


Abstract
The discovery of biocompatible or bioactive nanoparticles for medicinal applications is an expensive and time-consuming process that may be significantly facilitated by incorporating more rational approaches combining both experimental and computational methods. However, it is currently hindered by two limitations: (1) the lack of high-quality comprehensive data for computational modeling and (2) the lack of an effective modeling method for the complex nanomaterial structures. In this study, we tackled both issues by first synthesizing a large library of nanoparticles and obtained comprehensive data on their characterizations and bioactivities. Meanwhile, we virtually simulated each individual nanoparticle in this library by calculating their nanostructural characteristics and built models that correlate their nanostructure diversity to the corresponding biological activities. The resulting models were then used to predict and design nanoparticles with desired bioactivities. The experimental testing results of the designed nanoparticles were consistent with the model predictions. These findings demonstrate that rational design approaches combining high-quality nanoparticle libraries, big experimental data sets, and intelligent computational models can significantly reduce the efforts and costs of nanomaterial discovery.
Keywords: nanomaterial design, QNAR modeling, nanoparticle library, cellular uptake, model predictions, virtual simulations
Graphical abstract
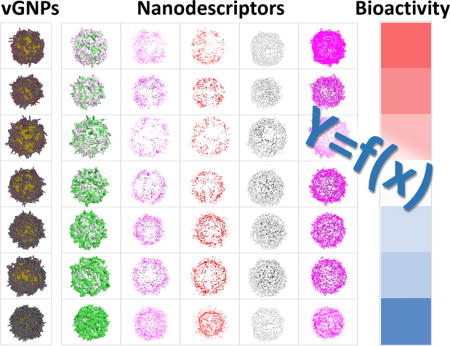
Nanoscience and nanotechnology have made significant impacts on modern medicine,1 various technology fields, and thousands of consumer products.2 For these applications, nanomaterials with desirable properties and low side effects are in high demand. However, the search for such nanomaterials depends heavily on traditional “trial and error” experimental protocols, which are time- and resource-consuming. Rational approaches that use in silico models to predict the bioactivities of nanomaterials before experimental testing would be an attractive approach for nanomaterial research.3 However, there are currently two key limitations to this advancement: (1) Most existing data available for modeling were based on limited numbers of nanomaterials with limited experimental characterization of chemical properties (e.g., basic physicochemical properties).4–6 This is due to the fact that the conventional “one-at-a-time” experimental approach has been practiced in most laboratories, allowing only limited numbers of nanoparticles to be made and tested. Furthermore, coming from different laboratories, even results for the same material may be contradictory due to poor characterization and different operations.7 (2) Despite significant efforts from various researchers, the available modeling approaches were designed and applicable only for a specified small set of nanomaterials and rarely used to design nanomaterials. One such effort is based on molecular dynamics (MD). The reaction behaviors of individual nanoparticles were investigated under certain conditions using MD, e.g., interactions with or passing through membranes, along with the effects of the size, density, position, distribution, length, and type of surface ligands on the biological properties of the nanomaterials.8–12 The advantage of MD simulations is that they can precisely simulate molecular structures. However, the clear disadvantages are that (1) modeling procedures are computationally expensive and cannot provide rapid predictions for big databases due to the current limitation of computational resources; (2) these simulations require extensive prior expertise knowledge; (3) MD simulations are inherently unsuitable for the predictions of end points with complex mechanisms, such as cytotoxicity. Thus, the usage of this approach in designing nanomaterials is limited. Another computational approach is to apply traditional quantitative structure–activity relationship (QSAR) modeling methods to nanomaterials. QSAR modeling for small molecules requires precisely calculated diverse chemical descriptors.13 The lack of suitable chemical descriptors for nanomaterials strongly limits the applicability and predictability of QSAR models. Although the descriptors calculated only from the surface ligands are useful in predicting certain properties of nanoparticles,14–16 the effects of the nanoparticle size, density, position, distribution, length, and type of surface ligands on the biological properties were not considered in these studies. Some other studies have incorporated descriptors derived from some nanoparticle-related properties (e.g., nanoparticle size)17–20 or testing results (e.g., proteomics data)6,21–23 for computational models. Efforts were also made to combine molecular simulations and QSAR modeling.24,25 Instead of simulating the nanoparticles, metal oxide substructures were used as substitution, which is only applicable to metal oxides within a specific size range. To date, there are no universal “nano-QSAR” models that can model all nanomaterials for complex bioactivities.26 Thus, a bottleneck to apply QSAR approaches for nanomaterial modeling is that nanostructure diversity is not accurately represented during the modeling process.
To address the above two limitations, we first assembled a large gold nanoparticle (GNP) library with comprehensive characterizations and bioactivity measurements. We then constructed a virtual gold nanoparticle (vGNP) library based on these experimental results and calculated a large set of nanodescriptors using precise surface chemistry simulations of each vGNP. Then predictive quantitative nanostructure activity relationship (QNAR) models were developed. With these QNAR models, we predicted and designed GNPs with different biological profiles, and these GNPs were then synthesized and confirmed experimentally.
RESULTS AND DISCUSSION
Workflow of Experimental Testing, QNAR Modeling, and Rational Nanomaterial Design
Figure 1 shows the workflow of this project, including two major parallel components, the GNP library synthesis/testing and vGNP library construction, which are the key steps of the modeling process. First the initial nanoparticle library was synthesized and tested for their cellular uptake potentials and relevant properties. The nanostructure diversity was modulated by changing the surface ligands on the GNPs. As a parallel step, the vGNP library was virtually constructed for the same nanoparticles by computationally (1) building a gold core with proper GNP size, (2) simulating the nanostructural diversity by attaching the corresponding surface ligands on the gold core, and (3) simulating the surface chemistry by calculating important physicochemical properties (Figure 1).
