

Abstract
For several thousand years, Ganoderma lucidum (Ling-Zhi in Chinese and Reishi in Japanese) has been widely used as a traditional medication for the prevention and treatment of various diseases in Asia. Its major biologically active components, ganoderic acids (GAs), exhibit significant medicinal value due to their anti-inflammatory effects. Dysregulation of microglial function may cause seizures or promote epileptogenesis through release of proinflammatory cytokines, including interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α. At present, only little information is available on the effects of GAs on microglia-mediated inflammation in vitro and/or in vivo. The present study aimed to investigate the role of GA-A on microglia-mediated inflammation in vitro. In addition, the effect of GA-A on lipopolysaccharide (LPS)-evoked alterations in mitochondrial metabolic activity of microglia was evaluated. The results of the present study demonstrated that GA-A significantly decreased LPS-induced IL-1β, IL-6 and TNF-α release from mouse-derived primary cortical microglial cells in a concentration-dependent manner. GA-A treatment reduced LPS-induced expression of nuclear factor (NF)-κB (p65) and its inhibitor, demonstrating that non-toxic suppression of IL-1β, IL-6 and TNF-α production by GA-A is, at least in part, due to suppression of the NF-κB signaling pathway. In addition, the LPS-induced stimulation of mitochondrial activity of microglial cells was abolished by co-treatment with GA-A. Thus, GA-A treatment may be a potential therapeutic strategy for epilepsy prevention by suppressing microglia-derived proinflammatory mediators.
Keywords: Ganoderma lucidum, ganoderic acid, epilepsy, inflammation, microglia
Introduction
For several thousand years, Ganoderma (G.) lucidum (Ling-Zhi in Chinese and Reishi in Japanese) has been widely used as a traditional medication for the prevention and treatment of various human diseases in Asia (1). The major biologically active components of G. lucidum are polysaccharides and the secondary metabolites ganoderic acids (GAs), including GA-A, -B, -C, -D, -E and -F. Most GAs have a significant pharmacological potential; they were demonstrated to exhibit significant medicinal value with activities including inhibition of histamine release and cholesterol synthesis as well as antitumor and anti-inflammatory effects (2).
It was demonstrated that inflammation participates in the mediation of acute and chronic neurological disorders, including epilepsy and seizure (3). In human epilepsy patients and in experimental models of epilepsy, inflammatory processes, including activation of microglia and release of proinflammatory cytokines, have been described (4–6). Emerging evidence thus supports the hypothesis that inflammation may contribute to epileptogenesis. Microglia are intimately associated with diverse neuronal functions, such as modulation of synaptic function and plasticity, regulation of the delivery of energy substrates and enforcement of cellular immunity in the brain to restore function and promote healing, which helps to maintain tissue homeostasis (7). However, dysregulation of microglial functions may cause seizures or promote epileptogenesis. Activation of microglia through increases of excitability and inflammation is a prominent feature of epileptic foci in the human brain and in experimental epilepsy models (8–10). Uncontrolled microglia-mediated immunity may cause sustained release of inflammatory cytokines, including interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α, to facilitate epileptogenesis (11).
GAs, the triterpenoid components of G. lucidum mushroom extracts, are attractive sources of anti-inflammation products. Although numerous studies have reported that GAs exhibit anti-inflammatory and antitumor properties in animal models mainly through the induction of cytokines such as IL-1, IL-6, interferon-γ and TNF-α in monocytes/macrophages and T lymphocytes (12–14), encouraging the potential use of GAs in combination therapy against inflammation and cancer, little information is available on their in vitro and in vivo effect on microglia-mediated inflammation. The present study aimed to investigate the role of GA-A on microglia-mediated inflammation in vitro. In addition, the effect of GA-A on lipopolysaccharide (LPS)-evoked alterations in mitochondrial metabolic activity of microglia was evaluated.
