

Abstract
Previous studies have demonstrated that ginkgol C17:1 significantly inhibits human liver cancer cells and enhances the anticancer activity of cisplatin in vivo and in vitro. However, the mechanism and biological function of ginkgol C17:1 on cells undergoing chemotherapy remain unclear. The aim of the present study was to determine the antitumor activity and mechanism of ginkgol C17:1 in combination with cisplatin in human hepatoblastoma HepG2 cells. The green fluorescent protein (GFP)-light chain 3 (LC3) adenovirus was transfected into HepG2 cells and autophagic flux was determined using fluorescence microscopy. Western blot analysis was also conducted to measure the expression of proteins associated with apoptosis, autophagy and their associated signaling pathways. Compared with the control group, autophagic flux and nucleus aberration rates were significantly increased (P<0.05), and the expression of proteins associated with autophagy and apoptosis were increased in the groups treated with cisplatin or ginkgol C17:1, respectively. However, following co-treatment with ginkgol C17:1 and cisplatin, the autophagic flux and the expression of autophagy proteins decreased; however, the nucleus aberration rate and apoptosis protein expression significantly increased (P<0.05) compared with the group treated with cisplatin alone. Additionally, the signaling pathways of autophagy and apoptosis were also activated following treatment with cisplatin, alone and in combination with ginkgol C17:1. Taken together, these results indicate that ginkgol C17:1 inhibits cisplatin-induced autophagy via AMP-activated protein kinase/ULK1signaling and increases cisplatin-induced apoptosis in HepG2 cells via the phosphoinositide 3-kinase/Akt/mechanistic target of rapamycin pathway.
Keywords: ginkgol C17:1, autophagy, apoptosis, cisplatin, HepG2 cells
Introduction
Liver cancer is a leading cause of cancer-associated mortality and morbidity worldwide, and consists of hepatocellular carcinoma (HCC) and hepatoblastoma (HB) (1,2). HCC and HB are two different types of liver cancer with their own distinctive cytological features (3). HepG2 was previously considered to be a HCC cell line; however, in 2009, López-Terrada et al (4) identified that HepG2 is a HB-derived cell line. The misidentification of HepG2 cells remains widespread. The properties of the HepG2 cell line significantly differ from those of HCC (5); thus, HepG2 is not an appropriate cell line to use to investigate HCC but can be used in liver cancer research as a HB cell line (6). Thus, the aim of the present study was to investigate the mechanism of HepG2 cells to develop a potential novel method of treating HB.
Although the overall incidence of HB is rare, it is the most common type of liver tumor diagnosed in children (7). Complete resection of the tumor via surgery often leads to remission in patients with liver cancer; however, the majority of patients are unable to undergo tumor resection at diagnosis, as certain tumors are accompanied by familial hepatoblastomas or adrenocortical tumors, for which surgical resection is not the best treatment option (8). In such situations, patients undergo chemotherapy as a first-line treatment. Cisplatin is commonly used as a chemotherapeutic drug to treat patients with cancer, as it is effective against cancer cells derived from solid tumors. It is a DNA-damaging anticancer agent and induces apoptosis in cancer cells (9). However, long-term chemotherapy with cisplatin may induce resistance and reduce the sensitivity of cancer cells to apoptosis, which is a major cause of the uncontrolled progression of various types of cancer (10). Due to the development of chemoresistance, the prognosis of patients with advanced-stage cancer is particularly poor (11). Current therapeutic strategies of treating intermediate and advanced liver cancer use a combination of cisplatin and other chemotherapy agents, including paclitaxel, gemcitabine and cyclophosphamide. However, the mechanism underlying their anticancer or apoptosis-promoting effects remains unclear and numerous studies have indicated that autophagy may serve a potential role in the anticancer effect of chemotherapeutic reagents (12,13).
Autophagy is a process of cell self-destruction that occurs in all eukaryotic cells. Damaged molecules and organelles are absorbed by autophagosomes and subsequently degraded by lysosomal hydrolases to recycle energy. Previous studies have demonstrated that autophagy acts as a double-edged sword in the initiation, development and metastasis of cancer, depending upon the stage of autophagy (14,15). For example, in early stages of autophagy, the initiation and development of cancer are stimulated, while cancer cell death begins to occur during later stages of autophagy. Autophagy serves an important antitumor role (16) but also protects tumor cells against stress as an adaptive response (17,18). Autophagy dysregulation has been associated with numerous diseases, including cancer (19); however, the role of autophagy in the development of cancer chemoresistance remains unknown. The inhibition of autophagy significantly enhances the cytocidal activity of combinatorial treatments, indicating that the induction of autophagy is a compensatory response to therapeutic stress (20).
The phosphoinositide 3-kinase (PI3K)/Akt/mechanistic target of rapamycin (mTOR) signaling pathway is a key regulator of the physiological cellular processes associated with proliferation, differentiation, autophagy, apoptosis, motility and metabolism (21). During energy and oxygen deficiency, the PI3K/Akt signaling pathway negatively regulates autophagy by mediating mTOR expression. Furthermore, AMP-activated protein kinase (AMPK), which is a serine/threonine protein kinase, positively regulates autophagy by phosphorylating ULK1 at specific sites (22). By contrast, the combination of ULK1 and mTOR inhibits ULK1 activity, which inhibits the interaction between ULK1 and AMPK. Furthermore, it has been demonstrated that AMPK phosphorylates the tuberous sclerosis complex, thus inhibiting the mTOR pathway (23). However, the detailed mechanisms of different anticancer drug treatments, all of which involve an association between autophagy and apoptosis, remain poorly understood.
Gingko is the only living species of the gymnospermae group and may exhibit considerable medicinal benefits (24). Ginkgo biloba extract (EGb) is one of the most commonly administered therapeutic agents and is primarily used to promote blood circulation and the dilation of blood vessels. Chen et al (25) and the results of our previous study (26), demonstrated that EGb effectively inhibits cell division and induces apoptosis in the hepatoma cell line SMMC-7721. However, to the best of our knowledge, the effect of ginkgol on cisplatin-induced autophagy in HB has not yet been investigated. The aim of the present study was to investigate the effect of ginkgol C17:1 combined with chemotherapy on autophagy and apoptosis in HepG2 cells.
