

Abstract
Improvements have been made to the safety and efficacy of bumped kinase inhibitors, and they are advancing toward human and animal use for treatment of cryptosporidiosis. As the understanding of bumped kinase inhibitor pharmacodynamics for cryptosporidiosis therapy has increased, it has become clear that better compounds for efficacy do not necessarily require substantial systemic exposure. We now have a bumped kinase inhibitor with reduced systemic exposure, acceptable safety parameters, and efficacy in both the mouse and newborn calf models of cryptosporidiosis. Potential cardiotoxicity is the limiting safety parameter to monitor for this bumped kinase inhibitor. This compound is a promising pre-clinical lead for cryptosporidiosis therapy in animals and humans.
Keywords: Cryptosporidiosis, Calcium-dependent protein kinase 1, Gatekeeper residue, Drug target, BKI 1369
1. Introduction
Cryptosporidium infections have been associated with mortality and morbidity from diarrhoea in malnourished 6–18 month old children in resource limited environments (Kotloff et al., 2013; Platts-Mills et al., 2015). The only US Food and Drug Administration (FDA) approved therapeutic drug for cryptosporidiosis, nitazoxanide, is only approximately 30% effective in 1–5 year old malnourished children with cryptosporidiosis (Amadi et al., 2002). Nitazoxanide is not approved for use in children under 12 months of age (package insert, nitazoxanide). Furthermore, Cryptosporidium infections can be life threatening in immunocompromised HIV-positive individuals, in whom nitazoxanide has also been shown to lack efficacy (Amadi et al., 2009). Clearly, new effective therapeutic methods are needed for cryptosporidiosis.
Bumped kinase inhibitors (BKIs) have been developed to selectively target Cryptosporidium parvum and Toxoplasma gondii calcium-dependent protein kinase 1 (CDPK1) (Murphy et al., 2010). These BKIs have excellent activity against C. parvum and T. gondii parasites in vitro and in vivo. A recently reported series of pyrazolo[2,3-d]pyrmidine (PP) BKIs are effective in experimental therapeutic treatments for cryptosporidiosis (Hulverson et al., 2017). Since that publication, additional data make it clear that one compound, BKI 1369, is emerging as a pre-clinical lead for cryptosporidiosis therapy.
The study presented here describes the pharmacokinetic, safety and efficacy parameters of BKI 1369. BKI 1369 is very active at low doses in the C. parvum-infected interferon-γ knockout (IFN-γ KO) mouse model. It is also efficacious in the C. parvum-infected new-born calf clinical model, abrogating diarrhoea and reducing parasite excretion. In addition, this compound is safe in that it lacks mutagenic activity, is not toxic to a variety of human cell lines, is safely administered orally to mice at up to 150 mg/kg of body weight for 7 days, and is safe for both mothers and fetuses in a mouse pregnancy model. One limitation of BKI 1369 is potential cardiotoxicity, both from ether-à-go-go (hERG) inhibition and negative inotropic activity. However, there is a sufficient safety window regarding cardiotoxicity to suggest BKI 1369 will be safe in humans and animals at therapeutic doses. BKI 1369 has low plasma exposure after oral administration, and has two major identified metabolites, each of which appears to be non-toxic to human cells. A pharmacodynamic model is presented here to further explain the therapeutic efficacy of BKI 1369.
