloaded from the UCSC Genome Browser (CRG mappability track). For a given read length k, a position is considered to be mappable if the k-mer starting at that position has only one mapped position (itself) in the genome while allowing for two mismatches. Supplementary Figure S1A shows the percentages of uniquely mappable positions in the human reference genome hg19 for k = 36, 50, 75 and 100. If only uniquely aligned reads are used, BIC-seq2 has a lower power for CNVs in repetitive DNA; if multiply-aligned reads are used, it has a higher false discovery rate.
Normalization of sequencing data
Statistical model
For every position in the mappability map, the number of reads mapped to this position is counted. In addition to the local copy number, this read count also depends on local genomic features. We employ a generalized additive model (GAM) to describe this dependence. At each genomic position  in the mappability map, we model
in the mappability map, we model  , the number of mapped reads starting at the position
, the number of mapped reads starting at the position  , as a quasi-Poisson model with its mean depending on local genomic features. The random variable
, as a quasi-Poisson model with its mean depending on local genomic features. The random variable  is assumed to have a mean
is assumed to have a mean  , and the mean
, and the mean  depends on the local GC-content and nucleotide compositions of the sequences around the reads. We assume
depends on the local GC-content and nucleotide compositions of the sequences around the reads. We assume
 |
(1) |
where  is the GC-proportion in a local genomic window of
is the GC-proportion in a local genomic window of  ,
,  is the nucleotide compositions around
is the nucleotide compositions around 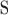 , and
, and  , g are unknown functions. The GC window is chosen as the mean fragment size since GC-bias is mainly introduced in the polymerase chain reaction (PCR) amplification step during library construction (18). This fragment size can be easily estimated with paired-end sequencing data. Nucleotide composition was introduced in the model because it was shown to influence read-depth of WGS, although to a lesser degree than for RNA-seq (17). We only consider nucleotide compositions near the ends of short reads (default 5 bp of the ends) because their influence is restricted to the ends of the reads (17) (also see Supplementary Figure S1B). The choice of the function form of
, g are unknown functions. The GC window is chosen as the mean fragment size since GC-bias is mainly introduced in the polymerase chain reaction (PCR) amplification step during library construction (18). This fragment size can be easily estimated with paired-end sequencing data. Nucleotide composition was introduced in the model because it was shown to influence read-depth of WGS, although to a lesser degree than for RNA-seq (17). We only consider nucleotide compositions near the ends of short reads (default 5 bp of the ends) because their influence is restricted to the ends of the reads (17) (also see Supplementary Figure S1B). The choice of the function form of  can have important effect on how well the regression model can explain the data. One choice is to assume a parametric form such as polynomials of fixed degree (11). The polynomial-based regression is advantageous in its simplicity and its ease of interpretation. However, we observe that the GC-dependence can take various forms (Supplementary Figure S2). If the function form is mis-specified, the model can be biased. Therefore, we choose to use splines, a non-parametric method, to estimate
can have important effect on how well the regression model can explain the data. One choice is to assume a parametric form such as polynomials of fixed degree (11). The polynomial-based regression is advantageous in its simplicity and its ease of interpretation. However, we observe that the GC-dependence can take various forms (Supplementary Figure S2). If the function form is mis-specified, the model can be biased. Therefore, we choose to use splines, a non-parametric method, to estimate  . Since nucleotide compositions are categorical variables, the function g is the summation of indicator functions. Details of the model are given in the Supplementary Text.
. Since nucleotide compositions are categorical variables, the function g is the summation of indicator functions. Details of the model are given in the Supplementary Text.
Model training and refinement
To estimate the parameters in the above GAM, we adopt the method as proposed by Woods (2008) (19). With the human reference genome having over 2 billion uniquely mappable positions, it is very difficult to estimate the parameters with all the data. Therefore, we choose to randomly select a small portion of the entire data set (e.g. 1%) and train the model only based on the randomly sampled data. After fitting the GAM, an expected read count can be calculated for each mappable position. However, we observe that the ratio between the observed and the expected number of reads still have some correlation with the GC-content (Supplementary Figure S3), possibly due to the insufficient sampling of regions with extreme GC-contents.
To remove this correlation, we introduce an additional step to refine the predicted value by the GAM. This refinement step captures the remaining GC-dependence on a relatively large bin level. Here the bins are variable-sized bins containing the same number of mappable positions. The log ratios of the observed and the expected number of reads in the bins are regressed against the proportions of GC in the bins. The regression is again based on a GAM. If the model (1) captures all GC-dependence, the fitted curve of the log ratio model should be a horizontal straight line at y = 0, i.e. for each GC-proportion, its fitted value of log ratio should be zero or close to zero. The deviance of the fitted value from zero is the uncaptured dependence on GC-content. We then adjust the expected number of reads given by model (1) using fitted values from this log-ratio-vs-GC regression model. Briefly, let 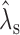 be the predicted read count at a mappable position
be the predicted read count at a mappable position  , and
, and  be its local GC-proportion. Suppose that
be its local GC-proportion. Suppose that  is the fitted value for
is the fitted value for 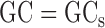 from the log-ratio-vs-GC regression model. We refine the expected read count at
from the log-ratio-vs-GC regression model. We refine the expected read count at  to be
to be  . Details of the refinement step are given in the Supplementary Text.
. Details of the refinement step are given in the Supplementary Text.
Segmentation and post-processing
After normalization, BIC-seq2 utilizes the BIC-seq segmentation algorithm to perform segmentation. BIC-seq detects somatic CNVs by comparing a tumor genome with its matched genome. Given an initial set of bins, BIC-seq performs segmentation based on each bin's read counts of the tumor and normal genomes, and the bins are iteratively merged to give the final segmentation. Specifically, suppose that at one merging step, the remaining bins are 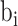 (
( ). Let
). Let  and
and  be the numbers of reads for the case and the control genome in the bin
be the numbers of reads for the case and the control genome in the bin  . The BIC is defined as
. The BIC is defined as
 |
(2) |
where 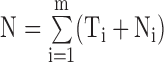 and
and  is a tuning parameter. BIC-seq merges the bin pair
is a tuning parameter. BIC-seq merges the bin pair 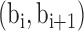 such that the merging can give the largest reduction of the BIC. BIC-seq stops merging if merging of any bin pair cannot further reduce the BIC. In BIC-seq2, let
such that the merging can give the largest reduction of the BIC. BIC-seq stops merging if merging of any bin pair cannot further reduce the BIC. In BIC-seq2, let  and
and  be the observed and expected numbers of reads in the bin
be the observed and expected numbers of reads in the bin  . BIC-seq2 sets
. BIC-seq2 sets 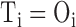 and
and 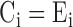 in (2) to perform the segmentation.
in (2) to perform the segmentation.