Materials and methods
Reagents
NH4Cl was purchased from Sinopharm Chemical reagent Co., Ltd. (Shanghai, China). MTT (cat. no. M5655), Hoechst 33342 (cat. no. B2261), cisplatin (cat. no. P4394) and 3-methyladenine (3-MA; cat. no. M9281) were purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Penicillin and streptomycin were purchased from Harbin Pharmaceutical Group Co., Ltd. (Harbin, China). The Ad-mRFP-GFP-LC3 adenovirus was obtained from the Hanheng Biotechnology Company (Shanghai, China). Mouse monoclonal antibody (mAb) against β-actin (cat. no. sc-47778) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Rabbit mAbs against Beclin-1 (cat. no. 3495), microtubule-associated protein 1 light chain 3 (LC3) I/II (cat. no. 12741), mTOR (cat. no. 2983), phosphorylated (p)-mTOR (Ser2448; cat. no. 5536), p-ULK1 (Ser555; cat. no. 5869), PI3K (cat. no. 4249), p-PI3K (Tyr458; cat. no. 4228), B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax; cat. no. 5023) and cleaved caspase-3 (cat. no. 9661) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Horseradish peroxidase (HRP)-labeledanti-mouse (cat. no. A0216) and HRP-conjugated anti-rabbit (cat. no. A0208) secondary antibodies were purchased from Beyotime Institute of Biotechnology (Haimen, China). Compound C (cat. no. ab120843), rapamycin (cat. no. ab120224), rabbit mAb SQSTM1/p62 (cat. no. ab91526) and rabbit anti-ULK1 (cat. no. ab128859) were purchased from Abcam (Cambridge, UK). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), trypsin-EDTA solution, rabbit anti-Akt (cat. no. IM001-0359), anti-p-Akt1/2/3 (Tyr315/316/312; cat. no. IM001-0270), anti-c-JUN N-terminal kinase (JNK; cat. no. IM001-0504), anti-p-JNK (Tyr185; cat no. IM001-0272) and anti-Bcl-2 (cat. no. IM001-0363) were purchased from Shanghai ExCell Biology, Inc. (Shanghai, China). Mouse anti-AMPKα1 (cat. no. RLM3361) and anti-p-AMPKα1/2 (Thr172; cat. no. RLM0575) were purchased from Suzhou Ruiying-Runze Trading Co., Ltd. (Suzhou, China). Skimmed milk was purchased from Bright Dairy & Food Co., Ltd. (Harbin, China). Ginkgol C17:1 (>96.5%, as determined by high-performance liquid chromatography) was obtained from the Laboratory of the Food and Biological Engineering School at Jiangsu University.
Cell line and culture
The human HB HepG2 cell line was obtained from the Institute of Cell Biology at the Chinese Academy of Sciences (Shanghai, China). HepG2 cells were cultured in DMEM supplemented with 10% FBS, penicillin and streptomycin (10 mg/l) at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. The medium was replenished every 2 days and cells were maintained at sub-confluence.
MTT assay
HepG2 cells were seeded in a 96-well plate (5×103 cells/well) in a humidified atmosphere with 5% CO2 at 37°C and treated with ginkgol C17:1 alone (0, 10, 20, 40, 80 and 160 µg/ml), cisplatin alone (0, 1, 2, 4, 8 and 16 µg/ml), or ginkgol (0, 20, 40 and 80 µg/ml) in combination with cisplatin (2 µg/ml) for 24 h. Subsequently, 10 µl MTT (5 mg/ml) was added to each well and cells were incubated for an additional 4–6 h. Following removal of the supernatant, dimethyl sulfoxide (100 µl/well) was added to dissolve the blue formazan crystals converted from MTT by living cells. Cell viability was assessed using a microplate reader at an optical density of 490 nm.
Analysis of cell autophagy
HepG2 cells were seeded in a 24-well plate (5×104 cells/well) in a humidified atmosphere with 5% CO2 at 37°C for 12 h. Cells were infected with Ad-mRFP-GFP-LC3 adenovirus for 12 h (MOI=50) and the DMEM medium was replaced every 2 h. This process was performed as the weakening of GFP expression is considered to indicate the formation of autophagy-lysosomes and is therefore able to reflect the level of cell autophagic flux. After 10–12 h, the cells were treated with ginkgol C17:1 (0, 20, 40 and 80 µg/ml) and/or cisplatin (2 µg/ml) for 24 h. Cell fixation was performed using 4% paraformaldehyde at room temperature for 15 min. Subsequent to fixation and washing to remove the excess water, sealing was performed using drops of Aqueous Mounting Medium (Beyotime Institute of Biotechnology; cat. no. P0126) were applied to each of the treated wells. The treated cells were immobilized for 20 min at room temperature. LC3 is a marker protein of autophagy, and LC3 proteins cluster together to form LC3 puncta. In the present study, LC3 puncta (fluorescing green) were observed under a fluorescence microscope at a magnification of ×200. The numbers of LC3 puncta (green, selected to demonstrate the intensity of autophagy, and not the autophagy flux) from five different wells that underwent the same treatment were calculated using ImageJ software 1.48u (National Institute of Health, Bethesda, MD, USA) (19,27).
Hoechst 33342 staining
HepG2 cells were seeded in a 24-well plate (5×104 cells/well) in a humidified atmosphere with 5% CO2 at 37°C. Following 24 h treatment, cells were fixed with 4% paraformaldehyde for 2 h at room temperature. Cells were counterstained with Hoechst 33342 reagent (5 µg/ml) for 15 min at room temperature and stained cell nuclei were observed using a fluorescence microscope. Subsequently, the amount of nuclei aberration from five different wells that underwent the same treatment was calculated using ImageJ software (National Institute of Health).