2. Materials and methods
2.1. Previously described methods
Methods for the propagation of C. parvum (IOWA-II strain) oocysts in calves (Riggs and Perryman, 1987; Riggs et al., 1989), propagation of transgenic Nanoluciferase (Nluc) expressing C. parvum (UGA1 strain) oocysts in mice (Vinayak et al., 2015; Hulverson et al., 2017), in vitro microsomal stability (Tatipaka et al., 2014), pharmacokinetic analysis of mouse plasma, faecal, and urine BKI concentrations by LC-MS/MS analysis (Ojo et al., 2012; Schaefer et al., 2016; Hulverson et al., 2017), in vitro protein binding using dialysis membranes (Tatipaka et al., 2014), Nluc expressing C. parvum in vitro growth inhibition in infected HCT-8 cells (Hulverson et al., 2017), in vitro cytotoxicity in CRL-8155 and HepG2 cells (Tatipaka et al., 2014), and in vivo mouse gastrointestinal (GI) tract tissue exposure by LC-MS/MS analysis (Arnold et al., 2017) have all been previously described. Detailed descriptions of these methods are included in Supplementary Data S1. Methods for high-content imaging of Cryptosporidium proliferation in HCT-8 cells (Love et al., 2017), QPatch hERG (Danker and Moller, 2014), rat cardiovascular screening (Banfor et al., 2016), in vitro micronucleus assay (Nicolette et al., 2011), the 24-well Ames screening assay, kinome profiling (Goedken et al., 2015), Nluc C. parvum in infected adult IFN-γ KO mouse efficacy (Hulverson et al., 2017), Gastroplus (Simulations Plus, Inc., Lancaster, CA, USA) modelling of efficacy and GI tissue and lumen exposures (Arnold et al., 2017), and IOWA-II strain C. parvum in infected neonatal calf efficacy (Schaefer et al., 2016) have all been previously described. All LC-MS/MS analytes were measured with an Acquity ultra performance liquid chro6 matography (UPLC) system in tandem with a Xevo TQ-S mass spectrometer (Waters, Milford, MA, USA). For all statistical analyses, P < 0.05 was considered significant. Methods for toxicity profiling in mice to determine the maximum tolerated single dose and toxicity related to multiple dosing were previously described (Vidadala et al., 2016). In toxicity profiling experiments, mice were monitored twice daily and dosing was halted after the first observable signs of toxicity. Necropsy, obtaining blood for complete blood counts (CBCs) and serum chemistries, and histology of organs (brain, heart, lungs, spleen, liver, stomach, small intestine, large intestine and kidney) were performed within 48 h of the final dose. Examination of organ histology included routine H&E and Oil-Red-O (lipid)-stained organs from the treated mice and a set of untreated matched controls. Pathological examination was performed by a board certified pathologist who was blinded to the treatment and control group identities.
2.2. Mouse pregnancy safety
Female BALB/c mice aged 8–10 weeks were mated for 3 days, after which the males were removed. Weights were monitored daily to confirm pregnancy. Starting at day 9 after mating, treated groups (n = 6 or 7) were dosed by oral gavage (PO) with 0.1 mL of BKI suspended in 100% corn oil and control groups were dosed PO with corn oil only for 5 days. On day 18 after mating, mice that were positively identified as pregnant were moved to individual housing and monitored 2–3 times daily. After birth, pups were observed for an additional 3 days, counted and dead pups were removed.
2.3. Metabolite identification, formation and excretion
Adult female BALB/c mice were given a single 0.2 mL oral dose of 60 mg/kg of body weight in 3% ethanol/7% Tween80/90% saline (ETS). Blood was taken by cardiac puncture from mice (n = 3) at time points of 1, 4, 8 and 24 h post dose. Control mice given ETS only were also sampled at 8 h post dose. A list of expected metabolites was generated by analysing the structure of BKI 1369. Plasma was analysed on LC-MS/MS to confirm the presence of these metabolites in vivo. Pure samples of two of the probable metabolites were synthesised for use as standards to quantify sample concentration by LC-MS/MS.
Additional adult female BALB/c mice (n = 3) were given a single oral BKI dose of 60 mg/kg of body weight in 0.2 mL of ETS and housed in metabolic cages to sample urine and faeces at 8 h intervals for a total of 48 h. Additional mice (n = 3) were given the same 60 mg/kg dose and blood was collected at 1, 4, 8 and 24 h post dose. Mice dosed with ETS only were used as controls. Plasma, urine and faeces were extracted with acetonitrile and analysed by LC-MS/MS to determine the concentrations of 1369 and its metabolites identified from previously obtained plasma samples.
2.4. Cytochrome P450 (CYP) inhibition
To determine the inhibition kinetics for the reversible inhibition of CYP3A4 and CYP2D6 by BKI 1369, experiments were performed in triplicate with either midazolam (CYP3A4) or dextromethorphan (CYP2D6) concentrations of 1 μM, which is fivefold below the reported Michaelis constant (Km) values for each of these substrates. The incubations were performed in 100 mM potassium phosphate buffer (pH 7.4), and final incubation volumes were 100 μL. BKI 1369 at concentrations of 0.01–100 μM, human liver microsomes (0.1 mg/mL) and substrate were pre-incubated at 37 °C before reactions were initiated by adding NADPH (1 mM, final concentration). The reactions were terminated after 4 min (midazolam) or 20 min (dextromethorphan) by addition of an equal volume of ice-cold acetonitrile containing internal standard. The samples were vortexed followed by centrifugation at 4,000g for 15 min. The supernatants were removed and analysed by LC-MS/MS. The samples were normalised by the no inhibitor controls to determine the percentage of activity remaining. Prism (Graph-Pad, La Jolla, CA, USA) was used to fit the normalised data to a log(dose)-response curve and calculate a half maximal inhibitory concentration IC50.
