

Abstract
Aneuploidy, a hallmark of cancer cells, poses an appealing opportunity for cancer treatment and prevention strategies. Using a cell-based screen to identify small molecules that could selectively kill aneuploid cells, we identified the compound N-[2-hydroxy-1-(4-morpholinylmethyl)-2-phenylethyl]-decanamide monohydrochloride (DL-PDMP), an antagonist of UDP-glucose ceramide glucosyltransferase. DL-PDMP selectively inhibited proliferation of aneuploid primary mouse embryonic fibroblasts and aneuploid colorectal cancer cells. Its selective cytotoxic effects were based on further accentuating the elevated levels of ceramide which characterize aneuploid cells, leading to increased apoptosis. We observed that DL-PDMP could also enhance the cytotoxic effects of paclitaxel, a standard of care chemotherapeutic agent that causes aneuploidy, in human colon cancer and mouse lymphoma cells. Our results offer pharmacological evidence that the aneuploid state in cancer cells can be targeted selectively for therapeutic purposes, or for reducing the toxicity of taxane-based drug regimens.
Keywords: Sphingolipid, aneuploid, Gastrointestinal cancers: colorectal
Introduction
Aneuploidy, an abnormal karyotype characterized by gains and losses of whole chromosomes has a profound impact on human health. Most constitutive autosome gains and all autosome losses cause embryonic lethality. The few trisomies that survive to term display severe abnormalities including developmental delays, cognitive and organ defects, as well as a shortened lifespan. Somatic aneuploidy is also associated with disease; it is a hallmark of cancer. More than 90% of solid tumors harbor an aberrant karyotype (1,2).
The adverse effects of aneuploidy have been characterized. Aneuploid mammalian and yeast cells exhibit proliferation defects (3–5) and genomic instability (6–9). In addition, cells harboring whole chromosome gains and losses display metabolic alterations (5,10) and experience proteotoxic stress, which is caused by aneuploidy-induced changes in protein abundance that place an increased demand on the cell’s protein folding and degradation machineries (3,4,11–13).
Because aneuploidy is a rare occurrence in normal tissues (14) and even selected against in vivo (15), aneuploidy-associated stresses represent a unique opportunity to specifically eliminate cancer cells. A previously conducted, small scale, targeted proof-of-principle screen showed that compounds indeed exist that preferentially inhibit the growth of aneuploid cells (11) and spurred the larger scale effort to identify aneuploidy selective compounds described here. Using trisomy 13 mouse embryonic fibroblasts (MEFs) we identified DL-PDMP, an UDP-glucose ceramide glucosyltransferase (UGCG) antagonist (16), to preferentially inhibit the growth of primary aneuploid cells and highly aneuploid colorectal cancer cells.
Ceramides belong to the sphingolipid family. These lipids play a critical role in eukaryotic membrane biology and cell signaling. Sphingolipids are synthesized de novo through the conjugation of serine and palmitoyl-CoA to produce dihydrosphingosine, which is then further condensed into dihydroceramide (Figure 1) (17). Desaturation of dihydroceramide by dihydroceramide desaturase facilitates the generation of ceramide (18). Ceramide serves as an essential substrate for several different modifications (Figure 1). The modifications include phosphorylation to produce ceramide-1-phosphate. Addition of a phosphocholine head group converts ceramide into sphingomyelin, the major sphingolipid species in mammalian membranes (Figure 1) (19). Ceramide is also converted into glucosylceramide through the addition of glucose by glucosylceramide synthase. This sphingolipid is critical for the production of more complex glycosphingolipids such as lactosylceramide and gangliosides employed for cell-cell communication. Importantly, the production of sphingolipids is highly dynamic, as members of this lipid family interconvert depending on the cell’s need. For example, sphingomyelin, glucosylceramide and sphingosine are inter-converted via a ceramide intermediate (Figure 1).
Figure 1. Ceramide biosynthesis pathways.
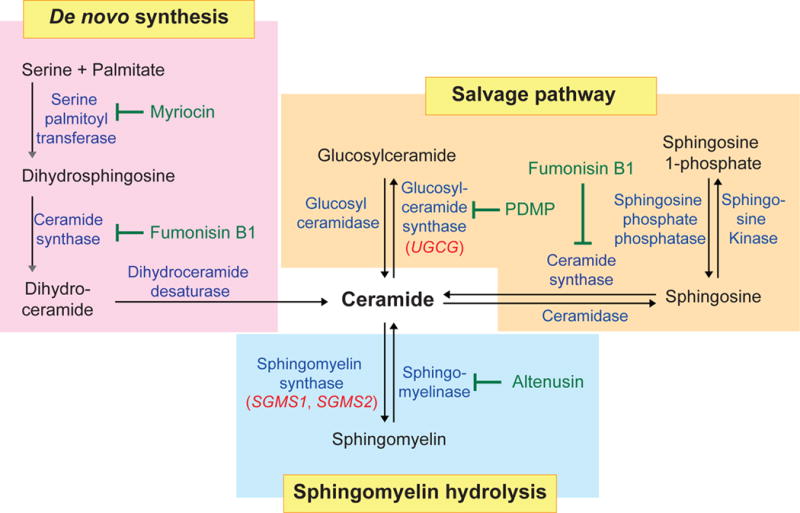
Ceramides are generated through de novo synthesis in the endoplasmic reticulum. In the de novo synthesis pathway, serine palmitoyltransferase converts serine and palmitate into dihydrosphingosine. In a series of reactions dihydrosphingosine is converted into ceramide. Complex sphingolipids can also be degraded into ceramide. In the salvage pathway, sphingosine is metabolized into ceramide by ceramide synthase, and glucosylceramide is degraded into ceramide by glucosyl ceramidase. In the sphingomyelin hydrolysis pathway, plasma membrane sphingomyelin is hydrolyzed into ceramide via sphingomyelinase. Compounds that inhibit various enzymes in the ceramide biosynthesis pathway are shown in green.
In addition to their crucial role in membrane function, many sphingolipids, such as ceramide, ceramide-1-phosphate (C1P), sphingosine, and sphingosine-1-phosphate (S1P) are bioactive signaling molecules that have been demonstrated to regulate apoptosis, senescence, differentiation, proliferation and inflammation (19). Owing to the central role of sphingolipids in membrane biology and cell signaling, sphingolipid pathways have been considered as therapeutic targets in many diseases, including obesity, type 2 diabetes, asthma, and Gaucher’s disease, which is caused by loss of glucosylceramidase GBA1 activity (20,21). Targeting sphingolipid metabolism through sphingosine kinase inhibitors has also been explored in the treatment of cancers, such as glioblastoma but off-target effects and side effects of these kinase inhibitors remain a concern (22).
Here we describe the identification of DL-PDMP, an UDP-glucose ceramide glucosyltransferase antagonist (16), as selectively inhibiting the proliferation of aneuploid primary cells and highly aneuploid colorectal cancer cells. We show that this selectivity is due to DL-PDMP further elevating already high levels of ceramide in aneuploid cells, which leads to apoptosis. Genetic manipulations that cause an increase in intracellular ceramide levels are also detrimental to aneuploid primary cells and aneuploid colorectal cancer cells. Finally, consistent with the idea that increasing ceramide levels is especially detrimental to aneuploid cells we find that in some cell types, DL-PDMP exhibits strong synergistic anti-proliferative effects with Taxol, a chemotherapeutic that causes chromosome mis-segregation and hence aneuploidy. Our results raise the exciting possibility that chemical interventions that lead to increased intracellular ceramide levels might not only represent a new broad-spectrum anti-cancer agent but could be combined with standard of care Taxane-based chemotherapy regimens to augment efficacy and mitigate toxicity.
Materials and Methods
Mouse strains
All mouse strains were obtained from the Jackson Laboratory. Strains used to generate trisomic embryos are: Rb(1.2)18Lub/J and Rb(1.3)1Ei/J for Ts1; Rb(11.13)4Bnr/J and Rb(13.16)1Mpl/J for Ts13; Rb(6.16)24Lub and Rb(16.17)7Bnr for Ts16; and Rb(5.19)1Wh/J and Rb(9.19)163H for Ts19. All male compound Robertsonian heterozygous mice were mated with C57BL/6J females and embryos were collected at specific stages of embryogenesis by timed matings as described (5). All animal studies and procedures were approved by MIT Institutional Animal Care and Use Committee.
Primary MEF cell lines
Littermate-derived euploid and trisomic primary MEFs were prepared as described previously (5,11). Experiments were performed in at least three independent trisomic cell lines and analyzed together with euploid littermates. MEFs were used at early passages (≤p5) to ensure that their karyotypes had not evolved. Two independent CDC20(AAA/AAA) MEF cell lines were kindly provided by Dr. Pumin Zhang and BUB1b(H/H) mice by Dr. Jan van Deursen. MEFs were cultured in DMEM (Invitrogen) supplemented with 10 % FBS, 2 mM L-glutamine and 100 U/ml penicillin/streptomycin. All cells were maintained at 37°C with 5 % CO2 in a humidified environment, and examined to ensure mycoplasma-free. All MEF cell lines were authenticated by genotyping.