Materials and methods
Reagents and chemicals
GA-A, poly-L-ornithine hydrobromide, dimethyl sulphoxide (DMSO), deoxyribonuclease I from bovine pancreas (DNaseI), penicillin-streptomycin, MTT and LPS were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Mouse monoclonal anti-CD68 antibody conjugated to phycoerythrin (PE) was purchased from eBioscience (San Diego, CA, USA; cat. no. 12-0681-82). Mouse monoclonal anti-CD11b antibody conjugated to fluorescein isothiocyanate was purchased from ProSpec-Tany Technogene Ltd. (East Brunswick, NJ, USA; cat. no. ANT-136). High-glucose Dulbecco's modified Eagle's medium (DMEM), PBS, 0.05% (w/v) trypsin-EDTA and fetal bovine serum (FBS) were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Isolation and culture of primary microglia
The primary microglia cultures were established as follows: Mixed glial cultures from male C57BL/6 mice, purchased from the Animal Centre of the Xiangya Third Hospital (Changsha, China) were established from neonatal cortices (postnatal day 0–1; n=5; mean weight, 1.4 g) (15). Cells were cultured in high-glucose DMEM supplemented with 10% FBS, penicillin and streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. The culture medium was replaced with fresh medium 24 h after the initial preparation and every 3 days thereafter. Following 1 week of culture, microglia were obtained by mechanical shaking of the mixed glial cell cultures for 1 h. Cells were routinely monitored for purity by fluorescence-activated cell sorting and the population of CD11b+ CD68+ cells was >80% (Fig. 1). All experimental procedures were approved by the Xiangya Third Hospital Ethics Committee for Experimentation on Animals (Changsha, China), where the cultures were established.
Figure 1.
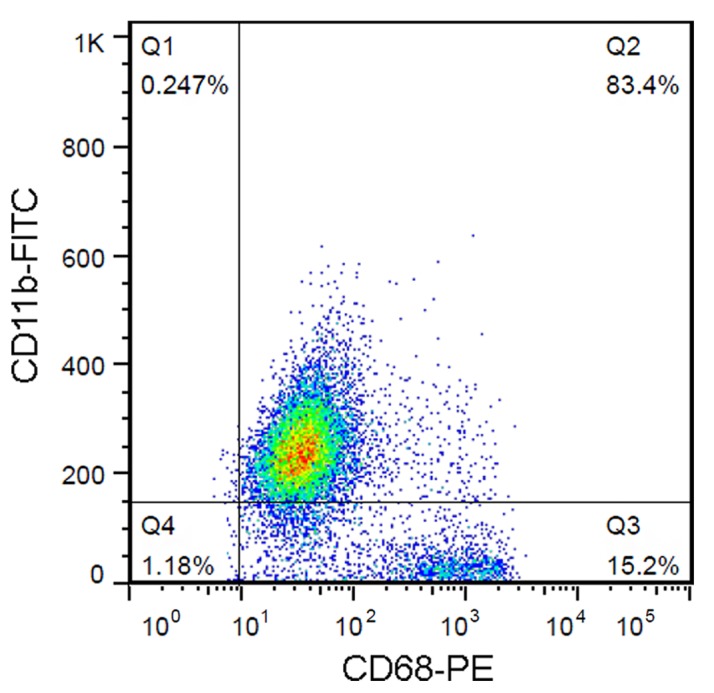
Primary microglial cells were isolated from mice cerebral cortex and identified by fluorescence-activated cell sorting following staining with anti-CD11b-FITC and CD68-PE antibodies. CD11b- and CD68-positive cells were indicated as microglial cells (~80%). FITC, fluorescein isothiocyanate; PE, phycoerythrin.
Cell treatment
To induce the release of proinflammatory cytokines, microglia were treated with DMEM containing 0.1 µg/ml LPS or vehicle for 24 h. To investigate the role of GA-A in LPS-induced release of cytokines, microglial cells were treated with a range of concentrations of GA-A (10, 20, 50 or 100 µg/ml), or co-treated with LPS. Cells treated with vehicle were used as a control.
RNA extraction and SYBR green quantitative polymerase chain reaction (qPCR) analysis
Total RNA was extracted from cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). RevertAid™ First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.) was used to reverse transcribe the mRNA to cDNA according to the manufacturer's protocol. Briefly, 0.5 ng of the template RNA, 1 µl Oligo(dT)18 Primer and 12 µl RNase free water were mixed, and incubated at 65°C for 5 min, then cooled down on ice. The mixture was added to Reaction Buffer (4 µl), RiboLock RNase inhibitor (1 µl), 10 mM dNTP mix (2 µl) and RevertAid Reverse Transcriptase (1 µl), and incubated at 42°C for 1 h. Finally, the mixture was heated to 70°C for 5 min to obtain the cDNA. The expression of IL-1β, IL-6 and TNF-α in cells was detected using a SYBR Fast qPCR mix (cat. no. RR430A; Takara, Dalian, China) at following condition: 95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 10 sec. Expression of β-actin was assessed as an endogenous control. The mRNA levels were quantified using the 2−ΔΔCq method (16).
Measurement of the release of IL-1β, IL-6 and TNF-α
Levels of IL-1β, IL-6 and TNF-α were measured in supernatants of the primary mouse microglia cell culture with commercially available ELISA kits (Sigma-Aldrich; Merck KGaA) according to the manufacturer's instructions. Measurements were performed in three independent experiments.