Because equal-sized bins can contain substantially different numbers of mappable positions, the number of reads in those bins tend to have higher variance, potentially masking real copy number changes (Figure 1A). Even if we remove the bins with low mappability, the number of reads in the remaining bins still tend to have high variance, resulting in ambiguous copy number states (Supplementary Figure S4). When we choose the initial bins as variable-sized bins with each bin containing the same number of mappable positions, the copy number states become much clearer (Figure 1B). Note that variable-sized bins contain equal numbers of mappable positions, but not equal number of mapped reads, as is done in some methods. After segmentation, the log2 copy ratio of each segment is defined as the log2 ratio of the observed read count and the expected read count of the segment. Finally, we assign P-values to the log2 copy ratios of all segments based on permutation. Specifically, given a segment and its log2 copy ratio  , suppose that the segment contains M initial bins. At each permutation, we randomly select M non-contiguous bins from the initial set of bins and we can calculate a log2 copy ratio based on these M bins. By repeating this process B times (e.g. 1000), we obtain B log2 copy ratios. Supposing that
, suppose that the segment contains M initial bins. At each permutation, we randomly select M non-contiguous bins from the initial set of bins and we can calculate a log2 copy ratio based on these M bins. By repeating this process B times (e.g. 1000), we obtain B log2 copy ratios. Supposing that  is the mean and
is the mean and 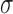 is the standard deviation of these B log2 copy ratios, the P-value of the segment is calculated as the probability
is the standard deviation of these B log2 copy ratios, the P-value of the segment is calculated as the probability  , with the random variable
, with the random variable  following the normal distribution
following the normal distribution  .
.
Figure 1.

GC bias revealed by fix-sized bins and variable-sized bins. (A) The low-coverage WGS data from a TCGA sample (TCGA-AG-3574) are binned to equal-sized bins (10 Kb). There is no clear pattern of GC-dependence. (B) The data are binned to variable-sized bins with equal number of mappable positions (10 K). This shows clear positive GC-dependence and bands corresponding to different copy number states. (C and D) Two lanes of the individual NA19240 in the same library show different GC dependence.
CNV detection for samples with data from multiple lanes and somatic CNV detection
To increase sequencing coverage, samples are often sequenced in multiple lanes or at multiple times. A typical strategy is simply to call CNVs after pooling all the data, implicitly assuming that the biases in all data sets are the same. We observe that this assumption may not be true (Figure 1C and D), suggesting that the simple pooling strategy could lead to a high false positive rate. Therefore, BIC-seq2 instead performs normalization for each data set separately and performs segmentation jointly using the multi-sample BIC-seq segmentation (13). Suppose that there are 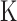 data sets for a sample. Let
data sets for a sample. Let  and
and  be the observed and the expected numbers of reads for the kth data set in the bin
be the observed and the expected numbers of reads for the kth data set in the bin  . BIC-seq2 then iteratively merges bin pairs by improving the following
. BIC-seq2 then iteratively merges bin pairs by improving the following
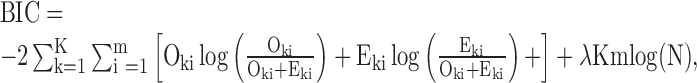 |
(3) |
where  . After segmentation, each segment will have
. After segmentation, each segment will have  estimates of log2 copy ratios corresponding to
estimates of log2 copy ratios corresponding to  data sets. BIC-seq2 defines the log2 copy ratio of a segment as the mean of its
data sets. BIC-seq2 defines the log2 copy ratio of a segment as the mean of its 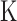 log2 copy ratios. For somatic CNV detection, BIC-seq2 first normalizes tumor and normal genomes separately and performs multi-sample BIC-seq joint segmentation (
log2 copy ratios. For somatic CNV detection, BIC-seq2 first normalizes tumor and normal genomes separately and performs multi-sample BIC-seq joint segmentation ( in Equation (3)). The log2 copy ratios for the tumor (normal) genome are defined as the log2 ratios between the observed and expected number of tumor (normal) reads in the segments. BIC-seq2 also filters germline CNV events by removing regions with log2 copy ratios for the normal genome <−0.2 or >0.2. In all of the following analyses, BWA (20) was used for alignment of the reads with read length <100 bp. If the read length is ≥100bp, BWA-MEM was used, but the hard-clipped reads were removed before analysis.
in Equation (3)). The log2 copy ratios for the tumor (normal) genome are defined as the log2 ratios between the observed and expected number of tumor (normal) reads in the segments. BIC-seq2 also filters germline CNV events by removing regions with log2 copy ratios for the normal genome <−0.2 or >0.2. In all of the following analyses, BWA (20) was used for alignment of the reads with read length <100 bp. If the read length is ≥100bp, BWA-MEM was used, but the hard-clipped reads were removed before analysis.
RESULTS
Characterization of GC bias
We applied the normalization procedure to a hundred genomes from the 1000 Genome Project to evaluate its performance (Supplementary Table S1; see the Supplementary Text for more descriptions). The 50-bp reads in these genomes were mapped to the human reference genome hg18. To assess GC bias, we computed the correlation between the read count and the GC-content (Figure 2A). We observe that the dependence of read count on GC-content varies significantly across the samples with the correlation spanning a wide range and negative in most cases. After normalization, we can see that GC-dependence has been effectively removed and the observed/expected ratio has almost no correlation with GC for all samples considered. Supplementary Figure S2 shows the effect of the normalization procedure for three other HapMap genomes from the 1000 Genomes. The read counts in two samples (Supplementary Figure S2A and B) show particularly strong non-linear GC-dependency, but the observed/expected ratio after normalization shows no evidence of GC-dependency (Supplementary Figure S2D and E). These results demonstrate that the normalization procedure can accurately capture and remove the non-linear GC-dependency. Even if we include the multiply-aligned reads, this normalization procedure can also effectively remove the GC biases (Supplementary Figure S5A). Figure 2B shows the effect of the normalization on the tumor genome that was shown in Figure 1B (the read length is 50 bp). In this example, the tumor genome has a strong positive correlation with GC-content and the dependence of the tumor read count on GC-content is nonlinear. After normalization, we can see the correlation with GC-content is successfully removed. We further compared the distribution of copy ratio estimates for variable-sized bins and equal-sized bins (Figure 2C top and middle panel). The variable-sized bins contain 10 Kb uniquely mappable positions and the equal-sized bins are 10 Kb bins. The distribution for the variable-sized bins shows a very clear separation of different copy number states, but the one for the equal-sized bins has no clear peaks. Since the biases in tumor and normal genomes are different (Figure 1B and Supplementary Figure S5B), the tumor/normal copy ratios in the variable-sized bins also have no clear separation among regions with different copy numbers (Figure 2C bottom panel). Supplementary Figure S5C and D show the effect of the normalization on a tumor genome with 100 bp data, which again shows that the normalization is effective in removing GC-biases.
Figure 2.

Effectiveness of the normalization procedure in BIC-seq2. (A) The normalization procedure was applied to a hundred genomes from the 1000 Genome project. The boxplots of Spearman's correlation with GC-content at three different levels before (the observed read count) and after (the ratio between the observed and the expected read counts) normalization are shown. This demonstrates that nearly all GC-dependency is removed. The bins with equal number of uniquely mappable positions were used. (B) For the TCGA sample in Figure 1 A and B, the GC-dependency is entirely removed after normalization. (C) The BIC-seq2 normalization in variable-sized bins containing 10 Kb mappable positions shows well-defined peaks corresponding to different copy number states (top panel), while the same normalization in 10 Kb equal-sized bins is diffuse with little separation of different copy number states (middle panel). A simple normalization by the matched normal (tumor/normal ratios) in variable-sized bins shows no clear separation of different copy number states (bottom panel).