Human cell lines
All cancer cell lines and primary cell lines were obtained from ATCC (May, 2011) and upon arrival, prepared early-passage cell stocks to ensure all cell line experiments were analyzed within 10 further passages. The cell lines are human colorectal MIN lines (HCT-116, HCT-15, DLD-1, SW48 and LoVo), human colorectal CIN lines (Caco2, HT29, SW403, SW480 and SW620), leukemia lines (Kasumi-1, Ku812, K562 and HL60), and two primary human cell lines (CCD112 CoN and CCD841 CoN). All cell lines were characterized by cellular morphology and DNA fingerprinting, and examined to ensure that they were free of mycoplasma with LookOut Mycoplasma PCR detection Kit (Sigma-Aldrich, latest test in June, 2017). Karyotype information was obtained from the NCI database and the COSMIC dataset at the Sanger Institute. All human cells were cultured in RPMI (Invitrogen) supplemented with 10 % FBS, 2 mM L-glutamine and 100 U/ml penicillin/streptomycin and maintained at 37°C with 5 % CO2 in a humidified environment.
Reagents
Chemicals applied to target sphingolipid pathways were: DL-PDMP (Cayman), an inhibitor for glucosylceramide synthase (UGCG); myriocin (Sigma-Aldrich), an inhibitor for serine palmitoyltransferase; fumonisin B1 (Biomol), an inhibitor for ceramide synthase; altenusin (Enzo), an inhibitor for neutral sphingomyelinases. C8-ceramide (Biomol) is a cell-permeable analog of ceramides. Taxol (TOCRIS) binds to β-tubulin and causes cell cycle arrest. Antibodies were obtained from the following sources: anti-UGCG (12869-1-AP, Proteintech), anti-SGMS1 (19050-1-AP, Proteintech), anti-SMPD1 (14609-1-AP, Proteintech) and anti-ASAH1 (11274-1-AP, Proteintech); anti-CERS1 (ARP49654_P050, Aviva Systems Biology); anti-GAPDH (60004, Proteintech). anti-mouse secondary antibody (A9044, Sigma-Aldrich), and anti-rabbit secondary antibody (A9169, Sigma-Aldrich).
Library screen
ICCB compound library test (Institute of Chemistry and Cell Biology, Harvard). The screen was carried out in a 384 well format, with approximately 1,000 MEFs seeded 24 hours before chemicals were applied. After 72 hours of treatment, relative cell number was determined with CellTiter-Glo® luminescent cell viability assay. Summary of compound effect in aneuploid Ts13 MEF compared to euploid wild-type MEF is listed in Table S1.
Cell viability assay
To determine the effect of various compounds on wild-type and trisomic MEFs, we performed initial tests using a 96-well alamarBlue viability assay (Invitrogen). Candidates were further analyzed using the trypan blue viability assay (Sigma-Aldrich) in a 6-well format. Briefly, cells were plated at a density of 2,000 cells per well in 96-well plates one day prior to drug treatment. After a 72 hour treatment, each medium was replaced with 100 μl of fresh medium containing 10 % alamarBlue. Viable cells were analyzed after 4-hour incubation according to the manufacturer’s instructions. For trypan blue staining, cells were plated at a density of 2×105 cells per well in 6-well plates one day prior to drug treatment. At the indicated time points, cells were trypsinized and mixed with an equal volume of 0.4% trypan blue and cell number was counted with the Cellometer Mini and AutoT4 cell counter (Nexcelom). Each analysis was performed at least three times. Standard deviations (SDs) are shown.
Apoptosis assay
1×106 cells were seeded on a 10 cm plate and exposed to the indicated treatments for 24 hours. Cells were resuspended in 1ml Annexin V buffer and further stained with propidium iodide and FITC-conjugated annexin V antibody (BD Pharmingen) for 15 minutes at room temperature prior to FACS analysis.
Cell proliferation assay
For EdU incorporation analysis, 1×104 wild type, trisomic or Bub1bH/H MEFs were plated onto coverslips coated with 10 μg/ml Fibronectin (Sigma-Aldrich) and allowed to attach to the coverslips for 12 hours. Cells were then treated with the indicated concentration of DL-PDMP for 48 hours and exposed to 10 μM EdU during the last 12 hours of treatment. Cells were fixed with 3.7% paraformaldehyde (in PBS) for 15 minutes at room temperature and EdU incorporation into DNA was visualized using the Click-iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen) following manufacturer’s instructions.
For Ki67 analysis, 2×105 MIN or CIN cells were seeded into 6-well plates for 24 hours, followed by treatment with the indicated concentration of DL-PDMP for 24 hours. Cells were harvested and incubated with anti-Ki67 antibody (BioLegend) for 1 hour at 4°C. Cells were washed with PBS before FACS analysis.
Xenograft assay
4×106 tumor cells were resuspended in 100 μl PBS and inoculated subcutaneously into single flanks of 6-week old female nude mice. Each nude mouse received inoculations of one MIN cell line in the left flank, and one CIN cell line in the right flank. Seven days after inoculation, animals were treated with daily i.p. injections of DL-PDMP (100 mg/kg body weight) or an equal volume of DMSO vehicle control. Tumor volumes (mm3) were recorded once a week using the formula π/6 × A × B2 (A is the larger diameter, B is the smaller) to score tumor progressions.
LC-MS analyses of sphingolipids
Approximately 106 cells were collected in 3 ml ice-cold methanol and lysed and stored at −80°C until analysis. Cell lysates were thawed and briefly centrifuged, then 75 μl cleared cell extracts were transferred to a vial with 25 μl aqueous solution containing internal standards (1.0 g/μl 17:0 LPC and 24:0 PC, Avanti, and 2.0 ng/μl PGE2-d4, Cayman). LC-MS analyses were conducted using an Open Accela 1250-Q Exactive hybrid quadrupole orbitrap LC-MS system (Thermo Fisher Scientific). Extracts (10 μl) were injected onto an ACQUITY BEH C8 column (1.7 μm, 2.1×100 mm, Waters) that was eluted isocratically at a flow rate of 450 μl/min for 1 min with 80% mobile phase A (5% methanol with 0.1% formic acid, v/v) and 20% mobile phase B (99.9% methanol with 0.1% formic acid, v/v), followed by a linear gradient to 80% mobile phase B over 2 minutes, then a linear gradient to 100% mobile phase B over 7 minutes. Full scan MS data were acquired over m/z 200-850 at 70,000 resolution in the negative ion mode to profile sphingosine 1-phosphate and over m/z 200-850 in the positive ionization mode for all other sphingolipids. Raw data were processed using TraceFinder software (Thermo) for peak detection and manual review of integrated peaks.
Immunofluorescence microscopy
Cells were first fixed with 4% ice-cold paraformaldehyde for 10 min, then permeabilized with 0.5% Triton X-100 for 5 min, and subsequently blocked with 5% BSA at room temperature for 1hour. Cells were incubated at 4 °C overnight with the primary antibody (anti-Ceramide, Sigma-Aldrich, 1:200), followed by a PBS wash. The secondary antibody (Donkey anti-mouse antibody conjugated to Alexa488, Thermo Fisher Scientific, 1:500) was applied for 2-hours at room temperature. Nuclei were stained with DAPI in antifade mounting medium (Vector Lab.). All fluorescence images were acquired using a Nikon A1 inverted confocal microscope. Single plane images were exported with the NIS-Element viewer 4.0 software and further analyzed using ImageJ. Fluorescence intensities were determined by subtracting background fluorescence.
Single cell sequencing analysis
Single MEFs were isolated by microaspiration, and genomic DNA was amplified using the GenomePlex Single Cell Whole Genome Amplification Kit (Sigma). Amplified DNA was purified, barcoded, pooled, and sequenced on an Illumina HiSeq2000 as described (14).
Plasmids
Doxycycline inducible shRNAs constructs for UGCG and SGMS1 were generated in pLVX-Tight-Puro plasmid (Clontech) via NotI and MluI sites (corresponding oligonucleotide sequences are listed in Table S2). The rtTA expressing construct pMA2640 (#25434); the helper plasmids for retroviral production, pUMVC (#8449) and pCMV-VSV-G (#8454); and the helper plasmids for lentiviral production, pMD2.G (#12259) and psPAX2 (#12260), were obtained from Addgene.
Protein and quantitative RT-PCR analysis
Cells were lysed and subjected to immunoblot analyses as described previously (11). Total RNA was extracted using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. 1μg RNA was used for cDNAs synthesis using Omniscript RT Kit (Qiagen). Quantitative RT-PCR was carried out in a LightCycler 480 II (Roche) using SYBR Green PCR Kit (Qiagen). The LightCycler software was used to determine gene expression levels and normalized to a ribosomal reference gene, RPL19. Primer sequences are listed in Table S3.
Retroviral and lentiviral transduction
For retrovirus production, 293T cells were transfected with the pMA2640 plasmid, together with the packaging plasmids pUMVC and pCMV-VSV-G using Fugene HD transfection reagent (Promega). Retroviral particles were collected 36-48 hours after transfection, and passed through a 0.45 μM filter to remove cell debris. After transduction, blasticidin (5 μg/ml) was used to select for rtTA-expressing stable MIN and CIN cells. Lentiviruses were generated by co-transfecting the pLVX-Tight-Puro shRNA vectors together with the packaging plasmids psPAX2 and pMD2.G into 293T cells using the Fugene HD transfection reagent. Lentiviruses were collected 36 h after transfection. After passing through 0.45 μM filters, the cleared supernatants were used to transduce retroviral-targeted MIN and CIN cells for doxycycline inducible assays. Puromycin (1 μg/ml) was used for stable cell line selection.
siRNA knockdown
Silencer oligonucleotides for UGCG (SASI_Hs01_00050364, 00050365, 00050366), SGMS1 (SASI_Hs01_00060990, 00060991, 00060992) and scramble controls were purchased from Sigma-Aldrich. MIN and CIN cells were transiently transfected with UGCG or SGMS1 siRNA oligonucleotides using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. mRNA levels were assessed 24 hours after transfection.