2.5. Animal ethics
All animal experiments conducted at the University of Washington, USA, and University of Arizona, USA, were approved by the Institutional Animal Care and Use Committees. Animal experiments conducted at the University of Bern, Switzerland, were approved by the Animal Welfare Committee of the Canton of Bern under the lisense BE115/14. All animals used in this study were handled in strict accordance with practices made to minimise suffering.
3. Results
3.1. Toxicity in mice
The relative efficacies of BKIs 1369, 1534 and 1649 (Supplementary Fig. S1) in mice were previously described (Hulverson et al., 2017). Due to the previously encountered hERG cardiotoxicity of BKI 1294 (Ojo et al., 2014), as well as the toxicity seen with PP BKI 1561 (Vidadala et al., 2016), BKIs 1369, 1534 and 1649 were tested in additional mouse safety models including escalated dosing, multiple dosing and pregnancy safety.
3.2. Toxicity profile, maximum tolerated dose and multiple dosing toxicity
BKIs 1369, 1534 and 1649 were further tested in mice for toxicity with escalating single doses and multiple doses administered PO as described (Vidadala et al., 2016). Escalating single doses were administered until signs of toxicity were observed or BKI solubility limits within the dosing vehicle were reached (Table 1). BKI 1369 showed signs of toxicity after a single oral 600 mg/kg dose, but not after a single oral 500 mg/kg dose. Mice treated with 600 mg/kg displayed hunched posture and visible discharge around squinted eyes. BKI 1534 did not show any signs of toxicity with a single dose up to 250 mg/kg; however, the solubility of this compound in the dosing vehicle prevented it from being used at any higher concentrations. Mice treated with a single dose of 100 mg/kg of BKI 1649 showed lethargy, hunched posture and a temporary lack of grooming.
Table 1.
Escalating single and multiple dosages of bumped kinase inhibitors administered orally to determine maximum tolerated doses and toxicity profiles.
| BKI – |
Determined maximum tolerated single oral dose (mg/kg) |
Multiple PO dose concentrations used mg/kg of body weight (dose frequency) |
Observed signs of toxicity – |
Plasma concentration at 2 h post last dose Avg ± S.D. (μM) |
|---|---|---|---|---|
| 1369 | 600 | 500 (QD) | After 3rd dose | 11.5 ±1.5 |
| 300 (QD) | After 5th dose | 8.3 ± 0.2 | ||
| 150 (QD) | None after 7th dose | 3.9 ± 1.5 | ||
| 1534 | 250a | 250 (QD) | After 4th dose | 2.9 ± 1.5 |
| 100 (QD) | After 5th dose | 1.8 ± 0.9 | ||
| 1649 | 100 | 50 (QD) | After 5th dose | 307 ± 26.8 |
| 30 (QoD) | None after 5th dose | 46.8 ±1.7 | ||
| 10 (QoD) | None after 5th dose | 17.9 ±1.0 |
QD, once per day; QoD, every other day; Avg, average.
No signs of toxicity observed after a single dose, however the BKI reached its solubility limit in vehicle and no higher concentration could be tested.
BKI 1369 was dosed at 150, 300 and 500 mg/kg once daily (QD) to check for signs of toxicity (Table 1). Mice dosed at 500 mg/kg began showing lethargy, hunched posture and discharge around the eyes after the third dose, and mice dosed at 300 mg/kg began showing lethargy and hunched posture after the fifth dose. Mice dosed with 150 mg/kg of BKI 1369 showed no signs of toxicity after being dosed for 7 days. Plasma concentrations sampled 2 h after the last dose averaged 3.9 μM for the 150 mg/kg doses, 8.3 μM for the 300 mg/kg doses, and 11.5 μM for the 500 mg/kg doses (Table 1). Histology on blood and organs sampled from all mice after the final dose showed no abnormalities. CBC and serum chemistry results were all within the normal ranges for mice. Although the mice dosed with 500 mg/kg and 300 mg/kg showed no abnormalities in the organ tissues, it was noted during necropsy that the stomachs were packed with undigested food and the intestinal tracts were filled with faecal matter throughout, suggesting a disruption to digestion and/or peristalsis at these higher dose concentrations.