Figure 1.

Schematic workflow of virtual GNP (vGNP) development, predictive modeling, and experimental validation.
In this study, GNPs 1–34 were synthesized and experimentally tested in the first step to form the modeling set (Figure 1). Up to two types of surface ligands with different properties (e.g., one hydrophobic and the other hydrophilic) were attached to a gold core with a sulfur–gold linkage, and the GNP properties were changed by varying the ratio and density of these two ligands as well as the size of the gold core. The corresponding vGNPs were created, and the structures of these vGNP were then optimized. Their surface chemistries were precisely simulated as the actually synthesized GNPs. Using the resulting optimized vGNPs, nanostructural descriptors were calculated, such as the surface area and potential energy. These nanodescriptors were then used to build QNAR models that quantitatively relate the nanostructures to their complex bioactivities (e.g., cellular uptake) that were determined experimentally. By screening the external vGNP library, which contains other vGNPs with various sizes, surface ligands, and density, using the resulting QNAR models, GNPs (e.g., GNPs with different surface ligands) with desired bioactivities (e.g., high or low cellular uptake potentials) can be designed and prioritized. Seven GNPs, 35–41, were designed and synthesized based on the prediction results for the experimental validation in this study.
Design and Synthesis of a Chemically and Biologically Diverse GNP Library
The library of GNPs used in this study was designed with diverse chemical and biological activities to simulate potential GNPs used in medicine. In our previous studies, we have shown that the physicochemical properties and other complex bioactivities of nanoparticles can be modulated by systematically changing the surface ligands.27–32 In this study, we designed and synthesized a total of seven GNP library series (GNPs 1–34), with GNP size ranging from 5 to 10 nm. For each series, different surface ligands were designed to gradually change GNP hydrophobicity (S1, GNPs 1–7, red), positive charge density (S2, GNPs 8–12, navy), negative charge density (S3, GNPs 8, 13–16, green), surface hydrogen bond acceptor density (S4, GNPs 8, 17–20, magenta), surface hydrogen bond donor density (S5, GNPs 8, 21–24, orange), surface pi-bond density (S6, GNPs 25–29, blue), and molecular geometry (S7, GNPs 30–34, purple), as indicated by the colors in Figure 2. With the exception of S7 (GNPs 30–34), these GNPs each have two surface ligands with different properties as shown in Figure 2. By gradually changing the ratio of the two ligands, the major physicochemical properties of these GNP series are altered. Specifically, GNP 8 belongs to four series (S2, S3, S4, and S5) as shown in Figure 2. The relevant information about the chemical synthesis and the resulting biological data are summarized in Supplementary Table SI. This table shows that the bioactivities of GNPs (e.g., cellular uptakes) can be modulated by changing these properties. In this study, a total of 34 GNPs, which made up these seven GNP library series, were synthesized and experimentally characterized. The relevant experimental data are also shown in Supplementary Table SI. These nine experimentally tested properties cannot be directly used to predict the properties of vGNPs yet to be synthesized and thus are not suitable for prioritizing GNPs with desirable biological activities. However, some properties (i.e., size, number of ligands per GNP) are critical structural parameters of GNPs affecting their bioactivities10,12 and should always be considered during computational modeling. Accordingly, the computational calculation of a precise and diverse set of descriptors is required in order to develop models for predicting external nanoparticles.
Figure 2.

Gold nanoparticle (GNP) data set. (a) Synthesis of the GNP libraries with a combination of surface ligands for each series. (b) Experimental data of (1) cellular uptake by A549 cells; (2) cellular uptake by HEK293 cells; (3) HO-1 level in A549 cells; and (4) the partition coefficient (logP). The first six series (GNPs 1–29) were designed as dual surface ligand GNPs, and the last series (GNPs 30–34) was designed with single surface ligands. Series are distinguished by colors. Error bars represent the standard deviations (n = 3).
Virtual GNP Construction and Structure Optimization
An in-house GNPrep program was created to batch-construct the GNPs virtually, namely, vGNPs, in the library by inputting three basic structural parameters: particle size, surface ligand structure, and ligand density (number of ligands per GNP). Briefly, the surface ligands were randomly attached to the spherical gold particle shell through sulfur–gold linkages at random angles and directions. To simulate the actual configuration of the GNP, the vGNP structures were then geometrically optimized with a minimized potential energy. Up to two types of surface ligands with different properties (e.g., one hydrophobic and one hydrophilic) could be attached to the gold core.
Virtual GNP Chemical Descriptor Calculation
Nanodescriptors that are specifically useful for representing GNP chemical structure information can be calculated from the optimized vGNP structures and be used directly for modeling. It was shown in a previous study that the size, shape, surface area, surface charge, energy, functional groups, ligands, hydrophobicity, and electrostatic interactions are among the main physicochemical features that influence the interactions between nanoparticles and biological systems.33 In this study, this nanostructural information can be calculated and served as the key to correlate nanostructures to biological activities. Thus, 86 nanodescriptors were characterized and calculated based on the simulated structures of vGNPs (for details about the 86 descriptors, see Supplementary Tables SII, SIII). These 86 descriptors provided massive information for the big vGNP library from diverse aspects, which can be used for QNAR modeling to predict complex biological activities.
