

Abstract
The brain has a high demand for energy, of which creatine (Cr) is an important regulator. Studies document neurocognitive benefits of oral Cr in mammals, yet little is known regarding their physiological basis. This study investigated the effects of Cr supplementation (3%, w/w) on hippocampal function in male C57BL/6 mice, including spatial learning and memory in the Morris water maze and oxygen consumption rates from isolated mitochondria in real time. Levels of transcription factors and related proteins (CREB, Egr1, and IκB to indicate NF-κB activity), proteins implicated in cognition (CaMKII, PSD-95, and Egr2), and mitochondrial proteins (electron transport chain Complex I, mitochondrial fission protein Drp1) were probed with Western blotting. Dietary Cr decreased escape latency/time to locate the platform (P < 0.05) and increased the time spent in the target quadrant (P < 0.01) in the Morris water maze. This was accompanied by increased coupled respiration (P < 0.05) in isolated hippocampal mitochondria. Protein levels of CaMKII, PSD-95, and Complex 1 were increased in Cr-fed mice, whereas IκB was decreased. These data demonstrate that dietary supplementation with Cr can improve learning, memory, and mitochondrial function and have important implications for the treatment of diseases affecting memory and energy homeostasis.
Neurons require high energy levels for various cellular processes related to normal function (e.g., high levels of adenosine triphosphate (ATP) required for establishing ion gradients, neurotransmitter exocytosis, synaptic function, etc.). An important molecule associated with energy production, creatine (Cr) is derived from the diet and from endogenous synthesis in the liver and brain from glycine, arginine, and S-adenosylmethionine (Wyss and Kaddurah-Daouk 2000). Cr and ATP are converted to phosphocreatine (pCr) and ADP in a reversible reaction that is catalyzed by the enzyme creatine kinase (CK) (Kenyon and Reed 1983). Cr and pCr function as high-energy molecules, with the pCr–Cr conversion capable of regenerating ATP significantly faster than with oxidative phosphorylation (OXPHOS) and glycolysis (Wallimann et al. 1992).
Studies have investigated the neurobehavioral consequences of Cr supplementation in humans, with several reports of cognitive enhancements (Turner et al. 2015). For example, Cr supplementation has been shown to boost attention under short-term hypoxic conditions (Turner et al. 2015), improve mood and psychomotor function among sleep-deprived subjects (McMorris et al. 2006), improve performance on working and long-term memory tasks in elderly subjects (McMorris et al. 2007), improve scores on general intellectual ability and working memory (Rae et al. 2003), and reduce mental fatigue as well as the task-dependent rise in cerebral oxygenated hemoglobin in young healthy subjects (Watanabe et al. 2002). Further, Cr supplementation in humans increases brain pools of both Cr (Dechent et al. 1999) and the pCr/ATP ratio (Pan and Takahashi 2007) at doses shown to enhance cognition. These data provide evidence that Cr supplementation can positively impact both brain physiology and function in humans.
Given that learning and memory formation are energy-demanding at the neurophysiological level, that certain types of memory formation have been shown to be dependent upon intact mitochondrial respiratory function (Tanaka et al. 2008), and that Cr is considered a key regulator of a cell's energy status, Cr may exert effects on learning and memory via alterations to brain bioenergetics. In vitro, Cr pretreatment elevated pCr and ATP levels (Brewer and Wallimann 2000) and increased OXPHOS in synaptosomes and isolated brain mitochondria (Monge et al. 2008). In hippocampal neuron cultures, Cr treatment stimulated mitochondrial activity, effects that were also seen with overexpression of the mitochondrial fission protein dynamin-related protein 1 (Drp1), a regulator of mitochondrial dynamics (Li et al. 2004). Outside of its role in regulating cellular energy systems, however, little is known regarding the mechanisms by which oral Cr could enhance neuronal and cognitive function.
The formation of spatial memory relies heavily on the hippocampus (Bannerman et al. 1999). In rats, intrahippocampal injection of Cr (2.5 nmol/hippocampus) in the CA1 subfield facilitated spatial memory formation in the Barnes maze and object exploration task in mice (Souza et al. 2012). These effects were blocked with coinjection of an inhibitor of calcium/calmodulin-dependent protein kinase II (CaMKII) (Souza et al. 2012), a kinase shown to be required for long-term memory formation and shown to be activated within dendritic spines during hippocampal long-term potentiation (LTP) (Frankland et al. 2001). Phosphorylated levels of both CaMKII and cAMP-response element binding protein (CREB), a key transcription factor extensively studied in the context of activity-dependent plasticity, learning, and memory (Alberini and Kandel 2014), were up-regulated 30 min after Cr injection (Souza et al. 2012). The mechanisms by which spatial memory is enhanced by chronic oral Cr, a more applicable route of administration for use as a potential neuroenhancer in humans, remain poorly understood.
In addition to CREB, the transcription factor nuclear factor kappa B (NF-κB) is also implicated in cognition and synaptic plasticity (Snow et al. 2014). Interestingly, Cr can modulate NF-κB signaling in vitro; in neuronal cultures, Cr down-regulates levels of the NF-κB inhibitor, IκB (Juravleva et al. 2005). Whether NF-κB-associated alterations occur in vivo with oral Cr, however, has not been examined. In an inactive state, the NF-κB complex consists of several isoforms in various dimer compositions bound by the tethering protein IκB. Upon activation, IκB is degraded, rendering the dimer free to translocate to the nucleus where it can bind to target genes are either initiate or suppress transcription (Ghosh and Baltimore 1990; Mincheva-Tasheva and Soler 2013).
Neuronal NF-κB regulates the expression of several genes shown to be important for cognition (30). Members of the CaMKII family serve as both activators of NF-κB (Meffert et al. 2003) and neuronal gene targets (Federman et al. 2013). Postsynaptic density protein-95 (PSD-95), an integral synaptic protein implicated in several neuronal functions, including receptor anchoring (Niethammer et al. 1996) and regulation of spine formation (Vickers et al. 2006), is also a gene target of NF-κB in neurons (Boersma et al. 2011). Genetic reductions in PSD-95 disrupt spatial learning, synaptic plasticity (Migaud et al. 1998; Park et al. 2008), and spine formation (Vickers et al. 2006). Cr treatment up-regulates PSD-95 and spine density in neuron cultures (Li et al. 2004). Members of the early growth response (Egr) factor family of proteins have also been associated with cognition and with NF-κB signaling. Egr1 (a.k.a. Krox24, Zif268), also a transcription factor, has been implicated in synaptic plasticity and cognition (Worley et al. 1993; Poirier et al. 2008). Hippocampal-dependent memory has been shown to require NF-κB-dependent Egr1 expression (Zalcman et al. 2015). In vitro, hippocampal LTP results in an NF-κB-dependent up-regulation of another transcriptional member of the Egr family, Egr2 (a.k.a. Krox20) (Nafez et al. 2015), a neuronal gene target of NF-κB (45). Behavioral studies, however, report a gain of function in Egr2-conditional knockout (KO) mice lacking forebrain expression, including enhanced motor skill learning and long-term object recognition memory (Poirier et al. 2007). Whether these memory-related NF-κB mediated proteins are affected with dietary Cr has not been examined.
To expand our current understanding of the effects of dietary Cr supplementation on cognitive function and putative mechanisms of Cr-induced enhancements, we investigated hippocampal-dependent effects of oral Cr in male C57BL/6 mice, including: (1) Cr levels; (2) spatial learning and memory in the Morris water maze (MWM), a hippocampal-dependent task; (3) levels of synaptic and plasticity proteins that are known activators and/or downstream targets of NF-κB (e.g., CaMKII, PSD-95, Egr2) and other transcriptional factors implicated in learning and memory (e.g., CREB, Egr1); (4) IκB protein expression; (5) oxygen consumption rates (OCR) in freshly isolated mitochondria; and (6) levels of proteins involved in mitochondrial dynamics and function, including a subunit of OXPHOS Complex I and the mitochondrial fission protein Drp1.
Materials and Methods
Animals, diet formulation, and experimental design
All experiments were carried out in C57BL/6 male mice that were randomly assigned to either control (n = 8) or Cr-supplemented (n = 10) groups. Dietary intervention began at 7 mo of age and continued for 8 wk prior to MWM training. Mice were maintained on their respective diets right up to the experimental endpoint (e.g., behavioral, functional mitochondrial, or molecular experiments). Due to logistics, molecular experiments and mitochondrial experiments were carried out 4 and 6 d after MWM training, respectively. Cr monohydrate (3%, w/w, Alfa Aesar) was added to the semisynthetic nutritionally complete diet (see Table 1). Mice were allowed food and water ad libitum. Food intake was measured twice/week by subtracting food remaining from food provided (g). Body weights were measured weekly. At the beginning of week 8, all mice underwent MWM testing. For mitochondrial experiments, mice were sacrificed by inhalation of isoflurane, brains extracted, hippocampi removed, and mitochondria isolated. For Western blotting experiments, mice were sacrificed under anesthetic (intraperitoneal injection of ketamine at 62.5 mg/kg and xylazine at 12.5 mg/kg), followed by decapitation and tissue extraction. All experiments reported herein were conducted at St. Boniface Hospital Albrechtsen Research Centre, where mice were single-housed in standard cages at the animal holding facility. Mice were exposed to a 12-h light/12-h dark schedule in a room maintained at 22°C and 40% humidity. All experiments were approved by the University of Manitoba Animal Care and Use Committee, which conforms to the Canadian Council for Animal Care's Guide to the Care and Use of Experimental Animals.