RNAi signatures
An eighth of a million cells in 250 μl B-cell medium (BCM) of each shRNA bearing Eμ-Myc p19arf−/− lymphoma cell line were seeded into 24-well plates. For wells that would remain untreated as a control, only 1/16th of a million cells were seeded. Next, 250 μl of medium containing several concentrations of the compound/s as indicated were added to cells. After 24 hours, 300 μl of cells from untreated wells were removed and replaced by 300 μl fresh BCM. All wells then received 500 μl BCM before being placed in the incubator for another 24 h. At 48 h, cells were checked for viability via flow cytometry on a FACScan flow cytometer (BD Biosciences) using propidium iodide (PI) as a live/dead marker. The LD was determined to be the dose at which that percent of cells remained PI negative. For single drug signatures, only doses that elicited an LD80-90 were further diluted with 1 ml of fresh BCM and analyzed for GFP+% the next day. However, for combination doses, the combination dose that achieved LD80-90 and the single doses that contributed to the combination were diluted and analyzed 24 hours later.
Relative enrichment and depletion values for each shRNA in response to drug were then analyzed via a modified K-nearest neighbors algorithm in order to classify compound mechanism of action (23,24).
Statistics
Results are presented as the mean ± SD. Statistical analyses were performed using Prism version 5.0 (GraphPad) using an unpaired two-tailed Student’s t test. Tumor growth was compared by ANOVA and patient survival analyses were performed using log-rank test. Statistical significances are labeled as ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
Results
An unbiased chemical screen identifies DL-PDMP as an aneuploidy-selective proliferation antagonist
Whole chromosome gains and losses cause a number of cellular stresses, which can be enhanced by further interference with the cellular pathways affected by aneuploidy (5,11,25). To identify novel vulnerabilities of aneuploid cells, we screened a library comprised of 2640 off-patent bioactive compounds for small molecules that preferentially impaired the proliferation of mouse embryonic fibroblasts (MEFs) trisomic for chromosome 13 (Ts13) compared to isogenic euploid control MEFs. We found DL-PDMP, a glucosylceramide synthase inhibitor, to significantly impair the proliferation of trisomy 13 MEFs, while only minimally affecting the growth of euploid control MEFs (Figure 2A; Table S1).
Figure 2. DL-PDMP inhibits the proliferation of primary aneuploidy cells.
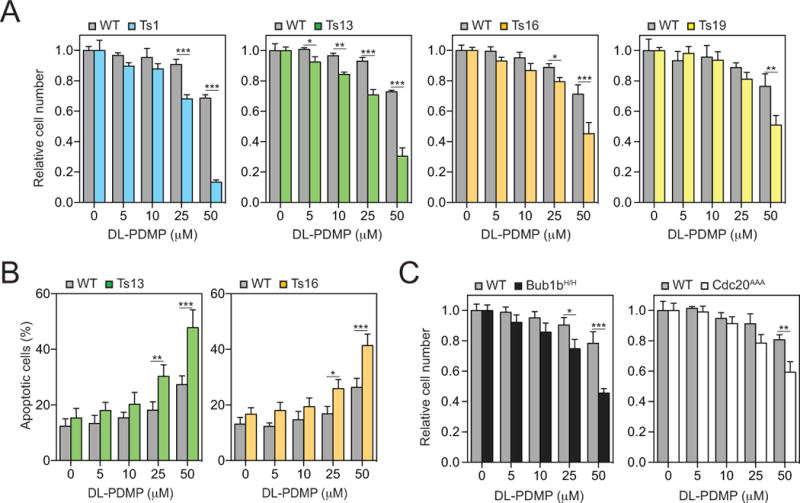
(A) Euploid wild-type (grey bars) and trisomic MEFs (colored bars) were treated with the indicated concentrations of DL-PDMP. Cell number was determined after 72 hours. In each experiment, aneuploid MEFs were compared to euploid littermate control MEFs. To specifically assess the effects of DL-PDMP on cell proliferation we normalized cell number of drug treated cells to that of the same cells treated with vehicle only. This was necessary because aneuploid cells proliferate poorly.
(B) Percentage of annexin V-FITC positive, PI negative cells was determined 24 hours after DL-PDMP treatment.
(C) Euploid wild-type (grey bars), BUB1bH/H cells (black bars) and CDC20AAA/AAA cells (open bars) were treated with the indicated concentrations of DL-PDMP. Cell number was determined after 72 hours. In each experiment, aneuploid MEFs were compared to euploid littermate control MEFs. To specifically assess the effects of DL-PDMP on cell proliferation we normalized cell number of drug treated cells to that of the same cells treated with vehicle only. This was necessary because aneuploid cells proliferate poorly.
The data are shown as the mean ± standard deviation. *P<0.05, **P<0.01, ***P<0.001, t test.
To determine whether this anti-proliferative effect of DL-PDMP was specific to trisomy 13 or whether the compound also inhibited the proliferation of other aneuploid cell lines we measured proliferative capacity of trisomy 1, 16 and 19 MEFs over a range of DL-PDMP concentrations. This analysis revealed that the proliferation of all trisomic cell lines was preferentially inhibited by the drug compared to euploid control lines (Figure 2A). The most dramatic effects were observed when high drug concentrations of DL-PDMP (25 and 50 μM) were applied. The anti-proliferative effects of DL-PDMP on trisomic cells were largely due to increased apoptosis as judged by an increase in annexin V positive -PI negative cells (Figure 2B). To determine whether DL-PDMP also interfered with cell proliferation in aneuploid cells we assessed degree of EdU incorporation. EdU incorporation was decreased in trisomic MEFs irrespective of whether they were treated with DL-PDMP or not, and similarly in another aneuploid model, Bub1bH/H (Figure S1A, B). We conclude that DL-PDMP induces apoptosis in multiple MEF lines harboring different trisomies, to a greater extent than in littermate euploid control MEFs. Colorectal MIN and CIN cell lines did not reveal cell cycle progression difference upon DL-PDMP treatment as judged by Ki67 staining (Figure S1C).
Next, we examined whether DL-PDMP also exhibited cytotoxicity towards MEF lines that continuously spawn aneuploid cells of different karyotypes because they carry mutations in genes essential for accurate chromosome segregation. The spindle assembly checkpoint (SAC) prevents chromosome segregation when chromosomes are not correctly attached to the mitotic spindle (reviewed in (26)). Mutations in genes encoding SAC components thus cause high levels of chromosome mis-segregation. We utilized MEFs harboring the hypomorphic BUB1bH/H allele (27) or that express a checkpoint resistant allele of CDC20, the chromosome segregation inducer that is inhibited by the SAC (Cdc20AAA; (28)). Both alleles cause chromosome mis-segregation and the accumulation of cells with single and multiple chromosome gains and losses in culture over time (27,28). We found that primary MEFs carrying these mutations were also sensitive to DL-PDMP (Figure 2C). However, the anti-proliferative effects of DL-PDMP were less dramatic in these cell lines compared to MEFs harboring constitutive trisomies. This is most likely due to the fact that only a fraction of Bub1bH/H and Cdc20AAA MEFs are in fact aneuploid. Single cell sequencing showed that 46% and 70% of the BUB1bH/H and CDC20AAA MEFs are aneuploid (Figure S2). It is also possible that some specific aneuploidies confer drug resistance dampening the cytotoxic effects of DL-PDMP (i.e. 29,30). We conclude that DL-PDMP impairs the accumulation of primary cells harboring constitutive aneuploidies and primary cells that are chromosomally unstable.
DL-PDMP inhibits the proliferation of highly aneuploid colorectal cancer cells
Are the cytotoxic effects of DL-PDMP restricted to aneuploid primary cells or does the compound also induce apoptosis in aneuploid human cancer cell lines? To address this question we compared the effects of the drug on colorectal cancer cell lines that harbored highly complex karyotypes (CIN cell lines) with that of colorectal cancer cell lines harboring low levels of aneuploidy (MIN cell lines)(31). Colorectal cancer cell lines that exhibit microsatellite instability (MIN) such as HCT-116, HCT-15, DLD-1, SW48 and LoVo maintain a close to euploid (pseudodiploid) karyotype (32). In contrast, colorectal cancer cell lines that display chromosome instability (CIN) such as Caco2, HT-29, SW403, SW480 and SW620 harbor chromosome numbers ranging from 50 to 96 (33). DL-PDMP preferentially inhibited the proliferation of CIN cell lines compared to MIN cell lines and euploid control lines (CCD112 CoN and CCD841 CoN; Figure 3A). As in aneuploid MEFs, increased apoptosis appeared to be a cause of the cytotoxic effect of DL-PDMP. Treatment with the drug dramatically increased apoptosis in CIN, but not MIN and euploid control lines (Figure 3B). As in aneuploid MEFs, DL-PDMP did not significantly antagonize cell cycle progression of MIN and CIN cell lines as judged by Ki67 staining (Figure S1C).
Figure 3. DL-PDMP antagonizes the growth of CIN colorectal cancer cell lines.