As an example, the S1 series (GNPs 1–7) shown in Figure 3 was designed specifically for changing the GNP hydrophobicity with different ratios of hydrophilic and hydrophobic ligands. In this study, for each vGNP in the constructed library, a specific descriptor was used to represent the hydrophobic potential, which can be visualized by the colored contours (i.e., green as the most hydrophobic and purple as the most hydrophilic) shown in the second column of Figure 3. Similarly, some other descriptor values of this GNP series (e.g., interaction potentials) can be visualized (e.g., third to sixth columns of Figure 3). For these four descriptors, the colored dots indicate the vGNP surface regions where the calculated descriptor values are above the original input threshold. For each surface property, there is a large range of values distributed along with the surface ligands on each vGNP. In order to make use of these multidimensional massive structure information data, we designed several algorithms to quantify and unify simulated surface property features into sets of descriptors that can be used for modeling (see “Methods” and Supplementary Table SIII).
Figure 3.

Simulated surface features of the vGNPs. First column: series 1 (GNPs 1–7); second: hydrophobic potentials; third: interaction potential with sodium cation; fourth: interaction potential with dry (hydrophobic) probe; fifth: electrostatic surface associated with hydrophobic interaction atom types; sixth: nonbonded contact preference with hydrophobic ligand atoms.
Nanostructure Diversity Visualization
On the basis of the calculated nanodescriptors of the 34 GNPs, we first visualized how these vGNPs were structurally differentiated from each other. After performing principal component analysis using the 90 descriptors (86 calculated descriptors, along with four experimentally determined basic properties: three surface ligand densities and GNP size), the two top-ranked principal components, covering 89% of the variance of all descriptors, were used to construct a GNP chemical space, which represents the distribution of vGNPs based on their structural diversities. As shown in Figure 4, within most vGNP series, individual vGNPs are structurally different from each other. However, the vGNPs within two series designed to have different positive and negative charge densities (S2 and S3: GNPs 8–16) showed relatively small structural differences in the current GNP chemical space (Figure 4). This issue may be due to the lack of suitable descriptors for describing their structural diversity and might negatively affect the model predictability for the external GNPs with similar surface ligands to these two series. This issue is further discussed in detail below.
Figure 4.

Principal component analysis of the 41 GNPs based on the 90 chemical descriptors. Dots are GNPs in the modeling set, and star points are those in the external validation set.
Predictive Computational Modeling
Among all 86 descriptors and their four physical properties (i.e., particle size and three types of ligand density), some descriptor values were highly correlated with each other. Highly correlated descriptors will induce issues during the modeling procedure, and normally one of two highly correlated descriptors needs to be removed.34–36 After removing the correlated descriptors, 29 descriptors remained, as shown in Figure 5. These descriptors were then used in the following modeling procedure.
Figure 5.

Heatmap of the chemical descriptors generated for 34 GNPs. Descriptor values were normalized between 0 and 1.
Using the 29 descriptors and the k-nearest-neighbor (kNN) algorithm, we developed QNAR models for cellular uptake in human lung and kidney cells (A549 and HEK293 cells), ability to induce oxidative stress (indicated by the HO-1 level in the A549 cells), and hydrophobicity (indicated by logP values). In each individual kNN model, up to 11 descriptors were used. The model performance was first shown by a 10-fold cross-validation process of the modeling set. The resulting four models showed high predictabilities (modeling set GNPs are shown as dots in Figure 6) with correlation coefficients (R2) of 0.995, 0.990, 0.967, and 0.988 and mean absolute error (MAE) values of 0.11 (× 107 GNPs/cell), 0.14 (× 107 GNPs/cell), 0.14, and 0.18, respectively.
Figure 6.

QNAR model performance in the 10-fold cross-validation (dots) and external validation (stars) results in (a) cellular uptake in A549 cells; (b) cellular uptake in HEK293 cells; (c) HO-1 level in A549 cells; and (d) logP.
Nanoparticle Discovery with the QNAR Models and Experimentation
The ultimate goal of any computational model is its applicability in prediction. To realize this goal, first we virtually designed and created seven vGNPs (shown in Supplementary Table SIV, GNPs 35–41) with different surface chemistries (i.e., sizes, surface ligand ratios, and densities) as shown in Figure 7a. Then the developed QNAR models were used to predict the physicochemical properties and bioactivities of these vGNPs (Figure 7b). These nanoparticles were intentionally designed with predicted diverse physicochemical properties and bioactivities. Experimental data convincingly confirmed most of the modeling predictions (Figure 7, Supplementary Tables SI, SII). The correlations between model predictions and the experimental results (Figure 7c) were reflected by R2 values (0.918, 0.919, 0.768, and 0.930) and MAE values (0.49 × 107 GNPs/cell, 0.46 × 107 GNPs/cell, 0.26, and 0.43) for each end point, respectively (this external validation set of GNPs is shown as star points in Figure 6).
Figure 7.

Computational profile, design, and experimental validation of seven external nanoparticles. (a) Computationally designed vGNPs; (b) predicted properties and bioactivities of the vGNPs; and (c) experimental validation results.