BKI 1534 was dosed at 100 and 250 mg/kg QD. Lethargy, hunched posture and dehydration were observed in mice dosed at 250 mg/kg after the fourth dose and in mice dosed at 100 mg/kg after the fifth dose. CBC and serum chemistry results were all within the normal ranges for mice and histology showed no abnormalities in the blood or organ tissues but, as observed with the highest doses of BKI 1369, necropsy revealed that digestion and/or peristalsis were disrupted, resulting in undigested food in the stomach and faecal matter filling the intestinal tract.
Dosing of BKI 1649 at 50 mg/kg QD for 5 days led to obvious signs of toxicity only after five doses, including lack of grooming, hunched posture, loss of activity, lethargy and dehydration. Plasma concentrations of BKI 1649 at 2 h after the first dose of 50 mg/kg averaged 146 μM, but rose to concentrations >300 μM at 2 h after the last dose (Table 1), suggesting that the accumulation of the compound in the plasma eventually reached toxic levels. Due to its extremely high systemic exposure, BKI 1649 was again multidosed to check for toxicity at lower systemic levels by dosing at 10 and 30 mg/kg QD on Monday, Wednesday and Friday for five doses. No signs of toxicity were observed with the lower doses and average plasma concentrations were substantially lower after the fifth dose (Table 1). CBC and safety chemistry results were all within the normal ranges for mice and histology and necropsy showed no abnormalities in the blood or organ tissues.
3.3. Mouse pregnancy safety
BKIs shown to be efficacious in IFN-γ KO mice were tested for safety in pregnant mice (Table 2). Of these compounds, only BKI 1369 had no negative effect on mouse pregnancy or faetal health. All confirmed pregnant dams treated with BKI 1369 delivered pups, with all 31 pups surviving and appearing healthy for the duration of the experiment. BKIs 1534 and 1649 dosed at therapeutic levels led to aborted pregnancies or stillbirths in most treated dams (Table 2).
Table 2.
Pregnancy safety in IFN-γ knockout mice dosed with a placebo or bumped kinase inhibitors (BKIs) at therapeutic levels.
| BKI | Dose (mg/kg of body weight in corn oil) |
Dams (confirmed pregnant/mated) |
Surviving pups
|
|
|---|---|---|---|---|
| Day 0 postpartum | Day 3 postpartum | |||
| Placebo | Corn oil | 6/6 | 36 (from all dams) | 35 |
| 1369 | 60 | 5/6 | 31 (from all pregnant dams) | 31 |
| 1534 | 50 | 4/7 | 5 (all from one dam, 2 other dams aborted) | 5 |
| 1649 | 6 | 4/6 | 4 (stillborn) | 0 |
3.4. In vivo dose response
BKI 1369 was prioritized for further evaluation based on the pregnancy safety and toxicity results. Efficacy testing of BKI 1369 previously showed a significant effect on C. parvum infection in IFN-γ KO mice with 60 mg/kg QD for 5 days (Hulverson et al., 2017). The efficacy of titrated doses at 5, 15 and 30 mg/kg QD, and 60 mg/kg QD for 5 days, was predicted using Gastroplus modelling (Arnold et al., 2017). The Gastroplus predictions (Fig. 1A) of the efficacy of a BKI 1369 dose response was tested by dosing C. parvum-infected adult IFN-γ KO mice. Predicted efficacy was confirmed or exceeded for all doses when tested in vivo (Fig. 1B), with the exception of the 5 mg/kg treatment group, which was underestimated. However, the difference between the predicted and actual efficacies for the 5 mg/kg of BKI 1369-treated group was not statistically significant (P > 0.1). All doses significantly reduced infection levels in mice compared with the control (P < 0.05), with 30 mg/kg QD for 5 days showing no significant difference (P > 0.1) compared with 60 mg/kg QD for 5 days. Plasma samples taken from treated mice 2 h after the last dose displayed BKI 1369 concentrations of 3.5 ±1.4 μM for the 60 mg/kg dose, 1.6 ± 0.2 μM for the 30 mg/kg dose, 1.2 ± 0.2 μM for the 15 mg/kg dose, and 0.7 ± 0.1 μM for the 5 mg/kg dose.
Fig. 1.