Western blot analysis
Immunoblotting was performed to detect the expression of IL-1β, IL-6 and TNF-α in the cell lysate, as well as the levels of nuclear factor (NF)-κB and phosphorylated inhibitor of NF-κB (p-IκBα) in microglial cells. Cultured or transfected cells were lysed in radioimmunoprecipitation assay buffer with 1% phenylmethane sulfonyl fluoride. The concentration of protein was determined using the BCA Protein Assay kit (cat. no. P0011; Beyotime Institute of Biotechnology, Haimen, China) according to the manufacturer's protocol. A total of 60 µg/lane of protein was loaded into a 12% SDS-PAGE minigel, electrophoresed and transferred onto a polyvinylidene difluoride membrane (Wuhan Boster Biological Technology, Ltd., Wuhan, China). The membranes were probed with antibodies directed against IL-1β (cat. no. I3767; 1:1,000), IL-6 (cat. no. SAB4301665; 1:1,000) and TNF-α (cat. no. SAB4502982; 1:2,000) from Sigma-Aldrich (Merck KGaA); p-IκBα (cat. no. 9246S; 1:1,000) and NF-κB (cat. no. 8242; 1:3,000) from Cell Signaling Technology, Inc. (Danvers, MA, USA); GAPDH (cat. no. ab9485; 1:3,000) from Abcam (Cambridge, UK) at 4°C overnight. The blots were subsequently incubated with horseradish peroxidase-conjugated secondary antibodies (cat. no. ab7090 and ab97040; 1:5,000; Abcam) for 1 h at 37°C. Signals were visualized using enhanced chemiluminescence substrate (EMD Millipore, Billerica, MA, USA). GAPDH was used as an endogenous protein for normalization.
Measurement of the mitochondrial metabolic activity
The mitochondrial activity of microglial cells was determined by measurement of MTT reduction to MTT formazan by cellular mitochondrial dehydrogenases. After incubation with GA-A for 24 h, MTT (0.5 mg/ml) was added to the medium, followed by incubation for 4 h at 37°C. Subsequently, dimethyl sulfoxide (150 µl) was added to dissolve the formazan crystals and the absorbance was measured at 570 nm using a microplate reader (Paradigm Detection Platform; Beckman Coulter, Brea, CA, USA); this value was proportional to the number of viable cells with intact mitochondria. Measurements were performed in three independent experiments.
Statistical analysis
Values are expressed as the mean ± standard error. Statistical analysis was performed using SPSS software (version 20; IBM Corp., Armonk, NY, USA). Student's t-test was performed for two-group comparisons, while one-way analysis of variance with Tukey's post-hoc test was performed for multiple-group comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of GA-A on LPS-induced release of IL-1β, IL-6 and TNF-α from cortical microglial cells in culture
To investigate the role of GA-A on LPS-induced release of IL-1β, IL-6 and TNF-α from mouse cortical microglial cells, the cells were treated with 0.1 µg/ml LPS for 24 h. Treatment with LPS resulted in a potent, 80-, 42- and 110-fold increase in IL-1β, IL-6 and TNF-α release, respectively (Fig. 2A). In addition, the microglial cells were treated with a range of concentrations of GA-A (10, 20, 50 and 100 µg/ml) for 24 h. The results indicated that GA-A treatment slightly but not significantly altered the release of IL-1β, IL-6 and TNF-α (Fig. 2B-D). GA-A increased IL-1β at 20 µg/ml, and IL-6 at 10 and 20 µg/ml; GA-A at 100 µg/ml decreased the release of IL-1β, IL-6 and TNF-α to ~75% of that in the control group, but this effect was not statistically significant (Fig. 2B-D).
Figure 2.

Effects of LPS on the release of IL-1β, IL-6 and TNF-α from mouse cortical microglial cells in culture. (A) The cells were treated with LPS (0.1 µg/ml) for 24 h, and ELISA were used to determine the release of IL-1β, IL-6 and TNF-α. (B-D) The cells were treated with GA (10, 20, 50 or 100 µg/ml) for 24 h and ELISA was used to determine the release of (B) IL-1β, (C) IL-6 and (D) TNF-α. Values are expressed as the mean ± standard error of the mean and relative to the control. Three independent experiments were performed. ***P<0.001 vs. control. IL, interleukin; TNF, tumor necrosis factor; LPS, lipopolysaccharide; GA, ganoderic acid A.
