

Abstract
Loss-of-function of succinate dehydrogenase-B (SDHB) is a predisposing factor of aerobic glycolysis and cancer progression. Adenosine monophosphate activated protein kinase (AMPK) is involved in the regulation of aerobic glycolysis and the diverse hallmarks of cancer. The present study investigated whether AMPK mediated the regulatory effects of SDHB in aerobic glycolysis and cancer growth. The expression of SDHB and AMPK in colorectal cancer (CRC) and normal tissues was assessed by western blotting. HT-29 CRC cells were used to establish in vitro models of ectopic overexpression and knockdown of SDHB. SDHB was downregulated, while AMPK and phosphorylated-AMPK (Thr172) were upregulated in CRC tissues. Experiments involving the loss- or gain-of-function of SDHB, revealed that this protein negatively regulated AMPK by influencing its expression and activity. However, SDHB and AMPK were identified to suppress lactic acid production in CRC cells, indicating that each had an inhibitory effect on aerobic glycolysis. Therefore, the regulation of aerobic glycolysis by SDHB is unlikely to be mediated via AMPK. SDHB knockdown promoted the viability, migration and invasion of HT-29 cells, whereas inhibition of AMPK demonstrated the opposite effect. SDHB overexpression impaired cell migration and invasion, and this effect was reversed following AMPK activation. These results indicate that AMPK may mediate the effects of SDHB in CRC cell proliferation and migration. In conclusion, SDHB downregulation in CRC cells may increase AMPK activity, which may subsequently facilitate the proliferation and invasion of these cancer cells. However, the regulation of aerobic glycolysis by SDHB may be independent of AMPK. Further studies are warranted to elucidate the mechanism by which SDHB regulates aerobic glycolysis.
Keywords: succinate dehydrogenase-B, colorectal cancer, adenosine monophosphate activated protein kinase, aerobic glycolysis
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-associated mortality in malignant tumors of the digestive tract worldwide (1). The rapid malignant transformation of cells, combined with the development of resistance in CRC tumors makes effective treatment difficult. Previous studies have demonstrated that CRC tumors undergo metabolic reprogramming to favor aerobic glycolysis, even following exposure to normoxic conditions; a phenomenon known as the Warburg effect (2,3). This effect is important, as it contributes to cancer proliferation and survival (2,3). Although this bio-energetic switch occurs at the expense of efficient glucose utilization, metabolic intermediates from aerobic glycolysis may be used as biosynthetic precursors for the generation of nucleic acids, phospholipids, fatty acids, cholesterol and porphyrins, which are essential to support the rapid proliferation of cancer cells (4). Aerobic glycolysis also confers survival benefits to CRC cells within the ischemic and acidic tumor microenvironment, and may function to facilitate immune system evasion and maintenance of the cancer stem cell state (5). Through aerobic glycolysis, glucose is metabolized to lactate, which is subsequently released from cells and leads to the development of an acidic extracellular microenvironment. The CRC cell metabolic shift affects cell signaling, histone acetylation and the production of reactive oxygen species, which are considered to be hallmarks of cancer (6). However, their precise influence requires further clarification (6). Therefore, understanding the mechanisms underlying the bio-energetic switch of CRC cells may be crucial for the development of effective cancer treatments.
Succinate dehydrogenase (SDH) is a metabolic enzyme located on the inner mitochondrial membrane. It is required for the oxidation of succinate to fumarate in the citric acid cycle; an essential stage of oxidative phosphorylation (7). It also facilitates the engagement of electron transportation (7). SDHB is the core catalytic subunit of the heterotetrameric complex of SDH. Previous studies have demonstrated that SDHB loss-of-function leads to SDH dysfunction, which alters the citric acid cycle and facilitates the development of different cancers, including bladder, neuroendrocrine neoplasms, renal cell carcinoma and gastrointestinal stromal tumors (8–10). SDHB dysfunction can also suppress the α-ketoglutarate-dependent synthesis of histone and DNA demethylases, which may influence the expression of tumor suppressor genes and oncogenes (7). Additionally, the accumulation of succinate as a consequence of SDHB dysfunction contributes to the stabilization of hypoxia-inducible factor (HIF) under normoxic conditions (7). This allows HIF to facilitate the anaerobic metabolism and angiogenesis of tumor cells. Therefore, SDHB loss-of-function may be implicated in the Warburg effect observed in CRC cells.
Adenosine monophosphate activated protein kinase (AMPK) is an important regulator of cellular metabolism and has been implicated in the development of cancer (11,12). The phosphorylation activity of AMPK is primarily, but not exclusively, regulated by the ratio of AMP/adenosine 5′-triphosphate (ATP) (11,12). AMPK activity is also affected by reactive oxygen species, nitrous oxide, hormones (including adiponectin, leptin, thyroid hormone, ghrelin and cannabinoids) and pharmacological agents (11). Previous studies have demonstrated that signaling pathways mediated by the LKB1-STE20-related kinase adaptor-MO25 complex and by calcium/calmodulin-activated protein kinase kinases, serve to control AMPK phosphorylation (11,12). In addition, AMPK has been implicated in the regulation of the Warburg effect. Li et al (13) reported that growth arrest-specific 1 activates AMPK and inhibits mechanistic target of rapamycin (mTOR)/p70S6K signaling in CRC, effectively inhibiting the Warburg effect, cell proliferation and invasion. Further results have demonstrated that the multi-tyrosine kinase inhibitors of anti-angiogenics initiates a cellular switch from glycolysis to mitochondrial respiration-dependent oxidative phosphorylation in spontaneous breast and lung tumor models (14). This metabolic switch is mediated by the downregulation of HIF1α and Akt and the upregulation of AMPK (14). When AMPK function is inhibited by AMPK inhibitors, AMPK small-interfering RNA (siRNA) or dominant negative AMPK, the inhibitory effects of 2-deoxyglucose on aerobic glycolysis is diminished (14). These studies therefore provide evidence to suggest that AMPK serves an inhibitory role in aerobic glycolysis.