Designed GNPs with Desired Bioactivities
As shown above, using the resulting QNAR models and important nanodescriptors, we predicted and selected seven external GNPs, which were then experimentally synthesized and experimentally confirmed (Figure 7). The advantage of this study is that the GNPs can be characterized by critical physicochemical properties (e.g., nanodescriptors) and bioactivities (e.g., the precisely predicted cellular uptake levels). This approach allowed us to cover most known factors for designing potential nanomedicines. These external GNPs were prioritized by QNAR models due to the diverse predicted bioactivities (e.g., low or high cellular uptake potentials). As shown in previous studies, GNPs with desired bioactivities can be designed by systematically changing the surface ligands.20 In this study, we not only successfully reached this goal by creating virtual nanoparticles and precisely simulating their surface chemistry but also predicted their target bioactivities before experimental synthesis. Those with optimal properties can be visualized and selected computationally upon requirements. For example, the biological profiles of vGNPs 35 and 40 were predicted to be relatively similar, aside from the size difference (Figure 7b). vGNPs 41 and 35 have similar cellular uptakes in both HEK293 and A549 cells. But vGNP 41 was predicted to have higher HO-1 activity and lower logP than 35 (Figure 7b). We may select the most suitable GNPs for future development by considering the whole biological profile. This way, we can precisely design nanomaterials that meet the therapeutic requirements of modern nanomedicines.
Elucidated Mechanisms of Cellular Uptake
The important mechanisms of GNP cellular uptakes can be obtained by analyzing modeling results and used to guide nanomaterial design. The results showed that there are several descriptors that are critical to the QNAR models. For example, the descriptor hydrophobic potential has clear and high linear correlations with the experimental hydrophobicity logP values (R2 = 0.76), the cellular uptake in A549 cells (R2 = 0.74), and the cellular uptake in HEK293 cells (R2 = 0.74). Indeed, not surprisingly, in the models built for these three end points, the hydrophobic potential is the most important descriptor that is mostly used in all the acceptable kNN models (87%, 75%, and 80% of all acceptable models for cellular uptake in A549 cells, cellular uptake in HEK293 cells, and logP, respectively). The other important descriptors for the cellular uptake models in the A549 cells are the partial charge, nonbonded hydrophobic contact preference, and particle size, while those important for cellular uptake models in the HEK293 cells are the nonbonded hydrophobic contact preference, partial charge, and surface area. For example, GNP 7, which has high cellular uptake potentials for both cells, was featured with a hydrophobic potential as high as 3.62 and a nonbonded hydrophobic contact preference as low as 0.49. Compared to the other three models, the top four descriptors that are most important to the oxidative stress induction model are the number of surface ligands, nonbonded hydrophobic contact preference, interaction potential with water molecules, and electrostatic positivity. This indicated that different mechanisms of action and extra interactions are involved in oxidative stress induction by GNPs compared to other nano–bio interactions, such as cellular uptake. These factors should be considered for the development of nanomaterials.
Advanced GNP Design by Applying Applicability Domain and Additional Experimental Testing
Although the current chemical descriptors have covered a variety of aspects of the GNP structural diversity and the resulting models yielded satisfactory predictability, more studies need to be conducted for GNP development. As shown in Figure 6, two external GNPs (36, the navy star, and 38, the magenta star) have relatively large prediction errors in at least two models. As shown in the GNP chemical space (Figure 4), the diversity of GNP series S2 (GNP 8–12) with changes in the positive charge density cannot be distinguished, and GNP 36 belongs to this series. In our previous QSAR modeling studies, the use of the applicability domain (AD) could improve the model predictivity.37 The definition of the AD was normally based on the structure similarity between the external compounds and their nearest neighbors in the modeling sets. In this study, a similar analysis was applied. As expected, 36 was identified as a structural outlier with a normalized Euclidean distance as large as 0.86 to the closest GNP in the modeling set. For this reason, the relatively larger prediction error in the models of cellular uptake of this GNP may be due to the diversity limitation of the GNPs distributed in this created GNP chemical space (i.e., a lack of representative descriptors describing the cellular uptake relatives). Without extensively expanding the current nano-structure landscape by experimentally testing more GNPs, the AD cannot be defined without enough external prediction results. However, this issue can be resolved by developing more chemical descriptors from the vGNP library to better represent their structure diversity. For example, the potential descriptors in the future can be derived by understanding biophysicochemical interactions at the nano–bio interface, such as receptor–ligand binding interaction potentials and nanomaterial conformational changes. Probing these various biophysicochemical interactions may improve the current QNAR models by including additional knowledge information on nanostructures.33 Meanwhile, the GNP 38 is shown to be structurally different from other GNPs in the current GNP chemical space. Its only nearest neighbor, GNP 33, has a high logP and cellular uptake, which is the opposite of those of GNP 38. This issue can be resolved by experimentally testing more GNPs within this series to generate more chemical nearest neighbors of GNP 38. For this reason, experimental testing is critical and needed when there is not enough data available to cover specific areas of the GNP chemical space.
Potential Pitfalls and Future Directions
Currently the technical issues of limited computational power and lack of software can limit studies involving large sets of nanomaterials. For example, in this study, we used nanoparticles with sizes ranging from 5 to 10 nm and number of surface ligands ranging from 100 to 900. Based on the GNP library, the constructed vGNPs have almost reached the upper limit of the protein database (PDB) format used to store the relevant nanostructures (i.e., up to 99 999 atoms for each vGNP).38 For more complicated nanostructures (e.g., larger GNPs with more surface ligands), the PDB format cannot be used. And there is no other generally acknowledged substitution file formats that can overcome this issue. To this end, we are designing other computational approaches to resolve this issue and make this strategy applicable for more complicated nanomaterials.