Furthermore, it was determined whether GA-A treatment was able to abolish the LPS-induced release of IL-1β, IL-6 and TNF-α from mouse cortical microglial cells. First, RT-qPCR was performed to detect whether the levels of IL-1β, IL-6 and TNF-α were affected at the transcriptional level, revealing that GA-A treatment (50 µg/ml) did not significantly alter the cellular mRNA expression of IL-1β, IL-6 and TNF-α (Fig. 3A). However, ELISAs indicated that GA-A treatment (10, 20, 50 or 100 µg/ml) for 24 h caused a statistically significant reduction of LPS-stimulated release of IL-1β from primary mouse microglial cells in a concentration-dependent manner (Fig. 3B). At lower concentrations (10 µg/ml), the drug did not cause any significant decrease of IL-6 and TNF-α release (Fig. 3C and D). However, IL-6 and TNF-α release were markedly reduced at higher concentrations of the drug (50 and 100 µg/ml; Fig. 3C and D). The most prominent effect was observed at a dose of 100 µg/ml (a decrease by 30, 32 and 22% for IL-1β, IL-6 and TNF-α, respectively). These results were confirmed by western blot analysis for IL-1β, IL-6 and TNF-α expression in cell lysate (Fig. 3E).
Figure 3.

Effects of GA on LPS-induced release of IL-1β, IL-6 and TNF-α from mouse cortical microglial cells in culture. (A) The cells were treated with GA (50 µg/ml) for 24 h, and total RNA was then extracted. Quantitative polymerase chain reaction was used for analysis of the mRNA levels of IL-1β, IL-6 and TNF-α. (B-D) The cells were treated with LPS alone or co-treated with LPS and GA (10, 20, 50 and 100 µg/ml) for 24 h, and ELISA was used to determine the release of (B) IL-1β, (C) IL-6 and (D) TNF-α. (E) Western blot was performed to analyze the expression of IL-1β, IL-6 and TNF-α in cell lysate after the indicated treatments (left) and quantification (right). Values are expressed as the mean ± standard error of the mean and relative to the control that was treated with LPS alone. Three independent experiments were performed. *P<0.05, **P<0.01 vs. control. IL, interleukin; TNF, tumor necrosis factor; LPS, lipopolysaccharide; GA, ganoderic acid A.
GA-A inhibits LPS-induced NF-κB pathway activation
Microglial cells were then treated with GA-A (0, 10 or 50 µg/ml) and stimulated with LPS (0.1 µg/ml). Western blot analysis of the total protein extract indicated that treatment with GA-A at 10 and 50 µg/ml reduced LPS-induced p-IκBα and NF-κB (p65) expression, with 50 µg/ml being more effective (Fig. 4A). These results revealed that GA-A inhibited LPS-induced NF-κB pathway activation, which may suggest that that non-toxic suppression of IL-1β, IL-6 and TNF-α production by GA-A is, at least in part, due to suppression of the NF-κB signaling pathway.
Figure 4.

Effect of GA on LPS (0.1 µg/ml)-induced changes in the NF-κB signaling pathway and mitochondrial activity of mouse cortical microglial cells in culture. (A) Western blot was performed to analyze the expression of p-IκB and p-p65 in microglial cells after the indicated treatments (left), and quantification (right). (B) The cells were treated with GA (10, 20, 50, 100 µg/ml), or vehicle (control) for 24 h, and MTT were used to determine the mitochondrial activity of microglia cells. (C) The cells were treated with LPS alone, or co-treated with LPS and GA (10, 20, 50 or 100 µg/ml), or vehicle (control) for 24 h, and the MTT assay was used to determine the mitochondrial activity of microglial cells. Values are expressed as the mean ± standard error of the mean and relative to the control treated with vehicle. Three independent experiments were performed. #P<0.05 vs. control, *P<0.05, **P<0.01 vs. LPS alone. GA, ganoderic acid A; LPS, lipopolysaccharide; NF-κB, nuclear factor κB; p-IκBα, phosphorylated inhibitor of NF-κB.
GA-A reduces LPS-induced increases in mitochondrial activity of mouse cortical microglia cells
GA-A treatment (10, 20, 50 or 100 µg/ml for 24 h) did not significantly alter the mitochondrial activity of microglial cells (Fig. 4B), while cells stimulated with LPS (0.1 µg/ml) for 24 h exhibited a significant increase in their mitochondrial activity by 50% (Fig. 4C). The increase of mitochondrial activity induced by LPS was markedly attenuated by treatment of the cells with GA-A used at 10 and 20 µg/ml, and was abolished by higher concentrations of the drug (50 and 100 µg/ml; Fig. 4C).
Discussion
The present study demonstrated, for the first time, to the best of our knowledge, the effects of GA-A on LPS-stimulated release of proinflammatory cytokines from primary mouse microglia cultures. To date, only a few studies have examined the effects of GA-A on microglia-mediated inflammation (17). Microglial cells are brain-resident macrophage-like cells that contribute to innate immune mechanisms (18). Microglia-mediated inflammation was reported to have an important role in the pathogenesis of epilepsy (19). In response to stressors, microglia are activated, leading to the release of proinflammatory mediators that may promote seizures and epileptogenesis, particularly when uncontrolled inflammation occurs (5). Of note, the extent of microglia activation correlates with the frequency and duration of seizures (20).