Simulation study
We first used simulation to study the performance of BIC-seq2 for CNV detection. In the simulation, we generated 100 pseudo-chromosomes harboring 42 CNVs (6 copy number levels × 7 CNV size levels) based on chromosome 22 of the human reference genome (hg18). The CNVs were randomly placed on the pseudo-chromosomes with a requirement that each CNV segment cannot have more than 20% Ns. The program metaSim (21) was used to simulate 50 bp reads from the pseudo-chromosomes, and GC-biases were further introduced (the Supplementary Text and Supplementary Figure S6). The reads were then mapped back to chromosome 22 with BWA allowing two mismatches. We generated 3 data sets for each pseudo-chromosome at 3 different sequencing coverage levels.
We considered two scenarios for BIC-seq2: using only uniquely mapped reads and using all reads including the multiply-aligned reads. We normalized data and set the initial bin size as 10 bp for segmentation. The penalty parameter  was chosen as 1.2. The candidate CNV regions were chosen as regions with P-values less than 0.01 and log2 copy ratio >0.2 or <−0.2. For comparison, we also applied CNVnator (10), FREEC (11) and ReadDepth (12) to the simulated data. For CNVnator, we chose the initial bin size to be 500 bp as it performed the best at this bin size. Default parameters were used for FREEC and ReadDepth. Of note, since CNVnator and FREEC often predict gaps in the reference genome as deletions, we removed their CNV predictions overlapping these gaps in the comparison. If we do not remove these predictions, CNVnator has significantly more false discoveries (Supplementary Figure S7). We also applied the same filtering to BIC-seq2, FREEC and ReadDepth predictions to make the comparison fair (though it is not necessary for BIC-seq2).
was chosen as 1.2. The candidate CNV regions were chosen as regions with P-values less than 0.01 and log2 copy ratio >0.2 or <−0.2. For comparison, we also applied CNVnator (10), FREEC (11) and ReadDepth (12) to the simulated data. For CNVnator, we chose the initial bin size to be 500 bp as it performed the best at this bin size. Default parameters were used for FREEC and ReadDepth. Of note, since CNVnator and FREEC often predict gaps in the reference genome as deletions, we removed their CNV predictions overlapping these gaps in the comparison. If we do not remove these predictions, CNVnator has significantly more false discoveries (Supplementary Figure S7). We also applied the same filtering to BIC-seq2, FREEC and ReadDepth predictions to make the comparison fair (though it is not necessary for BIC-seq2).
Figure 3 shows the sensitivity of the five algorithms for detecting the following groups of CNVs: 2-copy deletions, 1-copy deletions, 1-copy gains, 2-copy gains and 3-copy gains. The sensitivity plots for 4-copy gains are shown in Supplementary Figure S7A–C. Here, a predicted CNV is defined as a true positive if it overlaps at least 50% with a simulated CNV. Overall, we can see that the two versions of BIC-seq2 achieve the highest sensitivity at all sequencing depths, though BIC-seq2-Unique is a little less sensitive for detecting large CNVs (10 kb, 50 kb and 100 kb) compared with BIC-seq2-Nonunique, CNVnator and ReadDepth. This is because CNVs in non-uniquely mappable regions cannot be detected by only uniquely mappable reads. For smaller CNVs, both versions of BIC-seq2 generally are significantly more sensitive than other algorithms. For example, at 30x coverage, the powers of both BIC-seq2-Unique and BIC-seq2-Nonunique at detecting 1 kb homozygous deletions are over 90%, but the corresponding powers for FREEC, CNVnator and ReadDepth are only around 40%, 20% and 0%.The false discovery rates for BIC-seq2-Unique, CNVnator and FREEC are all very low (close to 0 on average) (Supplementary Figure S7D,E and F). The false discovery rates for BIC-seq2-Nonunique are a little higher than the three aforementioned algorithms, because some parts of the true copy gain regions are not uniquely mappable and reads from these regions may be randomly mapped to other non-CNV regions. Hence, these non-CNV regions will have increased sequencing depth and will be detected by BIC-seq2 as copy gains when the sequencing depth is high enough. For this reason, we will only use BIC-seq2-Unique in the following real data analyses (thus BIC-seq2 refers to BIC-seq2-Unique in the rest of the paper). The false positives for ReadDepth are significantly more than other algorithms. Taken together, we conclude that BIC-seq2 can predict more small CNVs with similar or fewer false discoveries compared with other algorithms.
Figure 3.

Performance of BIC-seq2, CNVnator, FREEC and ReadDepth for simulated data. BIC-seq2-Unqiue: only uniquely mapped reads; BIC-seq2-Nonunique: all mapped reads are used, with a randomly sampled position when the reads are multiply-aligned. (A1–A5) At 3X coverage, the sensitivities for (A1) 2-copy deletions, (A2) 1-copy deletions, (A3) 1-copy gains, (A4) 2-copy gains and (A5) 3-copy gains. (B1–B5) and (C1–C5) are similar plots but at 10X and 30X coverages.
CNV detection comparison on a normal genome
We applied BIC-seq2, CNVnator, FREEC and ReadDepth to the NA12878 sample from the 1000 Genome Project. Since this data set consists of data sequenced from four libraries, we normalized the data separately for each subset (although the biases were very similar in this case) and performed joint segmentation for CNV detection with BIC-seq2. For other algorithms, we applied the same parameter setup as in the simulation study. To compare the performance of these algorithms, we overlapped the copy losses predicted by the algorithms with the deletions reported in Mills et al. (22) where the authors catalogued experimentally-validated deletions of this individual after prediction by a set of algorithms. Specifically, we took all deletion predictions of NA12878 that were marked as validated and the deletions in the gold standard sets of the paper and merged overlapping deletions as deletion regions. The deletion predictions of the four algorithms are considered true positives if they overlap by 50% with any of these deletion regions. BIC-seq2 has the highest true positive rate (TPR) (83%), and then followed by FREEC (81%), ReadDepth (63%) and CNVnator (60%).
Next, we applied each algorithm separately to the four libraries of NA12878 to investigate replicability of CNV predictions. A CNV called from one library is marked as replicated by another library if it is covered by CNVs of the same type called from the second library. Figure 4A shows the percentages of replicable CNVs from all pair-wise comparisons. We clearly see that BIC-seq2 has the highest percentage (∼95%) of replicated CNVs. Similar to the above result, FREEC performs better than CNVnator and ReadDepth, but its percentage of replicated CNVs is consistently less that of BIC-seq2. Figure 4B shows the CNVs on the first half of chromosome 1 called based on each library given by BICseq2, FREEC, CNVnator and ReadDepth (See Supplementary Figure S8 for examples of other chromosomes). This plot clearly shows that BIC-seq2 gives consistent calls across the libraries, whereas other algorithms give many calls that are not stable across the libraries.