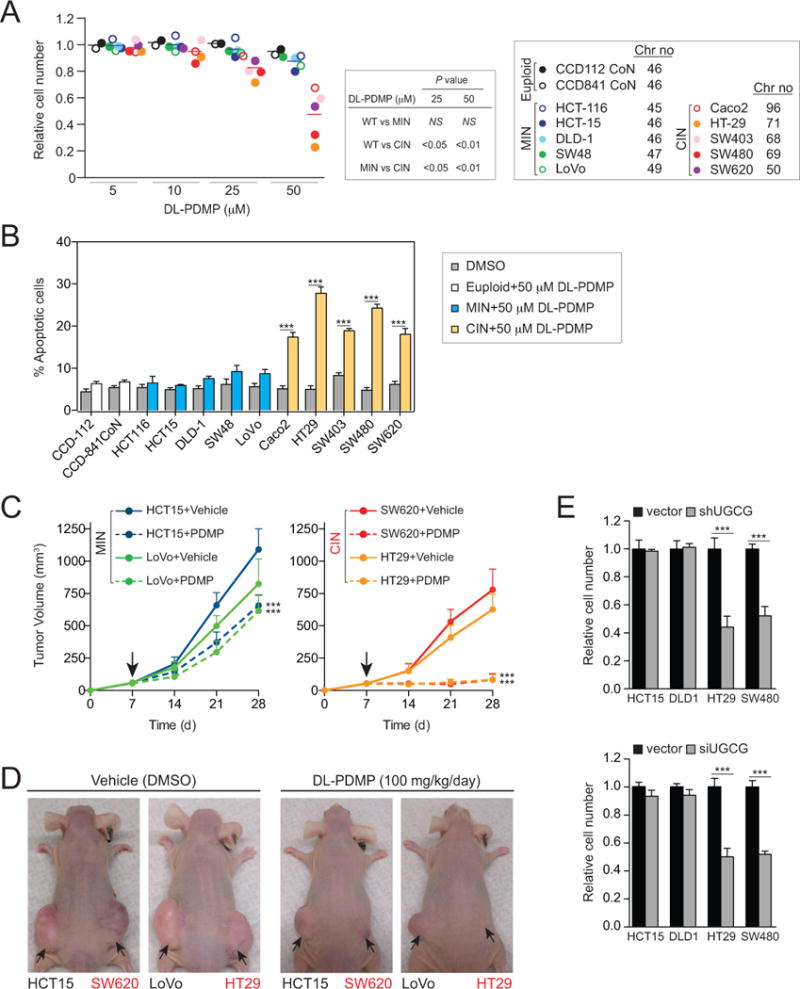
(A) Human colorectal cancer cell lines were treated with the indicated concentrations of DL-PDMP. Cell number was determined 72 hours after compound addition. To specifically assess the effects of DL-PDMP on cell proliferation we normalized cell number of drug treated cells to that of the same cells treated with vehicle only.
(B) Percentage of annexin V-FITC positive, PI negative cells was determined 24 hours after DL-PDMP treatment.
(C, D) Mice were inoculated with 4 ×106 MIN cells on the left flank and an equal number of CIN cells on the right flank. Seven days after injection (indicated by the arrow in C), mice were treated with daily i.p. injections of DL-PDMP or vehicle (n ≥ 6 per group). Tumor volume (mm3) was measured at the indicated time points and is shown as mean tumor volumes. Mice treated with vehicle (left) or DL-PDMP (right) 28 days after transplantation are shown in (D).
(E) MIN and CIN cancer cell lines were transfected with shRNA (upper) or siRNAs (lower) constructs targeting UGCG for 72 hours and cell number was determined. Cell number was normalized to vector control transfected cells.
The data are shown as the mean ± standard deviation. *P<0.05, **P<0.01, ***P<0.001, t test.
The cytotoxic effects of DL-PDMP were also observed in xenograft experiments. We injected MIN - CIN line pairs (HC15+SW620; LoVo+HT29) into the flanks of athymic nu/nu mice and administered DL-PDMP on a daily basis for four weeks. While the CIN cell lines SW620 and HT29 failed to form subcutaneous tumors, MIN cell line growth was only minimally inhibited by DL-PDMP (Figure 3C, D; S3).
To confirm that the cell death-inducing effects of DL-PDMP in CIN colorectal cancer cells were at least in part due to the drug’s inhibitory effects on glucosylceramide synthase we examined the consequences of knocking down expression of UGCG, the gene encoding glucosylceramide synthase. shRNA and siRNA mediated knock-down led to a significant decrease in UGCG mRNA levels (Figure S4A, B) and inhibited proliferation in highly aneuploid CIN colorectal cancer cell lines but not near-euploid MIN colorectal cancer cell lines (Figure 3E). Our results indicate that DL-PDMP impairs the proliferation of CIN colorectal cancer cells at least in part by inhibiting glucosylceramide synthase.
We also examined the effects of DL-PDMP on the myeloid lineage malignancies acute (AML) and chronic myeloid leukemia (CML). In these hematopoietic malignancies we did not observe a differential effect of DL-PDMP on aneuploid and near diploid/diploid cancer cell lines. Both Kasumi-1 and HL60 cell lines (diploid or near diploid) and KU812 and K562 cell lines (aneuploid) exhibited equally high sensitivity to the drug (Figure S5A). In contrast, CD34+ hematopoietic stem/progenitor cells (HSPCs) isolated from cord blood (34) were not sensitive to high doses of DL-PDMP (50μM; Figure S5B). These results indicate that leukemic cell lines are highly sensitive sphingolipid dysregulation irrespective of whether or not they are aneuploid and raise the possibility that DL-PDMP’s aneuploidy-selective cytotoxicity is cell-type specific.
Aneuploidy causes an increase in intracellular ceramide levels
A key question arising from our observations is why aneuploid MEFs and CIN colorectal cancer cells are more sensitive to DL-PDMP than euploid or pseudodiploid cells. DL-PDMP is a ceramide analog that inhibits glucosylceramide synthase, thus decreasing ceramide glycosylation and causing the accumulation of ceramides that inhibit proliferation and promote differentiation and apoptosis (reviewed in (19)). We reasoned that ceramide levels were already elevated in aneuploid MEFs and CIN cell lines making them more sensitive to compounds that increase intracellular ceramide levels. To test this hypothesis we quantified C16-ceramide, C18-ceramide, sphingosine, sphingosine 1-phosphate, dihydrosphingosine and sphingomyelins in aneuploid and euploid cells by mass spectrometry. C16-ceramide appeared to be especially responsive to DL-PDMP addition increasing significantly in both primary aneuploid MEFs and CIN colorectal cancer cell lines (Figure 4A, B; Figure S6). Importantly, C-16 ceramide levels were elevated in trisomic MEFs compared to euploid control MEFs even before DL-PDMP treatment (Figure 4A). This increase in basal C16-ceramide levels was even more dramatic in CIN colorectal cancer cells when compared to MIN lines (Figure 4B). Quantification of ceramide levels using an anti-ceramide antibody revealed similar results (Figure 4C). We conclude that C16-ceramide levels are elevated in aneuploid MEFs and CIN colorectal cancer cells and speculate that higher levels of aneuploidy are responsible for the stronger response of CIN cell lines to DL-PDMP compared to trisomic MEFs.
Figure 4. DL-PDMP causes C16-ceramide accumulation in aneuploid cells.
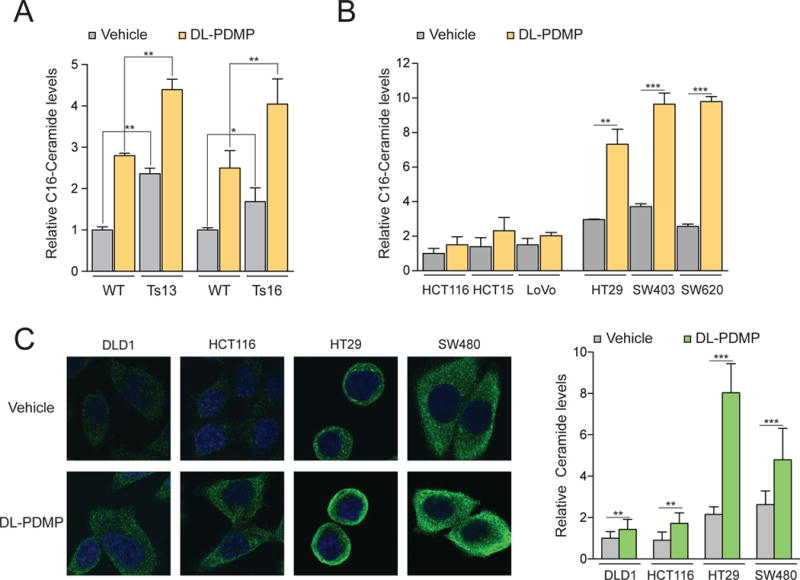
(A) Mass spectrometry based quantification of C16-ceramide levels in wild-type, trisomy 13 and trisomy 16 cultures following a 24-hour treatment with 50 μM DL-PDMP or vehicle. Ceramide levels were normalized to those of wild-type cells treated with vehicle only, which was set to 1.
(B) Mass spectrometry based quantification of the C16-ceramide levels in MIN and CIN cancer cell lines following a 24-hour treatment with 50 μM DL-PDMP or vehicle. Ceramide levels were normalized to HCT116 cells treated with vehicle only, which was set to 1.
(C) Immunofluorescence imaging of intracellular ceramide in MIN and CIN cancer cell lines following a 24-hour treatment with 50 μM DL-PDMP or vehicle. Quantifications of relative ceramide levels are shown on the right. Ceramide levels were normalized to DLD1 cells treated with vehicle only, which was set to 1.
The data are shown as the mean ± standard deviation. *P<0.05, **P<0.01, ***P<0.001, t test.
Increasing intracellular ceramide levels inhibits the proliferation of aneuploid cells
Our results raise the possibility that DL-PDMP impairs proliferation of aneuploid MEFs and CIN colorectal cancer cells by increasing intracellular ceramide levels. To test this idea we examined the effects of the cell-permeable C8-ceramide, on the growth of aneuploid cells. This analysis revealed that C8-ceramide, like DL-PDMP, preferentially inhibited the proliferation of trisomic MEFs and CIN colorectal cancer cells (Figure 5A, B; Note that C-8-ceramide was also identified to be aneuploidy selective in our compound screen, see Table S1).