Protein extraction
HepG2 cells (1×106 cells/well) were cultured with ginkgol C17:1 (0, 20, 40 or 80 µg/ml) or cisplatin (2 µg/ml) with/without NH4Cl (an upstream inhibitor of autophagy; 0.535 mg/ml) or 3-MA (a downstream inhibitor of autophagy; 0.75 mg/ml), rapamycin (an mTOR inhibitor; 100 ng/ml) or compound C (an AMPK inhibitor; 8 µg/ml) for 24 h in 6-well plates until they reached ~80% confluence. Subsequently, cells were lysed in lysis buffer [50 mM Tris, 150 mM NaCl, 1 mM EDTA and 1% Triton X-100 (pH 7.4)], washed 3 times with cold PBS and treated with 1 mM phenylmethylsulfonyl fluoride (Shanghai Bogoo Biotechnology Co., Ltd., Shanghai, China) for 30 min on ice. Following transfer into an Eppendorf tube (Corning Incorporated, Corning, NY, USA), proteins were centrifuged at 12,000 × g for 5 min at 4°C. Finally, the supernatant was obtained as the whole cell protein extract.
Western blot analysis
Total protein was quantified using a bicinchoninic acid assay and proteins (5 µg/lane) underwent electrophoresis on 10% (for proteins with a molecular weight <60 kDa) or 12% (for proteins with a molecular weight >60 kDa) SDS polyacrylamide gels. Subsequently, the proteins were transferred onto PVDF membranes (Bio-Rad Laboratories, Inc.). PVDF membranes were initially blocked with 5% skimmed milk for 1 h at room temperature. Subsequently, proteins were incubated with the following primary antibodies: β-actin, Beclin-1, LC3I/II, p62, mTOR, p-mTOR, ULK1, p-ULK1, PI3K, p-PI3K, Bcl-2, Bax and cleaved caspase-3 (dilution, 1:1,000), at 4°C overnight. They were subsequently incubated with horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies (dilution, 1:1,000) for 1 h at room temperature. Immobilon western chemiluminescent HRP substrates (EMD Millipore, Billerica, MA, USA) were used as visualization reagents and bands were imaged using a MiniChemi miniature chemiluminescence imager (Beijing Sage Creation Science Co., Ltd., Beijing, China).
Statistical analysis
All data are presented as the mean ± standard deviation (n=5). Independent sample one-way analysis of variance was used to assess the differences between experimental groups. Dunnett's test was used to perform multiple comparisons. All data were analyzed using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA) and P<0.05 was considered to indicate a statistically significant difference.
Results
Ginkgol C17:1 induces autophagy and apoptosis in HepG2 cells
Prior to investigating the effects of ginkgol C17:1 on the viability of HepG2 cells treated with cisplatin, the cytotoxicity of ginkgol C17:1 was initially analyzed by performing an MTT assay. It was revealed that ginkgol C17:1 significantly decreased the viability of HepG2 cells at doses of 40, 80 and 160 µg/ml (P<0.05; Fig. 1A). This inhibition of HepG2 cell viability occurred in a dose-dependent manner. Inhibition was higher following treatment with higher concentrations of ginkgol C17:1.
Figure 1.

Ginkgol C17:1 boosts autophagy and apoptosis in HepG2 cells. (A) Following treatment of HepG2 cells with the indicated concentrations of ginkgol C17:1 (0, 10, 20, 40, 80 and 160 µg/ml) for 24 h, cell viability was detected by an MTT assay. (B) The autophagy protein LC3 was examined by an immunofluorescence assay (magnification, ×200) following infection with GFP-RFP-LC3 adenovirus for 36 h and treatment with ginkgol C17:1 (40 µg/ml) for 24 h; (C) the relative rate of GFP-LC3 puncta was then quantified. (D) Following treatment of HepG2 cells with ginkgol C17:1 (40 µg/ml) with or without NH4Cl (0.535 mg/ml) for 24 h, the expression of Beclin-1, LC3I/II and p62 were analyzed by western blotting. Following treatment with ginkgol C17:1 (40 µg/ml) for 24 h, the morphology of HepG2 nuclei was observed by (E) immunofluorescence microscopy (magnification, ×200) and they were stained with Hoechst 33342. (F) The nucleus aberration rate was then analyzed and (G) western blot analysis was performed to determine the expression of cleaved caspase-3, Bax and Bcl-2. All values are presented as the mean ± standard deviation from three independent experiments (n=5). *P<0.05 and **P<0.01 vs. control. GFP, green fluorescent protein; RFP, red fluorescent protein; LC3, light chain 3; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein.
Autophagy and apoptosis are two different forms of programed cell death. To determine whether ginkgol C17:1 induces autophagy in HepG2 cells, cells were infected with Ad-mRFP-GFP-LC3 adenovirus. The results of the LC3 punctum assay revealed that ginkgol C17:1 enhanced autophagy as the LC3 punctarate increased (Fig. 1B and C). Furthermore, LC3 upregulation enhanced upstream autophagy and suppressed downstream degradation. During autophagy, NH4Cl, a downstream inhibitor of autophagy, may stimulate the accumulation of autophagy proteins. Following the treatment of HepG2 cells with ginkgol C17:1, the expression of Beclin-1 and LC3I/II autophagy proteins was increased and that of autophagy downstream protein, p62, was decreased through autophagic degradation, consequently increasing the level of autophagy. Following treatment with NH4Cl in combination with ginkgol C17:1, increased the expression of autophagy proteins compared with ginkgol C17:1 monotherapy (Fig. 1D), indicating that NH4Cl induces autophagy. To determine whether ginkgol C17:1 induces apoptosis in HepG2 cells, HepG2 cells that underwent Hoechst 33342 staining were treated with ginkgol C17:1 (40 µg/ml). Cells treated with ginkgol C17:1 exhibited nuclear shrinkage and rupture (Fig. 1E), along with a significantly increased nuclear aberration rate (Fig. 1F). In addition, treatment with ginkgol C17:1 increased the expression of the pro-apoptotic proteins Bax and cleaved caspase-3 but reduced the expression of the anti-apoptotic protein Bcl-2 (Fig. 1G). These results indicate that ginkgol C17:1 induced the apoptosis of HepG2 cells.