Table 1.
Experimental diets
Morris water maze
Hippocampal-dependent spatial learning and memory were assayed using the MWM, as previously described (Snow et al. 2015). A circular pool (100-cm diameter) was filled with tap water (24°C–25°C) to which nontoxic white paint was added for opacity. Visual cues were placed at equal distances above the water level surrounding the maze. Nonmaze cues were obscured with curtains. The escape platform (10 cm) was submerged ∼5 mm below the water level in the target quadrant. The acquisition (e.g., learning) phase occurred over 5 d, with three trials/day. Mice were given 90 sec to locate the platform and trained to stay on the platform for 10 sec before being returned to their home cage, where a heating lamp was temporarily provided. Parameters assessed during the spatial learning phase included escape latency (time to reach the platform) and swim speed.
The frequency of use of various search strategies in the MWM changes across training days, with an increase in the use of spatial versus nonspatial, random search strategies that is thought to be associated with formation of a spatial map (Janus 2004; Brody and Holtzman 2006; Oikawa et al. 2012). To more fully characterize the effect of oral Cr on spatial learning and memory, χ2 analyses were conducted on the percentage of use of various search strategies as a function of training and diet. In brief, the path traces were assigned as one of nine strategies under three broader categories (Brody and Holtzman 2006): (1) Repetitive Looping: consisting of (a) Peripheral Looping: swimming along the outer edge of the pool adjacent to the pool wall; (b) Chaining: swimming around the pool at a somewhat fixed distance more interior from the pool wall; (c) Circling: swimming in tight circular patterns; (2) Nonspatial Systemic: consisting of (a) Scanning: swimming within the interior portion of the pool with no obvious spatial focus; (b) Random: swimming the entire pool with no obvious spatial focus; and (c) focal incorrect: swimming within a defined but incorrect quadrant; and (3) Spatial: consisting of (a) Spatial Direct: swimming directly to the platform; (b) Spatial Indirect: swimming to the platform with no more than one loop; and (c) Focal Correct: direct swim to and search of the correct quadrant. In cases where more than one search strategy was used in any given trial, classification was based on the predominant strategy used for the duration of the trial.
Memory retention was measured by a probe trial on the last (sixth) day, in which case the platform was removed, and mice were given 90 sec to search for the missing platform. The time spent in the target quadrant as well as the number of passes into the target quadrant and over the platform area were calculated as indices of memory retention. Videos were captured using a digital camera, and tracking software (Videomex, Columbus Instruments) was used to collect behavioral data.
Protein extraction
After MWM training, hippocampal tissue was dissected out, snap-frozen in liquid nitrogen, and stored at −80°C prior to Western blotting and prepared as previously described (Snow et al. 2015). In brief, homogenates were prepared from frozen tissue in ice-cold RIPA buffer (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate (SDS), and 50 mM Tris, pH 8.0) with 1% protease inhibitor cocktail (Amresco), and 1% phosphatase inhibitor cocktail (Sigma-Aldrich) and exposed to constant agitation for 60 min at 4°C. Tissue lysates were centrifuged (10,000 rpm for 10 min at 4°C) and supernatants collected. Protein concentrations were calculated using the DC Protein Assay (Bio-Rad) as per manufacturer instructions. Samples were then diluted to equal concentrations in RIPA buffer.
Creatine assay
Hippocampal Cr levels were measured in homogenates (10 µL of homogenate diluted to 4 mg/mL of protein) using the Creatine Colorimetric Assay Kit (BioVision Inc.) as per manufacturer's instructions.
Western blotting
A 4× Laemmli buffer (16% SDS, 40% glycerol, 20% β-mercaptoethanol, 0.01% bromophenol blue, and 0.25 M Tris, pH 6.8) was added to hippocampal homogenates, followed by denaturing at 50°C for 8 min. Equivalent amounts of protein (10–20 µg) from each sample were loaded and subjected to SDS-PAGE at 200 V for 45 min in polyacrylamide gels (Bio-Rad). Proteins were transferred to nitrocellulose membranes (Bio-Rad) with the Trans-Blot Turbo Transfer System (Bio-Rad) or wet transfer for detection of heat-sensitive OXPHOS protein Complex I subunit. Total protein was detected by reversible Ponceau S staining or by TGX Stain-Free gels (Bio-Rad) before primary antibody incubation. Membranes were blocked for 1 h (room temperature; RT) in 1× Tris-buffered saline with 0.1% Tween-20 (TBS-T) with 5% skim milk, with the exception of membranes detecting phosphoCREB (pCREB), where nonspecific binding was reduced with 5% bovine serum albumin in place of milk. Membranes were incubated with primary antibodies in TBS-T with 5% skim milk at 4°C overnight, washed with 1× TBS-T, and incubated with either peroxidase-conjugated AffiniPure goat anti-rabbit or goat anti-mouse IgG (H+L) antibody (1:5,000 dilution, Jackson ImmunoResearch Laboratories) for 1.5 h at 4°C. Antibody binding was estimated with enhanced chemiluminescence (ECL) using Bio-Rad Clarity Western ECL Blotting Substrate (Bio-Rad) and imaged with the ChemiDoc MP (Bio-Rad). Band intensities were quantified with ImageLab software. Densitometry values were normalized to total protein loaded per lane. All primary antibodies were used at a dilution of 1:1,000 unless otherwise stated. The following primary antibodies were used: rabbit polyclonal anti-Egr1 (cat. no. sc-189, Santa Cruz; 1:200 dilution), rabbit monoclonal anti-IκBα (cat. no. ab32518, Abcam), rabbit polyclonal anti-CaMKII (cat. no. M-176, Santa Cruz; 1:2,000 dilution), rabbit monoclonal anti-Egr2 (a.k.a. Krox20) (abcam108399, Abcam; 1:7,500 dilution), rabbit monoclonal anti-PSD-95 (cat. no. ab76115, Abcam; 1:2,000 dilution), mouse monoclonal anti-Drp1 (cat. no. ab156951, Abcam), rabbit polyclonal anti-actin (cat. no. A5060, Sigma-Aldrich), total OXPHOS rodent WB antibody cocktail for Complex I detection (against NDUFB8 subunit; cat. no. ab110413, Abcam), mouse monoclonal anti-porin (cat. no. ab14734; Abcam), and rabbit monoclonal anti-pCREB (detects CREB phosphorylated at Serine 133; cat. no. ab32096, Abcam). In addition, membranes probed for pCREB were stripped and reprobed with the rabbit monoclonal anti-CREB primary antibody (cat. no. ab32515, Abcam).
Mitochondrial oxygen consumption rates from freshly isolated hippocampi
Mitochondria were freshly isolated from the hippocampus in a subset of animals after MWM training using a differential centrifugation method (Frezza et al. 2007). Briefly, hippocampal tissue was dissected out and rinsed in ice-cold mitochondrial isolation buffer (70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, and 0.5% (w/v) fatty acid free bovine serum albumin (BSA), pH 7.2), followed by manual homogenization. The homogenate was centrifuged (800g for 10 min, 4°C). After removal of the pellet, the supernatant was spun again (twice at 800g for 10 min at 4°C, once at 8000g for 15 min at 4°C). The pellet was resuspended in mitochondrial isolation buffer and spun (8000g for 15 min at 4°C). The final pellet was resuspended in phosphate-buffered saline and protein concentrations determined using the DC Protein Assay (Bio-Rad) as per manufacturer instructions.
Complex-I-dependent mitochondrial respiration was evaluated by measuring OCR in real time using the Seahorse XF Analyzer (Agilent Technologies). Isolated mitochondrial protein (10 µg/well) was diluted in mitochondrial assay solution containing 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, and 0.2% BSA and plated onto the Seahorse plate (final volume of 450 µL). Basal respiration was measured in the presence of pyruvate (10 mM) and malate (2 mM). Coupled respiration was measured after the addition of adenosine-diphosphate (ADP; 4 mM) as a substrate for ATP synthase and calculated after subtracting basal respiration rates. Oligomycin (2 µM) was then added to terminate coupled respiration through inhibition of ATP synthase. The protonophore carbonyl cyanide p-triflouromethoxy-phenylhydrazone (FCCP) was added (2 µM) to stimulate uncoupled respiration and allow for the measurement of maximal respiration. Finally, rotenone (Complex I inhibitor) and antimycin (Complex III inhibitor) were added (2 µM each) to block mitochondrial respiration. OCR data were calculated with subtraction of nonmitochondrial respiration rates (after the addition of rotenone and antimycin). In addition to basal, coupled, and maximal respiration, coupling efficiency was calculated by dividing OCR after the addition of ADP by basal OCR. The maximal respiratory control ratio (RCR) was calculated by dividing OCR after the addition of FCCP by OCR after the addition of oligomycin. Coupled RCR was calculated by dividing OCR after the addition of ADP by OCR after the addition of oligomycin.