Figure 5. C8-ceramide limits the proliferation of aneuploid cells.
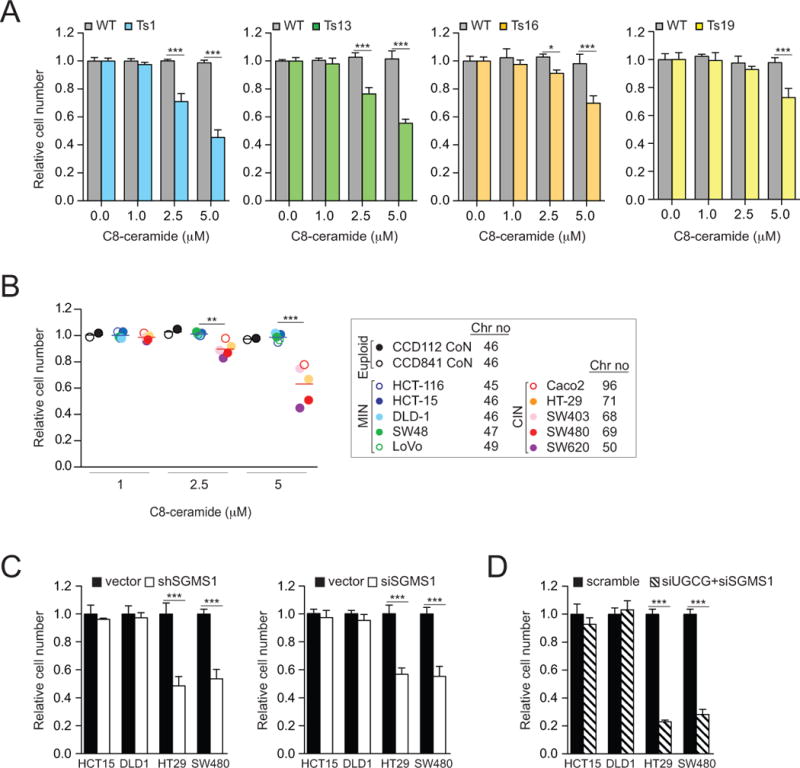
(A) Euploid wild-type (grey bars) and trisomic MEFs (colored bars) were treated with the indicated concentrations of C8-ceramide. Cell number was determined after 72 hours. In each experiment, aneuploid MEFs were compared to euploid littermate control lines. To specifically assess the effects of C8-ceramide on cell proliferation we normalized cell number of drug treated cells to that of the same cells treated with vehicle only. This was necessary because aneuploid cells proliferate poorly.
(B) Human colorectal cancer cells were treated with indicated concentrations of C8-ceramide. Cell number was determined 72 hours after compound addition. Cell number of drug treated cells was normalized to that of the same cell line treated with vehicle only.
(C) MIN and CIN cancer cell lines were transfected with shRNA (left) or siRNAs (right) constructs targeting SGMS1 for 72 hours, and cell number was determined.
(D) Cells were transfected with pooled siRNAs targeting UGCG and SGMS1, and cell number was determined after 72 hours. Cell number was normalized to vector control transfected cells. The data are shown as the mean ± standard deviation. *P<0.05, **P<0.01, ***P<0.001, t test.
Down-regulation of enzymes that metabolize ceramide into other sphingolipids revealed similar results. Knocking down expression of SGMS1, the gene encoding sphingomyelin synthase which metabolizes ceramide into sphingomyelin (35) led to a significant decrease in SGMS1 mRNA levels (Figure S4C) and inhibited proliferation of highly aneuploid CIN colorectal cancer cell lines but not of near-euploid MIN colorectal cancer cell lines (Figure 5C). Inhibition of both, sphingomyelin synthase and glucosylceramide synthase had an even more dramatic effect on the proliferation of CIN colorectal cancer cell lines (Figure 5D). We conclude that C16-ceramide levels are elevated in trisomic MEFs and CIN colorectal cancer cells and that this sensitizes aneuploid cells to chemical perturbations that elevate intracellular ceramide levels.
The ceramide salvage and sphingomyelin hydrolysis pathways contribute to high ceramide levels in aneuploid cells
Why are ceramide levels increased in aneuploid MEFs and CIN colorectal cancer cells? To begin to address this question we examined the consequences of inhibiting various ceramide synthesis pathways. Ceramides are synthesized by three major pathways. In the de novo synthesis pathway, ceramide is produced via the condensation of serine and palmitate through serine palmitoyltransferase and ceramide synthase (Figure 1). The salvage pathway either reutilizes long-chain sphingoid bases to form ceramide through ceramide synthase or by the hydrolysis of glucosylceramide via glucosyl ceramidase (Figure 1). In the sphingomyelin hydrolysis pathway, ceramide is produced by the hydrolysis of sphingomyelin through sphingomyelinase. Inhibitors have been developed for all the major enzymes in the three ceramide biosynthesis pathways.
To determine which ceramide synthesis route(s) is responsible for the accumulation of C16-ceramide in aneuploid cells, we first examined the consequences of inhibiting de novo ceramide synthesis by treating cells with myriocin, an inhibitor of serine palmitoyltransferase (36). We found that, addition of myriocin caused a decrease in C16-ceramide levels in trisomic MEFs and CIN colorectal cancer cell lines (Figure 6A, B). However, treatment of trisomic MEFs and CIN colorectal cancer cell lines with the palmitoyltransferase inhibitor did not have a significant effect on the proliferation of the cell lines (Figure 6C). In contrast, treatment of cells with fumonisin B1, which interferes with both de novo synthesis of ceramides and production of ceramides via the salvage pathway provided a slight growth advantage to trisomic MEFs and CIN colorectal cancer cells (Figure 6D). Inhibiting the sphingomyelin hydrolysis pathway by treating cells with the sphingomyelinase inhibitor altenusin also subtly improved the growth of aneuploid cells (Figure 6E). At high concentrations (50μM), CIN colorectal cancer cells are notably less sensitive than their near-diploid MIN colorectal cancer cell counterparts.
Figure 6. Effects of altering sphingolipid metabolism on DL-PDMP sensitivity in aneuploid cells.
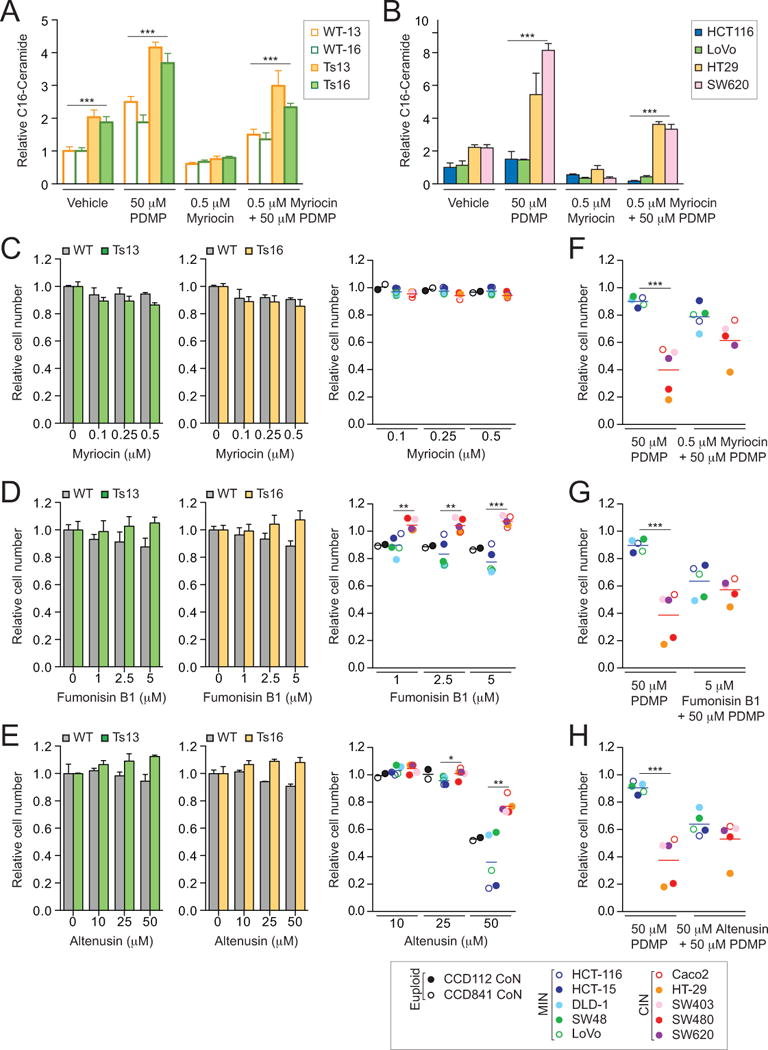
(A) Mass spectrometry based quantification of C16-ceramide levels in wild-type, trisomy 13 and trisomy 16 cells following a 24-hour treatment with the indicated compounds. Ceramide levels were normalized to those of wild-type cells treated with vehicle only, which was set to 1.
(B) Mass spectrometry based quantification of C16-ceramide levels in MIN and CIN cancer cell lines following a 24-hour treatment with the indicated compounds. Ceramide levels were normalized to those of HCT116 cells treated with vehicle only, which was set to 1.