Figure 4.

Replicability and profile comparison. (A) The boxplots of the percentages of replicable CNVs detected from the four libraries of NA12878. Each pair of libraries was compared. (B) The CNVs in the first half of chromosome 1 detected by BIC-seq2, FREEC, CNVnator and ReadDepth in the 4 libraries.
CNV detection in colorectal cancer genomes
Comparison with microarray data
To further evaluate the performance of BIC-seq2, we applied it to a hundred pairs of low coverage colorectal tumor and matched control (blood) genomes available from TCGA (Supplementary Table S2). The parameter setup was set as before. The CNV predictions are taken as regions with log2 tumor/expected ratio >0.2 or <−0.2. These cutoffs are often used by other TCGA studies. We also filtered regions harboring likely germline variations, i.e. regions with log2 control/expected ratios >0.2 and <−0.2. Since array-based copy number measurements on the same samples have been widely used (23), we sought to compare the copy number calls from the array and sequencing platforms. We found that the log2 tumor/expected ratio highly correlated with corresponding log2 copy number ratios of the array data (the ‘seg.mean’ values; Spearman's correlation: 0.95; Figure 5A). Under the criterion that a copy gain (loss) is defined as a true positive if its corresponding log2 copy ratio of the array data is greater (less) than 0.2 (−0.2), the true positive rate of BIC-seq2 is 90.8%. We further performed linear median regression with the log2 copy number ratios of the array data as the response variable and the log2 tumor/expected ratio as the predictor (Figure 5A). The estimated intercept is −0.041 (sd = 0.002) and the estimated slope is 1.038 (sd = 0.002). Since we would expect the intercept of this regression model to be 0 and the slope to be 1 under best circumstances, this indicates that the normalization procedure performed very well. For comparison, we also applied the original BIC-seq to these colorectal data and compared the results with those from microarray data. Analogous analysis showed that the correlation of log2 tumor/normal ratio with log2 copy number ratios of the array data is only 0.44 (also see Supplementary Figure S9A). The main reason for this low correlation is that a substantial proportion of the tumor/normal genome pairs have different GC-biases, similar to what was observed for the sample TCGA-AG-3574 (Figure 1B and Supplementary Figure S5B), and this violates the assumption of BIC-seq.
Figure 5.

Comparison with arrays and identification of CNVs with expression differences. (A) Comparison with Illumina SNP 6.0 array data. The tumor/expected-ratio versus seg.mean-value scatter plot for a hundred colorectal genomes is shown. The red line is the fitted median regression line; the black diagonal line corresponds to y = x. The true positive rate of BIC-seq2 compared to arrays is 90.8% if we treat copy gains (copy losses) with corresponding seg.mean values greater (less) than 0.2 (−0.2) as true positives. (B) Differences in expression levels in samples with or without the PIK4K2B amplification and RERE and NPM2 deletions.
Recurrent CNVs and correlation with expression data
To detect recurrent CNVs in these patients, we applied the GISTIC algorithm (24) to the segmentation given by BIC-seq2. Several known arm level alterations have been identified previously (25), including gains of 7p/q, 8p/q, 13q, 20p/q, deletions of 1p, 4q, 14q, 15q, 17p (including TP53), 21q. We identified 16 focal deletions and 12 focal amplifications with q-values < 0.1 (Supplementary Figure S9B and C; Supplementary Tables S3 and S4), containing 447 and 165 genes, respectively. For each gene in the focal deletion (amplification) peaks, we identified genes for which the samples harboring the deletion/amplification have concordant down-regulation/up-regulation of expression compared with the samples without the deletion/amplification. This resulted in 177 (out of 447) significant genes (P-value < 0.01; Wilcoxon's signed rank sum test) in the deletion peaks and 95 (out of 165) genes in the amplification peaks (Supplementary Tables S5 and S6). Fisher's test shows that these genes are significantly (P-value 2.5 × 10−5) enriched with cancer census genes (26). Pathway analysis using DAVID (27–29) finds that the most significantly altered pathway for the significant amplification genes is Phosphatidylinositol (PI) signaling system (P-value = 7.8 × 10−5), a pathway critical for cancer development (30). The most significantly altered pathway for deletion genes is the apoptosis pathway (P-value = 1.5 × 10−6) (Supplementary Table S7).
The genes with concordant transcript up-regulation and chromosomal amplification include known oncogenes MYC, IGF2, ERBB2 and HNF4A (31) (Supplementary Figure S10A). We also identified novel genes that may be important in colorectal cancers. In particular, PIP4K2B (phosphatidylinositol-5-phosphate 4-kinase, type II, beta), whose average expression in the amplified and non-amplified cases are shown in Figure 5B, is a member of the phosphatidylinositol-5-phosphate 4-kinase family and has been shown to be frequently amplified in breast cancer. Overexpression of PIP4K2B can confer proliferation advantage to tumor cells and it may therefore serve as a drug target for preventing and treating cancers with mutations in TP53 (32,33). Genes whose transcripts show down-regulation coupled with chromosomal deletions include cancer-related genes such as ARID1A, SDHB, MAP2K4, FLCN (Supplementary Figure S10B). ARID1A encodes a protein involved in chromatin remodeling. It was previously shown to be frequently mutated and proposed as a tumor suppressor in a number of cancers (25,34–36). Here, we found that it is also frequently deleted in colorectal cancers. In addition, we also identified two additional genes, chromatin remodelers RERE and NPM2, as frequently deleted (Figure 5B). Over-expression of RERE was shown to trigger apoptosis (37) and frequent methylation of NPM2 was observed in melanoma (38) and leukemia (39).
Germline CNVs with cancer susceptibility
Most importantly, we applied BIC-seq2 to 969 low coverage (4X–6X) genomes of peripheral blood samples from the TCGA cancer patients (these samples are mostly distinct from the smaller number of high-coverage (30–60X) TCGA WGS samples). These samples were sequenced mainly to identify somatic structural variants and the fusions identified in these samples have been highlighted in several TCGA papers. Here, we applied our algorithm to identify germline CNVs that may confer cancer susceptibility. These genomes span 11 types of cancers including bladder urothelial carcinoma (BLCA), brain lower grade glioma (LGG), breast invasive carcinoma (BRCA), colon and rectum adenocarcinoma (COAD-READ), head and neck squamous cell carcinoma (HNSC), lung adenocarcinoma (LUAD), prostate adenocarcinoma (PRAD), skin cutaneous melanoma (SKCM), stomach adenocarcinoma (STAD), thyroid carcinoma (THCA) and uterine corpus endometrial carcinoma (UCEC) (Supplementary Table S8).
One major difficulty in identifying cancer-related CNVs in these data is to remove population polymorphic sites using a proper control data set. In this work, we used two control data sets. The first is the 71090 CNVs we identified in the above 100 HapMap genomes from the 1000 Genome Project. The second is the CNVs from the database of genomic variants (DGV). For the DGV CNVs, we only considered the CNVs that were called in studies with sequencing data since CNVs called by array-based technologies tend to have a lower resolution and their sizes tend to be overestimated (40,41). Thus, using only those studies with sequencing data would make the case and control more comparable. This gave us 6213 DGV control samples. The 1000 Genome Project data are helpful for removing CNVs that are enriched in the case data sets due to algorithm difference. The second data are much larger and thus gives us more power to detect and filter rare events.