CONCLUSION
Nanoparticle discovery by experimental data and intelligent computer modeling approaches is the method of choice to overcome the current bottleneck in nanomaterial research. The performance of QNAR models, like the conventional QSAR models, depends heavily on the availability and the amount of high-quality data. Only with big and comprehensive databases can models yield comprehensive and accurate prediction powers for nanomaterials with a wider range of applicability. Meanwhile, the modeling approaches need to be able to intelligently represent the real nanostructures’ diversity. By taking advantage of the precise simulation approaches that focus on understanding the individual actions of specific GNPs, the proposed method can virtually create a diverse collection of vGNPs from various aspects by simulating and calculating a broad set of surface features. Additionally, compared to previous QSAR studies on GNPs, this QNAR modeling approach has the advantage to not only rapidly screen big GNP data sets but also more accurately predict the properties of nanoparticles, which could help design or prioritize GNPs with desirable biological properties. Furthermore, the current workflow of QNAR modeling may be extended to other nanomaterials, such as other spherical nanoparticles or nanomaterials of various shapes, sizes, and surface coatings.
EXPERIMENTAL AND COMPUTATIONAL METHODS
GNP Library Synthesis
Each surface-modified member of the GNP library was made in a one-pot synthesis. Hydrogen tetrachloroaurate(III) (HAuCl4) trihydrate solutions (0.05 mol/L) were stirred with ligands at room temperature. Then, sodium tetrahydroborate was added dropwise to the mixture. The mixture was stirred for 4 h at room temperature. After the reaction is finished, the mixture was centrifuged, and the supernatant was discarded. The precipitate was resuspended in deionized water. The centrifugation– dissolution cycle was repeated five times.
GNP Library Characterization
The number of ligands on each GNP was characterized as described in our previous article.20,39 Briefly, the ligands on GNPs were first cleaved by I2. Then, the ligands was quantitatively analyzed by LC/MS to get the number of ligand molecules per nanoparticle. The diameters of the GNPs were analyzed by transmission electron microscopy observations (JEM-1011, JEOL, Tokyo, Japan). The hydrodynamic diameter and zeta potential were analyzed using a laser particle size analyzer (Malvern Nano ZS, Malvern, UK) in ultrapure water (18.2 MΩ) or in 10% fetal bovine serum.
Experimental logP Measurement
The experimental logP values of all the GNPs were determined using a modified “shaking flask” method as described in our previous paper.20 Briefly, GNPs were mixed with octanol-saturated water and water-saturated octanol. The mixture was shaken for 24 h. Then, the mixture was kept still for 3 h to separate the organic and water phases. The GNPs in both phases were quantitatively determined by ICP-MS. logP values were then calculated using the following equation:
where CGNP(octanol) is the concentration of GNPs in octanol and CGNP(water) is the concentration of GNPs in water.
Quantification of HO-1 Level
A549 cells were treated with GNPs (50 µg/mL) for 24 h. Then, the cells were harvested and proteins were extracted after cell lysis. HO-1 protein was quantitatively determined by Western blot. The band intensity was quantified by ImageJ 1.47v (National Institutes of Health, USA).
Cellular Uptake
GNPs (50 µg/mL) were incubated with A549 or HEK293 cells for 24 h. After washing cells three times with phosphate-buffered saline, we detached the cells from the flask by trypsin–EDTA solution. The cells were counted and then lysed overnight in aqua regia. ICP-MS was used to quantify the concentration of GNPs.
Virtual GNP Construction and Structure Optimization
The construction of vGNPs was accomplished by the in-house GNPrep program coded in Python 3.5, which takes input information on both the gold core and surface ligands and generates individual vGNPs in PDB format. First, according to the input size of the GNP, it forms a spherical gold core. In this study, only the gold shell (i.e., Au atoms on the core surface) was generated for each vGNP since (1) the atoms in the gold core are stable and compact, (2) the conformation of the gold core is unlikely to change, and (3) the simulation focuses mostly on the surface chemistry. Then, the surface ligands were connected to the shell by randomly attaching their sulfur–sulfur linkers to the surface Au atoms. Originally, the surface ligands were set at random angles and directions. To simulate the actual conformations of the GNPs under experimental conditions, the structures of the constructed vGNPs were refined and optimized under the Amber10:EHT force field,40–42 a function provided by Molecular Operating Environment (MOE version 2015.10).42 Since the structure optimization using different force fields did not significant affect the descriptor calculation and the model development, this structure optimization method was chosen arbitrarily.
Virtual GNP Chemical Descriptor Calculation
To simulate the surface chemistry of a GNP, two types of surfaces were identified and isolated using MOE:42,43 the interaction surface (also called the van der Waals accessible surface) and the electron density surface. From the interaction surface, the total surface area of the vGNP and the average surface area per surface ligand were calculated. Furthermore, several types of potential vGNP–target interactions were simulated on the interaction surface: hydrophobicity, electrostatic features, nonbonded contact preferences, and interaction potentials with certain fragmental structures. Then, the resulting interaction potentials obtained for each above interaction were quantified. Specifically, since the interaction potentials were calculated for each grid point on the vGNP surface, we calculated overall interaction potential scores of the vGNP. To calculate the scores, we (1) simply averaged the interaction values of all grid points or (2) counted the number of points that are above an interaction threshold, which is determined based on all the vGNPs in the modeling set. Meanwhile, the electron density surface, which represents the electron density distribution in a grid unit cell, was also calculated for the vGNP as described above. The surface simulation was initially realized in MOE,42 while the quantification was accomplished by in-house codes written in Python 3.5. The quantified features were then used as nanodescriptors in the following modeling procedures. For more information about the descriptors, please refer to Supplementary Table SIII.