Based on a previous report indicating a close association between SDHB and AMPK (15), it was hypothesized that AMPK may mediate the aerobic glycolysis stimulated by SDHB deficiency. The present study investigated the association between SDHB and the diverse hallmarks of CRC cells, and further explored the regulatory effect of SDHB on AMPK activity. The conclusions drawn from the present study may provide some insight regarding how the Warburg effect is generated in CRC.
Materials and methods
Tissue samples
CRC and corresponding para-carcinoma tissues were obtained from a total of 23 patients (age, 43–51 years old; 16 males and 7 females) who were diagnosed between January 2014 and May 2016 and underwent surgical resection in The Third Xiangya Hospital of Central South University (Changsha, China). A pair of CRC and para-carcinoma tissues were obtained from each patient. All these patients included in the study did not receive radiotherapy and chemotherapy treatments prior to the surgery. All tissue samples were assessed via histopathological evaluation, and stored in liquid nitrogen until utilization. The present study was reviewed and approved by the Ethics Committee of Xiangya School of Medicine (Changsha, China) and all patients enrolled in the study had signed legally effective informed consent forms.
Cell culture and treatments
The human malignant colonic carcinoma cell line, HT-29, was obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were incubated with Eagle's medium (Hyclone, Logan, UT, USA) containing 10% fetal bovine serum (FBS, Hyclone) at 37°C and 5% CO2. A total of four short-hairpin RNAs (shRNAs), including SDHB-homo-305 (shRNA1), SDHB-homo-447 (shRNA2), SDHB-homo-498 (shRNA3) and SDHB-homo-779 (shRNA4) targeting SDHB, were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China). Following growth to 60–80% confluence, 1 µmol/ml shRNAs and 1 µmol/ml scrambled siRNA (Shanghai GenePharma Co., Ltd.) were transfected into HT-29 cells using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer's protocol. Transfection was performed for 12 h at 37°C. An Olympus FluoView 1000 confocal microscope was used to determine transfection efficiency and a western blot analysis was performed to detect the inhibitory effects of shRNAs on SDHB expression. A clone of the human SDHB gene open reading frame was inserted into the enhanced green fluorescent protein plasmid-C1 vector (Shanghai GenePharma Co., Ltd.) to construct an overexpression vector containing SDHB. Transfection of HT-29 cells with overexpression vectors or empty vectors was achieved using Lipofectamine® 2000 (Invitrogen: Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. 5-aminoimidazole-4-carboxamide 1-β-ribofuranoside (AICAR; an AMPK activator) and Compound C (an AMPK inhibitor) were purchased from Selleck Chemicals (Houston, Texas, USA). Cells were treated with 0.1 mM AICAR and 20 um/l Compound C for 12 h to activate and inhibit the activity of AMPK, respectively.
Adenosine 5′-diphosphate (ADP)/ATP ratio assay
Changes in the ADP/ATP ratio were evaluated using the ApoSENSOR™ ADP/ATP ratio assay kit (BioVision Inc., Milpitas, CA, USA) according to the manufacturer's protocol. The luciferase enzyme included in the assay was used to catalyze the generation of light from ATP and luciferin. ADP levels were measured by its conversion to ATP, which was detected using the same reaction. In the present study, the cells (1×105 cells/well) were plated onto the 96-well plate. The reaction mix (100 µl, provided by the assay kit) was added to each well of a 96-well plate. The reaction mix was incubated for 3 h at room temperature. Following removal, Nucleotide Releasing Buffer (50 µl) was added, and the plate was gently agitated for 5 min at room temperature. Light was measured using a luminometer.
Lactic acid measurement
Intracellular lactic acid concentrations were determined using a Lactic Acid assay kit (cat. No. K607-100; BioVision Inc.), according to the manufacturer's protocol. The kit provides a high-sensitive probe that generates fluorescence (excitation/emission wavelength, 535/587 nm) following a reaction with lactic acid. Fluorescence was detected with an Infinite F500 Microplate Reader (Tecan Group, Ltd., Männedorf, Switzerland).
Analysis of cell viability
Cell viability was evaluated using an MTT assay. HT-29 cells (1×105 cells/well) were first seeded in 96-well plates with 100 µl complete medium. Immediately after the transfection and/or treatments with AMPK inhibitor or activator, 20 µl (at 5 mg/ml) MTT reagent (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) was added to each well at 37°C. The medium containing MTT was removed following 4 h, and 150 µl dimethyl sulfoxide was subsequently added to terminate the reaction. A microplate reader was used to examine the absorbance of each well at 570 nm.
Flow cytometry analysis
HT-29 cells (1×106 cells/tube) were stained with Annexin-V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) prior to flow cytometry analysis. The HT-29 cells were washed twice with cold PBS, and incubated with Annexin V-FITC and PI (Beyotime Institute of Biotechnology, Nanjin, China) at room temperature for 15 min in the dark. The collected cells were analyzed using BD FACSCanto™ II clinical software and a BD FACSCanto II flow cytometer (BD Biosciences, Franklin lakes, NJ, USA) to evaluate the apoptosis rate.