We first merged the CNV segments discovered in more than two cancer patients to form ‘CNV regions’ (CNVR). Since the predicted CNVs in different samples can have slightly different boundaries, this merging process gives us a single region on which we can perform statistical test. We focused on CNVRs overlapping with protein-coding sequences. Since the population structures in the case data set and the two control data sets can be different and the majority of cancers are not related to heritable factors (42), we filtered out CNVRs with a frequency >20%. Since the first control data set is small and a statistical test based on this control data would remove rare but true cancer predisposing CNVs due to insufficient power, we instead filter the remaining CNVRs with a frequency cutoff: a CNVR is filtered out if more than 5 samples in the first control data set have CNVs overlapping with the CNVR. Lastly, Fisher's test was used to compare the case CNVs with the DGV CNVs. Details of the analysis procedure are presented in the Supplementary Text. Table 1 shows the CNVRs covering protein-coding regions with a P-value less than 1.0 × 10−7 as well as significant CNVRs (P-value < 0.05) overlapping with known cancer genes (See Supplementary Table S9 for all of the significant CNVRs).
Table 1. Highly significant CNVRs. First eight rows: all genes that overlap with the highly significant CNVRs (P-value < 1E-7); Last three rows: known cancer-related genes overlapping with significant CNVRs (P-value < 0.05).
| Cytoband | Start End | PC/1KG/DGV* | CNV Type | Gene | P-value |
|---|---|---|---|---|---|
| 7q36.1 | 15195749 151990501 | 68/3/16 7.0%/3.0%/0.3% | Amp | KMT2C | 6.55E-45 |
| 5p13.3 | 32105400 32168040 | 41/3/0 4.2%/3.0%/0.0% | Amp | PDZD2,GOLPH3 | 1.02E-36 |
| 7p14.1 | 38283144 38416157 | 192/1/397 19.8%/1.0%/6.4% | Del | TARP | 3.08E-36 |
| 2q36.3 | 228240991 228258967 | 23/0/11 2.4%/0.0%/0.2% | Amp | TM4SF20 | 5.05E-13 |
| 3q12.2 | 100334650 100446463 | 16/0/2 1.6%/0.0%/0.03% | Amp | GPR128,TFG | 1.27E-12 |
| 7q34 | 142824400 142894023 | 20/0/11 2.1%/0.0%/0.2% | Del | PIP,TAS2R39 | 6.48E-11 |
| 12p12.3 | 19467296 19580473 | 11/0/0 1.1%/0.0%/0.0% | Amp | PLEKHA5 | 2.57E-10 |
| 6q27 | 168332692 168598263 | 15/1/9 1.5%/1.0%/0.1% | Amp | KIF25,MLLT4,HGC6.3,FRMD1 | 3.21E-08 |
| 12q24.13 | 112180349 112316664 | 5/0/0 0.5%/0.0%/0.0% | Amp | ALDH2 | 4.43E-05 |
| 8q12.1 | 57050810 57098666 | 4/0/0 0.4%/0.0%/0.0% | Amp | PLAG1 | 0.00033 |
| 17q12 | 37671717 37918575 | 4/0/5 0.4%/0.0%/0.08% | Amp | ERBB2,CDK12 | 0.023654 |
*PC: Pan-Cancer Patients; 1KG: 1000 Genome Project; DGV: Database of Genomic Variants; Numbers shown on the top and on the bottom are the numbers and the percentages of individuals in each category that have CNVs overlapping with this CNVR.
We identified known as well as novel cancer-predisposing CNVRs. Among the known CNVRs, the most significant is the 7p14.1 deletion CNVR overlapping with the gene TARP (Supplementary Figure S11A). This TARP CNVR was found to be the most significant germline CNVR in neuroblastoma (1), and here we found that it is also enriched in other tumor types (P-values < 0.01) including COAD-READ (39/94), HNSC (16/105), LGG (12/48), PRAD (24/79), SKCM (34/118), STAD (24/114) and UCEC (25/103). The 7q34 deletion CNVR (Supplementary Figure S11B) harboring the gene PIP was previously identified as a potential cancer-predisposing variation by screening of high-risk cancer patients (43). It was also shown that the expression of PIP is significantly associated with good prognosis factors of breast cancer such as lower tumor grade and lower pN stage (44). The 6q37 amplification CNVR (Supplementary Figure S11C) is also known and is significantly enriched in Li–Fraumeni symdrome (45). The gene MLLT4 in this CNVR is well known to be the fusion partner of KMT2A (ALL-1) in acute myeloid leukemias (46).
Novel CNVRs include the most significant CNVR located at 7q36.1 overlapping with KMT2C (MLL3) (Supplementary Figure S12A). KMT2C is a well-known cancer-related gene and it is frequently mutated in many cancers such as gastric cancer (47), hepatocellular carcinoma (48) and cholangiocarcinoma (49). Recent studies showed that germline mutations in KMT2C might be associated with ovarian cancer, colorectal cancer and acute myeloid leukemia (50,51). Here, we find that the CNVR in KMT2C is enriched (P-values < 0.01) in BLCA (5/106), BRCA (6/20), HNSC (5/105), LGG(11/48), PRAD (7/79), SKCM (9/118), STAD (18/114) and UCEC (4/103). The novel 5p13.3 CNVR (Figure 6A) overlaps with the gene GOLPH3. High frequency of GOLPH3 amplification was observed in several solid tumor types such as lung, ovarian, breast, pancreatic, prostate and skin cancers (52). Extensive studies have validated that GOLPH3 is an oncogene (52,53), and over-expression of GOLPH3 is correlated with poor prognosis in multiple tumor types (54,55). This CNVR also overlaps with the gene PDZD2, which was shown to be highly expressed in prostate cancers and to be associated with initiation or early promotion of prostate cancer (56). Although we found three individuals in the 1000 Genome Project having duplications covering this CNVR, the duplication of one individual is in fact much larger than this CNVR (Figure 6A). This CNVR was previously identified as a risk factor for recurrent miscarriage (57). The occurrence frequency of the 5p13.3 CNVR in the Estonian Biobank control set used in this work (57) is ∼1% (9/1000), while the occurrence frequency in our tumor data is ∼4% (41/969). Fisher's test based on this control data gives a P-value of 2.0 × 10−6. Furthermore, we found that 30 out of 41 cancer patients have at least 2 discordant reads supporting an intra-chromosomal translocation with two breakpoints involving GOLPH3 and PDZD2, indicating that this CNVR may be related with the translocation. GOLPH3 was shown to play an important role in the mTOR signaling pathway (52) and mTOR was known to be an essential component of mammalian reproductive function (58,59). Thus, alteration of the mTOR pathway might provide an explanation for conferring both cancer and recurrent miscarriage with this GOLPH3 amplification. The 12p12.3 amplification CNVR overlaps with the gene PLEKHA5; expression of PLEKHA5 in melanoma was shown to be associated with early development of brain metastasis (60).