Statistical analysis
To account for variation in food intake as a function of body weight, food intake data (measured twice per week) were analyzed by averaging the grams of food eaten per week as grams per day. This value was then divided by the animal's body weight at the start of the week to yield a value of mean grams per day per gram of body weight. Body weight data were available for 7 wk of the 8-wk treatment. Therefore, food intake was analyzed using a 2 (diet) × 7 (week) mixed model two-way ANOVA, with repeated measures on the last factor. MWM acquisition data were analyzed using 2 (diet) × 5 (day) ANOVAs, with day as a repeated measure. Post hoc comparisons were conducted using Fisher's least significant difference (LSD) tests. Spatial strategy use data were analyzed by χ2 tests. Data obtained in the 1-d retention phase, mitochondrial data, and Western blot data were analyzed by Student's t-test or Mann–Whitney U (MWU), where appropriate. Pearson correlations coefficients were computed between hippocampal Cr levels and densitometry values from western blots and memory parameters from the probe trial in the MWM. Significance was set at P < 0.05, and analyses were two-tailed. Data were analyzed using SPSS V20 (IBM Corp.).
Results
Food intake and weight gain
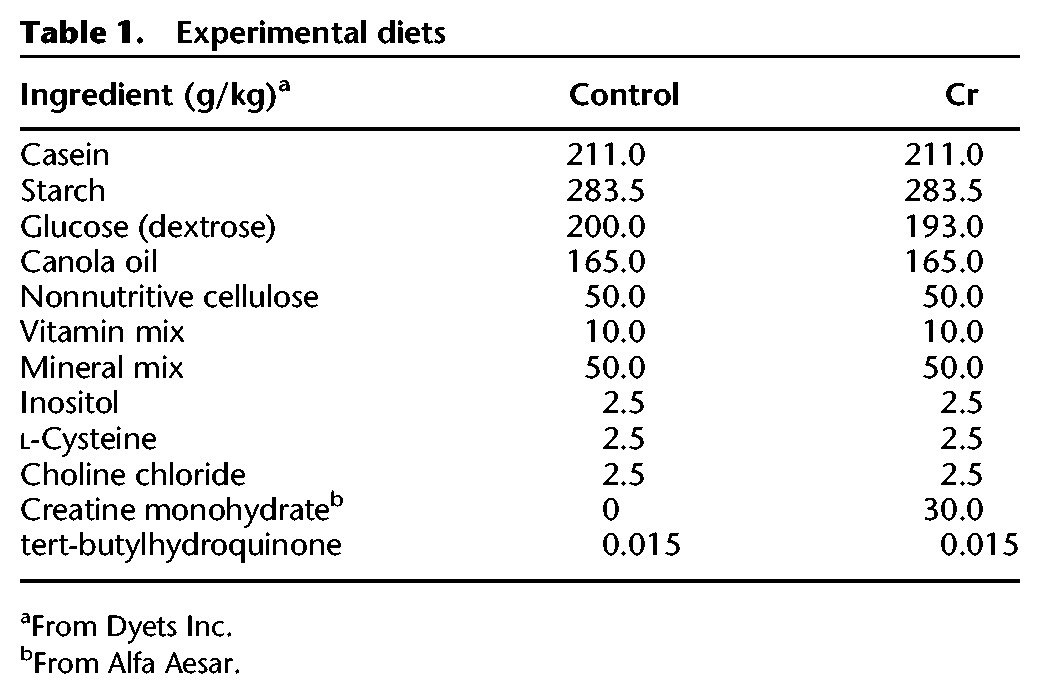
To confirm that food intake was not affected by supplementation with Cr, as caloric restriction has been associated with improved learning and memory in mice (Martin et al. 2007), food intake data (mean g/day/g body weight) were analyzed across weeks and between groups. ANOVA revealed a significant main effect of week (F(6,96) = 24.26, P < 0.001), with no influence of Cr consumption, as no main effect of diet (F(1,16) = 0.18, P = 0.7) or diet × week interaction (F(6,96) = 0.32, P = 0.9) was detected. Irrespective of diet, mice ate significantly more during Week 1 as compared to all other weeks (P < 0.001 for Weeks 3, 4, 5, 6, and 7; P < 0.01 for Week 8; no data for Week 2). There was a small but significant decline in food intake between Weeks 3 and 4 (P < 0.05), after which time intake remained fairly stable (Fig. 1). In addition to food intake, weight gain over the 8-wk treatment was similar between groups (control: 6.64 ± 0.82; Cr: 6.5 ± 0.88; P = 0.9).
Figure 1.
Food intake as a function of diet. Plot depicting food intake in adult (7-mo-old) control mice and mice supplemented with Cr. Food intake (g) was measured twice/week over the 8–9-wk dietary intervention, averaged across the week, and expressed as g/day (mean ± SEM depicted). The g/day value was divided by the animal's body weight to account for variations in food intake based on body weight. Data were analyzed by two-way ANOVA, followed by Fisher's LSD post hoc comparisons for the 7 wk for which intake data were available (no data for week 2). Control: n = 8; Cr: n = 10; (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
MWM acquisition phase—spatial learning
Although four trials/day is typical of most training regimes using the MWM (Vorhees and Williams 2006), reduced trial number can increase the cognitive demand of the task as well as the acquisition period needed to demonstrate plateaus in performance (Mandel et al. 1989; Hebda-Bauer et al. 2005, 2007), thereby yielding measurably greater differences in learning acquisition with experimental manipulations (Mandel et al. 1989). In efforts to avoid possible ceiling effects that may impair the ability to detect enhancements with Cr supplementation, a training regime of three trials/day was chosen for the present study.
Mixed model two-way ANOVA of escape latencies (time to locate the platform) revealed a significant main effect of day (F(4,64) = 16.75, P < 0.001) and diet (F(1,16) = 5.42, P < 0.05), with no interaction effect between the two factors (F(4,64) = 0.37, P = 0.8). Simple main effects analysis of day revealed that irrespective of diet, escape latencies significantly decreased from Day 1 to Day 2 (P < 0.001), Day 3 (P < 0.001), Day 4 (P < 0.001), and Day 5 (P < 0.001). Latencies did not significantly decrease from Day 2 to 3 (P = 0.15) or from Day 3 to Day 4 (P = 0.8) but were significantly decreased from Day 4 to 5 (P = 0.03). By the end of the acquisition phase, latencies were significantly lower relative to all other training days (versus Day 5: Day 1: P < 0.001; Day 2: P = 0.002; Day 3: P = 0.04; Day 4: P = 0.03). Simple main effects analysis of diet found that across training days, oral Cr decreased the time to locate the platform (P < 0.05; Fig. 2A). To ensure mice started the testing session at similar levels, the effect of Diet was analyzed on the first two trials on Day 1. As data failed tests of normality, nonparametric MWU tests were performed. No significant differences were found on Trial 1 (P = 0.4) or Trial 2 (P = 0.4), confirming that mice did not exhibit prior differences that could confound interpretation of performance in the MWM as it relates to learning and memory retention (Fig. 2A).
Figure 2.

MWM as a function of diet. (A–D) Spatial learning in the acquisition phase. In male mice, oral Cr significantly decreased escape latency (time to locate the platform; mean ± SEM) overall across training days, indicating enhanced learning of the spatial task. n = 8–10; P < 0.05; two-way ANOVA. (B) Swim speed (mean ± SEM) was assessed by two-way ANOVA to rule out enhanced motor performance as a contributing to factor to decreased latency. Swim speed decreased from the first day of training (P < 0.05) overall, as diet did not influence swim speed. (C) The frequency of use (% of total) of various search strategies (operationally defined as per Brody and Holtzman 2006) was calculated across training days for control and (D) Cr-fed mice and analyzed using Chi-square tests. In both groups, the frequency of use of various strategies differed across training days ((***) P < 0.001). Spatial strategies increased across training days, with the frequency of use peaking earlier in Cr-fed mice, although the difference fell short of significance (p = 0.06). Control: n = 8; Cr: n = 10. (E–G). Memory retention on the sixth day of the MWM after removal of the platform. The platform was removed, and mice were allowed 90 sec to swim. Memory retention (mean ± SEM) was evaluated in mice with and without oral Cr by measuring (E) time in target quadrant, (F) passes into the target quadrant, and (G) passes over the platform area. Student's t-test; dotted line indicates chance level. Control: n = 8; Cr: n = 9; (*) P < 0.05; (**) P < 0.01.
To rule out any potential influence of Cr supplementation, a well-known muscle enhancer (Archer 2004), on improvements in motor function as a confounding factor in interpreting escape latency data, swim speeds were analyzed. Mixed model two-way ANOVA (2 diet × 5 Day; day as a repeated measure) of swim speed data detected differences across days (F(4,64) = 4.98, P = 0.001), with no main effect of diet (F(1,16) = 0.28, P = 0.6) or interaction effect (F(4,64) = 1.24, P = 0.3), indicating that although swim speed changed across training days, diet did not influence this effect. Irrespective of diet, swim speed decreased significantly from Day 1 to Day 2 (P < 0.01), Day 3 (P < 0.05), Day 4 (P = 0.001), and Day 5 (P = 0.02; Fig. 2B).