Wound healing assay
HT-29 cells (3×105 cells/well) were cultured in 6-well plates at 37°C for ~48 h until 100% cell confluence. Immediately after the transfection and/or treatments with AMPK inhibitor or activator, a 1-ml pipette tip was used to aggregate a ‘scratch-wound’ on the cell monolayer. The culture medium was then replaced with FBS-free medium. Microscope images of the cells were captured immediately following scratching and following 24 h. The cell migration rate was calculated based on the movement of cells from initial placement to the final distance travelled following 24 h. Cell migration rate was calculated using the following equation: (initial distance-final distance)/initial distance ×100.
Cell invasion assay
HT-29 cells (2×105 cells) were seeded in the upper portion of Transwell chambers (Corning Inc., Corning, NY, USA). The upper chamber contained an 8-µm pore and inserts coated with Matrigel (Sigma-Aldrich; Merck KGaA). Complete medium was subsequently added to the lower chamber. Following culture for 24 h, non-invading cells on the upper surface of the membrane were removed with a cotton-tipped swab, while the invading cells on the surface of the lower chamber were stained with 0.1% crystal violet at room temperature for 20 min. The invading cells were quantified by counting 10 random fields of view at ×200 magnification using a light microscope (E200; Nikon Corporation, Tokyo, Japan).
Western blot assay
The tissues and cells used in the present study were lysed with radioimmunoprecipitation assay buffer (Beyotime institute of Biotechnology, Haimen, China) containing phosphatase inhibitors (okadaic acid; cat. no. ab120375; Abcam, Cambridge, UK). Equal quantities (20 µg) of total protein were separated on 10–12% SDS-PAGE gels and then transferred to nitrocellulose membranes. Following blocking with 5% non-fat milk overnight at 4°C, membranes were immunoblotted with primary antibodies overnight at 4°C. These included the anti-SDHB antibody (1:500 dilution; cat. no. ab14714; Abcam), the anti-AMPKα antibody (1:1,000 dilution; cat. no. 5831; CST Biological Reagent, Co., Ltd., Shanghai, China), the anti-phosphorylated (p)-AMPKα (Thr172) antibody (1:1,000 dilution; cat. no. 2535, CST Biological Reagent, Co., Ltd.), anti-p-acetyl-CoA carboxylase (ACC; 1:1,000 dilution; cat. no. sc-271965; Santa Cruz Biotechnology, Inc., Dallas, TX USA), the anti-p21 antibody (1:1,000 dilution; cat. no. ab109199; Abcam), the anti-active caspase-3 antibody (1:1,000 dilution; cat. no. ab109199; Abcam) and the anti-β-actin antibody (1:1,000 dilution; cat. no. ab2302; Abcam). Membranes were washed with Tris-buffered saline-Tween-20, and then incubated with horseradish peroxidase-conjugated anti-IgG secondary antibodies (Sigma-Aldrich; Merck KGaA) at 37°C for 1 h. Blots were visualized using a Beyotime Enhanced Chemiluminescence Kit (Beyotime Institute of Biotechnology) and Fuji medical X-ray film (Fujifilm, Tokyo, Japan).
Tumor xenografts in nude mice
The animal experiments performed in the present study were approved by the Ethics Committee for Animal Research of The Southern Medical University (protocol number: 2011–020; Changsha, China). A total of 9 male nude mice (age, 4–5 weeks; weight, ~25 g) were purchased from the Central Animal Facility of The Southern Medical University. All animals were maintained under environmentally controlled conditions (12-h light/dark cycles, 20–23°C and 50% relative humidity) with access to food and water ad libitum. Animals were divided into three groups: Control group, OV group and OV+Act group (3 mice per group). In the OV group, HT-29 cells (1×106 cells in 20 ml medium), were transfected with SDHB overexpression vectors for 24 h. These cells were subsequently injected into the left side on the back of each mouse. For the OV+Act group, at 12 days following the cell injection, 5 mg/kg (body weight) of AICAR was subcutaneously injected once a day for 6 days. Control mice were injected with HT-29 cells that were transfected with empty vectors. Subsequently, control mice were injected with saline solution instead of AICAR. The tumors were obtained and removed 20 days following subcutaneous injection of AICAR. Tumor volume (V) was calculated as V=(length × width2)/2.
Statistical analysis
The results were analyzed using SPSS 12.0 software (SPSS, Inc., Chicago, IL, USA). Significant differences were calculated using a one-way analysis of variance followed by Scheffe's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Expression profiles of SDHB and AMPK in CRC tissues
Western blotting analysis demonstrated a decrease in SDHB protein expression in CRC tissues when compared with that of para-carcinoma tissues (P<0.01), while AMPK and p-AMPK(Thr172) expression were increased in CRC tissues (both P<0.05; Fig. 1). No significant differences in the ratio of p-AMPK(Thr172)/AMPK were identified between CRC and para-carcinoma tissues (data not shown).
Figure 1.

Expression profiles of SDHB and AMPK in CRC tissues. A total of 23 CRC and 23 para-carcinoma tissues were obtained from patients. Western blotting was performed to detect the protein expression levels of SDHB, AMPK, and p-AMPK. Representative western blotting images are presented. *P<0.05, **P<0.01 vs. para-carcinoma tissues. SDHB, succinate dehydrogenase-B; p-AMPK, phosphorylated adenosine monophosphate activated protein kinase α; CRC, colorectal cancer; T172, Thr172.