Figure 6.

Examples of novel potential cancer-predisposing germline CNVs. (A) GOLPH3, PDZD2 amplification, (B) PLAG1 amplification, (C) ERBB2 amplification and (D) TFG amplification.
In addition, we also identified less significant but novel CNVRs that overlap with known cancer genes. For example, the 8q12.1 CNVR (amplification; P-value: 3.3 × 10−4) covers the gene PLAG1 (Figure 6B), which is frequently altered in pleomorphic adenomas of the salivary glands as well as other types of tumors (61). Another is the amplification CNVR overlapping ERBB2 (HER2). Although 4 patients have CNVs overlapping with this CNVR, only 3 of them cover ERBB2 (Figure 6C). Interestingly, all three patients are stomach cancer patients. If we only consider stomach cancer patients, the P-value of this CNVR is 3.0 × 10−4. ERBB2 is well-known to be frequently amplified in breast cancer and has also been shown to be frequently amplified in other types of cancer such as gastric and gastroesophageal cancers (62). While somatic amplification and overexpression of ERBB2 has served as druggable targets with monoclonal antibody (Herceptin), the germline copy number changes have not been well recognized. It may be associated with germline susceptibility of the disease as well as the druggability with the ERBB2-targeting agents. Germline mutations of ERBB2 that may confer cancer risk were also identified previously, such as in familial lung adenocarcinomas (63).
DISCUSSION
GC bias has been noted previously in many applications of high-throughput sequencing. Here, we first show that this problem is pervasive in WGS data, GC-dependence can take various linear or nonlinear forms and the bias is experiment-dependent. This makes it difficult to identify CNVs in a single sample (e.g. germline analysis) or to simply use a matched control to remove germline variants in somatic variant analysis. To overcome this problem, we developed a statistical method that can be used to normalize high-throughput WGS data at a nucleotide level. Our analysis demonstrates that this normalization procedure can successfully capture and remove GC and related biases. Combining this step with a robust segmentation method, our BIC-seq2 algorithm is capable of detecting CNVs at high resolution. Although we have shown that BIC-seq2 has better performance than other methods, it can be further improved by incorporating the information from the discordant reads (defined above) and split reads (reads that span a breakpoint).
In the normalization step, BIC-seq2 treats mappability differently from other methods (11,16). Available methods often bin the data to equal-sized bins first and treat mappability (e.g. as the percentage of uniquely mappable positions in a bin) essentially as a covariate in a regression model to remove the mappability bias. BIC-seq2 does not first bin the data, and mappability is not treated as a covariate of the regression model for the normalization. Instead, BIC-seq2 performs its normalization at each single uniquely mappable position, i.e. the expected number of reads is calculated at each mappable position. After normalization, we typically bin the nucleotide-level normalized data to larger variable-sized bins (e.g. 10 bp or 100 bp) in the segmentation step for computational efficiency. We note that nucleotide-level normalization is advantageous because it allows detection of CNVs with high-resolution. In particular, PEM-methods often generate small CNV calls, and a nucleotide-level read-depth normalization method can be used effectively to remove false positives from PEM-methods.
Application of our method to 969 blood genomes of cancer patients allowed us to confirm known and cancer-predisposing CNVRs and identify new ones. The novel CNVRs cover known cancer related genes such as GOLPH3, PLAG1 and ERBB2. The CNVRs containing PLAG1 and ERBB2 are very likely to be true cancer risk amplifications, even though they are less significant. Especially, the ERBB2 CNVR covers entire ERBB2 gene and the 3 patients are all stomach cancer patients. A larger data size is needed to confirm them being true cancer susceptibility variation. The main difficulty in identifying cancer predisposing CNVR using TCGA data is the lack of proper control data. Here, we used two different control data to filter the common CNVs. Although the first control data are small in size, it helped to effectively filter the CNVs that are enriched in the case but not in the DGV control data due to algorithmic difference. The threshold 5 used in the first control data can be restrictive. True but low penetration cancer predisposing CNVs can be filtered by this threshold. In fact, the germline deletion of APOBEC3A and APOBEC3B is known to be associated with modestly increased cancer risk (64), but it was filtered because 6 individuals in the 1000 Genome control data have this deletion (Supplementary Figure S12B).
The method used in this work has allowed us discover both known and novel cancer predisposing CNVRs, but the highly significant CNVRs might still contain copy number polymorphism. For example, the CNVR overlapping with the gene TFG might be a polymorphism in normal population (Figure 6D). The gene TFG is listed as a cancer gene in the COSMIC (Catalogue of Somatic Mutations in Cancer) database (65). There are several documented oncoproteins encoded by fusion genes involving this gene, such as the TFG-NTRK1 gene fusion (66), the TFG-ALK gene fusion (67) and the TFG- NR4A3 gene fusion (68). We found that 14 out of 16 patients have at least 2 paired-end reads supporting a gene fusion TFG-GPR128 and so this copy number change is related to the gene fusion. This gene fusion was identified in typical myeloproliferative neoplasms but was also found in healthy individuals (69). This CNVR thus might be a rare polymorphism that we could not properly filter. On the other hand, among the 16 patients with the amplification at TFG, 5 of them (2 BLCA patients, 1 LGG patient and 2 STAD patients) had data on family history in their clinical annotations. The 2 BLCA patients and the LGG patient had a family history of cancer; the 2 STAD patients did not have family history of stomach cancer, but data were not available on whether there was a history of any other cancers. These data suggest the possibility that the TFG CNVR might have a role in cancer development. Further investigation will be required to elucidate the role of this CNVR in cancer. Lastly, we emphasize that in addition to the discussed potential germline CNVs, the CNVs of cancer patients profiled in this work provide a valuable resource for future analyses.
AVAILABILITY
BICseq2 is available at http://compbio.med.harvard.edu/BIC-seq
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01EY024230 to P.J.P.]; Ludwig Cancer Center (to PJP); National Natural Science Foundation of China [11471022 and 71532001 to R.X.]; National Key Basic Research Program of China [2015CB856000 to R.X.]; Recruitment Program of Global Youth Experts of China (to R.X.). Funding for open access charge: NIH [R01EY024230]; Ludwig Cancer Center; NSFC; National Key Basic Research Program of China.
Conflict of interest statement. None declared.