(C - E) Wild-type and trisomic cells (left and middle panels) and human colorectal cancer cells (right panel) were treated with the indicated concentrations of myriocin (C), fumonisin B1 (D), or altenusin (E). Cell number was determined after 72 hours. Cell number of drug treated cells was normalized to that of the same cell line treated with vehicle only.
(F - H) MIN and CIN cancer cell lines were treated with 50 μM DL-PDMP either alone or in combination with myriocin (0.5 μM; F), fumonisin B1 (5 μM; G), or altenusin (50 μM; H). Cell number was determined after 72 hours. Cell number of drug treated cells was normalized to that of the same cell line treated with vehicle only.
The data are shown as the mean ± standard deviation. *P<0.05, **P<0.01, ***P<0.001, t test.
To further determine whether a particular ceramide biosynthesis pathway was responsible for DL-PDMP cytotoxicity in aneuploid cells, we examined the consequences of inhibiting the three individual ceramide biosynthesis pathways on survival when combined with a DL-PDMP treatment. We found that inactivation of each of the three pathways subtly relieved the anti-proliferative effects of DL-PDMP in CIN colon cancer cell lines (Figure 6F-H), and concluded that all ceramide synthesis pathways are required for the anti-proliferative effects of DL-PDMP. Based on the finding that inhibition of the salvage and sphingomyelin hydrolysis pathways subtly improves cell growth of aneuploid cells, we conclude that these two pathways play a more substantial role in increasing ceramide levels in aneuploid cells.
Sphingomyelin synthase levels decrease in CIN colorectal cancer cell lines upon DL-PDMP treatment
Our data suggest that the salvage and sphingomyelin hydrolysis pathways are in part responsible for the sensitivity of aneuploid cells to DL-PDMP. We therefore tested the possibility that changes in the expression of enzymes involved in these pathways occur in CIN colorectal cancer cells but not MIN colorectal cancer cells. Sphingomyelin synthase (SGMS1 and SGMS2), glucosylceramide synthase (UGCG) and ceramidase (ASAH1, ASAH2, ACER1 and ACER3) are the major classes of enzymes that metabolize ceramide into other bioactive sphingolipids (Figure 1). RNA levels of these enzymes were not evidently lowered in aneuploid cells compared to euploid cells, and expression did not change, or changed only marginally in response to DL-PDMP treatment in CIN and MIN colorectal cancer cell lines (Figure S7A). However, SGMS1 and to some extent UGCG protein levels decreased in CIN colorectal cancer cell lines (HT29, SW48) upon DL-PDMP treatment compared to MIN colorectal cancer cell lines (HCT15, DLD1; Figure 7A). Expression of enzymes that produce ceramides were not significantly changed in CIN colorectal cancer cell lines (Figure S7B, C). Together, these observations raise the possibility that in CIN cancer cells sphingomyelin synthase and to a lesser extent glucosylceramide synthase are down-regulated by post-transcriptional mechanisms and that this down-regulation contributes to the increased accumulation of ceramide in these cells upon DL-PDMP treatment. We conclude that decreased expression of enzymes involved in metabolizing ceramide into other sphingolipids may be one reason for the increased sensitivity of CIN colorectal cancer cells to compounds that elevate intracellular ceramide levels. We note however that changes in protein levels of these enzymes do not appear to be altered in untreated cells, indicating that other mechanisms must be responsible for increased basal ceramide levels in CIN colorectal cancer cell lines.
Figure 7. Effects of altering ceramide levels on cancer development and treatment.
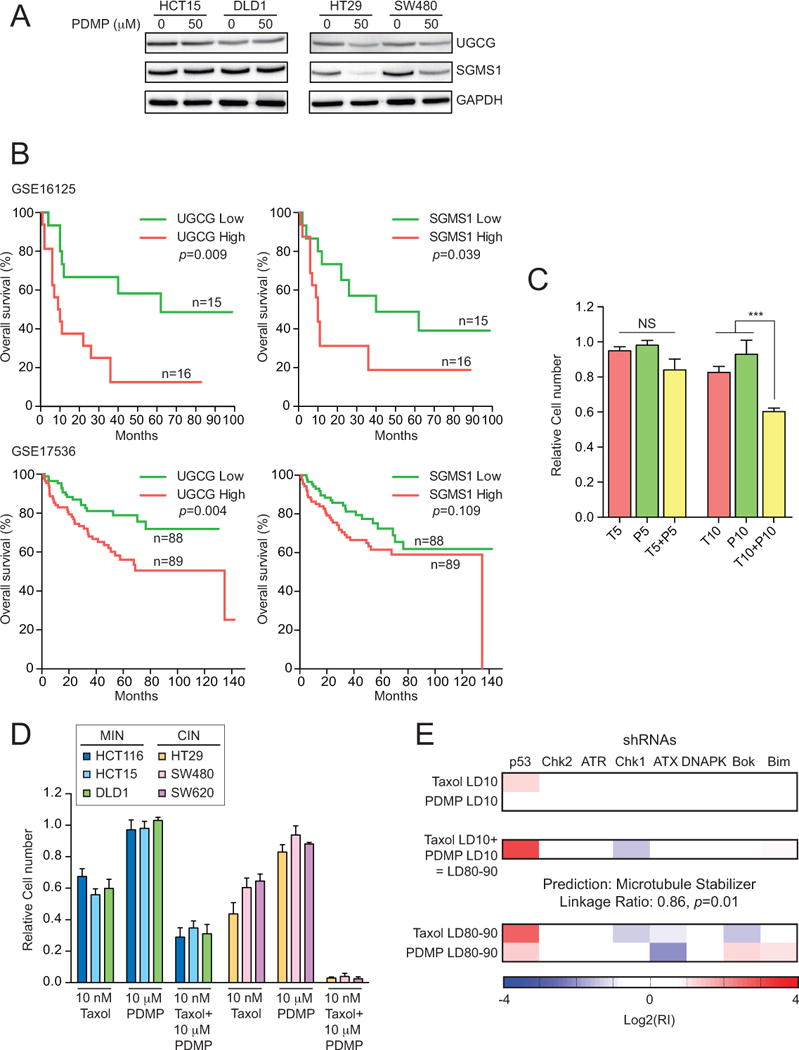
(A) MIN and CIN colorectal cancer cells were treated with DL-PDMP at the indicated doses for 24 hours and UGCG and SGMS1 levels were determine by immunoblot analysis. GAPDH served as a loading control.
(B) Survival analysis (log-rank test) of colon cancer patients expressing high or low levels of UGCG (left) or SGMS1 (right). Data set GSE16125 is shown on the top, data set GSE17536 below.
(C, D) Wild-type MEFs (C) or MIN and CIN colorectal cancer cell lines (D) were treated with the indicated concentrations of DL-PDMP (P) and Taxol (T). Cell number was determined after 72 hours. Cell number of drug treated cells was normalized to that of cells treated with vehicle only.
(E) Eμ-Mycp19arf−/− lymphoma cells were treated with Taxol and DL-PDMP. The shRNA signatures generated at LD10 single drug doses, which produce LD80–90 signatures when combined, are shown at the top. The signature that is produced when the two drugs are combined to yield an LD of 80-90 is shown in the middle panel. LD80-90 signatures for the single drugs are shown at the bottom. The data are shown as the mean ± standard deviation. ***P<0.001, t test.
An alternative way to assess the importance of ceramide metabolizing enzymes in colorectal cancer cell proliferation is to ask whether expression of these enzymes predicts patient survival. Specifically, upregulation of UGCG and SGMS1 ought to cause a decrease in intracellular ceramide levels and hence increased tumor growth which would lead to decreased survival. To test this possibility we correlated expression of UGCG and SGMS1 with survival in two colorectal cancer studies (37,38). Analyses of clinical datasets GSE16125 and GSE17536 revealed that high levels of UGCG and to some extent SGMS1 are associated with decreased survival (Figure 7B). This observation raises the possibility that up-regulation of either or both genes could promote the survival of aneuploid cancer cells and thus affect morbidity. We further note that targeting either or both enzymes may have a therapeutic benefit.
DL-PDMP enhances the cytotoxic effects of the aneuploidy inducing chemotherapeutic Taxol
Taxol is the most commonly used chemotherapeutic agent in the treatment of both solid tumors and hematopoietic malignancies (39). Taxol is a microtubule poison that arrests cells in mitosis, but induces chromosome mis-segregation at clinically relevant concentrations (40). If DL-PDMP indeed preferentially induces apoptosis in aneuploid cells, we predict that this ceramide analog ought to enhance the cytotoxic effects of Taxol.
We treated wild-type MEFs with low concentrations of Taxol, DL-PDMP or a combination thereof and found DL-PDMP to subtly enhance the toxicity of Taxol (Figure 7C). The effects of combining Taxol and DL-PDMP were more striking in MIN colorectal cancer cell lines and especially evident in CIN colorectal cancer cell lines (Figure 7D).
To further characterize the enhancement of Taxol’s cytotoxicity by DL-PDMP we employed a previously developed method to examine anti-cancer drug combination synergy and mechanism of action (23,41). In this assay, murine Eμ-Mycp19arf−/− lymphoma cells harboring eight different shRNA-GFP constructs are treated at equipotent doses with either a single compound or compound combinations. The pattern of resistance and sensitivity of these shRNA harboring cells to a drug or drug combination is then used to bioinformatically classify the mechanism of action of the drugs being tested.