Ginkgol C17:1 reduces cisplatin-induced autophagy and enhances cisplatin-induced apoptosis in HepG2 cells
Following treatment of cells with various concentrations of cisplatin, cell viability was measured using an MTT assay (Fig. 2A). The results revealed that 2, 4, 8 and 16 µg/ml cisplatin significantly decreased the viability of HepG2 cells; therefore, 2 µg/ml cisplatin was used to treat HepG2 cells in subsequent experiments. Combination therapy of 2 µg/ml cisplatin with various concentrations of ginkgol C17:1 significantly inhibited the viability of HepG2 cells in a dose-dependent manner (P<0.05; Fig. 2B). Furthermore, the results of the LC3 punctum assay revealed that cisplatin significantly enhanced autophagy (P<0.01); however, treatment with all concentrations of ginkgol C17:1 significantly reduced it (P<0.05; Fig. 2C). Additionally, the results of western blotting demonstrated that ginkgol C17:1 inhibits cisplatin-induced autophagy (Fig. 2D). Fig. 2D demonstrates that the expression of autophagy-associated proteins is decreased following treatment with ginkgol C17:1, thereby indicating that this drug inhibited cisplatin-induced autophagy. Hoechst 33342 staining revealed that ginkgol C17:1 significantly enhanced the nucleus aberration rate associated with cisplatin-induced apoptosis in a dose-dependent manner (P<0.05; Fig. 2E) and the results of the western blot analysis revealed that ginkgol C17:1 and cisplatin increased the expression of cleaved caspase-3 and the ratio of Bax/Bcl-2 (Fig. 2F), indicating that ginkgol C17:1 increases the rate of HepG2 cell apoptosis.
Figure 2.

Effects of ginkgol C17:1 on cisplatin-induced autophagy and apoptosis in HepG2 cells. (A) Following treatment with cisplatin (0, 1, 2, 4, 8 and 16 µg/ml) for 24 h, HepG2 cell viability was determined via an MTT assay. (B) Cell viability was detected by MTT assay following treatment with cisplatin (2 µg/ml) and ginkgol C17:1 (0, 20, 40 and 80 µg/ml) for 24 h. Following co-treatment with cisplatin and ginkgol C17:1, LC3 autophagosomes were detected by an (C) immunofluorescence assay (magnification, ×200) and the expression of Beclin-1, LC3I/II and p62 were analyzed by (D) a western blot assay. Under the same conditions, the morphology of HepG2 nuclei was observed by (E) immunofluorescence microscopy (magnification, ×200) staining with Hoechst 33342 and the expression of cleaved caspase-3, Bax and Bcl-2 were analyzed by (F) western blot analysis. Data are presented as the mean ± standard deviation from three independent experiments. *P<0.05 and **P<0.01 vs. control group, and ##P<0.01. LC3, light chain 3; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; GFP, green fluorescent protein.
Inhibition of autophagy can enhance apoptosis during chemotherapy
To determine the association between autophagy and apoptosis in HepG2 cells treated with cisplatin, cells were incubated with cisplatin, 3-MA (an upstream inhibitor of autophagy) or ginkgol C17:1. LC3 punctum and western blot assays revealed that 3-MA significantly inhibited the induction of autophagy by cisplatin (P<0.05; Fig. 3A and B). Furthermore, Hoechst 33342 staining indicated that 3-MA significantly enhanced the nuclear aberration rate of cells treated with cisplatin (P<0.05; Fig. 3C) and the ratio of Bax/Bcl-2 (Fig. 3D). The effects of ginkgol C17:1 on cisplatin-induced autophagy and apoptosis were similar to 3-MA (Fig. 3E and F). The present study revealed that ginkgol C17:1 has a similar function to that of 3-MA in regulating cisplatin-induced autophagy and apoptosis in HepG2 cells; ginkgol C17:1 and 3-MA enhance cisplatin-induced apoptosis and inhibit cisplatin-induced autophagy.
Figure 3.

Ginkgol C17:1 enhances apoptosis and inhibits the autophagy of cells, similar to 3MA. Following infection of HepG2 cells with the GFP-LC3 virus and treatment with or without cisplatin and 3-MA for 24 h, (A) LC3 autophagosomes were detected by an immunofluorescence assay and the average rate of LC3 puncta was valued (magnification, ×200). (B) Western blot analysis measuring the expression of Beclin-1 and p62 was performed. Following treatment with or without cisplatin and 3MA for 24 h, (C) nuclei morphology were observed by immunofluorescence microscopy (magnification, ×200) following staining with Hoechst 33342 and (D) the expression of Beclin-1, LC3 and p62 were measured by western blotting. Following treatment of HepG2 with cisplatin with or without ginkgol C17:1 and 3MA for 24 h, western blot analysis investigating the expression of (E) Beclin-1 and p62 and (F) Bax and Bcl-2 was performed. Data are presented as the mean ± standard deviation from five independent experiments. *P<0.05 and ##P<0.01. Cisplatin, 2 µg/ml; 3MA, 0.75 mg/ml and ginkgol C17:1, 40 µg/ml. GFP, green fluorescent protein; LC3, light chain 3; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; 3-MA, 3-methyladenine.
Ginkgol C17:1 regulates cisplatin-induced autophagy by inhibiting the AMPK/ULK1 pathway
AMPK is one of the primary stress-sensing enzymes and actively regulates cell metabolism and proliferation. The AMPK/ULK1 pathway serves an important role in regulating autophagy. A western blot assay was therefore performed to investigate the effect of ginkgol C17:1 on the AMPK/ULK1 pathway in HepG2 cells. The expression of p-AMPK was increased in cells treated with cisplatin, indicating that cisplatin induces autophagy (Fig. 4A). However, treatment with ginkgol C17:1 decreased the expression of p-AMPK (Thr172) and p-ULK1 (Ser555) in cells treated with cisplatin in a dose-dependent manner. In addition, the AMPK inhibitor compound C reduced the expression of p-AMPK and p-ULK1 (Fig. 4B). The expression of Beclin-1 and LC3 I/II were increased following treatment with cisplatin and this was inhibited following treatment with ginkgol C17:1 (Fig. 4B). However, the effects of ginkgol C17:1 were reversed following the inhibition of AMPK by compound C. By contrast, the expression of Bax and Bcl-2 remained unchanged following treatment with compound C, indicating that ginkgol C17:1 still induced a pro-apoptotic effect in cells treated with compound C and ginkgol C17:1 (Fig. 4B). These data imply that the AMPK/ULK1 signaling pathway is a vital regulatory pathway involved in cisplatin-induced autophagy on HepG2 cells.