Activated microglial cells release cytokines that induce transcriptional and post-transcriptional signaling. For instance, microglia release a variety of pro-inflammatory and cytotoxic soluble factors, including IL-1β and IL-6, and subsequent activation of the proinflammatory IL-1 receptor/Toll-like receptor (IL1R/TLR) system (21). In epilepsy models, this IL1R/TLR signaling is activated, which promotes the onset and recurrence of seizures (22). Of note, pharmacological blockade or genetic inactivation of the IL1R/TLR system drastically reduces seizure activity (23). IL-1β increases glutamate release via TNF-α production, resulting in elevated extracellular glutamate levels and hyperexcitability (24). Furthermore, IL-1β also stimulates IL-6 release (25). After a febrile seizure, children had significantly higher serum IL-6 levels than a healthy control group (26), and IL-6 levels had a descending trend during the time of recovery from the seizure in the intractable epilepsy group and the non-intractable epilepsy group (27), indicating that high IL-6 levels may be a pathogenetic factor in epilepsy.
LPS is a component of the wall of Gram-negative bacteria, which may induce immediate focal epileptic-type discharges in the mouse neocortex mediated by IL-1β release (28). LPS activates microglia via TLR4. Endogenous ligands of TLR4, including IL-1β, may be generated by microglia following brain injury, mimicking the effect of LPS (29). An animal study indicated that activated microglia release proinflammatory molecules to decrease the seizure threshold (30). Consequently, microglia may help generate seizures by releasing, and responding to, endogenous inflammatory mediators, including IL-1β and TNF-α (31). For instance, chemokine-activated microglia cooperated with astrocytes to release TNF-α and other cytokines, thereby contributing to cell loss and seizures (32).
Anti-inflammatory molecules then help to resolve the inflammatory tissue response. Clinical anti-inflammatory or immunosuppressive treatments may control seizures in certain epileptic syndromes. For instance, intravenous immunoglobulin (IVIG) increases circulating levels of IL-1 receptor antagonist and blocks IL-1β signaling (33,34). IVIG suppresses seizures, which may be partially mediated by the reduction of proinflammatory cytokines and suppression of microglia activation (35).
Numerous studies have indicated that GAs enhance the immune system. Akihisa et al (36) identified four GAs isolated from the fruiting bodies of the fungus G. lucidum and identified that these GAs inhibit 12-O-tetradecanoylphorbol-13-acetate-induced inflammation in mice. The triterpene extract from G. lucidum markedly suppressed the secretion of the inflammatory cytokines TNF-α and IL-6, as well as the inflammatory mediator nitric oxide from LPS-stimulated murine RAW264.7 cells (37). In addition, natural killer cell activity was significantly enhanced by intraperitoneal administration of GA-Me (38). The anti-inflammatory properties of purified GAs encourage the potential use of GAs in combination therapy against inflammation.
The present study demonstrated that GA-A significantly decreased LPS-induced IL-1β, IL-6 and TNF-α release from primary mouse cortical microglial cells in a concentration-dependent manner. Furthermore, GA treatment did not alter the mRNA expression of IL-1β, IL-6 and TNF-α, but decreased the release of IL-1β, IL-6 and TNF-α in the cell culture medium, indicating that GA may target the release of cytokines. LPS induces proinflammatory cytokines, including IL-1β, IL-6 and TNF-α, through the activation of several intracellular signaling pathways such as NF-κB (39). A previous study demonstrated that G. lucidum extracts significantly and non-toxically suppressed TNF-α production by murine macrophages induced by peripheral blood mononuclear cells from asthma patients (12). Furthermore, the inhibitory effect of G. lucidum extracts on LPS-induced TNF-α production by macrophages was associated with the suppression of NF-κB signaling. In line with these previous studies, the results of the present study indicated that GA treatment reduced LPS-induced p-IκBα and NF-κB (p65) expression, suggesting that non-toxic suppression of IL-1β, IL-6 and TNF-α production by GA is, at least in part, due to suppression of NF-κB signaling. Although the results of the present study indicate GA regulates the IL-1β, IL-6 and TNF-α production through NF-κB at transcriptional level, other mechanism by which GA control the cytokines protein production cannot be excluded, such as the ubiquitin pathway (40).