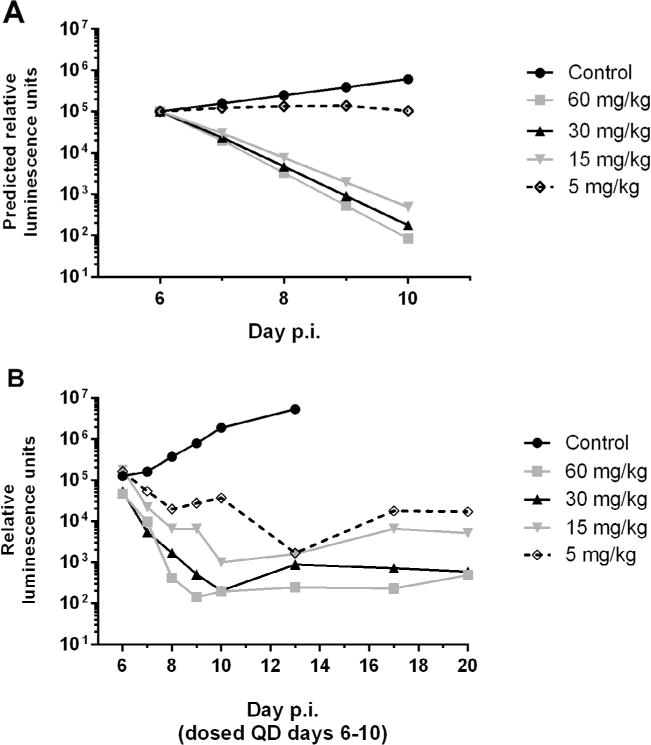
Predicted and observed bumpked kinase inhibitor efficacy against Cryptosporidium parvum in adult female IFN-γ knockout mice. (A) Gastroplus predicted values for titrated oral (PO) doses of BKI 1369 given once daily (QD) on days 6–10 p.i. (B) Observed in vivo efficacy of titrated PO doses of BKI 1369 given QD on days 6–10 p.i.
3.5. In vitro potency, metabolic stability and plasma protein binding
Plasma protein binding of BKI 1369 was determined for multiple species and found to be somewhat variable, with mouse plasma binding BKI 1369 the least (37% bound) and calf plasma binding 1369 the most (93% bound) (Table 3). BKI 1369 was separately tested for stability against CYP enzymes in pooled liver microsomes from humans, mice, rats, dogs, cattle (adult Bos indicus (Zebu)), cats, and monkeys (Table 3). All species’ microsome assays yielded a half-life of >120 min. Additionally, BKI 1369 was assayed for CYP inhibition against CYP3A4 and CYP2D6. CYP3A4 was not significantly inhibited up to 100 μM, while CYP2D6 was inhibited with an IC50 of 18.6 μM (Fig. 2A, B). BKI 1369 was found to be 20% bound to microsomal protein, giving it an IC50unbound value of 14.9 μM against CYP2D6. BKI 1369 in vitro efficacy against Cryptosporidium was assayed using high-content imaging against additional strains and species of Cryptosporidium (Table 4). Results showed a lower half maximal effective concentration (EC50) against Cryptosporidium spp. than nitazoxanide on both C. parvum and Cryptosporidium hominis for the strains tested.
Table 3.
Microsomal stability and plasma protein binding of bumped kinase inhibitor 1369.
| Species | Microsomal stability Time (min) |
Plasma protein binding (% bound) |
|---|---|---|
| Human | >120 | 77 |
| Mouse | >120 | 37 |
| Rat | >120 | 76 |
| Cattle | >120 | 93 |
| Dog | >120 | 40 |
| Cat | >120 | ND |
| Monkey | >120 | ND |
ND, not determined.
Fig. 2.
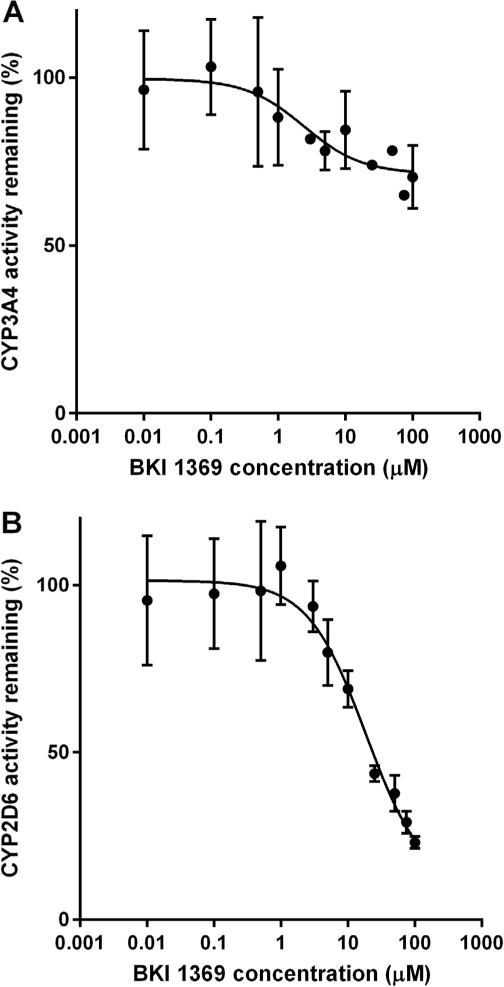
Bumped kinase inhibitor 1369 inhibition of Cytochrome P450 enzymes. (A) Inhibition of CYP3A4. (B) Reversible inhibition of CYP2D6. Half maximal inhibitory concentration (IC50)unbound = 14.9 μM.