SDHB regulates AMPK expression and activity in CRC cells
SDHB in HT-29 CRC cells was overexpressed or silenced in order to assess its regulatory effect on AMPK activity. Compared with the control, cells transfected with the SDHB expression vector exhibited an increase in SDHB protein expression (P<0.01 vs. control; Fig. 2). A total of four shRNAs targeting SDHB were transfected into HT-29 cells individually. SDHB protein levels were significantly decreased following transfection with shRNA2 (P<0.01), shRNA3 (P<0.05) and shRNA4 (P<0.05; Fig. 2) when compared with controls. shRNA2 was selected for SDHB knockdown in further experiments.
Figure 2.
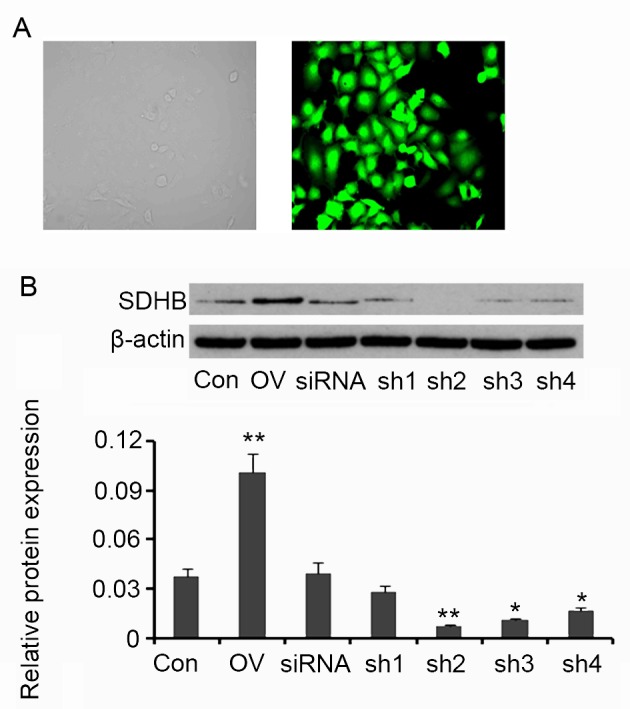
Ectopic overexpression and knockdown of SDHB in CRC HT-29 cells. Four shRNAs targeting SDHB and a scrambled siRNA were transfected into HT-29 cells The human SDHB open reading frame was inserted into an enhanced green fluorescent protein plasmid-C1 vector to construct SDHB overexpression vectors. (A) Fluorescent photographs indicate the transfection efficiency (magnification, ×400). (B) Western blot analyses were performed to detect SDHB protein expression in HT-29 cells. *P<0.05, **P<0.01 vs. control group. SDHB, succinate dehydrogenase-B; CRC, colorectal cancer; shRNA, short-hairpin RNA; Con, control; OV, SDHB expression vector; sh1, SDHB-homo-305; sh2, SDHB-homo-447; sh3, SDHB-homo-498; sh4, SDHB-homo-779.
Western blotting analysis demonstrated that upregulation of SDHB using the overexpression vector was associated with reduced AMPK and p-AMPK(Thr172) protein levels, when compared with the group that received no treatment (both P<0.05; Fig. 3). The subsequent addition of AICAR increased the protein level of p-AMPK(Thr172) but did not affect AMPK levels. HT-29 cells treated with AICAR alone exhibited an increase in p-AMPK(Thr172) expression when compared with the control (P<0.05). shRNA-mediated suppression of SDHB stimulated the upregulation of AMPK and p-AMPK(Thr172) when compared with controls (both P<0.05). Treatment of HT-29 cells with Compound C alone resulted in a decrease in p-AMPK(Thr172) expression (P<0.05) compared with control. The p-AMPK(Thr172)/AMPK ratio was not affected by the ectopic over-expression of SDHB. The addition of AICAR, alone or in combination with SDHB overexpression, significantly elevated the p-AMPK(Thr172)/AMPK ratio (P<0.05), indicating an increase in AMPK activity. SDHB knockdown also increased the p-AMPK(Thr172)/AMPK ratio (P<0.05 vs. controls; Fig. 3). Cells treated with Compound C demonstrated a significant reduction in the p-AMPK(Thr172)/AMPK ratio, irrespective of SDHB knockdown (P<0.05). ACC is a direct downstream target of AMPK; therefore, the level of ACC phosphorylation is used to evaluate AMPK function (16). The overexpression of SDHB or the inhibition of AMPK activity resulted in a significant decrease in p-ACC protein expression levels compared with the controls (both P<0.05). Conversely, SDHB knockdown or an enhanced AMPK activity demonstrated a significant increase in p-ACC levels (P<0.05 vs. controls; Fig. 3).
Figure 3.

Inhibitory effect of SDHB on AMPK in CRC cells. SDHB was overexpressed or knocked down in HT-29 CRC cells. A total of 0.1 mM AICAR (an AMPK activator) and 20 µm/l Compound C (an AMPK inhibitor) were added to activate and inhibit the activity of AMPK, respectively. Western blot analysis was performed to detect the protein levels of SDHB, AMPK p-AMPK(Thr172) and p-ACC in HT-29 cells. *P<0.05 and #P<0.05 vs. the respective control group. SDHB, succinate dehydrogenase-B; p-AMPK, phosphorylated adenosine monophosphate activated protein kinase α; CRC, colorectal cancer; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; OV, SDHB expression vector; Act, AMPK activator; shR, shRNA targeting SDHB; Inh, AMPK inhibitor; ACC, anti-p-acetyl-CoA carboxylase.