Figure 4.
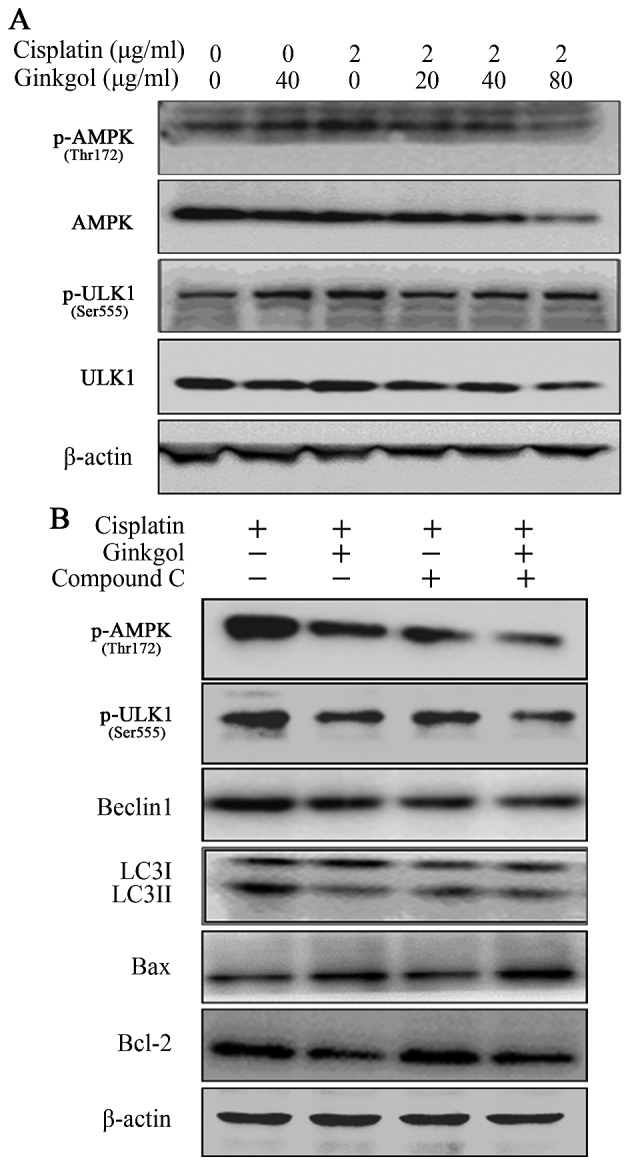
Effect of ginkgol C17:1 on the AMPK/ULK1 pathway. (A) Following co-treatment of HepG2 cells with cisplatin (2 µg/ml) and ginkgol C17:1 (0, 20, 40 and 80 µg/ml) for 24 h, western blot analysis was performed to determine the expression of AMPK, p-AMPK (Thr172), ULK1 and p-ULK1 (Ser555). (B) Following treatment of cells with cisplatin (2 µg/ml) with or without ginkgol C17:1 (40 µg/ml) and compound C (8 µg/ml) for 24 h, western blot analysis was performed to measure the expression of p-AMPK (Thr172), p-ULK1 (Ser555), Beclin-1, LC3I/II, Bax and Bcl-2. AMPK, AMP-activated protein kinase; p-, phosphorylated; LC3, light chain 3; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein.
Ginkgol C17:1 regulates cisplatin-induced apoptosis by inhibiting the PI3K/Akt/mTOR pathway
The PI3K/Akt/mTOR signaling pathway is involved in the molecular biological mechanisms by which autophagy or apoptosis occur (21). The expression of p-PI3K (Tyr458), p-Akt (Tyr315) and p-mTOR (Ser2448) decreased in a dose-dependent manner following the co-culture of HepG2 cells with cisplatin and ginkgol C17:1 (Fig. 5A). To determine whether ginkgol C17:1 affects the autophagy and apoptosis induced by cisplatin via the PI3K/Akt/mTOR signaling pathway, HepG2 cells were treated with rapamycin (an mTOR inhibitor). The effect of ginkgol C17:1 on the expression of Bax and Bcl-2 activated by cisplatin was attenuated following rapamycin treatment, however, the expression of the autophagy proteins Beclin-1 and LC3I/II remained evidently unchanged following co-treatment with or without ginkgol C17:1, and following the inhibition of mTOR signaling by rapamycin (Fig. 5B). These data imply that ginkgol C17:1 stimulated cisplatin-induced apoptosis by inhibiting the PI3K/Akt/mTOR signaling pathway in HepG2 cells, but did not affect cisplatin-induced autophagy.
Figure 5.