For this classification we first needed to determine the drug concentrations required to kill 80-90% (LD80-90) of the lymphoma cells. We found that the LD80-90 of DL-PDMP was between 61μM and 85μM. The same degree of growth inhibition was accomplished at Taxol concentrations of 17nM to 20nM. Notably, the LD80-90 of the DL-PDMP and Taxol combination was achieved at concentrations of 7-14μM DL-PDMP and 5-6.25nM Taxol. When applied individually at these concentrations, the compounds reduced proliferation by only 10%. The combination of DL-PDMP and Taxol was the most synergistic combination tested in this system relative to the Bliss Independence additivity model (41).
To characterize the mechanism of action of the synergistic DL-PDMP/Taxol combination, we obtained the eight shRNA signature of cells treated with either compound alone or in combination. As expected, Taxol classified as a microtubule stabilizer signature (Taxol LD80-90; Figure 7E). This signature is characterized by increased resistance to the drug upon p53 knock-down and increased sensitivity upon knock-down of Chk1 and Bok (Figure 7E; (41)). DL-PDMP classified as a new previously uncharacterized mechanism of action, as judged by its eight shRNA signature. This novel signature is characterized by increased sensitivity to DL-PDMP upon knock-down of the PI3K-like ATX (Figure 7E). The signature of the combination of DL-PDMP and Taxol classified as a microtubule stabilizer indicating that DL-PDMP enhances the effects of Taxol. Our results raise the exciting possibility that compounds that like DL-PDMP lead to an increase in intracellular ceramide levels could significantly improve the efficacy of Taxane-based chemotherapeutics.
Discussion
Previous studies have identified both gene-specific effects of aneuploidy as well as phenotypes seen in many different aneuploid cells (42–44). The latter category includes proliferation defects, proteotoxic and energy stress, as well as genomic instability. Here we describe another broadly observed aneuploidy-associated phenotype – dysregulation of sphingolipid metabolism that results in elevated levels of ceramide. An unbiased chemical genetic approach identified two compounds that increase intracellular ceramide levels, DL-PDMP and C8-ceramide, to induce apoptosis in aneuploid primary and cancer cell lines. Our data further indicate that one reason for this increased sensitivity is that aneuploid cells already harbor elevated levels of this pro-apoptotic lipid. Importantly the adverse effects of ceramide on the proliferation of aneuploid cells appear conserved across species. In an unbiased genetic selection Torres and coworkers identified mutations that cause a decrease in intracellular ceramide levels to improve the fitness of aneuploid budding yeast cells ((45); E. M. Torres, personal communication). Thus, two non-hypothesis driven approaches in different experimental systems have uncovered the same biological process as being deregulated and causing a fitness defect in aneuploid cells. These observations lend support to the idea that large-scale changes in a cell’s karyotype and hence proteome impact cell physiology in fundamentally the same way in all eukaryotes.
A key question posed by our results is why ceramide levels are elevated in aneuploid cells. We find that inhibition of all three ceramide synthesis pathways, the de novo synthesis pathway, the salvage pathway and the sphingomyelin hydrolysis pathway, are required for DL-PDMP induced apoptosis in aneuploid cells. This finding indicates that all three pathways contribute to high levels of ceramide in aneuploid cells. However, the fact that chemical inhibition of the salvage pathway and the sphingomyelin hydrolysis pathway but not of the de novo synthesis pathway improves the proliferation of trisomic cells, suggests that these two pathways are particularly important for the accumulation of ceramides in aneuploid cells.
Why baseline ceramide levels are higher in aneuploid cell lines remains unknown. The abundance of enzymes involved in the biosynthesis and metabolism of ceramides is not noticeably altered in aneuploid cells. This finding indicates that enzyme activities rather than levels are altered in aneuploid cells. Consistent with this conclusion are previous studies that showed DNA damage stress to cause ceramide accumulation by affecting the activity of ceramide producing and metabolizing enzymes. In HeLa and U937 cells, UV irradiation triggers the hydrolysis of sphingomyelin by acid sphingomyelinase thereby raising intracellular ceramide levels (46). The DNA damaging agent daunorubicin has been shown to stimulate de novo synthesis of ceramide via activation of ceramide synthase (47). Aneuploid cells experience a number of stresses including DNA damage (8). We propose that it is these aneuploidy-associated stresses that alter the activity of ceramide biosynthesis and metabolizing enzymes thereby causing an increase in intracellular ceramide levels. When aneuploid cells are treated with DL-PDMP or C8-ceramide the ceramides accumulate to even higher levels and induce apoptosis by previously described mechanisms (48). It is also possible that DL-PDMP induces yet further alterations in ceramide metabolism in aneuploid cells. In this regard we note that levels of two enzymes critical for the metabolism of ceramides into other lipids, UGCG and SGMS1, do decrease upon treatment of CIN colorectal cancer cells with DL-PDMP.
Our results indicate that aneuploid primary cells are more sensitive to increased ceramide levels than euploid cells. A similar differential sensitivity is seen in colorectal cancer. Highly aneuploid CIN cancer cell lines were more sensitive to DL-PDMP and C8-ceramide than pseudo-diploid MIN cancer cell lines. This differential sensitivity of aneuploid and pseudo-diploid cell lines does, however, not exist in all cancer types. Myeloid leukemia cell lines are very sensitive to DL-PDMP treatment irrespective of whether or not they are aneuploid. The absence of a differential effect in this cancer may be due to the fact that all myeloid leukemias are sensitive to ceramides for additional reasons. Metabolizing ceramide into other lipids is central for myeloid leukemia cells to proliferate (49). Consistent with this finding, SGMS1 activity has been shown to be up regulated by the BCR-ABL oncogene, the cause of CML (50). In the AML cell line HL-60 SGMS1 activity is also upregulated (50). Finally, mutations in the neutral sphingomyelinase (nSMase) gene SMPD3, which encodes a primary ceramide biosynthesis enzyme, were identified in 5 of 92 AMLs and 8 of 131 acute lymphoid leukemias (ALLs), but not in other tumor types (51). Together our findings and that of others suggest that aneuploidy sensitizes some cells types to increasing intracellular ceramide levels, in other cell types ceramide sensitivity may already be so high that karyotype no longer matters.
Ceramide biosynthesis regulation has been shown to be clinically relevant in cancer. The first report of a role of sphingolipid metabolism in tumorigenesis was published more than 30 years ago (52). Since then it has become clear that this lipid synthesis pathway contributes to disease progression and outcome in many different cancers. In human gliomas low levels of ceramide are associated with poor outcome (53). A similar correlation was described in ovarian cancer. In this tumor type ceramide levels are reduced compared to normal ovarian tissues and tumors harbor biologically inactive dihydroceramide instead (54). Our analyses of two colorectal cancer data sets also support the idea that low levels of ceramide are associated with aggressive disease.
Targeting sphingolipid metabolism has been explored in a variety of clinical contexts including obesity, type 2 diabetes, Gaucher’s disease and cancer (55). In the case of glioblastoma, for example, increasing ceramide levels has been suggested as a therapy following relapse after chemo- or radiation therapy. Recurrent glioblastomas harbor high levels of sphingosine kinase activity. Targeting sphingosine kinase (SK) would prevent conversion of ceramide into sphingosine 1-phosphate (S1P) hence causing an increase in ceramide levels and apoptosis (22). Our study suggests that the aneuploid state causes an increase in intracellular ceramide levels. Given that most solid human tumors are aneuploid, treatments that elevate intracellular ceramide to levels where the lipid induces apoptosis could represent a new cancer therapy with ideal properties – broad spectrum efficacy and selectivity. To explore this avenue it is important to understand how DL-PDMP increases intracellular ceramide levels. DL-PDMP is a ceramide analog that is thought to inhibit glucosylceramide synthase (16) thereby preventing the conversion of ceramide into glucosylceramide. However, DL-PDMP may also interfere with the function of other ceramide metabolizing enzymes: DL-PDMP has been suggested to partially inhibit SGMS as well (56). It has not escaped our attention that DL-PDMP potentially targeting multiple enzymes would make the development of high affinity inhibitors with DL-PDMP’s properties challenging.
We are nevertheless intrigued by the possibility that increasing intracellular ceramide levels synergizes with the chemotherapeutic Taxol, which at clinically relevant concentrations causes chromosome mis-segregation (40). The effects of combining Taxol with DL-PDMP are not uniform across cell lines. We find that combining Taxol and DL-PDMP dramatically reduces the effective dose of Taxol in CIN cell lines and Eμ-Mycp19arf−/− lymphoma cells, but augmentation was modest in MEFs and MIN cell lines. Why the response varies between cell types is not yet clear. We speculate that degree of aneuploidy could at least in part dictate sensitivity to combination treatment. Recall, primary cell lines in which only a fraction of cells are aneuploid (BUB1bH/H and CDC20AAA MEFs) show limited sensitivity to DL-PDMP presumably because a good fraction of cells in the population is euploid. A similar logic could apply to euploid or pseudodiploid cells treated with low doses of Taxol. Low doses of Taxol cause aneuploidy in only a fraction of cells, limiting the impact of DL-PDMP treatment.
In closing we note that reduction in effective dose is not only observed when Taxol was combined with DL-PDMP but also when Taxol is combined with C8-ceramide. This finding indicates that it is indeed elevated levels of ceramide that make Taxol more cytotoxic. This observation raises the exciting possibility that combining compounds that increase the intracellular ceramide levels with Taxane-based chemotherapeutics could not only create highly effective treatment regimens but may also serve as an excellent way to mitigate the serious side effects associated with taxane based chemotherapies.
Supplementary Material
Acknowledgments
We thank H.-C. Chang and E. M. Torres for discussions, J. M. van Deursen for Bub1bH/H mice, and P. Zhang for Cdc20AAA MEF cells. We are grateful to members of the Tang and Amon labs for their critical reading of the manuscript.