Effect of SDHB and AMPK on aerobic glycolysis in CRC cells
Lactic acid is produced as a by-product of aerobic glycolysis, but not aerobic respiration. Therefore, the yield of lactic acid can be used to evaluate the level of glycolysis activity (17,18). The results of the present study demonstrated that treatment with an AMPK activator inhibited lactic acid production in cells when compared with controls (P<0.05; Fig. 4). SDHB overexpression was also identified to exert the same effect (P<0.05 vs. controls). The suppression of SDHB and/or AMPK activity resulted in a significant increase in the production of lactic acid in cells (all P<0.05; Fig. 4). The combination of SDHB overexpression and AMPK inhibition did not influence the production of lactic acid. Treatment with an AMPK agonist did not abrogate the effect of SDHB knockdown on lactic acid production, as the levels remained significantly higher when compared with the controls (P<0.05; Fig. 4).
Figure 4.

Effect of SDHB and AMPK on aerobic glycolysis in CRC cells. SDHB was overexpressed or knocked down in HT-29 CRC cells. A total of 0.1 mM AICAR (an AMPK activator) and 20 µm/l Compound C (an AMPK inhibitor) were added to cells to activate and inhibit the activity of AMPK, respectively. Intracellular lactic acid levels were assessed using a lactic acid assay kit. *P<0.05 vs. the control group. SDHB, succinate dehydrogenase-B; AMPK, adenosine monophosphate activated protein kinase; CRC, colorectal cancer; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; OV, SDHB expression vector; shR, shRNA targeting SDHB; Act, AMPK activator; Inh, AMPK inhibitor.
Regulation of the ADP/ATP ratio in CRC cells by SDHB
The ADP/ATP ratio significantly influences AMPK activity in cells (17,18). Therefore, the present study investigated whether the regulation of AMPK activity via SDHB may be dependent on the ADP/ATP ratio. As demonstrated in Fig. 5, SDHB overexpression and knockdown resulted in a significant reduction and elevation of the ADP/ATP ratio, respectively when compared with controls (both P<0.05).
Figure 5.

Effect of SDHB on the ADP/ATP ratio in CRC cells. SDHB was overexpressed or knocked down in CRC HT-29 cells. The ADP/ATP ratio was assessed using the ApoSENSOR™ ADP/ATP ratio assay kit. *P<0.05 vs. control group. SDHB, succinate dehydrogenase-B; ATP, adenosine 5′-triphosphate; ADP, adenosine 5′-diphosphate; CRC, colorectal cancer.
Regulation of the diverse hallmarks of CRC cells through SDHB-mediated AMPK modulation
Results from the MTT assay demonstrated that overexpression of SDHB in HT-29 cells is associated with a significant decrease in cell viability (P<0.05 vs. controls; Fig. 6A), and that the subsequent addition of AICAR reverses this effect. SDHB silencing or the addition of AICAR significantly increased the viability of HT-29 cells when compared with the control (both P<0.05). SDHB knockdown in combination with Compound C treatment exerted no effect on cell viability. However, treatment with Compound C alone significantly inhibited cell viability when compared with controls (P<0.05; Fig. 6A). The effect of SDHB on the apoptosis rate of HT-29 cells using flow cytometry analysis was then determined. The apoptosis rate was significantly increased when HT-29 cells overexpressed SDHB compared with controls (P<0.01; Fig. 6B). Subsequent treatment with AICAR reversed the increase in apoptosis rate; however, the rate remained significantly greater than that observed in controls (P<0.05). AICAR also significantly suppressed the apoptosis rate of HT-29 cells that were not transfected with the SDHB expression vector (P<0.05 vs. controls). SDHB silencing in HT-29 cells significantly reduced the apoptosis rate (P<0.05); however co-treatment with Compound C abrogated this effect. The addition of Compound C in HT-29 cells, in which SDHB was not silenced, significantly enhanced the rate of apoptosis (P<0.05 vs. controls; Fig. 6B).
Figure 6.

SDHB-mediated regulation of the diverse hallmarks of CRC cells through AMPK modulation. SDHB was overexpressed or knocked down in CRC HT-29 cells. A total of 0.1 mM AICAR (an AMPK activator) and 20 µm/l Compound C (an AMPK inhibitor) were added to activate and inhibit the activity of AMPK, respectively. Cell viability and apoptosis were evaluated using (A) MTT and (B) flow cytometry analysis, respectively. (C) Western blot analysis was performed to detect p21/WAF1 and caspase-3 expression. Cell migration and invasion were evaluated using (D) wound healing (magnification, ×200; Con indicated the 0 h time point) and (E) invasion (magnification, ×400) assays. *P<0.05, **P<0.01, #P<0.05 and ##P<0.01 vs. the respective control group. SDHB, succinate dehydrogenase-B; CRC, colorectal cancer; AMPK, adenosine monophosphate activated protein kinase; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; OV, SDHB expression vector; Act, AMPK activator; shR, shRNA targeting SDHB; Inh, AMPK inhibitor; FITC, Annexin-V-fluorescein isothiocyanate.