REFERENCES
- 1.Diskin S.J., Hou C., Glessner J.T., Attiyeh E.F., Laudenslager M., Bosse K., Cole K., Mosse Y.P., Wood A., Lynch J.E., et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009;459:987–991. doi: 10.1038/nature08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuiper R.P., Ligtenberg M.J., Hoogerbrugge N., Geurts van K.A. Germline copy number variation and cancer risk. Curr. Opin. Genet. Dev. 2010;20:282–289. doi: 10.1016/j.gde.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Yoshihara K., Tajima A., Adachi S., Quan J., Sekine M., Kase H., Yahata T., Inoue I., Tanaka K. Germline copy number variations in BRCA1-associated ovarian cancer patients. Genes Chromosomes Cancer. 2011;50:167–177. doi: 10.1002/gcc.20841. [DOI] [PubMed] [Google Scholar]
- 4.Stefansson H., Rujescu D., Cichon S., Pietilainen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanciulli M., Norsworthy P.J., Petretto E., Dong R., Harper L., Kamesh L., Heward J.M., Gough S.C., de S.A., Blakemore A.I., et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat. Genet. 2007;39:721–723. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stratton M.R., Campbell P.J., Futreal P.A. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen K., Wallis J.W., McLellan M.D., Larson D.E., Kalicki J.M., Pohl C.S., McGrath S.D., Wendl M.C., Zhang Q., Locke D.P., et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hormozdiari F., Alkan C., Eichler E.E., Sahinalp S.C. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korbel J.O., Abyzov A., Mu X.J., Carriero N., Cayting P., Zhang Z., Snyder M., Gerstein M.B. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abyzov A., Urban A.E., Snyder M., Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boeva V., Zinovyev A., Bleakley K., Vert J.P., Janoueix-Lerosey I., Delattre O., Barillot E. Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics. 2011;27:268–269. doi: 10.1093/bioinformatics/btq635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller C.A., Hampton O., Coarfa C., Milosavljevic A. ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS. One. 2011;6:e16327. doi: 10.1371/journal.pone.0016327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xi R., Hadjipanayis A.G., Luquette L.J., Kim T.M., Lee E., Zhang J., Johnson M.D., Muzny D.M., Wheeler D.A., Gibbs R.A., et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc. Natl. Acad. Sci. U.S.A. 2011;108:E1128–E1136. doi: 10.1073/pnas.1110574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiang D.Y., Getz G., Jaffe D.B., O'Kelly M.J., Zhao X., Carter S.L., Russ C., Nusbaum C., Meyerson M., Lander E.S. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szatkiewicz J.P., Wang W., Sullivan P.F., Wang W., Sun W. Improving detection of copy-number variation by simultaneous bias correction and read-depth segmentation. Nucleic. Acids Res. 2013;41:1519–1532. doi: 10.1093/nar/gks1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheinin I., Sie D., Bengtsson H., van de Wiel M.A., Olshen A.B., van Thuijl H.F., van Essen H.F., Eijk P.P., Rustenburg F., Meijer G.A., et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome. Res. 2014;24:2022–2032. doi: 10.1101/gr.175141.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen K.D., Brenner S.E., Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38:e131. doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aird D., Ross M.G., Chen W.S., Danielsson M., Fennell T., Russ C., Jaffe D.B., Nusbaum C., Gnirke A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome. Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood S.N. Fast stable direct fitting and smoothness selection for generalized additive models. J. R. Stat. Soc. B. 2008;70:495–518. [Google Scholar]
- 20.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richter D.C., Ott F., Auch A.F., Schmid R., Huson D.H. MetaSim: a sequencing simulator for genomics and metagenomics. PLoS One. 2008;3:e3373. doi: 10.1371/journal.pone.0003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mills R.E., Walter K., Stewart C., Handsaker R.E., Chen K., Alkan C., Abyzov A., Yoon S.C., Ye K., Cheetham R.K., et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhang C.Z., Wala J., Mermel C.H., et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beroukhim R., Getz G., Nghiemphu L., Barretina J., Hsueh T., Linhart D., Vivanco I., Lee J.C., Huang J.H., Alexander S., et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl. Acad. Sci. U.S.A. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Network Cancer Genome Atlas Research. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higgins M.E., Claremont M., Major J.E., Sander C., Lash A.E. CancerGenes: a gene selection resource for cancer genome projects. Nucleic. Acids Res. 2007;35:D721–D726. doi: 10.1093/nar/gkl811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Handsaker R.E., Korn J.M., Nemesh J., McCarroll S.A. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 2011;43:269–276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang d.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 29.Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic. Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bunney T.D., Katan M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat. Rev. Cancer. 2010;10:342–352. doi: 10.1038/nrc2842. [DOI] [PubMed] [Google Scholar]
- 31.Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M.C., Zimmerman L.J., Shaddox K.F., Kim S., et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emerling B.M., Hurov J.B., Poulogiannis G., Tsukazawa K.S., Choo-Wing R., Wulf G.M., Bell E.L., Shim H.S., Lamia K.A., Rameh L.E., et al. Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell. 2013;155:844–857. doi: 10.1016/j.cell.2013.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luoh S.W., Venkatesan N., Tripathi R. Overexpression of the amplified Pip4k2beta gene from 17q11-12 in breast cancer cells confers proliferation advantage. Oncogene. 2004;23:1354–1363. doi: 10.1038/sj.onc.1207251. [DOI] [PubMed] [Google Scholar]
- 34.Jones S., Wang T.L., Shih I., Mao T.L., Nakayama K., Roden R., Glas R., Slamon D., Diaz L.A., Jr, Vogelstein B. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang K., Yuen S.T., Xu J., Lee S.P., Yan H.H., Shi S.T., Siu H.C., Deng S., Chu K.M., Law S., et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014;46:573–582. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 36.Wu R.C., Wang T.L., Shih I. The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther. 2014;15:655–664. doi: 10.4161/cbt.28411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waerner T., Gardellin P., Pfizenmaier K., Weith A., Kraut N. Human RERE is localized to nuclear promyelocytic leukemia oncogenic domains and enhances apoptosis. Cell Growth. Differ. 2001;12:201–210. [PubMed] [Google Scholar]