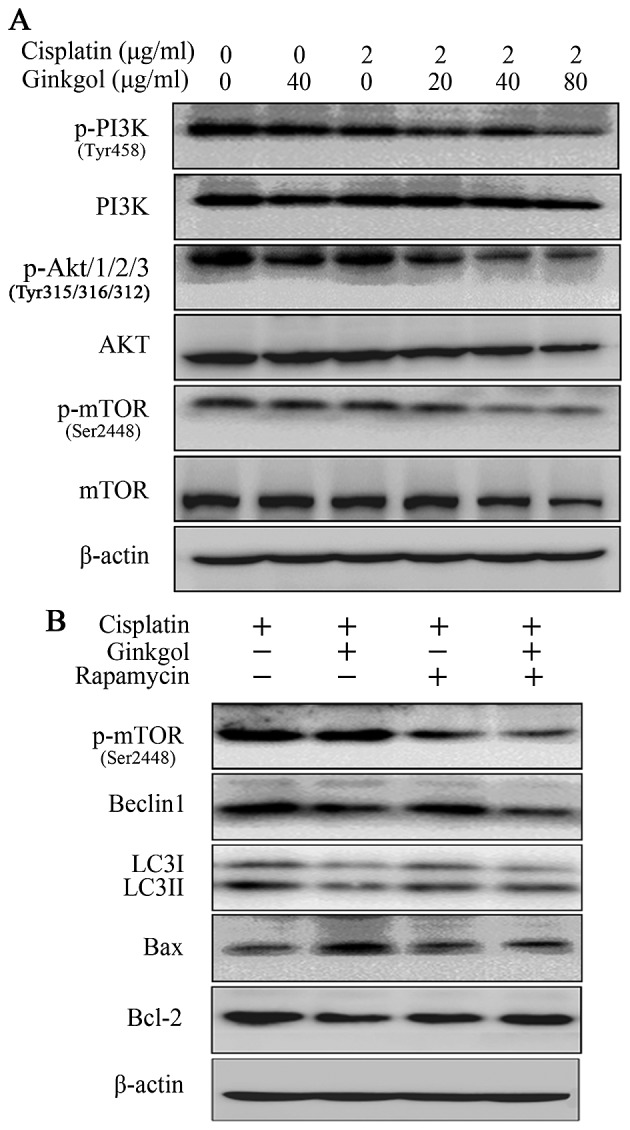
Influence of ginkgol C17:1 on the PI3K/Akt/mTOR pathway. (A) Following co-treatment of HepG2 cells with cisplatin (2 µg/ml) and ginkgol C17:1 (0, 20, 40 and 80 µg/ml) for 24 h, western blot analysis was performed to measure the expression of proteins in the upstream pathway: PI3K and p-PI3K (Tyr458), Akt and p-Akt (Tyr315), mTOR and p-mTOR (Ser2448), JNK and p-JNK (Tyr185). (B) Following treatment of cells were with cisplatin (2 µg/ml), with or without ginkgol C17:1 (40 µg/ml) and rapamycin (100 ng/ml) for 24 h, western blotting was performed to measure the expression of p-mTOR (Ser2448), Beclin-1, LC3I/II, Bax and Bcl-2. p-, phosphorylated; PI3K, phosphoinositide 3-kinase; mTOR, mechanistic target of rapamycin; LC3, light chain 3; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein.
Discussion
The theory of substrate selectivity in autophagy has received increasing recognition. This differs from the original understanding of autophagy, which was that autophagy was a non-specific process of self-destruction (28). In its ‘active form’, LC3II assists in elongating the phagophore membrane and in the recruitment of damaged or degraded organelles to the phagophore (29). Beclin-1 is sometimes essential for autophagy (27). However, it has been suggested that Beclin-1 expression is only necessary for autophagy occurring in certain types of cells and that it may not be required for autophagy induced by cytotoxic compounds, including resveratrol, staurosporin or gossypol (30). To the best of our knowledge, the impact of autophagy on antitumor therapy has not yet been demonstrated. It has been demonstrated that autophagy and apoptosis are distinctive; however, the two processes are associated and cross-talk also occurs between them, where by individual proteins may serve functions in both processes (31). The cross-talk that occurs between them is partly complicated by the fact that they share numerous common regulatory molecules, including Bcl-2 and Beclin-1, as well as the PI3K/Akt/mTOR signaling pathway (32,33).
Cisplatin is commonly used in chemotherapeutic regimes to treat patients with cancer. However, the efficacy of cisplatin is limited due to the development of drug resistance in many patients and the fact that it causes multiple side effects; therefore, it is important to improve the pharmacological effect of cisplatin (34). It has been demonstrated that cisplatin induces autophagy and apoptosis in cancer cells (35,36), which has also been determined in the present study. Apoptosis directly causes the death of cancer cells; by contrast, autophagy may have two effects on tumors; an antitumor or protective effect (14,37). Therefore, autophagy may beinvolved in the regulation of tumor growth. A previous study demonstrated that ginkgol ultimately induced apoptosis by activating the expression of caspases via inhibition ofthe PI3K/Akt pathway (38). In the present study, ginkgol C17:1 not only induced apoptosis in HepG2 cells but also induced autophagy. Following co-treatment with cisplatin, ginkgol C17:1 stimulated cisplatin-induced apoptosis, however, it also inhibited cisplatin-induced autophagy. Previous studies have demonstrated that inhibitors of autophagy administered in combination with anti-cancer agents may enhance chemosensitization in human cancer cells (39–41). The resultsof the present study indicating the effects of ginkgol C17:1 on cisplatin-induced autophagy are in accordance with the results of a study by Bao et al (33); they suggest that ginkgol C17:1 may overcome cisplatin resistance.
The present study revealed that ginkgol C17:1 and cisplatin may affect the PI3K/Akt/mTOR and AMPK/ULK1 signaling pathways, which are two primary methods of regulating autophagy and apoptosis (42–44). Co-treatment with ginkgol C17:1 and cisplatin inhibited these pathways, while treatment with ginkgol C17:1 or cisplatin alone caused the AMPK/ULK1 pathway to become activated. However, monotherapy with either drug resulted in the inhibition of the PI3K/Akt/mTOR pathway.
AMPK is a multi-functional protein kinase; thus, its activation may promote apoptosis by inducing the phosphorylation of p53, activating the nuclear factor-κB and c-myc, or the c-Jun pathway (45–47). Based on this function, activation of the AMPK pathway may block tumor cell proliferation and regulate tumor development. Thus activating the AMPK pathway may be a novel method of treating patients with cancer.
It has been demonstrated that AMPK activates autophagy by directly and indirectly activating ULK1 (48). AMPK indirectly activates ULK1 by inhibiting mTOR, which phosphorylates and inhibits ULK1 to disrupt the interaction between AMPK and ULK1 (49). However, as feedback, activated ULK1 also phosphorylates AMPK and inhibits its activation, providing a potential negative-feedback loop resulting in autophagy induction (50). Following co-treatment with ginkgol C17:1 and cisplatin, the AMPK/ULK1 pathway was inhibited and the negative-feedback loop was activated by ginkgol C17:1.