MWM acquisition phase—search strategy analysis
χ2 analyses of the frequency of use of various search strategies revealed significant differences across training days for each group (Cr χ2 = 30.97, P < 0.001; Control χ2 = 25.75, P < 0.001; Fig. 2C,D). Overall, in both groups, the reliance on nonspatial strategies decreased, whereas spatial strategies usage increased during training. In Cr-fed mice, the use of spatial strategies peaked on Day 3 (30%); in control-fed mice, comparable levels (33.3%) were not reached until Day 5, suggestive of advanced reliance on spatial strategies in Cr-fed mice. χ2 analysis comparing the frequency of strategy use between groups collapsing the first three training days, by which time peak spatial strategy use was achieved in Cr-fed mice, fell just short of significance (P = 0.057).
MWM probe phase—spatial memory retention
Time in target quadrant during the probe trial, considered a particularly sensitive indicator of memory in this behavioral paradigm (Crawley 2000), was significantly increased in Cr-fed mice when compared with control-fed mice (P = 0.04). Further examination of the data revealed one mouse in the Cr-fed group that performed poorly on all retention parameters relative to group means and met the criterion of an outlier (±2.5 SD from the mean). Reanalysis of probe data after removal of the outlier revealed a significant increase in the time spent in the target quadrant (P = 0.008) with oral Cr, with only Cr-fed mice surpassing chance levels (Fig. 2E). The number of passes into the target quadrant was not significantly different as a function of diet (P = 0.6; Fig. 2F), although mice on the Cr diet did exhibit significantly more passes specifically over the platform area (P = 0.03) relative to those on the control diet (Fig. 2G).
Hippocampal Cr levels
Cr concentration was measured in hippocampal homogenates after MWM training in a subset of animals. Although absolute values indicate an 8.7% increase in hippocampal Cr concentration with dietary Cr, this increase was nonsignificant (P = 0.19; Fig. 3).
Figure 3.
Hippocampal Cr levels after 8–9 wk of dietary intervention. Cr levels were evaluated in hippocampal homogenates using a colorimetric kit (BioVision Inc). Control: n = 4; Cr: n = 5; P > 0.05; Student's t-test.
Hippocampal mitochondrial function
Mitochondrial function was measured in the hippocampi in a subset of animals after MWM training. All OCR data were corrected for nonmitochondrial respiration rates (after rotenone and antimycin). At baseline, OCR were similar in wells containing isolated mitochondria from control and Cr-fed mice (P = 0.4). After application of oligomycin, OCR associated with coupled respiration were significantly increased in mitochondria isolated from Cr-fed mice versus controls (P = 0.04). After correcting for basal rates, OCR indicative of maximal respiration capacity (measured after application of the uncoupler FCCP), were not significantly different as a function of diet (P = 0.7). Coupling efficiency (coupled respiration relative to basal levels) was significantly increased with oral Cr (P = 0.01). Consistent with changes in parameters associated with coupling, coupled RCR was significantly higher in hippocampal mitochondria isolated from Cr-fed versus control-fed mice (P < 0.001), whereas maximal RCR was not significantly different as a function of diet (P = 0.5). Therefore, Cr consumption selectively enhanced functional parameters associated with coupled respiration in mitochondria isolated from the hippocampus in trained mice (Fig. 4).
Figure 4.

OCR in mitochondria isolated from the hippocampus after MWM training in control and Cr-fed mice. (A) Kinetics graphing indicating real-time OCR at baseline and after addition of substrates ADP, oligomycin, FCCP, and rotenone/antimycin (R/A) in 10 µg of mitochondrial protein per well. (B) Basal respiration, (C) coupled respiration, and (D) maximal respiration (mean ± SEM) were calculated after subtracting nonmitochondrial respiration (after R/A) and compared between control and Cr-fed mice. (E) Coupling efficiency was calculated by dividing coupled respiration (after addition of ADP) by basal respiration. (F) Coupled RCR was calculated by dividing coupled respiration rates (after ADP) by OCR after oligomycin. (G) Maximal RCR was calculated by dividing maximal respiration rates (after FCCP) by OCR after oligomycin. Student's t-test; n = 9–10 wells per group; (*) P < 0.05; (***) P < 0.001.
Hippocampal protein levels—Western blotting
To further investigate putative molecular mechanisms underpinning the enhanced mitochondrial function and cognition with dietary Cr supplementation, various proteins in the hippocampus were semi-quantified with Western blotting (expressed as percentage change in densitometry values from control mean of 100% ±SEM).
Transcription factors and related proteins
Several transcription factors that have been implicated in learning, memory, and experience-dependent plasticity were evaluated after MWM training as a function of Cr intake. Western blotting of hippocampal homogenates with anti-Egr1 revealed two bands at ∼60 and 55 kDa, presumably representing different phosphorylated forms of the protein. Mean densitometry values of either band were not significantly different between Cr-supplemented and control mice (∼60 kDa: P = 0.8, ∼55 kDa: P = 0.1). Levels of total CREB were also unchanged by dietary Cr (normalized to total protein; P = 0.9), as were levels of pCREB (normalized to total CREB; P = 0.3). In contrast, IκBα protein levels used as a measure of NF-κB involvement, significantly reduced with oral Cr in the hippocampus (P = 0.01), suggestive of enhanced NF-κB-mediated signaling after chronic supplementation in trained mice (Fig. 5).
Figure 5.

Relative levels of transcription factors and related proteins in the hippocampus of MWM-trained mice as a function of diet. Representative Western blots detecting (A) Egr1, (D) CREB, and (G) IκBα. Total protein bands were visualized with Ponceau S staining (in A and G) or with TGX Stain-Free gels (Bio-Rad) in D; representative bands shown to indicate loading across samples. Samples from each group were immunoblotted on the same gels for comparison. Bar graphs depict densitometry values of (B) ∼60 kDa (C) and ∼55 kDa bands detected with anti-Egr1 as well as (E) CREB and (H) IκBα after normalizing to total protein bands (expressed as the percentage change from control means of 100% ±SEM. (F) Bar graph depicting densitometry values of pCREB, normalized to total CREB band (% change from control mean set at 100% ±SEM). Student's t-test; n = 4–5; (**) P ≤ 0.01.
NF-κB-associated plasticity proteins
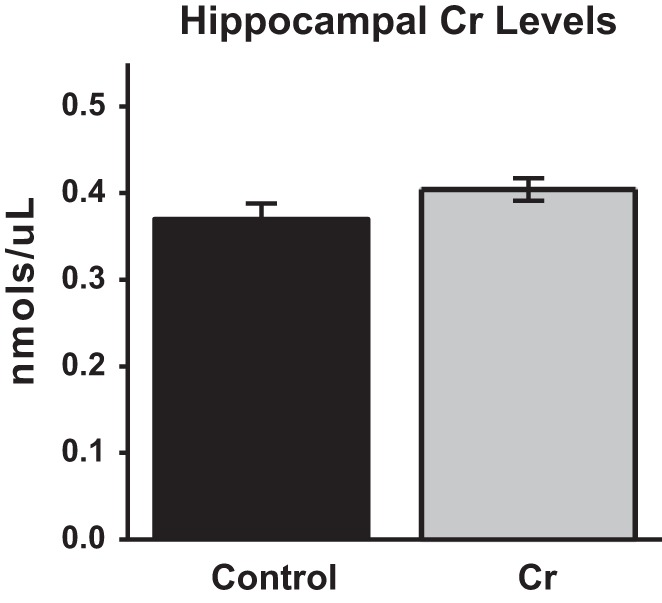
Oral Cr significantly up-regulated hippocampal CaMKII levels after MWM training relative to levels in control-fed mice (P = 0. 008). Levels of PSD-95, a downstream target of NF-κB in neurons, were also significantly elevated with oral Cr (P = 0.004). In contrast, levels of Egr2, another neuronal NF-κB target implicated in memory, were not altered with oral Cr in hippocampal homogenates after MWW training (p = 0.3). To rule out an increase in brain cellular material through Cr (e.g., increased dendritic arborization, neurogenesis) as a possible cause for up-regulation in CaMKII and PSD-95 levels found with Western blotting, actin levels were evaluated between groups as a measure of cellular material. No significant differences (P = 0.98) were found in hippocampal actin levels (normalized to total protein) between control and Cr-fed mice (Fig. 6).
Figure 6.
Relative levels of NF-κB-associated plasticity proteins in the hippocampus of MWM-trained mice as a function of diet. (A,B) Representative Western blot and bar graph (percent change from control mean of 100% ±SEM) depicting CaMKII levels in trained mice after normalizing to total protein (Ponceau S staining). (C,D) Representative Western blot and bar graph (% change from control mean of 100% ±SEM) depicting PSD-95 levels in trained mice after normalizing to total protein (Ponceau S staining). (E,F) Representative Western blot and bar graph (% change from control mean of 100% ±SEM) depicting Egr2 levels in trained mice after normalizing to total protein (Ponceau S staining). (G,H) Representative Western blot and bar graph (% change from control mean of 100% ±SEM) depicting actin levels in trained mice after normalizing to total protein (TGX Stain-Free gels; Bio-Rad). Student's t-test; n = 3–5; (**) P < 0.01.