p21/WAF1 serves an inhibitory role in the regulation of cell proliferation through the induction of cell cycle arrest (19). A key step in the apoptosis signaling pathway is the activation of caspase-3. This process is dependent on the cleavage of the inactive form of caspase-3 to the active form. Increased levels of activated caspase-3 are therefore associated with the promotion of apoptosis (19). In the present study, the levels of p21/WAF1 and activated caspase-3 in HT-29 cells were assessed to investigate the effect of SDHB on CRC cell proliferation and apoptosis. Induced expression of SDHB in HT-29 cells significantly upregulated p21/WAF1 (P<0.05) and activated caspase-3 (P<0.01) expression when compared with controls (Fig. 6C). Despite reducing the effects of SDHB overexpression on cell proliferation, the levels of p21/WAF1 and activated caspase-3 remained significantly higher following AICAR treatment compared with controls (both P<0.05; Fig. 6C). Cells that were not subjected to SDHB overexpression, demonstrated a significant reduction in p21/WAF1 levels following treatment with AICAR (P<0.05 vs. controls). However, the level of activated caspase-3 remained the same. Silencing of SDHB expression with shRNA resulted in a significant decrease in p21/WAF1 (P<0.05) and activated caspase-3 (P<0.05) expression, regardless of additional treatment with Compound C. Compound C treatment alone significantly upregulated p21/WAF1 (P<0.05) and activated caspase-3 levels (P<0.05) in HT-29 cells compared with controls (Fig. 6C).
HT-29 cell migration and invasion were then evaluated by wound healing and cell invasion assays, respectively. SDHB upregulation significantly attenuated cell migration (P<0.05 vs. controls; Fig. 6D); however, subsequent AICAR treatment reversed the observed inhibitory effects. AICAR treatment or SDHB knockdown significantly promoted cell migration (both P<0.05 vs. controls). Compound C moderately attenuated the increase in migration of cells treated with SDHB shRNA (Fig. 6D). Cell invasion was also significantly attenuated by the upregulation of SDHB (P<0.05 vs. controls; Fig. 6E). Subsequent AICAR treatment did not restore the invasiveness of the cells. AICAR treatment and/or SDHB knockdown instead promoted cell invasion (all P<0.05 vs. controls). The subsequent addition of Compound C led to a moderate decrease in the cell invasiveness following knockdown of SDHB (Fig. 6E).
SDHB inhibits CRC growth in nude mice through AMPK downregulation
A tumor xenograft model involving nude mice was developed to assess the inhibitory effect of SDHB on CRC growth in vivo. Western blotting demonstrated that SDHB upregulation in xenografted tumors was associated with a significant decrease in AMPK and p-AMPK(Thr172) expression (P<0.05 vs. controls; Fig. 7A). The subsequent injection of AICAR restored protein levels to that of the controls. Upregulation of SDHB significantly decreased the volume of xenografted tumors (P<0.05 vs. controls), while the injection of AICAR abrogated this effect (Fig. 7B).
Figure 7.

Effect of SDHB on CRC growth in nude mice and the role of AMPK. HT29 cells (1×106) were transfected with SDHB overexpression vectors or empty vectors, and were then injected into the left side on the back of each mouse (3 mice per group). At 12 days following injection, 5 mg/kg (body weight) AICAR (an AMPK inhibitor) was subcutaneously injected once a day for 6 days. The tumors were obtained and removed 20 days following the injection of cells. (A) Western blotting was performed to detect SDHB, AMPK and p-AMPK(Thr172) expression. (B) Images of the xenografted tumors are presented and tumor volumes were ascertained. *P<0.05, #P<0.05 and &P<0.05 vs. the respective control group. SDHB, succinate dehydrogenase-B; CRC, colorectal cancer; AMPK, adenosine monophosphate activated protein kinase; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; OV, SDHB expression vector; Act, AMPK activator.
Discussion
SDHB dysfunction and downregulation has been closely associated with the development of cancer. Individuals with germline mutations in the genes encoding SDHB are predisposed to paraganglioma, pheochromocytoma, renal cell carcinoma and gastrointestinal stromal tumors (8,9). In patients with recurrent nasopharyngeal carcinoma, low SDHB expression rates are associated with reduced overall survival (20). A previous study involving SDHB-transfected cells and cDNA microarray assays, demonstrated that the majority of differentially expressed genes are associated with cell cycle control and cell proliferation (21). The present study demonstrated that SDHB was downregulated in CRC tissues when compared to para-carcinoma tissues, indicating that SDHB may be involved in the development of CRC. Cardaci et al (7) investigated the changes in metabolism following SDHB ablation in immortalized mouse kidney cells. Through metabolomics screening and isotope-labeling, it was established that cellular loss of SDHB is sufficient to inhibit the tricarboxylic acid (TCA) cycle and promote the Warburg-like bioenergetic features of aerobic glycolysis in proliferating cells (7). In addition, SDHB silencing induced via siRNA promoted the occurrence of characteristic features of the tumor phenotype, such as the shift from respiration to glycolysis (22). Microarray analysis identified >400 genes that are influenced by SDHB silencing, and several of these genes were associated with the TCA cycle and tumor development (22). The results of these studies therefore suggest that a decrease in SDHB in CRC tissues may facilitate aerobic glycolysis and cancer progression.
AMPK, an established regulator of metabolic pathways, serves an inhibitory role in aerobic glycolysis and participates in the regulation of the diverse hallmarks of cancer (13,14,23,24). The present study identified that AMPK and p-AMPK(Thr172) were increased in CRC tissues; however, the ratio of p-AMPK(Thr172)/AMPK remained unchanged. The authors hypothesized that AMPK exerts an inhibitory effect on aerobic glycolysis in CRC. In accordance with this, in vitro experiments demonstrated that AMPK negatively regulated lactic acid production in CRC cells, further indicating that aerobic glycolysis was affected.