Since the alterations of microglial function have an impact on neuronal excitability and epileptic activity, the present study also examined the effect of GA-A on the metabolic activity in mitochondria. In line with other studies (41,42), the results of the present study indicated that LPS stimulation resulted in significant increase in mitochondrial activity of microglial cells, and this effect was abolished by co-treatment with GA-A. A previous study identified that GA-T treatment resulted in a reduction of mitochondrial membrane potential and release of cytochrome C, as well as subsequent apoptosis in lung cancer cells, suggesting that the apoptosis induction of GA-T may be mediated by mitochondrial dysfunction (43). However, incubation in GAC2-conditioned media attenuated mitochondrial defects in a 3-nitropropionic acid (3-NP) cell model, and G. lucidum treatment therapeutically restored neuronal loss in mice with 3-NP-induced behavioral impairment and striatal degeneration (44). Whether the effects of GA-A on LPS-induced changes in mitochondrial activity of microglial cells are involved in the drug's action on the release of proinflammatory cytokines remains to be elucidated.
In conclusion, the results of the present study demonstrated that GA-A reduced the LPS-induced release of proinflammatory cytokines, IL-1β, IL-6 and TNF-α from mouse cortical microglial cells in culture at least in part via suppression of the NF-κB signaling pathway, and suppressed the LPS-stimulated increase in mitochondrial metabolic activity of the cells. Thus, treatment with GA-A may be potential therapeutic strategy for epilepsy via suppression of microglia-derived proinflammatory mediators.
Acknowledgements
This study was supported by the Natural Science Foundation of Heilongjiang province, China (grant no. B2015013) and the Youth Fund of Jiamusi University (grant no. Sq2013-025).
References
- 1.Bishop KS, Kao CH, Xu Y, Glucina MP, Paterson RR, Ferguson LR. From 2000 years of Ganoderma lucidum to recent developments in nutraceuticals. Phytochemistry. 2015;114:56–65. doi: 10.1016/j.phytochem.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira IC, Heleno SA, Reis FS, Stojkovic D, Queiroz MJ, Vasconcelos MH, Sokovic M. Chemical features of Ganoderma polysaccharides with antioxidant, antitumor and antimicrobial activities. Phytochemistry. 2015;114:38–55. doi: 10.1016/j.phytochem.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Dey A, Kang X, Qiu J, Du Y, Jiang J. Anti-inflammatory small molecules to treat seizures and epilepsy: From bench to bedside. Trends Pharmacol Sci. 2016;37:463–484. doi: 10.1016/j.tips.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang B, Zou J, Han L, Rensing N, Wong M. Microglial activation during epileptogenesis in a mouse model of tuberous sclerosis complex. Epilepsia. 2016;57:1317–1325. doi: 10.1111/epi.13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfluger P, Viau CM, Coelho VR, Berwig NA, Staub RB, Pereira P, Saffi J. Gamma-decanolactone inhibits iNOS and TNF-alpha production by lipopolysaccharide-activated microglia in N9 cells. Eur J Pharmacol. 2016;780:38–45. doi: 10.1016/j.ejphar.2016.03.029. [DOI] [PubMed] [Google Scholar]
- 6.Avignone E, Lepleux M, Angibaud J, Nägerl UV. Altered morphological dynamics of activated microglia after induction of status epilepticus. J Neuroinflammation. 2015;12:202. doi: 10.1186/s12974-015-0421-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mecha M, Carrillo-Salinas FJ, Feliú A, Mestre L, Guaza C. Microglia activation states and cannabinoid system: Therapeutic implications. Pharmacol Ther. 2016;166:40–55. doi: 10.1016/j.pharmthera.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Kiyomoto M, Shinoda M, Honda K, Nakaya Y, Dezawa K, Katagiri A, Kamakura S, Inoue T, Iwata K. p38 phosphorylation in medullary microglia mediates ectopic orofacial inflammatory pain in rats. Mol Pain. 2015;11:48. doi: 10.1186/s12990-015-0053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jebelli J, Su W, Hopkins S, Pocock J, Garden GA. Glia: Guardians, gluttons, or guides for the maintenance of neuronal connectivity? Ann N Y Acad Sci. 2015;1351:1–10. doi: 10.1111/nyas.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation; Proc Natl Acad Sci USA; 2008; pp. 17151–17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dambach H, Hinkerohe D, Prochnow N, Stienen MN, Moinfar Z, Haase CG, Hufnagel A, Faustmann PM. Glia and epilepsy: Experimental investigation of antiepileptic drugs in an astroglia/microglia co-culture model of inflammation. Epilepsia. 