Mitochondrial proteins
Porin, an abundant protein of the outer mitochondrial membrane, was used as a marker of mitochondrial mass (Hanson et al. 2001). No significant differences were detected in porin when normalized to total protein (P = 0.8; Fig. 7A,B), indicating that Cr did not induce hippocampal mitochondrial biogenesis. As well, no significant differences were noted for levels of the fission protein Drp1 relative to total protein (P = 0.2; Fig. 7A,C). In contrast, levels of OXPHOS protein Complex I were significantly increased in the hippocampus of Cr-fed mice (P = 0.04; Fig. 7D,E), in accordance with the functional Complex-I-dependent mitochondrial assay.
Figure 7.
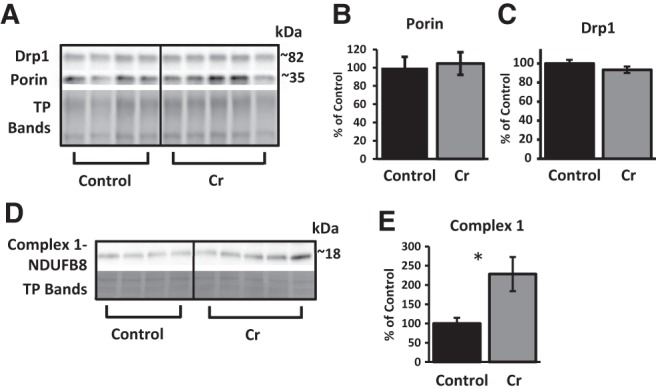
Relative levels of mitochondrial proteins in the hippocampus of MWM-trained mice as a function of diet. (A) Representative Western blot detecting Drp1, porin, and total protein (with TGX Stain-Free gels; representative bands from total proteins shown to indicate loading across samples). Bar graphs depict densitometry values of (B) porin and (C) Drp1 after normalizing to total protein bands and expressed as the percentage change from control means (100%) ±SEM. (D) Representative Western blot and (E) bar graph (percent change from control mean of 100% ±SEM) depicting levels of NDUFB8, a subunit of the electron-transport chain protein Complex I, in trained mice after normalizing to total protein (TP; Ponceau S staining). Student's t-test; n = 4–5; (*) P < 0.05.
Correlations between hippocampal Cr levels and densitometry values and MWM probe phase
To further investigate relationships between Cr and molecular and functional parameters in the hippocampus, Pearson correlation coefficients were measured (1) between hippocampal Cr levels and probe trial measures (as indices of spatial memory formation) in the MWM, and (2) between hippocampal Cr levels and relative normalized densitometry values from Western blot experiments. Despite between-group effects on measures of spatial memory with dietary Cr, no significant correlations were found between hippocampal Cr levels and any of the probe trial parameters. Of the proteins probed, only levels of CaMKII (r = 0.83; P = 0.006) and actin (r = 0.67; P < 0.05) were significantly correlated with hippocampal Cr; Fig. 8).
Figure 8.

Correlational analyses between Cr, memory, and protein levels. Pearson correlation coefficients between hippocampal Cr levels and (A–C) MWM probe trial parameters and relative normalized densitometry values from western blot experiments measuring (D–H) transcription factor, (I–L) plasticity-related, and (M–O) mitochondrial proteins in control (▪) and Cr-supplemented (▪) mice. n = 7–9, (*) P < 0.05; (**) P < 0.01.
Discussion
The present study demonstrated significant enhancements in hippocampal function and cognition with dietary Cr that were accompanied by alterations to the NF-κB transcription factor complex, indicating that under physiological conditions, Cr is capable of regulating several parameters, including brain bioenergetics, learning, memory, and the expression of plasticity-relevant proteins in vivo. In the MWM, Cr supplementation resulted in enhanced acquisition of a spatial learning task that was not explained by increased swimming endurance, group differences in food intake, or enhanced capabilities at the start of the training. Spatial memory was also significantly improved with Cr supplementation. Importantly, the time spent in the target quadrant reached above-chance levels only in mice on the Cr diet, demonstrating a recall for the platform location in supplemented mice only. These data indicate increased cognitive demand with the training regime provided, which was sufficient to impede acquisition and memory consolidation in control mice but not Cr-fed mice.
Previous studies have reported neurobehavioral enhancements with Cr in typical mice, including improved spatial learning and memory (Allahyar et al. 2016), object recognition memory (Bender et al. 2008), and increased lifespan (Bender et al. 2008), however these data were collected in females. Data regarding whether such differences persist in male mice, however, are lacking. Sex-dependent differences have been noted in regards to the effects of Cr on other physiological parameters (Ellery et al. 2016), which prompted our investigation of the effect of oral Cr on hippocampal function specifically in males.
Although classically considered a key amino acid for proper muscle function, aberrations to the Cr system in the brain have long been known to result in learning and memory impairments (for review, see Hanna-El-Daher and Braissant 2016). For example, genetic mutations of the SLC6A8 gene encoding the Cr transporter are associated with intellectual disability (van de Kamp et al. 2013) that is refractory to oral Cr (Salomons et al. 2001; van de Kamp et al. 2012). In contrast, deficiency syndromes associated with genetic mutations affecting Cr-synthesizing enzymes arginine–glycine amidinotransferase and guanidinoacetate methyltransferase also result in the absence or severe down-regulation of brain Cr levels and significant cognitive deficits but are responsive to dietary Cr supplementation (Rae and Bröer 2015). Conversely, memory training can alter metabolite levels in the hippocampi of elderly subjects, including increases in Cr and choline levels (Valenzuela et al. 2003). Levels of mitochondrial-localized CK are regulated by neuronal activity (Boero et al. 2003), reduced after traumatic brain injury, and up-regulated with environmental enrichment in both cortex and hippocampus (Briones et al. 2013). Tissue-specific cytoplasmic isoforms of CK are found in muscle (CK-M) and brain (CK-B) (Wallimann et al. 1992), both high-energy tissues with fluctuating energetic demands. Interestingly, CK-B localization is particularly abundant in highly plastic brain regions implicated in learning and memory, including the hippocampus and cerebellum (Kaldis et al. 1996). These data suggest a reciprocal relationship between the brain Cr system and experience-dependent plasticity in brain regions important for learning and memory and indicate cognitive dysfunction with aberrations to this system.
The present data, for the first time, implicates the IκB-NF-κB signaling pathway in the neurocognitive benefits of dietary Cr. Prior work has demonstrated modulation of this pathway with Cr in vitro in neurons (Juravleva et al. 2005), findings that are corroborated by the present results with dietary Cr indicating decreased protein levels of the most common inhibitory subunit, IκBα (Mattson and Meffert 2006), as well as elevations to both activators (e.g., CaMKII) and downstream neuronal targets (e.g., PSD-95) of NF-κB. In hippocampal cultured neurons, calcium-driven activation of NF-κB is dependent upon CaMKII (Meffert et al. 2003). CaMKII-mediated activation of NF-κB could further affect CaMKII signaling, given both upstream (Meffert et al. 2003) and downstream (Federman et al. 2013) associations for CaMKII with NF-κB.
Although CaMKII can also phosphorylate CREB (Sun et al. 1994), neither CREB nor Egr1, another key transcription factor implicated in learning and memory (Veyrac et al. 2014), was altered. The level of hippocampal Egr2, a recently identified target of neuronal NF-κB (Nafez et al. 2015) associated with memory (Poirier et al. 2007), was also unchanged with oral Cr. These data are in contrast to previous work implicating the CaMKII-CREB pathway in improved memory after an acute intrahippocampal injection of Cr (Souza et al. 2012). These disparate findings related to CREB up-regulation with Cr between the present study and that of Souza et al. could be due to several factors, including the method of Cr delivery (supplementation versus intrahippocampal injection), the extent of the intervention (chronic versus acute), and the interval between training and brain collection (days versus minutes). Given the time delay between training and tissue extraction, we cannot rule out the possibility that alterations to CREB or other transcription factors (e.g., Egrs) occurred prior to tissue collection.
In mice, CaMKIIα is essential for the formation of memory at long delays (Frankland et al. 2001). In mice with mitochondrial DNA damage, deficits were found in mitochondrial respiration in conjunction with down-regulation of hippocampal CaMKIIα (Tanaka et al. 2008). Further, mutant mice exhibited impaired remote spatial memory, whereas learning and recent memory were intact (Tanaka et al. 2008). Such data highlight the importance of preserved mitochondrial function in certain types of memory and further suggest involvement of CaMKII in both memory and in mitochondrial regulation. Alterations to NF-κB pathways with oral Cr, therefore, may ultimately influence memory and mitochondrial function through CaMKII-dependent pathways. The strong positive correlation between hippocampal Cr and CaMKII protein levels is also in agreement with a strong contribution of this kinase to the neurobeneficial effects of oral Cr. Future studies with oral Cr in which NF-κB and/or CaMKII are deficient would further elucidate the molecular pathways involved in Cr-mediated cognitive benefits.
In line with the present in vivo hippocampal data with oral Cr, treatment of hippocampal neurons in vitro increased expression of PSD-95 and stimulated mitochondrial function (Li et al. 2004). As PSD-95 is a marker of excitatory synapses (Li et al. 2004), PSD-95 up-regulation indicates a synaptogenic effect of oral Cr. Behavioral studies demonstrate a potent role for this synaptic protein in hippocampal-dependent spatial learning and cognition (Migaud et al. 1998), as evidenced by impairments in PSD-95-KO mice and up-regulation of PSD-95 after behavioral training (Pollak et al. 2005) and environmental enrichment (Nithianantharajah et al. 2004). The current data demonstrate potentiation of training-induced up-regulation in this well-known synaptic marker with oral Cr and suggest involvement of the CaMKII-NF-κB-PSD-95 pathway in the noted cognitive enhancements.