Further in vitro experiments demonstrated that knockdown of SDHB increased AMPK and p-AMPK expression, while overexpression of SDHB decreased the expression of AMPK alone. This indicated that AMPK and p-AMPK(Thr172) protein levels may be negatively regulated by SDHB. However, the regulatory effect of SDHB on AMPK activity may be condition-dependent as SDHB knockdown increased the expression of p-AMPK, whereas SDHB overexpression demonstrated no effect. The present study demonstrated that the level of p-ACC was negatively correlated with the level of SDHB. This suggested that SDHB exerted a negative effect on AMPK, as ACC is a direct downstream target (16). It is well established that AMPKα function is influenced by AMP/ATP and ADP/ATP ratios. AMPKα is activated in response to a shortage of ATP in conditions of metabolic stress (11–13). As SDH is an essential enzyme involved in the citric acid cycle, SDHB dysfunction inhibits the production of ATP (25,26). Previous studies have determined that a lower concentration of ATP is present in tumor cells than in their normal cellular counterparts (25,26). The present study demonstrated that SDHB knockdown increased the ADP/ATP ratio, whereas ectopic overexpression of SDHB was associated with a decreased ADP/ATP ratio. These results indicate that SDHB negatively regulates the ADP/ATP ratio in CRC cells, which may be important for the SDHB-mediated regulation of AMPK expression and activity.
The results of the current study demonstrated that oncogenic actions triggered by SDHB deficiency may be closely associated with an increase in AMPK function. Suppression of AMPKα activity inhibited the promoting effects of SDHB downregulation on CRC proliferation, migration and invasion. Although SDHB upregulation inhibited the proliferation and invasion of CRC cells and decreased AMPK expression, the addition of AICAR reversed this effect. This phenomenon was also observed in xenograft tumors in nude mice. Therefore, downregulation of SDHB in CRC may be dependent on AMPK activation. However, this is contradictory to the inhibitory role of AMPK in aerobic glycolysis. The stimulatory effects of SDHB deficiency on CRC proliferation and invasion may therefore be associated with AMPK functions that exclude those associated with the modulation of aerobic glycolysis. The regulatory roles of AMPKα in cancer are diverse. In the majority of cases, increased AMPKα activity is associated with a poor prognosis, as AMPK provides cells with a selective advantage to proliferate and survive by activating a number of signaling molecules, including eukaryotic elongation factor 2 kinase, NUAK1/2 and microtubule affinity regulating kinases (23,24). However, AMPKα may also function as a tumor suppressor through the negative regulation of aerobic glycolysis, mTOR and HIF-1α signaling pathways (12). Collectively, AMPK exerts a duality of functions that are either pro- or anti-tumorigenic, depending upon the context. Further studies are required to investigate the mechanisms by which AMPK mediates SDHB deficiency-induced proliferation and invasion of CRC cells.
In conclusion, the present study demonstrated that SDHB may function as a negative regulator of AMPKα in CRC. AMPKα mediated SDHB deficiency-induced proliferation, migration and invasion of CRC cells. It was also determined that the utilization of aerobic glycolysis, that arises from the deficiency of SDHB, is unlikely to be a result of AMPK function. Further studies are warranted to elucidate the mechanism by which SDHB regulates aerobic glycolysis.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant no. 81402010).
References
- 1.Li M, Song LH, Yue GG, Lee JK, Zhao LM, Li L, Zhou X, Tsui SK, Ng SS, Fung KP, et al. Bigelovin triggered apoptosis in colorectal cancer in vitro and in vivo via upregulating death receptor 5 and reactive oxidative species. Sci Rep. 2017;7:42176. doi: 10.1038/srep42176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sprowl-Tanio S, Habowski AN, Pate KT, McQuade MM, Wang K, Edwards RA, Grun F, Lyou Y, Waterman ML. Lactate/pyruvate transporter MCT-1 is a direct Wnt target that confers sensitivity to 3-bromopyruvate in colon cancer. Cancer Metab. 2016;4:20. doi: 10.1186/s40170-016-0159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taniguchi K, Sugito N, Kumazaki M, Shinohara H, Yamada N, Matsuhashi N, Futamura M, Ito Y, Otsuki Y, Yoshida K, et al. Positive feedback of DDX6/c-Myc/PTB1 regulated by miR-124 contributes to maintenance of the Warburg effect in colon cancer cells. Biochim Biophys Acta. 2015;1852:1971–1980. doi: 10.1016/j.bbadis.2015.06.022. [DOI] [PubMed] [Google Scholar]