2014;55:184–192. doi: 10.1111/epi.12473. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Yang N, Song Y, Wang L, Zi J, Zhang S, Dunkin D, Busse P, Weir D, Tversky J, et al. Ganoderic acid C1 isolated from the anti-asthma formula, ASHMI™ suppresses TNF-α production by mouse macrophages and peripheral blood mononuclear cells from asthma patients. Int Immunopharmacol. 2015;27:224–231. doi: 10.1016/j.intimp.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Dunkin D, Lai J, Song Y, Ceballos C, Benkov K, Li XM. Anti-inflammatory effects of Ganoderma lucidum triterpenoid in human crohn's disease associated with downregulation of NF-κB signaling. Inflamm Bowel Dis. 2015;21:1918–1925. doi: 10.1097/MIB.0000000000000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin X, Ruiz BJ, Sze DM, Chan GC. Ganoderma lucidum (Reishi mushroom) for cancer treatment. Cochrane Database Syst Rev. 2016;4:CD007731. doi: 10.1002/14651858.CD007731.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon R, Hogan CE, Neal ML, Anantharam V, Kanthasamy AG, Kanthasamy A. A simple magnetic separation method for high-yield isolation of pure primary microglia. J Neurosci Methods. 2011;194:287–296. doi: 10.1016/j.jneumeth.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Yoon HM, Jang KJ, Han MS, Jeong JW, Kim GY, Lee JH, Choi YH. Ganoderma lucidum ethanol extract inhibits the inflammatory response by suppressing the NF-κB and toll-like receptor pathways in lipopolysaccharide-stimulated BV2 microglial cells. Exp Ther Med. 2013;5:957–963. doi: 10.3892/etm.2013.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rangarajan P, Karthikeyan A, Dheen ST. Role of dietary phenols in mitigating microglia-mediated neuroinflammation. Neuromolecular Med. 2016;18:453–464. doi: 10.1007/s12017-016-8430-x. [DOI] [PubMed] [Google Scholar]
- 19.Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, Fritschy JM. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci. 2011;31:4037–4050. doi: 10.1523/JNEUROSCI.6210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson AM, Sugo E, Barreto D, Hiew CC, Lawson JA, Connolly AM, Somerville E, Hasic E, Bye AM, Cunningham AM. The severity of gliosis in hippocampal sclerosis correlates with pre-operative seizure burden and outcome after temporal lobectomy. Mol Neurobiol. 2016;53:5446–5456. doi: 10.1007/s12035-015-9465-y. [DOI] [PubMed] [Google Scholar]
- 21.Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun. 2011;25:1281–1289. doi: 10.1016/j.bbi.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 22.Maroso M, Balosso S, Ravizza T, Liu J, Bianchi ME, Vezzani A. Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: The importance of IL-1beta and high-mobility group box 1. J Intern Med. 2011;270:319–326. doi: 10.1111/j.1365-2796.2011.02431.x. [DOI] [PubMed] [Google Scholar]
- 23.Noe FM, Polascheck N, Frigerio F, Bankstahl M, Ravizza T, Marchini S, Beltrame L, Banderó CR, Löscher W, Vezzani A. Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol Dis. 2013;59:183–193. doi: 10.1016/j.nbd.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 24.Dolga AM, Granic I, Blank T, Knaus HG, Spiess J, Luiten PG, Eisel UL, Nijholt IM. TNF-alpha-mediates neuroprotection against glutamate-induced excitotoxicity via NF-kappaB-dependent up-regulation of K2.2 channels. J Neurochem. 2008;107:1158–1167. doi: 10.1111/j.1471-4159.2008.05701.x. [DOI] [PubMed] [Google Scholar]
- 25.Hwang JS, Jung EH, Kwon MY, Han IO. Glioma-secreted soluble factors stimulate microglial activation: The role of interleukin-1β and tumor necrosis factor-α. J Neuroimmunol. 2016;298:165–171. doi: 10.1016/j.jneuroim.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Azab SF, Abdalhady MA, Almalky MA, Amin EK, Sarhan DT, Elhindawy EM, Allah MA, Elhewala AA, Salam MM, Hashem MI, et al. Serum and CSF adiponectin, leptin, and interleukin 6 levels as adipocytokinesin Egyptian children with febrile seizures: A cross-sectional study. Ital J Pediatr. 2016;42:38. doi: 10.1186/s13052-016-0250-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei HY, Yang DQ, Li YX, Wang LQ, Zheng M. Association between human cytomegalovirus and onset of epilepsy. Int J Clin Exp Med. 2015;8:20556–20564. [PMC free article] [PubMed] [Google Scholar]