Despite up-regulation of PSD-95, its protein levels were not correlated with hippocampal Cr levels. In contrast, supplementation did not alter actin, although actin densitometry values were significantly correlated with hippocampal Cr levels. This correlation is consistent with an interpretation of increased demand for Cr in hippocampi with increased cellular material, as measured by actin. This essential element of the neuronal cytoskeleton has been implicated in activity-dependent plasticity, learning, and memory (Motanis and Maroun 2012). Actin regulates the growth and retraction of dendritic spines and, thereby synaptogenesis (Hotulainen and Hoogenraad 2010) and exists as six isoforms in mammalian cells, with β-actin being the major brain isoform (Cheever and Ervasti 2013). Alterations to brain β-actin specifically have been shown to alter MWM performance (Cheever et al. 2012). Given up-regulation of PSD-95 is indicative of synaptogenesis, one could expect alterations to actin as well with oral Cr, which were not found. We did not, however, evaluate actin levels specifically in synaptic compartments but rather whole hippocampal homogenates, nor did we specifically detect the major brain isoform, β-actin, but instead used a pan-actin antibody as a general marker of cellular material.
At the synapse, mitochondria are particularly sensitive to depletions in Complex I activity, with a 25% reduction in activity required to decrease ATP production and mitochondrial respiration rates (Davey et al. 1998). In contrast, >70% inhibition is required to see such effects with Complex III and IV (Davey et al. 1998). As such, a significant portion of OXPHOS in synaptic mitochondria is under the control of Complex I activity. Western blot data indicating Complex I up-regulation in the present study is in accordance with up-regulation of PSD-95 as an indicator of synaptogenesis, whereby increased levels of Complex I would be expected to facilitate the activity of newly generated synapses.
After 1 week of Cr supplementation in humans, increases in both Cr and the phosphoCr/ATP ratio were detectable in vivo in the hippocampus using high-field magnetic resonance 31P and 1H spectroscopic imaging (Pan and Takahashi 2007). The authors speculated that oral Cr may affect neuronal bioenergetics by increasing the coupling efficiency between mitochondrial production and cellular consumption. The results presented here of enhanced coupling efficiency in hippocampal mitochondria of Cr-fed mice support this hypothesis and provide evidence for the involvement of the Cr system in mitochondrial regulation.
The present results have important implications for treatment of disorders affected by mitochondrial dysfunction and cognitive impairment, such as Alzheimer's disease (AD). Published clinical studies investigating oral Cr in AD patients are lacking, despite evidence of dysfunction in the Cr system in the disease. For example, AD patients exhibit lower levels of pCr (Pettegrew et al. 1994). In addition, CK-B is reduced by 80% in human AD brain homogenates (Aksenov et al. 1997), consistent with reports of decreased pCr (Pettegrew et al. 1994). In neurons from CK-B-deficient mice, the proportion of motile mitochondria was significantly increased (Kuiper et al. 2008). In the AD brain, activity of OXPHOS complexes is reduced (Parker et al. 1994), including Complex I (Giachin et al. 2016). Several studies also document alterations in NF-κB in AD (for review, see Snow and Albensi 2016). Given these data and the findings of early and severe hippocampal pathology (Henneman et al. 2009) and spatial memory impairments in the disease (Cherrier et al. 2001), the present results of enhanced hippocampal function with Cr argue for a therapeutic, possibly preventative role for Cr supplementation in AD.
Studies document a therapeutic role for chronic oral Cr in other pathological states. For example, oral Cr improved locomotor function and performance in the MWM in neonatally induced ischemia in both female (Allah Yar et al. 2015) and male (Iqbal et al. 2015) mice. A single oral dose of Cr reduced depressive-like symptoms in a mouse model of the disease in females, an effect that was accompanied by up-regulation of hippocampal PSD-95 (Pazini et al. 2016). In the forced swim test model of depression, oral Cr exerted anti-depressant properties in female rats, whereas male rats supplemented with oral Cr were more prone to depressive-like symptoms (Allen et al. 2010), indicating sex-specific effects of dietary Cr under pathological states.
Although data indicate sex differences with dietary Cr in cases of disease, whether such differences extended to cognition in healthy animals has not been well-studied. In healthy female mice, long-term oral Cr increased life span and improved object recognition memory (Bender et al. 2008) as well as spatial memory (Allahyar et al. 2016). The present findings of improved performance in the MWM in male supplemented mice, however, suggest that neuroenhancements of dietary Cr may be sex-independent in healthy mice.
As in humans, Cr supplementation has been shown to up-regulate brain Cr levels in mice (Ipsiroglu et al. 2001; Choi et al. 2009). Using high performance liquid chromatography, increased brain Cr stores were detected in female mice fed a dose of 2 g/kg body weight (Ipsiroglu et al. 2001). In male mice, Cr supplementation for 3 wk resulted in a dose-dependent increase in brain Cr stores measured by proton magnetic resonance spectroscopy (Matthews et al. 1999), with 2% being the upper dosage. In the present study, although hippocampal Cr levels were 8.7% higher in absolute terms in supplemented mice, this did not reach statistical significance. Given that other studies found increased levels of brain Cr with Cr supplementation in mice at lower doses and shorter durations than used in the current study (average intake of 4.2 g/kg/day), the lack of current significance effects may be due to decreased sensitivity with the method used here relative to other techniques and/or may be due to an insufficient sample size; as the mitochondrial experiments required different tissue preparation methods, Cr levels were measured in a subset of the trained mice. Nonetheless, oral Cr had beneficial effects on several parameters of hippocampal function. In addition to previous supplementation studies, the strong positive correlation with CaMKII, in which between-group effects were noted based on diet, argue for reduced power/sensitivity as primary factors in the nonsignificant increase in hippocampal Cr levels rather than a lack of diet-induced augmentation of Cr stores.
Although Cr-induced enhanced learning and memory was associated with up-regulation of plasticity-related proteins and enhanced mitochondrial function, shown to contribute to memory (Tanaka et al. 2008), Cr is purported to exert influences on several physiological parameters, including redox regulation (Lawler et al. 2002), neuroprotection (Brewer and Wallimann 2000; Sullivan et al. 2000; Peña-Altamira et al. 2005), NMDA-mediated neural transmission (Royes et al. 2008), and the regulation of ion channel gradients that facilitate proper neuronal firing (Rambo et al. 2012). Therefore, dietary Cr may have altered other neurophysiological parameters that may have contributed to the cognitive enhancements noted herein. In any case, the present data add to the growing list of nonenergetic roles for Cr in the mammalian brain that may contribute to the regulation of activity-dependent plasticity.
In conclusion, the results of this report further posit the Cr system as part of the physiological machinery underlying neuronal information storage, learning, and memory and indicate that one mechanism by which it exerts such an effect is via its actions on a key transcriptional regulator once primarily thought of in terms of its role in inflammation, NF-κB, and associated proteins implicated in brain plasticity. These data further highlight the importance of NF-κB in cognition, including memory. Moreover, these data contribute to our understanding of the role of mitochondrial function in cognition by demonstrating Cr-induced enhancement of cognitive capacity that is accompanied by increased Complex-I-dependent mitochondrial respiration.
Competing interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funding from the Natural Sciences and Engineering Research Council (to B.C.A.; grant number RGPIN/04742-2014), the St. Boniface Hospital Research Foundation (to B.C.A.; grant numbers 1406-3216, 1403-3131, and 1410-3216), Research Manitoba (to B.C.A.; Postdoctoral Fellowship to W.M.S.), and the Alzheimer's Society of Manitoba (to B.C.A.). B.C.A. is a Research Affiliate at the University of Manitoba's Centre on Aging, a member of the Children's Hospital Research Institute of Manitoba, and the Honorable Douglas Everett, Patricia Everett, and the Royal Canadian Properties Endowment Fund Chair. B.C.A. is also the Manitoba Dementia Research Chair.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.046284.117.