All descriptors were normalized in the range of zero to one. Then, if two descriptors showed redundant results in the modeling set (correlation coefficient R2 > 0.99), one of them was removed. The descriptors with low variance (standard deviation <0.01 or less than three different values) were removed as well. This effort resulted in a set of 29 descriptors, which was used in the modeling process. As shown in Figure 5, these 29 descriptor values of the modeling set were shown as a clustered heatmap using the pHeatmap package44 in R version 3.1.1.
QNAR Modeling
Using the remaining 29 descriptors and the kNN algorithm, we developed QNAR models for the cellular uptakes in the A549 cell line and the HEK293 cell line, the HO-1 level in the A549 cell line, and the logP values. The kNN method45 uses the bioactivities of each GNP’s k nearest neighbors, which have the lowest Euclidean distances between GNPs in multidimensional GNP chemical space as its prediction, and employs optimized selection of variables to define neighbors. It was developed using our in-house program implementation (also available at chembench.mml.unc.edu).46 All models were validated using a 10-fold cross-validation within the modeling set. Briefly, the modeling set was randomly divided into 10 equivalent subsets. Nine subsets (90% of the modeling set GNPs) were used as the training set, as the remaining one served as the test set (10% of the modeling set GNPs). The training set was used to develop the QNAR models, and the resulting models were validated by predicting the excluded test set. This procedure was repeated 10 times so that each GNP was left out in the test set once. Then, seven external GNPs were synthesized and tested for the above four bioassays using the same experimental protocols. This experimental validation procedure was used to further validate the predictability of the resulting models and the whole modeling workflow. Details regarding the kNN modeling and validation procedure can be found in our previous publications.34,47
Supplementary Material
Acknowledgments
We thank the academic support of the Chemical Computing Group for their generous technique support; Q. Wu, Y. Liu, J. Wu, Q. Jiao, W. Diao, and C. Guo for assistance in GNP synthesis and characterization; S. Murlidaran, R. Lohia, and R. Ma for assistance and discussion on the PDB file format; and H. Ciallela for assistance in editing the manuscript. This research was supported in part by National Institutes of Health (NIH) grants P30ES005022 and R15ES023148, the Johns Hopkins Center for Alternatives to Animal Testing (CAAT) research grant, the National Key Research and Development Program of China (2016YFA0203103), the National Natural Science Foundation of China (91543204, 91643204, and 21137002), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB14030401).
Footnotes
ASSOCIATED CONTENT
- Details of the 41-GNP modeling set (Table SI), nanodescriptor calculation results (Table SII), nanodescriptor descriptions (Table SIII), and seven external GNPs (Table SIV) (PDF)
The authors declare no competing financial interest.
References
- 1.Roco MC. Nanotechnology: Convergence with Modern Biology and Medicine. Curr. Opin. Biotechnol. 2003;14:337–346. doi: 10.1016/s0958-1669(03)00068-5. [DOI] [PubMed] [Google Scholar]
- 2.Jones R. Nanotechnology, Energy and Markets. Nat. Nanotechnol. 2009;4:75–75. doi: 10.1038/nnano.2008.420. [DOI] [PubMed] [Google Scholar]
- 3.Winkler DA, Mombelli E, Pietroiusti A, Tran L, Worth A, Fadeel B, McCall MJ. Applying Quantitative Structure-Activity Relationship Approaches to Nanotoxicology: Current Status and Future Potential. Toxicology. 2013;313:15–23. doi: 10.1016/j.tox.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Fourches D, Pu D, Tropsha A. Exploring Quantitative Nanostructure-Activity Relationships (QNAR) Modeling as a Tool for Predicting Biological Effects of Manufactured Nanoparticles. Comb. Chem. High Throughput Screening. 2011;14:217–225. doi: 10.2174/138620711794728743. [DOI] [PubMed] [Google Scholar]
- 5.Hansen SF, Larsen BH, Olsen SI, Baun A. Categorization Framework to Aid Hazard Identification of Nanomaterials. Nanotoxicology. 2007;1:243–250. [Google Scholar]
- 6.Walkey CD, Olsen JB, Song F, Liu R, Guo H, Olsen DWH, Cohen Y, Emili A, Chan WCW. Protein Corona Fingerprinting Predicts the Cellular Interaction of Gold and Silver Nanoparticles. ACS Nano. 2014;8:2439–2455. doi: 10.1021/nn406018q. [DOI] [PubMed] [Google Scholar]
- 7.Krug HF. Nanosafety Research-Are We on the Right Track? Angew. Chem., Int. Ed. 2014;53:12304–12319. doi: 10.1002/anie.201403367. [DOI] [PubMed] [Google Scholar]
- 8.Heikkilä E, Martinez-Seara H, Gurtovenko AA, Javanainen M, Häkkinen H, Vattulainen I, Akola J. Cationic Au Nanoparticle Binding with Plasma Membrane-like Lipid Bilayers: Potential Mechanism for Spontaneous Permeation to Cells Revealed by Atomistic Simulations. J. Phys. Chem. C. 2014;118:11131–11141. [Google Scholar]