Due to the multifaceted role of autophagy in cancer cells, manipulating the activation and inhibition of AMPK may be developed as an additional therapeutic strategy for cancer treatment. Enhancing AMPK activity may be a novel method of sensitizing tumor cells to radiotherapy and chemotherapy (51). However, previous studies have demonstrated that autophagy also provides a survival advantage against cancer therapies by inducing AMPK and suppressing the apoptosis pathway (52,53). Thus, suppressing autophagy enhances cell death by inhibiting AMPK. In the present study, ginkgol C17:1 reduced autophagy by inhibiting the activity of AMPK to induce cell death.
When the two signaling pathways were interrupted, it was revealed that rapamycin (mTOR inhibitor) blocked the action of ginkgol C17:1 in cisplatin-induced apoptosis rather than autophagy. This indicates that ginkgol C17:1 enhances the apoptosis induced by cisplatin via the PI3K/Akt/mTOR signaling pathway. Additionally, compound C (an AMPK inhibitor) blocked the action of ginkgol C17:1 in cisplatin-induced autophagy rather than apoptosis, indicating that the AMPK/ULK1 pathway is a vital regulatory pathway involved in cisplatin-induced autophagy in HepG2 cells. These signaling pathways are associated with autophagy and apoptosis; however, studies highlight that different stimuli may induce autophagy via different mechanisms under different conditions (41,54). This may have therapeutic implications in the treatment of HB.
In conclusion, autophagy and apoptosis are two important mechanisms involved in cell regulation. In the present study, treatment with ginkgol C17:1 affected cancer cells primarily through autophagy and apoptosis. Furthermore, the present study indicated that ginkgol C17:1 inhibits cisplatin-induced autophagy via AMPK/ULK1 signaling and increases cisplatin-induced apoptosis via the PI3K/Akt/mTOR pathway. Ultimately, ginkgol C17:1 significantly inhibited human liver cancer cells and enhanced the anticancer activity of cisplatin. Furthermore, the present study provides an important theoretical basis for the future of antitumor research. To further assess the antitumor effect of ginkgol C17:1 and potentially develop it as a novel cancer treatment, further studies are required to identify the antitumor effects of ginkgol C17:1.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant no. 81372404); the College Students' Scientific Research Project of Jiangsu University (grant no. 14A056) and the Zhenjiang Social Development Project (grant no. SH2015072).
References
- 1.Farazi PA, Depinho RA. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 2.Khaderi S, Guiteau J, Cotton RT, O'Mahony C, Rana A, Goss JA. Role of liver transplantation in the management of hepatoblastoma in the pediatric population. World J Transplant. 2014;4:294–298. doi: 10.5500/wjt.v4.i4.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdul HA, Lai ML, Phaik LC. Tissue microarray immunohistochemical profiles of p53 and pRB in hepatocellular carcinoma and hepatoblastoma. Asian Pac J Cancer Prev. 2014;15:3959–3963. doi: 10.7314/APJCP.2014.15.9.3959. [DOI] [PubMed] [Google Scholar]
- 4.López-Terrada D, Cheung SW, Finegold MJ, Knowles BB. Hep G2 is a hepatoblastoma-derived cell line. Hum Pathol. 2009;40:1512–1517. doi: 10.1016/j.humpath.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Pang RTK, Poon TCW, Wong N, Lai PSB, Wong NLY, Chan CML, Yu JWS, Chan ATC, Sung JJY. Comparison of protein expression patterns between hepatocellular carcinoma cell lines and a hepatoblastoma cell line. Clin Proteomics. 2004;1:313–331. doi: 10.1385/CP:1:3-4:313. [DOI] [Google Scholar]
- 6.Rishi RR, Kimberlee KS, Ruth IH, Michael SB, Max B, Lisa EH. Hepatoblastoma: A need for cell lines and tissue banks to develop targeted drug therapies. Front Pediatr. 2016;4:22. doi: 10.3389/fped.2016.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pateva IB, Egler RA, Stearns DS. Hepatoblastoma in an 11-year-old: Case report and a review of the literature. Medicine (Baltimore) 2017;96:e5858. doi: 10.1097/MD.0000000000005858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomlinson GE, Kappler R. Genetics and epigenetics of hepatoblastoma. Pediatr Blood Cancer. 2012;59:785–792. doi: 10.1002/pbc.24213. [DOI] [PubMed] [Google Scholar]
- 9.Boulikas T, Vougiouka M. Cisplatin and platinum drugs at the molecular level (Review) Oncol Rep. 2003;10:1663–1682. [PubMed] [Google Scholar]
- 10.Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27–36. doi: 10.1016/j.phrs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Muggia F. Platinum compounds 30 years after the introduction of cisplatin: Implications for the treatment of ovarian cancer. Gynecol Oncol. 2009;112:275–281. doi: 10.1016/j.ygyno.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 12.Zhang HQ, Fang N, Liu XM, Xiong SP, Liao YQ, Jin WJ, Song RF, Wan YY. Antitumor activity of chloroquine in combination with Cisplatin in human gastric cancer xenografts. Asian Pac J Cancer Prev. 2015;16:3907–3912. doi: 10.7314/APJCP.2015.16.9.3907. [DOI] [PubMed] [Google Scholar]
- 13.García-Cano J, Ambroise G, Pascual-Serra R, Carrión MC, Serrano-Oviedo L, Ortega-Muelas M, Cimas FJ, Sabater S, Ruiz-Hidalgo MJ, Sanchez Perez I, et al. Exploiting the potential of autophagy in cisplatin therapy: A new strategy to overcome resistance. Oncotarget. 2015;6:15551–15565. doi: 10.18632/oncotarget.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu SZ, Harrison-Findik DD. Autophagy and cancer. World J Biol Chem. 2013;4:64–70. doi: 10.4331/wjbc.v4.i3.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu M, Ma S, Liu M, Hou Y, Liang B, Su X, Liu X. Synergistic killing of lung cancer cells by cisplatin and radiation via autophagy and apoptosis. Oncol Lett. 2014;7:1903–1910. doi: 10.3892/ol.2014.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakamoto A, Iwamoto Y. Current status and perspectives regarding the treatment of osteo-sarcoma: Chemotherapy. Rev Recent Clin Trials. 2008;3:228–231. doi: 10.2174/157488708785700267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Wang Q, Yin FQ, Zhang W, Yan LH, Li L. MTRR silencing inhibits growth and cisplatin resistance of ovarian carcinoma via inducing apoptosis and reducing autophagy. Am J Transl Res. 2015;7:1510–1527. [PMC free article] [PubMed] [Google Scholar]