- 38.Koga Y., Pelizzola M., Cheng E., Krauthammer M., Sznol M., Ariyan S., Narayan D., Molinaro A.M., Halaban R., Weissman S.M. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome. Res. 2009;19:1462–1470. doi: 10.1101/gr.091447.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kroeger H., Jelinek J., Estecio M.R., He R., Kondo K., Chung W., Zhang L., Shen L., Kantarjian H.M., Bueso-Ramos C.E., et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood. 2008;112:1366–1373. doi: 10.1182/blood-2007-11-126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinto D., Darvishi K., Shi X., Rajan D., Rigler D., Fitzgerald T., Lionel A.C., Thiruvahindrapuram B., Macdonald J.R., Mills R., et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011;29:512–520. doi: 10.1038/nbt.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lichtenstein P., Holm N.V., Verkasalo P.K., Iliadou A., Kaprio J., Koskenvuo M., Pukkala E., Skytthe A., Hemminki K. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 43.Silva A.G., Krepischi A.C., Torrezan G.T., Capelli L.P., Carraro D.M., D'Angelo C.S., Koiffmann C.P., Zatz M., Naslavsky M.S., Masotti C., et al. Does germ-line deletion of the PIP gene constitute a widespread risk for cancer? Eur. J. Hum. Genet. 2014;22:307–309. doi: 10.1038/ejhg.2013.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo M.H., Huang Y.H., Ni Y.B., Tsang J.Y., Chan S.K., Shao M.M., Tse G.M. Expression of mammaglobin and gross cystic disease fluid protein-15 in breast carcinomas. Hum. Pathol. 2013;44:1241–1250. doi: 10.1016/j.humpath.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 45.Shlien A., Tabori U., Marshall C.R., Pienkowska M., Feuk L., Novokmet A., Nanda S., Druker H., Scherer S.W., Malkin D. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proc. Natl. Acad. Sci. U.S.A. 2008;105:11264–11269. doi: 10.1073/pnas.0802970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prasad R., Gu Y., Alder H., Nakamura T., Canaani O., Saito H., Huebner K., Gale R.P., Nowell P.C., Kuriyama K., et al. Cloning of the ALL-1 fusion partner, the AF-6 gene, involved in acute myeloid leukemias with the t(6;11) chromosome translocation. Cancer Res. 1993;53:5624–5628. [PubMed] [Google Scholar]
- 47.Zang Z.J., Cutcutache I., Poon S.L., Zhang S.L., McPherson J.R., Tao J., Rajasegaran V., Heng H.L., Deng N., Gan A., et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 48.Fujimoto A., Totoki Y., Abe T., Boroevich K.A., Hosoda F., Nguyen H.H., Aoki M., Hosono N., Kubo M., Miya F., et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 49.Ong C.K., Subimerb C., Pairojkul C., Wongkham S., Cutcutache I., Yu W., McPherson J.R., Allen G.E., Ng C.C., Wong B.H., et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012;44:690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 50.Kanchi K.L., Johnson K.J., Lu C., McLellan M.D., Leiserson M.D., Wendl M.C., Zhang Q., Koboldt D.C., Xie M., Kandoth C., et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 2014;5:3156. doi: 10.1038/ncomms4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li W.D., Li Q.R., Xu S.N., Wei F.J., Ye Z.J., Cheng J.K., Chen J.P. Exome sequencing identifies an MLL3 gene germ line mutation in a pedigree of colorectal cancer and acute myeloid leukemia. Blood. 2013;121:1478–1479. doi: 10.1182/blood-2012-12-470559. [DOI] [PubMed] [Google Scholar]
- 52.Scott K.L., Kabbarah O., Liang M.C., Ivanova E., Anagnostou V., Wu J., Dhakal S., Wu M., Chen S., Feinberg T., et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature. 2009;459:1085–1090. doi: 10.1038/nature08109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buschman M.D., Rahajeng J., Field S.J. GOLPH3 links the Golgi, DNA damage, and cancer. Cancer Res. 2015;75:624–627. doi: 10.1158/0008-5472.CAN-14-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeng Z., Lin H., Zhao X., Liu G., Wang X., Xu R., Chen K., Li J., Song L. Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin. Cancer Res. 2012;18:4059–4069. doi: 10.1158/1078-0432.CCR-11-3156. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y., Ma M., Han B. GOLPH3 high expression predicts poor prognosis in patients with resected non-small cell lung cancer: an immunohistochemical analysis. Tumour Biol. 2014;35:10833–10839. doi: 10.1007/s13277-014-2357-3. [DOI] [PubMed] [Google Scholar]
- 56.Chaib H., Rubin M.A., Mucci N.R., Li L., Taylor J.M.G., Day M.L., Rhim J.S., Macoska J.A. Activated in prostate cancer: a PDZ domain-containing protein highly expressed in human primary prostate tumors. Cancer Res. 2001;61:2390–2394. [PubMed] [Google Scholar]
- 57.Nagirnaja L., Palta P., Kasak L., Rull K., Christiansen O.B., Nielsen H.S., Steffensen R., Esko T., Remm M., Laan M. Structural genomic variation as risk factor for idiopathic recurrent miscarriage. Hum. Mutat. 2014;35:972–982. doi: 10.1002/humu.22589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanwar P.S., Kaneko-Tarui T., Zhang L., Teixeira J.M. Altered LKB1/AMPK/TSC1/TSC2/mTOR signaling causes disruption of Sertoli cell polarity and spermatogenesis. Hum. Mol. Genet. 2012;21:4394–4405. doi: 10.1093/hmg/dds272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wen H.Y., Abbasi S., Kellems R.E., Xia Y. mTOR: a placental growth signaling sensor. Placenta. 2005;26(Suppl A):S63–S69. doi: 10.1016/j.placenta.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 60.Jilaveanu L.B., Parisi F., Barr M.L., Zito C.R., Cruz-Munoz W., Kerbel R.S., Rimm D.L., Bosenberg M.W., Halaban R., Kluger Y., et al. PLEKHA5 as a Biomarker and Potential Mediator of Melanoma Brain Metastasis. Clin. Cancer Res. 2015;21:2138–2147. doi: 10.1158/1078-0432.CCR-14-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Declercq J., Van D.F., Braem C.V., Van Valckenborgh I.C., Voz M., Wassef M., Schoonjans L., Van D.B., Fiette L., Van de Ven W.J. Salivary gland tumors in transgenic mice with targeted PLAG1 proto-oncogene overexpression. Cancer Res. 2005;65:4544–4553. doi: 10.1158/0008-5472.CAN-04-4041. [DOI] [PubMed] [Google Scholar]
- 62.Janjigian Y.Y., Werner D., Pauligk C., Steinmetz K., Kelsen D.P., Jager E., Altmannsberger H.M., Robinson E., Tafe L.J., Tang L.H., et al. Prognosis of metastatic gastric and gastroesophageal junction cancer by HER2 status: a European and USA International collaborative analysis. Ann. Oncol. 2012;23:2656–2662. doi: 10.1093/annonc/mds104. [DOI] [PubMed] [Google Scholar]
- 63.Yamamoto H., Higasa K., Sakaguchi M., Shien K., Soh J., Ichimura K., Furukawa M., Hashida S., Tsukuda K., Takigawa N., et al. Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J. Natl. Cancer Inst. 2014;106:djt338. doi: 10.1093/jnci/djt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nik-Zainal S., Wedge D.C., Alexandrov L.B., Petljak M., Butler A.P., Bolli N., Davies H.R., Knappskog S., Martin S., Papaemmanuil E., et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 2014;46:487–491. doi: 10.1038/ng.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Greco A., Mariani C., Miranda C., Lupas A., Pagliardini S., Pomati M., Pierotti M.A. The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol. Cell Biol. 1995;15:6118–6127. doi: 10.1128/mcb.15.11.6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hernandez L., Pinyol M., Hernandez S., Bea S., Pulford K., Rosenwald A., Lamant L., Falini B., Ott G., Mason D.Y., et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood. 1999;94:3265–3268. [PubMed] [Google Scholar]
- 68.Hisaoka M., Ishida T., Imamura T., Hashimoto H. TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes. Chromosomes Cancer. 2004;40:325–328. doi: 10.1002/gcc.20044. [DOI] [PubMed] [Google Scholar]
- 69.Chase A., Ernst T., Fiebig A., Collins A., Grand F., Erben P., Reiter A., Schreiber S., Cross N.C. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica. 2010;95:20–26. doi: 10.3324/haematol.2009.011536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.