- 9.Kyrychenko A, Korsun OM, Gubin II, Kovalenko SM, Kalugin ON. Atomistic Simulations of Coating of Silver Nanoparticles with Poly(vinylpyrrolidone) Oligomers: Effect of Oligomer Chain Length. J. Phys. Chem. C. 2015;119:7888–7899. [Google Scholar]
- 10.Ndoro TVM, Voyiatzis E, Ghanbari A, Theodorou DN, Böhm MC, Müller-Plathe F. Interface of Grafted and Ungrafted Silica Nanoparticles with a Polystyrene Matrix: Atomistic Molecular Dynamics Simulations. Macromolecules. 2011;44:2316–2327. [Google Scholar]
- 11.Van Lehn RC, Alexander-Katz A. Pathway for Insertion of Amphiphilic Nanoparticles into Defect-Free Lipid Bilayers from Atomistic Molecular Dynamics Simulations. Soft Matter. 2015;11:3165–3175. doi: 10.1039/c5sm00287g. [DOI] [PubMed] [Google Scholar]
- 12.Liu W, Wu Y, Wang C, Li HC, Wang T, Liao CY, Cui L, Zhou QF, Yan B, Jiang GB. Impact of Silver Nanoparticles on Human Cells: Effect of Particle Size. Nanotoxicology. 2010;4:319–330. doi: 10.3109/17435390.2010.483745. [DOI] [PubMed] [Google Scholar]
- 13.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’Min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014;57:4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toropov AA, Toropova AP, Puzyn T, Benfenati E, Gini G, Leszczynska D, Leszczynski J. QSAR as a Random Event: Modeling of Nanoparticles Uptake in PaCa2 Cancer Cells. Chemosphere. 2013;92:31–37. doi: 10.1016/j.chemosphere.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 15.Fourches D, Pu D, Li L, Zhou H, Mu Q, Su G, Yan B, Tropsha A. Computer-Aided Design of Carbon Nanotubes with the Desired Bioactivity and Safety Profiles. Nanotoxicology. 2016;10:374–383. doi: 10.3109/17435390.2015.1073397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luan F, Tang L, Zhang L, Zhang S, Monteagudo MC, Cordeiro MNDSA. Further Development of the QNAR Model to Predict the Cellular Uptake of Nanoparticles by Pancreatic Cancer Cells. Food Chem. Toxicol. 2016 doi: 10.1016/j.fct.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Epa VC, Burden FR, Tassa C, Weissleder R, Shaw S, Winkler DA. Modeling Biological Activities of Nanoparticles. Nano Lett. 2012;12:5808–5812. doi: 10.1021/nl303144k. [DOI] [PubMed] [Google Scholar]
- 18.Shaw SY, Westly EC, Pittet MJ, Subramanian A, Schreiber SL, Weissleder R. Perturbational Profiling of Nanomaterial Biologic Activity. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7387–7392. doi: 10.1073/pnas.0802878105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu R, Rallo R, George S, Ji Z, Nair S, Nel AE, Cohen Y. Classification NanoSAR Development for Cytotoxicity of Metal Oxide Nanoparticles. Small. 2011;7:1118–1126. doi: 10.1002/smll.201002366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li S, Zhai S, Liu Y, Zhou H, Wu J, Jiao Q, Zhang B, Zhu H, Yan B. Experimental Modulation and Computational Model of Nano-Hydrophobicity. Biomaterials. 2015;52:312–317. doi: 10.1016/j.biomaterials.2015.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen R, Zhang Y, Monteiro-Riviere NA, Riviere JE. Quantification of Nanoparticle Pesticide Adsorption: Computational Approaches Based on Experimental Data. Nanotoxicology. 2016;10:1118–1128. doi: 10.1080/17435390.2016.1177745. [DOI] [PubMed] [Google Scholar]
- 22.Pathakoti K, Huang MJ, Watts JD, He X, Hwang HM. Using Experimental Data of Escherichia Coli to Develop a QSAR Model for Predicting the Photo-Induced Cytotoxicity of Metal Oxide Nanoparticles. J. Photochem. Photobiol. B. 2014;130:234–240. doi: 10.1016/j.jphotobiol.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 23.Fourches D, Pu D, Tassa C, Weissleder R, Shaw SY, Mumper RJ, Tropsha A. Quantitative Nanostructure-Activity Relationship Modeling. ACS Nano. 2010;4:5703–5712. doi: 10.1021/nn1013484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puzyn T, Rasulev B, Gajewicz A, Hu X, Dasari TP, Michalkova A, Hwang H-M, Toropov A, Leszczynska D, Leszczynski J. Using Nano-QSAR to Predict the Cytotoxicity of Metal Oxide Nanoparticles. Nat. Nanotechnol. 2011;6:175–178. doi: 10.1038/nnano.2011.10. [DOI] [PubMed] [Google Scholar]
- 25.Gajewicz A, Schaeublin N, Rasulev B, Hussain S, Leszczynska D, Puzyn T, Leszczynski J. Towards Understanding Mechanisms Governing Cytotoxicity of Metal Oxides Nanoparticles: Hints from Nano-QSAR Studies. Nanotoxicology. 2015;9:313–325. doi: 10.3109/17435390.2014.930195. [DOI] [PubMed] [Google Scholar]
- 26.Puzyn T, Leszczynska D, Leszczynski J. Toward the Development of “Nano-QSARs”: Advances and Challenges. Small. 2009;5:2494–2509. doi: 10.1002/smll.200900179. [DOI] [PubMed] [Google Scholar]