- 28.Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci. 2008;28:6904–6913. doi: 10.1523/JNEUROSCI.1901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodgers KM, Hutchinson MR, Northcutt A, Maier SF, Watkins LR, Barth DS. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132:2478–2486. doi: 10.1093/brain/awp177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Han X, Bao J, Liu Y, Ye A, Thakur M, Liu H. Nicotine increases eclampsia-like seizure threshold and attenuates microglial activity in rat hippocampus through the α7 nicotinic acetylcholine receptor. Brain Res. 2016;1642:487–496. doi: 10.1016/j.brainres.2016.04.043. [DOI] [PubMed] [Google Scholar]
- 31.Kosonowska E, Janeczko K, Setkowicz Z. Inflammation induced at different developmental stages affects differently the range of microglial reactivity and the course of seizures evoked in the adult rat. Epilepsy Behav. 2015;49:66–70. doi: 10.1016/j.yebeh.2015.04.063. [DOI] [PubMed] [Google Scholar]
- 32.Zhang F, Liu J, Shi JS. Anti-inflammatory activities of resveratrol in the brain: Role of resveratrol in microglial activation. Eur J Pharmacol. 2010;636:1–7. doi: 10.1016/j.ejphar.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 33.Cattepoel S, Schaub A, Ender M, Gaida A, Kropf A, Guggisberg U, Nolte MW, Fabri L, Adlard PA, Finkelstein DI, et al. Intravenous immunglobulin binds beta amyloid and modifies its aggregation, neurotoxicity and microglial phagocytosis in vitro. PLoS One. 2013;8:e63162. doi: 10.1371/journal.pone.0063162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novak P, Williams A, Ravin P, Zurkiya O, Abduljalil A, Novak V. Treatment of multiple system atrophy using intravenous immunoglobulin. BMC Neurol. 2012;12:131. doi: 10.1186/1471-2377-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janke AD, Yong VW. Impact of IVIg on the interaction between activated T cells and microglia. Neurol Res. 2006;28:270–274. doi: 10.1179/016164106X98143. [DOI] [PubMed] [Google Scholar]
- 36.Akihisa T, Nakamura Y, Tagata M, Tokuda H, Yasukawa K, Uchiyama E, Suzuki T, Kimura Y. Anti-inflammatory and anti-tumor-promoting effects of triterpene acids and sterols from the fungus Ganoderma lucidum. Chem Biodivers. 2007;4:224–231. doi: 10.1002/cbdv.200790027. [DOI] [PubMed] [Google Scholar]
- 37.Dudhgaonkar S, Thyagarajan A, Sliva D. Suppression of the inflammatory response by triterpenes isolated from the mushroom Ganoderma lucidum. Int Immunopharmacol. 2009;9:1272–1280. doi: 10.1016/j.intimp.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 38.Wang G, Zhao J, Liu J, Huang Y, Zhong JJ, Tang W. Enhancement of IL-2 and IFN-gamma expression and NK cells activity involved in the anti-tumor effect of ganoderic acid Me in vivo. Int Immunopharmacol. 2007;7:864–870. doi: 10.1016/j.intimp.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Rummel C. Inflammatory transcription factors as activation markers and functional readouts in immune-to-brain communication. Brain BehavImmun. 2016;54:1–14. doi: 10.1016/j.bbi.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Lin TY, Hsu HY. Ling Zhi-8 reduces lung cancer mobility and metastasis through disruption of focal adhesion and induction of MDM2-mediated Slug degradation. Cancer Lett. 2016;375:340–348. doi: 10.1016/j.canlet.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 41.Ma B, Yu J, Xie C, Sun L, Lin S, Ding J, Luo J, Cai H. Toll-like receptors promote mitochondrial translocation of nuclear transcription factor nuclear factor of activated T-cells in prolonged microglial activation. J Neurosci. 2015;35:10799–10814. doi: 10.1523/JNEUROSCI.2455-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen H, Wang C, Wei X, Ding X, Ying W. Malate-aspartate shuttle inhibitor aminooxyacetate acid induces apoptosis and impairs energy metabolism of both resting microglia and LPS-activated microglia. Neurochem Res. 2015;40:1311–1318. doi: 10.1007/s11064-015-1589-y. [DOI] [PubMed] [Google Scholar]
- 43.Tang W, Liu JW, Zhao WM, Wei DZ, Zhong JJ. Ganoderic acid T from Ganoderma lucidum mycelia induces mitochondria mediated apoptosis in lung cancer cells. Life Sci. 2006;80:205–211. doi: 10.1016/j.lfs.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 44.Chen LW, Horng LY, Wu CL, Sung HC, Wu RT. Activating mitochondrial regulator PGC-1α expression by astrocytic NGF is a therapeutic strategy for Huntington's disease. Neuropharmacology. 2012;63:719–732. doi: 10.1016/j.neuropharm.2012.05.019. [DOI] [PubMed] [Google Scholar]