References
- Aksenov MY, Aksenova MV, Carney JM, Butterfield DA. 1997. Oxidative modification of glutamine synthetase by amyloid β peptide. Free Radic Res 27: 267–281. [DOI] [PubMed] [Google Scholar]
- Alberini CM, Kandel ER. 2014. The regulation of transcription in memory consolidation. Cold Spring Harb Perspect Biol 7: a021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahyar R, Akbar A, Iqbal F. 2015. Creatine monohydrate supplementation for 10 weeks mediates neuroprotection and improves learning/memory following neonatal hypoxia ischemia encephalopathy in female albino mice. Brain Res 1595: 92–100. [DOI] [PubMed] [Google Scholar]
- Allahyar R, Akbar A, Iqbal F. 2016. Effect of creatine monohydrate supplementation on learning, memory and neuromuscular coordination in female albino mice. Acta Neuropsychiatr 29: 27–34. [DOI] [PubMed] [Google Scholar]
- Allen PJ, D'Anci KE, Kanarek RB, Renshaw PF. 2010. Chronic creatine supplementation alters depression-like behavior in rodents in a sex-dependent manner. Neuropsychopharmacology 35: 534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer MC. 2004. The use of creatine to enhance athletic performance. Nutrition 20: 841. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Yee BK, Good MA, Heupel MJ, Iversen SD, Rawlins JN. 1999. Double dissociation of function within the hippocampus: a comparison of dorsal, ventral, and complete hippocampal cytotoxic lesions. Behav Neurosci 113: 1170–1188. [DOI] [PubMed] [Google Scholar]
- Bender A, Beckers J, Schneider I, Hölter SM, Haack T, Ruthsatz T, Vogt-Weisenhorn DM, Becker L, Genius J, Rujescu D, et al. 2008. Creatine improves health and survival of mice. Neurobiol Aging 29: 1404–1411. [DOI] [PubMed] [Google Scholar]
- Boero J, Qin W, Cheng J, Woolsey TA, Strauss AW, Khuchua Z. 2003. Restricted neuronal expression of ubiquitous mitochondrial creatine kinase: changing patterns in development and with increased activity. Mol Cell Biochem 244: 69–76. [PubMed] [Google Scholar]
- Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK. 2011. A requirement for nuclear factor-κB in developmental and plasticity-associated synaptogenesis. J Neurosci 31: 5414–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Wallimann TW. 2000. Protective effect of the energy precursor creatine against toxicity of glutamate and β-amyloid in rat hippocampal neurons. J Neurochem 74: 1968–1978. [DOI] [PubMed] [Google Scholar]
- Briones TL, Woods J, Rogozinska M. 2013. Decreased neuroinflammation and increased brain energy homeostasis following environmental enrichment after mild traumatic brain injury is associated with improvement in cognitive function. Acta Neuropathol Commun 1: 57. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brody DL, Holtzman DM. 2006. Morris water maze search strategy analysis in PDAPP mice before and after experimental traumatic brain injury. Exp Neurol 197: 330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever TR, Ervasti JM. 2013. Actin isoforms in neuronal development and function. Int Rev Cell Mol Biol 301: 157–213. [DOI] [PubMed] [Google Scholar]
- Cheever TR, Li B, Ervasti JM. 2012. Restricted morphological and behavioral abnormalities following ablation of β-actin in the brain. PLoS One 7: e32970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrier MM, Mendez M, Perryman K. 2001. Route learning performance in Alzheimer disease patients. Neuropsychiatry Neuropsychol Behav Neurol 14: 159–168. [PubMed] [Google Scholar]
- Choi JK, Küstermann E, Dedeoglu A, Jenkins BG. 2009. Magnetic resonance spectroscopy of regional brain metabolite markers in FALS mice and the effects of dietary creatine supplementation. Eur J Neurosci 30: 2143–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Ed. 2000. What's wrong with my mouse? Behavioral phenotyping of transgenic and knockout mice. Wiley-Liss, New York. [Google Scholar]
- Davey GP, Peuchen S, Clark JB. 1998. Energy thresholds in brain mitochondria--potential involvement in neurodegeneration. J Biol Chem 273: 12753–12757. [DOI] [PubMed] [Google Scholar]
- Dechent P, Pouwels PJ, Wilken B, Hanefeld F, Frahm J. 1999. Increase of total creatine in human brain after oral supplementation of creatine-monohydrate. Am J Physiol 277: R698–R704. [DOI] [PubMed] [Google Scholar]
- Ellery SJ, Walker DW, Dickinson H. 2016. Creatine for women: a review of the relationship between creatine and the reproductive cycle and female-specific benefits of creatine therapy. Amino Acids 48: 1807–1817. [DOI] [PubMed] [Google Scholar]
- Federman N, de la Fuente V, Zalcman G, Corbi N, Onori A, Passananti C, Romano A. 2013. Nuclear factor κB-dependent histone acetylation is specifically involved in persistent forms of memory. J Neurosci 33: 7603–7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, O'Brien C, Ohno M, Kirkwood A, Silva AJ. 2001. α-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature 411: 309–313. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. 2007. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2: 287–295. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Baltimore D. 1990. Activation in vitro of NF-κ B by phosphorylation of its inhibitor I κ B. Nature 344: 678–682. [DOI] [PubMed] [Google Scholar]
- Giachin G, Bouverot R, Acajjaoui S, Pantalone S, Soler-López M. 2016. Dynamics of human mitochondrial complex I assembly: implications for neurodegenerative diseases. Front Mol Biosci 3: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna-El-Daher L, Braissant O. 2016. Creatine synthesis and exchanges between brain cells: what can be learned from human creatine deficiencies and various experimental models? Amino Acids 48: 1877–1895. [DOI] [PubMed] [Google Scholar]
- Hanson BJ, Carrozzo R, Piemonte F, Tessa A, Robinson BH, Capaldi RA. 2001. Cytochrome c oxidase-deficient patients have distinct subunit assembly profiles. J Biol Chem 276: 16296–16301. [DOI] [PubMed] [Google Scholar]
- Hebda-Bauer EK, Watson SJ, Akil H. 2005. Cognitive performance is highly sensitive to prior experience in mice with a learning and memory deficit: failure leads to more failure. Learn Mem 12: 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebda-Bauer EK, Luo J, Watson SJ, Akil H. 2007. Female CREBαΔ- deficient mice show earlier age-related cognitive deficits than males. Neuroscience 150: 260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneman WJ, Sluimer JD, Barnes J, van der Flier WM, Sluimer IC, Fox NC, Scheltens P, Vrenken H, Barkhof F. 2009. Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology 72: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. 2010. Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189: 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsiroglu OS, Stromberger C, Ilas J, Höger H, Mühl A, Stöckler-Ipsiroglu S. 2001. Changes of tissue creatine concentrations upon oral supplementation of creatine-monohydrate in various animal species. Life Sci 69: 1805–1815. [DOI] [PubMed] [Google Scholar]
- Iqbal S, Ali M, Iqbal F. 2015. Long term creatine monohydrate supplementation, following neonatal hypoxic ischemic insult, improves neuromuscular coordination and spatial learning in male albino mouse. Brain Res 1603: 76–83. [DOI] [PubMed] [Google Scholar]
- Janus C. 2004. Search strategies used by APP transgenic mice during navigation in the morris water maze. Learn Mem 11: 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juravleva E, Barbakadze T, Mikeladze D, Kekelidze T. 2005. Creatine enhances survival of glutamate-treated neuronal/glial cells, modulates Ras/NF-κB signaling, and increases the generation of reactive oxygen species. J Neurosci Res 79: 224–230. [DOI] [PubMed] [Google Scholar]
- Kaldis P, Hemmer W, Zanolla E, Holtzman D, Wallimann T. 1996. ‘Hot spots’ of creatine kinase localization in brain: cerebellum, hippocampus and choroid plexus. Dev Neurosci 18: 542–554. [DOI] [PubMed] [Google Scholar]
- Kenyon GL, Reed GH. 1983. Creatine kinase: structure-activity relationships. Adv Enzymol Relat Areas Mol Biol 54: 367–426. [DOI] [PubMed] [Google Scholar]
- Kuiper JW, Oerlemans FT, Fransen JA, Wieringa B. 2008. Creatine kinase B deficient neurons exhibit an increased fraction of motile mitochondria. BMC Neurosci 9: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler JM, Barnes WS, Wu G, Song W, Demaree S. 2002. Direct antioxidant properties of creatine. Biochem Biophys Res Commun 290: 47–52. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. 2004. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119: 873–887. [DOI] [PubMed] [Google Scholar]
- Mandel RJ, Gage FH, Thal LJ. 1989. Enhanced detection of nucleus basalis magnocellularis lesion-induced spatial learning deficit in rats by modification of training regimen. Behav Brain Res 31: 221–229. [DOI] [PubMed] [Google Scholar]
- Martin B, Pearson M, Kebejian L, Golden E, Keselman A, Bender M, Carlson O, Egan J, Ladenheim B, Cadet JL, et al. 2007. Sex-dependent metabolic, neuroendocrine, and cognitive responses to dietary energy restriction and excess. Endocrinology 148: 4318–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF. 1999. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol 157: 142–149. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Meffert MK. 2006. Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ 13: 852–860. [DOI] [PubMed] [Google Scholar]