- 27.Wu L, Zhang Y, Zhang C, Cui X, Zhai S, Liu Y, Li C, Zhu H, Qu G, Jiang G, Yan B. Tuning Cell Autophagy by Diversifying Carbon Nanotube Surface Chemistry. ACS Nano. 2014;8:2087–2099. doi: 10.1021/nn500376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H, Mu Q, Gao N, Liu A, Xing Y, Gao S, Zhang Q, Qu G, Chen Y, Liu G, Zhang B, Yan B. A Nano-Combinatorial Library Strategy for the Discovery of Nanotubes with Reduced Protein-Binding, Cytotoxicity, and Immune Response. Nano Lett. 2008;8:859–865. doi: 10.1021/nl0730155. [DOI] [PubMed] [Google Scholar]
- 29.Zhang B, Xing Y, Li Z, Zhou H, Mu Q, Yan B. Functionalized Carbon Nanotubes Specifically Bind to α-Chymotrypsin’s Catalytic Site and Regulate Its Enzymatic Function. Nano Lett. 2009;9:2280–2284. doi: 10.1021/nl900437n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H, Jiao P, Yang L, Li X, Yan B. Enhancing Cell Recognition by Scrutinizing Cell Surfaces with a Nanoparticle Array. J. Am. Chem. Soc. 2011;133:680–682. doi: 10.1021/ja108527y. [DOI] [PubMed] [Google Scholar]
- 31.Gao N, Zhang Q, Mu Q, Bai Y, Li L, Zhou H, Butch ER, Powell TB, Snyder SE, Jiang G, Yan B. Steering Carbon Nanotubes to Scavenger Receptor Recognition by Nanotube Surface Chemistry Modification Partially Alleviates NFκB Activation and Reduces Its Immunotoxicity. ACS Nano. 2011;5:4581–4591. doi: 10.1021/nn200283g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Wang Y, Liu A, Xu SL, Zhao B, Zhang Y, Zou H, Wang W, Zhu H, Yan B. Modulation of Carbon Nanotubes’ Perturbation to the Metabolic Activity of CYP3A4 in the Liver. Adv. Funct. Mater. 2016;26:841–850. [Google Scholar]
- 33.Nel AE, Mädler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding Biophysicochemical Interactions at the Nano–bio Interface. Nat. Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 34.Wang W, Kim MT, Sedykh A, Zhu H. Developing Enhanced Blood-Brain Barrier Permeability Models: Integrating External Bio-Assay Data in QSAR Modeling. Pharm. Res. 2015;32:3055–3065. doi: 10.1007/s11095-015-1687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim MT, Huang R, Sedykh A, Wang W, Xia M, Zhu H. Mechanism Profiling of Hepatotoxicity Caused by Oxidative Stress Using Antioxidant Response Element Reporter Gene Assay Models and Big Data. Environ. Health Perspect. 2016;124:634–641. doi: 10.1289/ehp.1509763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solimeo R, Zhang J, Kim M, Sedykh A, Zhu H. Predicting Chemical Ocular Toxicity Using a Combinatorial QSAR Approach. Chem. Res. Toxicol. 2012;25:2763–2769. doi: 10.1021/tx300393v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu H, Martin TM, Ye L, Sedykh A, Young DM, Tropsha A. Quantitative Structure-Activity Relationship Modeling of Rat Acute Toxicity by Oral Exposure. Chem. Res. Toxicol. 2009;22:1913–1921. doi: 10.1021/tx900189p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sussman JL, Lin D, Jiang J, Manning NO, Prilusky J, Ritter O, Abola EE. Protein Data Bank (PDB): Database of Three- Dimensional Structural Information of Biological Macromolecules. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998;54:1078–1084. doi: 10.1107/S0907444998009378. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Zhou H, Yang L, Du G, Pai-Panandiker AS, Huang X, Yan B. Enhancement of Cell Recognition in Vitro by Dual-Ligand Cancer Targeting Gold Nanoparticles. Biomaterials. 2011;32:2540–2545. doi: 10.1016/j.biomaterials.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerber PR, Müller K. MAB, a Generally Applicable Molecular Force Field for Structure Modelling in Medicinal Chemistry. J. Comput.-Aided Mol. Des. 1995;9:251–268. doi: 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
- 41.Case D, Darden T, Cheatham T, Simmerling C. Amber 10. University of California; San Francisco: 2008. [Google Scholar]
- 42.Lambot S, Slob EC, Van den Bosch I, Stockbroeckx B, Vanclooster M. Modeling of Ground-Penetrating Radar for Accurate Characterization of Subsurface Electric Properties. IEEE Trans. Geosci. Remote Sens. 2004;42:2555–2568. [Google Scholar]
- 43.Wang XF, Huang DS, Xu H. An Efficient Local Chan-Vese Model for Image Segmentation. Pattern Recognit. 2010;43:603–618. [Google Scholar]
- 44.Kolde R. pHeatmap 0.7.7: Pretty Heatmaps. https://cran.r-project.org/web/packages/pheatmap/index.html.
- 45.Zheng W, Tropsha A. Novel Variable Selection Quantitative Structure-Property Relationship Approach Based on the K-Nearest-Neighbor Principle. J. Chem. Inf. Model. 2000;40:185–194. doi: 10.1021/ci980033m. [DOI] [PubMed] [Google Scholar]
- 46.Walker T, Grulke CM, Pozefsky D, Tropsha A. Chembench: A Cheminformatics Workbench. Bioinformatics. 2010;26:3000–3001. doi: 10.1093/bioinformatics/btq556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim MT, Sedykh A, Chakravarti SK, Saiakhov RD, Zhu H. Critical Evaluation of Human Oral Bioavailability for Pharmaceutical Drugs by Using Various Cheminformatics Approaches. Pharm. Res. 2014;31:1002–1014. doi: 10.1007/s11095-013-1222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.