- 4.Parra-Bonilla G, Alvarez DF, Al-Mehdi AB, Alexeyev M, Stevens T. Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2010;299:513–522. doi: 10.1152/ajplung.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee N, Kim D. Cancer metabolism: Fueling more than just growth. Mol Cells. 2016;39:847–854. doi: 10.14348/molcells.2016.0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu Z, Xia J, Xu H, Frech I, Tricot G, Zhan F. NEK2 promotes aerobic glycolysis in multiple myeloma through regulating splicing of pyruvate kinase. J Hematol Oncol. 2017;10:17. doi: 10.1186/s13045-017-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cardaci S, Zheng L, MacKay G, van den Broek NJ, MacKenzie ED, Nixon C, Stevenson D, Tumanov S, Bulusu V, Kamphorst JJ, et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat Cell Biol. 2015;17:1317–1326. doi: 10.1038/ncb3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saxena N, Maio N, Crooks DR, Ricketts CJ, Yang Y, Wei MH, Fan TW, Lane AN, Sourbier C, Singh A, et al. SDHB-deficient cancers: The role of mutations that impair iron sulfur cluster delivery. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuroda N, Yorita K, Nagasaki M, Harada Y, Ohe C, Jeruc J, Raspollini MR, Michal M, Hes O, Amin MB. Review of succinate dehydrogenase-deficient renal cell carcinoma with focus on clinical and pathobiological aspects. Pol J Pathol. 2016;67:3–7. doi: 10.5114/pjp.2016.59227. [DOI] [PubMed] [Google Scholar]
- 10.Mason EF, Sadow PM, Wagner AJ, Remillard SP, Flood TA, Belanger EC, Hornick JL, Barletta JA. Identification of succinate dehydrogenase-deficient bladder paragangliomas. Am J Surg Pathol. 2013;37:1612–1618. doi: 10.1097/PAS.0b013e318293d83c. [DOI] [PubMed] [Google Scholar]
- 11.Marín-Aguilar F, Pavillard LE, Giampieri F, Bullón P, Cordero MD. Adenosine Monophosphate (AMP)-activated protein kinase: A new target for nutraceutical compounds. Int J Mol Sci. 2017;18:pii: E288. doi: 10.3390/ijms18020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ata R, Antonescu CN. Integrins and cell metabolism: An intimate relationship impacting cancer. Int J Mol Sci. 2017;18:pii: E189. doi: 10.3390/ijms18010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q, Qin Y, Wei P, Lian P, Li Y, Xu Y, Li X, Li D, Cai S. Gas1 inhibits metastatic and metabolic phenotypes in colorectal carcinoma. Mol Cancer Res. 2016;14:830–840. doi: 10.1158/1541-7786.MCR-16-0032. [DOI] [PubMed] [Google Scholar]
- 14.Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, Mourón S, Muñoz J, Gómez-López G, Jimenez-Renard V, Mulero F, et al. Targeting tumor mitochondrial metabolism overcomes resistance to antiangiogenics. Cell Rep. 2016;15:2705–2718. doi: 10.1016/j.celrep.2016.05.052. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Liu T, Zhang S, Zhou J, Wang Y, Di W. Succinate dehydrogenase subunit B inhibits the AMPK-HIF-1α pathway in human ovarian cancer in vitro. J Ovarian Res. 2014;7:115. doi: 10.1186/PREACCEPT-8784219211432789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang YY, Attané C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V, et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight. 2017;2:e87489. doi: 10.1172/jci.insight.87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devic S. Warburg effect - a consequence or the cause of carcinogenesis? J Cancer. 2016;7:817–822. doi: 10.7150/jca.14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang D, Fei Q, Li J, Zhang C, Sun Y, Zhu C, Wang F, Sun Y. 2-deoxyglucose reverses the promoting effect of insulin on colorectal cancer cells in vitro. PLoS One. 2016;11:e0151115. doi: 10.1371/journal.pone.0151115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Festuccia C, Gravina GL, D'Alessandro AM, Muzi P, Millimaggi D, Dolo V, Ricevuto E, Vicentini C, Bologna M. Azacitidine improves antitumor effects of docetaxel and cisplatin in aggressive prostate cancer models. Endocr Relat Cancer. 2009;16:401–413. doi: 10.1677/ERC-08-0130. [DOI] [PubMed] [Google Scholar]
- 20.Dai Z, Pan S, Chen C, Cao L, Li X, Chen X, Su X, Lin S. Down-regulation of succinate dehydrogenase subunit B and up-regulation of pyruvate dehydrogenase kinase 1 predicts poor prognosis in recurrent nasopharyngeal carcinoma. Tumour Biol. 2016;37:5145–5152. doi: 10.1007/s13277-015-4107-6. [DOI] [PubMed] [Google Scholar]
- 21.Zhang D, Wang W, Xiang B, Li N, Huang S, Zhou W, Sun Y, Wang X, Ma J, Li G, et al. Reduced succinate dehydrogenase B expression is associated with growth and de-differentiation of colorectal cancer cells. Tumour Biol. 2013;34:2337–2347. doi: 10.1007/s13277-013-0781-4. [DOI] [PubMed] [Google Scholar]
- 22.Cervera AM, Apostolova N, Crespo FL, Mata M, McCreath KJ. Cells silenced for SDHB expression display characteristic features of the tumor phenotype. Cancer Res. 2008;68:4058–4067. doi: 10.1158/0008-5472.CAN-07-5580. [DOI] [PubMed] [Google Scholar]
- 23.Monteverde T, Muthalagu N, Port J, Murphy DJ. Evidence of cancer-promoting roles for AMPK and related kinases. FEBS J. 2015;282:4658–4671. doi: 10.1111/febs.13534. [DOI] [PubMed] [Google Scholar]
- 24.Lipovka Y, Konhilas JP. AMP-activated protein kinase signalling in cancer and cardiac hypertrophy. Cardiovasc Pharm Open Access. 2015;4:pii: 154. doi: 10.4172/2329-6607.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreira JD, Hamraz M, Abolhassani M, Bigan E, Pérès S, Paulevé L, Nogueira ML, Steyaert JM, Schwartz L. The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect. Metabolites. 2016;6:pii: E33. doi: 10.3390/metabo6040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lustgarten MS, Jang YC, Liu Y, Qi W, Qin Y, Dahia PL, Shi Y, Bhattacharya A, Muller FL, Shimizu T, et al. MnSOD deficiency results in elevated oxidative stress and decreased mitochondrial function but does not lead to muscle atrophy during aging. Aging Cell. 2011;10:493–505. doi: 10.1111/j.1474-9726.2011.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]