Grant Support
This work was supported by the National Key Research and Development Program of China (2014CB542003 and 2017YFA0503600), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000), General Program of National Natural Science Foundation of China (31471342 and 31671408) and National Thousand Talents Program for Distinguished Young Scholars to Y.-C. Tang; and the Curt W. and Kathy Marble Cancer Research Fund, by the Ludwig Institute for Cancer Research and the Koch Institute Support Grant P30-CA14051 to A.B. Amon, who is also an investigator of the Howard Hughes Medical Institute and of the Glenn Foundation for Biomedical Research. S. Santaguida was supported by the American Italian Cancer Foundation (AICF), by a Fellowship in Cancer Research from Marie Curie Actions and the Italian Association for Cancer Research (AIRC) and by a KI Quinquennial Cancer Research Fellowship. M. Trakala was supported by an EMBO long-term postdoctoral fellowship.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interests.
References
- 1.Duijf PH, Benezra R. The cancer biology of whole-chromosome instability. Oncogene. 2013;32:4727–36. doi: 10.1038/onc.2012.616. [DOI] [PubMed] [Google Scholar]
- 2.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–41. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 3.Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–24. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 4.Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol. 2012;8:608. doi: 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–9. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicholson JM, Macedo JC, Mattingly AJ, Wangsa D, Camps J, Lima V, et al. Chromosome mis-segregation and cytokinesis failure in trisomic human cells. eLife. 2015:4. doi: 10.7554/eLife.05068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blank HM, Sheltzer JM, Meehl CM, Amon A. Mitotic entry in the presence of DNA damage is a widespread property of aneuploidy in yeast. Mol Biol Cell. 2015;26:1440–51. doi: 10.1091/mbc.E14-10-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohashi A, Ohori M, Iwai K, Nakayama Y, Nambu T, Morishita D, et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat Commun. 2015;6:7668. doi: 10.1038/ncomms8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, et al. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–30. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes & development. 2012;26:2696–708. doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santaguida S, Vasile E, White E, Amon A. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 2015;29:2010–21. doi: 10.1101/gad.269118.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knouse KA, Wu J, Whittaker CA, Amon A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:13409–14. doi: 10.1073/pnas.1415287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfau SJ, Silberman RE, Knouse KA, Amon A. Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes Dev. 2016;30:1395–408. doi: 10.1101/gad.278820.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radin NS. Rationales for cancer chemotherapy with PDMP, a specific inhibitor of glucosylceramide synthase. Molecular and chemical neuropathology / sponsored by the International Society for Neurochemistry and the World Federation of Neurology and research groups on neurochemistry and cerebrospinal fluid. 1994;21:111–27. doi: 10.1007/BF02815346. [DOI] [PubMed] [Google Scholar]
- 17.Hannun YA, Obeid LM. The Ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem. 2002;277:25847–50. doi: 10.1074/jbc.R200008200. [DOI] [PubMed] [Google Scholar]
- 18.Causeret C, Geeraert L, Van der Hoeven G, Mannaerts GP, Van Veldhoven PP. Further characterization of rat dihydroceramide desaturase: tissue distribution, subcellular localization, and substrate specificity. Lipids. 2000;35:1117–25. doi: 10.1007/s11745-000-0627-6. [DOI] [PubMed] [Google Scholar]
- 19.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 20.Aburasayn H, Al Batran R, Ussher JR. Targeting ceramide metabolism in obesity. Am J Physiol Endocrinol Metab. 2016;311:E423–35. doi: 10.1152/ajpendo.00133.2016. [DOI] [PubMed] [Google Scholar]
- 21.Schauberger E, Peinhaupt M, Cazares T, Lindsley AW. Lipid Mediators of Allergic Disease: Pathways, Treatments, and Emerging Therapeutic Targets. Current allergy and asthma reports. 2016;16:48. doi: 10.1007/s11882-016-0628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sordillo LA, Sordillo PP, Helson L. Sphingosine Kinase Inhibitors as Maintenance Therapy of Glioblastoma After Ceramide-Induced Response. Anticancer Res. 2016;36:2085–95. [PubMed] [Google Scholar]
- 23.Jiang H, Pritchard JR, Williams RT, Lauffenburger DA, Hemann MT. A mammalian functional-genetic approach to characterizing cancer therapeutics. Nat Chem Biol. 2011;7:92–100. doi: 10.1038/nchembio.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pritchard JR, Bruno PM, Hemann MT, Lauffenburger DA. Predicting cancer drug mechanisms of action using molecular network signatures. Molecular bioSystems. 2013;9:1604–19. doi: 10.1039/c2mb25459j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol. 2013;14:25–37. doi: 10.1038/nrm3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–9. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 28.Li M, Fang X, Wei Z, York JP, Zhang P. Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol. 2009;185:983–94. doi: 10.1083/jcb.200904020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McClelland SE, Burrell RA, Swanton C. Chromosomal instability: a composite phenotype that influences sensitivity to chemotherapy. Cell Cycle. 2009;8:3262–6. doi: 10.4161/cc.8.20.9690. [DOI] [PubMed] [Google Scholar]
- 30.Swanton C, Nicke B, Schuett M, Eklund AC, Ng C, Li Q, et al. Chromosomal instability determines taxane response. Proc Natl Acad Sci U S A. 2009;106:8671–6. doi: 10.1073/pnas.0811835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, et al. Colorectal cancer. Lancet. 2010;375:1030–47. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 32.Bhattacharyya NP, Skandalis A, Ganesh A, Groden J, Meuth M. Mutator phenotypes in human colorectal carcinoma cell lines. Proc Natl Acad Sci U S A. 1994;91:6319–23. doi: 10.1073/pnas.91.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science. 2011;333:765–9. doi: 10.1126/science.1201662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slotte JP. Biological functions of sphingomyelins. Progress in lipid research. 2013;52:424–37. doi: 10.1016/j.plipres.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Miyake Y, Kozutsumi Y, Nakamura S, Fujita T, Kawasaki T. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem Biophys Res Commun. 1995;211:396–403. doi: 10.1006/bbrc.1995.1827. [DOI] [PubMed] [Google Scholar]
- 37.Reid JF, Gariboldi M, Sokolova V, Capobianco P, Lampis A, Perrone F, et al. Integrative approach for prioritizing cancer genes in sporadic colon cancer. Genes Chromosomes Cancer. 2009;48:953–62. doi: 10.1002/gcc.20697. [DOI] [PubMed] [Google Scholar]
- 38.Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010;138:958–68. doi: 10.1053/j.gastro.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rowinsky EK. Paclitaxel pharmacology and other tumor types. Semin Oncol. 1997;24:S19-1–S2. [PubMed] [Google Scholar]
- 40.Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Science translational medicine. 2014;6:229ra43. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pritchard JR, Bruno PM, Gilbert LA, Capron KL, Lauffenburger DA, Hemann MT. Defining principles of combination drug mechanisms of action. Proc Natl Acad Sci U S A. 2013;110:E170–9. doi: 10.1073/pnas.1210419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO reports. 2012;13:515–27. doi: 10.1038/embor.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol. 2015;16:473–85. doi: 10.1038/nrm4025. [DOI] [PubMed] [Google Scholar]
- 44.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nature reviews Genetics. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- 45.Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, et al. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai Q, Liu J, Chen J, Durrant D, McIntyre TM, Lee RM. Mitochondrial ceramide increases in UV-irradiated HeLa cells and is mainly derived from hydrolysis of sphingomyelin. Oncogene. 2004;23:3650–8. doi: 10.1038/sj.onc.1207430. [DOI] [PubMed] [Google Scholar]
- 47.Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82:405–14. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 48.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–9. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 49.Baran Y, Bielawski J, Gunduz U, Ogretmen B. Targeting glucosylceramide synthase sensitizes imatinib-resistant chronic myeloid leukemia cells via endogenous ceramide accumulation. Journal of cancer research and clinical oncology. 2011;137:1535–44. doi: 10.1007/s00432-011-1016-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burns TA, Subathra M, Signorelli P, Choi Y, Yang X, Wang Y, et al. Sphingomyelin synthase 1 activity is regulated by the BCR-ABL oncogene. J Lipid Res. 2013;54:794–805. doi: 10.1194/jlr.M033985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim WJ, Okimoto RA, Purton LE, Goodwin M, Haserlat SM, Dayyani F, et al. Mutations in the neutral sphingomyelinase gene SMPD3 implicate the ceramide pathway in human leukemias. Blood. 2008;111:4716–22. doi: 10.1182/blood-2007-10-113068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–16. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- 53.Riboni L, Campanella R, Bassi R, Villani R, Gaini SM, Martinelli-Boneschi F, et al. Ceramide levels are inversely associated with malignant progression of human glial tumors. Glia. 2002;39:105–13. doi: 10.1002/glia.10087. [DOI] [PubMed] [Google Scholar]
- 54.Rylova SN, Somova OG, Dyatlovitskaya EV. Comparative investigation of sphingoid bases and fatty acids in ceramides and sphingomyelins from human ovarian malignant tumors and normal ovary. Biochemistry Biokhimiia. 1998;63:1057–60. [PubMed] [Google Scholar]
- 55.Meikle PJ, Summers SA. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nature reviews Endocrinology. 2016 doi: 10.1038/nrendo.2016.169. [DOI] [PubMed] [Google Scholar]
- 56.Sprocati T, Ronchi P, Raimondi A, Francolini M, Borgese N. Dynamic and reversible restructuring of the ER induced by PDMP in cultured cells. J Cell Sci. 2006;119:3249–60. doi: 10.1242/jcs.03058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.