- 21.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: The path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 22.Tsai JP, Lee CH, Ying TH, Lin CL, Lin CL, Hsueh JT, Hsieh YH. Licochalcone A induces autophagy through PI3K/Akt/mTOR inactivation and autophagy suppression enhances Licochalcone A-induced apoptosis of human cervical cancer cells. Oncotarget. 2015;6:28851–28866. doi: 10.18632/oncotarget.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nash KM, Shah ZA. Current perspectives on the beneficial role of ginkgo biloba in neurological and cerebrovascular disorders. Integr Med InsIghts. 2015;10:1–9. doi: 10.4137/IMI.S25054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Q, Yang GW, An LG. Apoptosis of hepatoma cells SMMC-7721 induced by ginkgo biloba seed polysaccharide. World J Gastroenterol. 2002;8:832–836. doi: 10.3748/wjg.v8.i5.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang XM, Wang YF, Li YY, Ma HL. Thermal stability of ginkgolic acids from ginkgo biloba and the effects of ginkgol C17:1 on the apoptosis and migration of SMMMC7721 cells. Fitoterapia. 2014;98:66–76. doi: 10.1016/j.fitote.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Ma X, Liu H, Foyi SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reggiori F, Komatsu M, Finley K, Simonsen A. Autophagy: More than a nonselective pathway. Int J Cell Biol. 2012;2012:219625. doi: 10.1155/2012/219625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindqvist LM, Simon AK, Baehrecke EH. Current questions and possible controversies in autophagy. Cell Death Discov. 2015;1:pii: 15036. doi: 10.1038/cddiscovery.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Shao Z, Xiong L, Yang S. Inhibition of autophagy enhances cisplatin-induced apoptosis in the MG63 human osteosarcoma cell line. Oncol Lett. 2015;10:2941–2946. doi: 10.3892/ol.2015.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–7279. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 33.Bao L, Jaramillo MC, Zhang Z, Zheng Y, Yao M, Zhang DD, Yi X. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol Med Rep. 2015;11:91–98. doi: 10.3892/mmr.2014.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Grazie M, Biagini MR, Tarocchi M, Polvani S, Galli A. Chemotherapy for hepatocellular carcinoma: The present and the future. World J Hepatol. 2017;9:907–920. doi: 10.4254/wjh.v9.i21.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cavallo F, Feldman DR, Barchi M. Revisiting DNA damage repair, p53-mediated apoptosis and cisplatin sensitivity in germ cell tumors. Int J Dev Biol. 2013;57:273–280. doi: 10.1387/ijdb.130135mb. [DOI] [PubMed] [Google Scholar]
- 36.Zhang HQ, He B, Fang N, Lu S, Liao YQ, Wan YY. Autophagy inhibition sensitizes cisplatin cytotoxicity in human gastric cancer cell line SGC7901. Asian Pac J Cancer Prev. 2013;14:4685–4688. doi: 10.7314/APJCP.2013.14.8.4685. [DOI] [PubMed] [Google Scholar]
- 37.Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 38.Li YY, Liu J, Yang XM, Dong Y, Liu YL, Chen M. Ginkgol C17:1 inhibits tumor growth by blunting the EGF-PI3K/Akt signaling pathway. J Biomed Res. 2017;31:232–239. doi: 10.7555/JBR.31.20160039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang JY, Dai T, Zhang H, Xiong WJ, Xu MZ, Wang XJ, Tang QH, Chen B, Xu M. GDC-0980-induced apoptosis is enhanced by autophagy inhibition in human pancreatic cancer cells. Biochem Biophys Res Commun. 2014;453:533–538. doi: 10.1016/j.bbrc.2014.09.115. [DOI] [PubMed] [Google Scholar]
- 40.Wu HM, Jiang ZF, Ding PS, Shao LJ, Liu RY. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci Rep. 2015;5:12291. doi: 10.1038/srep12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan MM, Ni JD, Song D, Ding M, Huang J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol Lett. 2015;10:1959–1969. doi: 10.3892/ol.2015.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: Rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/S1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 43.Li F, Zeng J, Gao Y, Guan Z, Ma Z, Shi Q, Du C, Jia J, Xu S, Wang X, et al. G9a inhibition induces autophagic cell death via AMPK/mTOR pathway in bladder transitional cell carcinoma. PLoS One. 2015;10:e0138390. doi: 10.1371/journal.pone.0138390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 46.Kefas BA, Cai Y, Ling Z, Heimberg H, Hue L, Pipeleers D, Van de Casteele M. AMP-activated protein kinase can induce apoptosis of insulin-producing MIN6 cells through stimulation of c-Jun-N-terminal kinase. J Mol Endocrinol. 2003;30:151–161. doi: 10.1677/jme.0.0300151. [DOI] [PubMed] [Google Scholar]
- 47.Nieminen AI, Eskelinen VM, Haikala HM, Tervonen TA, Yan Y, Partanen JI, Klefström J. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis; Proc Natl AcadSci USA; 2013; pp. E1839–E1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48:e245. doi: 10.1038/emm.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, Campbell DG, Wesselborg S, Alessi DR, Stork B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7:696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- 51.Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: A novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2014;15:156–169. doi: 10.4161/cbt.26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 53.Xie BS, Zhao HC, Yao SK, Zhuo DX, Jin B, Lv DC, Wu CL, Ma DL, Gao C, Shu XM, Ai ZL. Autophagy inhibition enhances etoposide-induced cell death in human hepatoma G2 cells. Int J Mol Med. 2011;27:599–606. doi: 10.3892/ijmm.2011.607. [DOI] [PubMed] [Google Scholar]
- 54.Wu T, Wang MC, Jing L, Liu ZY, Guo H, Liu Y, Bai YY, Cheng YZ, Nan KJ, Liang X. Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway. Drug Des Devel Ther. 2015;9:6421–6431. doi: 10.2147/DDDT.S95606. [DOI] [PMC free article] [PubMed] [Google Scholar]