- McMorris T, Harris RC, Swain J, Corbett J, Collard K, Dyson RJ, Dye L, Hodgson C, Draper N. 2006. Effect of creatine supplementation and sleep deprivation, with mild exercise, on cognitive and psychomotor performance, mood state, and plasma concentrations of catecholamines and cortisol. Psychopharmacology (Berl) 185: 93–103. [DOI] [PubMed] [Google Scholar]
- McMorris T, Mielcarz G, Harris RC, Swain JP, Howard A. 2007. Creatine supplementation and cognitive performance in elderly individuals. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn 14: 517–528. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. 2003. NF-κ B functions in synaptic signaling and behavior. Nat Neurosci 6: 1072–1078. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, et al. 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396: 433–439. [DOI] [PubMed] [Google Scholar]
- Mincheva-Tasheva S, Soler RM. 2013. NF-κB signaling pathways: role in nervous system physiology and pathology. Neuroscientist 19: 175–194. [DOI] [PubMed] [Google Scholar]
- Monge C, Beraud N, Kuznetsov AV, Rostovtseva T, Sackett D, Schlattner U, Vendelin M, Saks VA. 2008. Regulation of respiration in brain mitochondria and synaptosomes: restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol Cell Biochem 318: 147–165. [DOI] [PubMed] [Google Scholar]
- Motanis H, Maroun M. 2012. Differential involvement of protein synthesis and actin rearrangement in the reacquisition of contextual fear conditioning. Hippocampus 22: 494–500. [DOI] [PubMed] [Google Scholar]
- Nafez S, Oikawa K, Odero GL, Sproule M, Ge N, Schapansky J, Abrenica B, Hatherell A, Cadonic C, Zhang S, et al. 2015. Early growth response 2 (egr-2) expression is triggered by NF-κB activation. Mol Cell Neurosci 64: 95–103. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. 1996. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci 16: 2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithianantharajah J, Levis H, Murphy M. 2004. Environmental enrichment results in cortical and subcortical changes in levels of synaptophysin and PSD-95 proteins. Neurobiol Learn Mem 81: 200–210. [DOI] [PubMed] [Google Scholar]
- Oikawa K, Odero GL, Platt E, Neuendorff M, Hatherell A, Bernstein MJ, Albensi BC. 2012. NF-κB p50 subunit knockout impairs late LTP and alters long term memory in the mouse hippocampus. BMC Neurosci 13: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JW, Takahashi K. 2007. Cerebral energetic effects of creatine supplementation in humans. Am J Physiol Regul Integr Comp Physiol 292: R1745–R1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CS, Elgersma Y, Grant SG, Morrison JH. 2008. α-isoform of calcium-calmodulin-dependent protein kinase II and postsynaptic density protein 95 differentially regulate synaptic expression of NR2A- and NR2B-containing N-methyl-d-aspartate receptors in hippocampus. Neuroscience 151: 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker WD Jr, Parks J, Filley CM, Kleinschmidt-DeMasters BK. 1994. Electron transport chain defects in Alzheimer's disease brain. Neurology 44: 1090–1096. [DOI] [PubMed] [Google Scholar]
- Pazini FL, Cunha MP, Rosa JM, Colla AR, Lieberknecht V, Oliveira Á, Rodrigues AL. 2016. Creatine, similar to ketamine, counteracts depressive-like behavior induced by corticosterone via PI3K/Akt/mTOR pathway. Mol Neurobiol 53: 6818–6834. [DOI] [PubMed] [Google Scholar]
- Peña-Altamira E, Crochemore C, Virgili M, Contestabile A. 2005. Neurochemical correlates of differential neuroprotection by long-term dietary creatine supplementation. Brain Res 1058: 183–188. [DOI] [PubMed] [Google Scholar]
- Pettegrew JW, Panchalingam K, Klunk WE, McClure RJ, Muenz LR. 1994. Alterations of cerebral metabolism in probable Alzheimer's disease: a preliminary study. Neurobiol Aging 15: 117–132. [DOI] [PubMed] [Google Scholar]
- Poirier R, Cheval H, Mailhes C, Charnay P, Davis S, Laroche S. 2007. Paradoxical role of an egr transcription factor family member, Egr2/Krox20, in learning and memory. Front Behav Neurosci 1: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier R, Cheval H, Mailhes C, Garel S, Charnay P, Davis S, Laroche S. 2008. Distinct functions of egr gene family members in cognitive processes. Front Neurosci 2: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak DD, Herkner K, Hoeger H, Lubec G. 2005. Behavioral testing upregulates pCaMKII, BDNF, PSD-95 and egr-1 in hippocampus of FVB/N mice. Behav Brain Res 163: 128–135. [DOI] [PubMed] [Google Scholar]
- Rae CD, Bröer S. 2015. Creatine as a booster for human brain function. how might it work? Neurochem Int 89: 249–259. [DOI] [PubMed] [Google Scholar]
- Rae C, Digney AL, McEwan SR, Bates TC. 2003. Oral creatine monohydrate supplementation improves brain performance: a double-blind, placebo-controlled, cross-over trial. Proc Biol Sci 270: 2147–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo LM, Ribeiro LR, Schramm VG, Berch AM, Stamm DN, Della-Pace ID, Silva LF, Furian AF, Oliveira MS, Fighera MR, et al. 2012. Creatine increases hippocampal Na+,K+-ATPase activity via NMDA-calcineurin pathway. Brain Res Bull 88: 553–559. [DOI] [PubMed] [Google Scholar]
- Royes LF, Fighera MR, Furian AF, Oliveira MS, Fiorenza NG, Ferreira J, da Silva AC, Priel MR, Ueda ES, Calixto JB, et al. 2008. Neuromodulatory effect of creatine on extracellular action potentials in rat hippocampus: role of NMDA receptors. Neurochem Int 53: 33–37. [DOI] [PubMed] [Google Scholar]
- Salomons GS, van Dooren SJ, Verhoeven NM, Cecil KM, Ball WS, Degrauw TJ, Jakobs C. 2001. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet 68: 1497–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow WM, Albensi BC. 2016. Neuronal gene targets of NF-κB and their dysregulation in Alzheimer's disease. Front Mol Neurosci 9: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow WM, Pahlavan PS, Djordjevic J, McAllister D, Platt EE, Alashmali S, Bernstein MJ, Suh M, Albensi BC. 2015. Morris water maze training in mice elevates hippocampal levels of transcription factors nuclear factor (erythroid-derived 2)-like 2 and nuclear factor κ B p65. Front Mol Neurosci 8: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow WM, Stoesz BM, Kelly DM, Albensi BC. 2014. Roles for NF-κB and gene targets of NF-κB in synaptic plasticity, memory, and navigation. Mol Neurobiol 49: 757–770. [DOI] [PubMed] [Google Scholar]
- Souza MA, Magni DV, Guerra GP, Oliveira MS, Furian AF, Pereira L, Marquez SV, Ferreira J, Fighera MR, Royes LF. 2012. Involvement of hippocampal CAMKII/CREB signaling in the spatial memory retention induced by creatine. Amino Acids 43: 2491–2503. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Geiger JD, Mattson MP, Scheff SW. 2000. Dietary supplement creatine protects against traumatic brain injury. Ann Neurol 48: 723–729. [PubMed] [Google Scholar]
- Sun P, Enslen H, Myung PS, Maurer RA. 1994. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev 8: 2527–2539. [DOI] [PubMed] [Google Scholar]
- Tanaka D, Nakada K, Takao K, Ogasawara E, Kasahara A, Sato A, Yonekawa H, Miyakawa T, Hayashi J. 2008. Normal mitochondrial respiratory function is essential for spatial remote memory in mice. Mol Brain 1: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CE, Byblow WD, Gant N. 2015. Creatine supplementation enhances corticomotor excitability and cognitive performance during oxygen deprivation. J Neurosci 35: 1773–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela MJ, Jones M, Wen W, Rae C, Graham S, Shnier R, Sachdev P. 2003. Memory training alters hippocampal neurochemistry in healthy elderly. Neuroreport 14: 1333–1337. [DOI] [PubMed] [Google Scholar]
- van de Kamp JM, Pouwels PJ, Aarsen FK, ten Hoopen LW, Knol DL, de Klerk JB, de Coo IF, Huijmans JG, Jakobs C, van der Knaap MS, et al. 2012. Long-term follow-up and treatment in nine boys with X-linked creatine transporter defect. J Inherit Metab Dis 35: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, Abulhoul L, Grünewald S, Anselm I, Azzouz H, Bratkovic D, de Brouwer A, Hamel B, et al. 2013. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet 50: 463–472. [DOI] [PubMed] [Google Scholar]
- Veyrac A, Besnard A, Caboche J, Davis S, Laroche S. 2014. The transcription factor Zif268/Egr1, brain plasticity, and memory. Prog Mol Biol Transl Sci 122: 89–129. [DOI] [PubMed] [Google Scholar]
- Vickers CA, Stephens B, Bowen J, Arbuthnott GW, Grant SG, Ingham CA. 2006. Neurone specific regulation of dendritic spines in vivo by post synaptic density 95 protein (PSD-95). Brain Res 1090: 89–98. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. 2006. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1: 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. 1992. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J 281(Pt 1):21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Kato N, Kato T. 2002. Effects of creatine on mental fatigue and cerebral hemoglobin oxygenation. Neurosci Res 42: 279–285. [DOI] [PubMed] [Google Scholar]
- Worley PF, Bhat RV, Baraban JM, Erickson CA, McNaughton BL, Barnes CA. 1993. Thresholds for synaptic activation of transcription factors in hippocampus: correlation with long-term enhancement. J Neurosci 13: 4776–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss M, Kaddurah-Daouk R. 2000. Creatine and creatinine metabolism. Physiol Rev 80: 1107–1213. [DOI] [PubMed] [Google Scholar]
- Zalcman G, Federman N, de la Fuente V, Romano A. 2015. Nuclear factor κ B-dependent Zif268 expression in hippocampus is required for recognition memory in mice. Neurobiol Learn Mem 119: 10–17. [DOI] [PubMed] [Google Scholar]