Table 4.
In vitro potency of bumped kinase inhibitor 1369 against various Cyptosporidium strains.
| Cryptosporidium strain | EC50 (mM)
|
|
|---|---|---|
| BKI 1369 | Nitazoxanide | |
| UGA1 N-luciferase expressing C. parvuma | 2.4 | 3.1 |
| University of Arizonab C. parvum | 0.78 | 1.68 |
| Bunch Grass Farm C. parvum c | 1.11 | 1.90 |
| Tufts University TU502 C. hominisc | 0.28 | 2.78 |
Assayed using luminiscence signal to determine Cryptosporidium proliferation.
University of Arizona Cryptosporidium Propagation Laboratory.
Assayed using high-content imaging for Cryptosporidium proliferation.
3.6. Pharmacokinetics
Adult BALB/c mice were given a single oral dose of BKI 1369 at 60 mg/kg in three different vehicles to determine vehicle effects on plasma exposure (Fig. 3). The vehicle consisting of 60% phosal 53 MCT/30% polyethylene glycol 400/10% ethanol (phosal) showed similar maximum concentrations (Cmax) and areas under the curve (AUC) to those observed when dosing in ETS. The 100% corn oil vehicle showed a slightly lower Cmax and AUC, but slightly longer half-life. All three vehicles had a similar plasma concentration of approximately 1 μM at 24 h post dose in all mice.
Fig. 3.
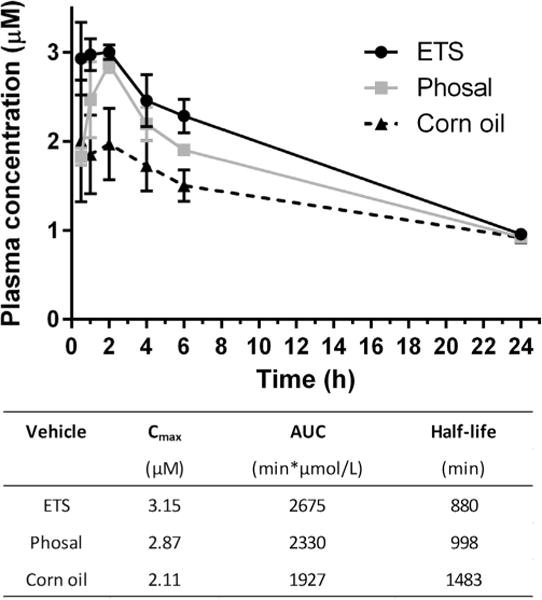
Plasma concentrations in mice after a single oral dose of bumped kinase inhibitor 1369 (60 mg/kg of body weight) in various vehicles. ETS, 3% ethanol/7% Tween 80/90% sal; Phosal, 60% Phosal 53 MCT/30% polyethylene glycol/10% ethanol.
A single dose of 30 mg/kg in mice was also used to determine exposure in tissues of the duodenum, jejunum and ileum, cecum and ascending colon, as previously described (Arnold et al., 2017), and in the plasma and brain for insights into possible CNS penetration (Vidadala et al., 2016). The resulting tissue concentrations were compared with predicted values from Gastroplus modelling (Fig. 4A–C). As predicted, experimentally determined concentrations of BKI 1369 quickly rose in the duodenum, and jejunum and ileum at 0.5–2 h, before falling to lower levels and dissipating over time. Gastoplus predicted a Cmax to be achieved in the duodenum at <0.5 h, so the actual Cmax was likely missed in the in vivo experiment (Fig. 4A, B). Conversely, the cecum and ascending colon concentrations rose more gradually and maintained a lower, yet more level, concentration over the observed 12 h time period (Fig. 4B). BKI 1369 penetrated the blood brain barrier, with brain tissue concentrations reaching a 1:1 ratio with plasma concentration and maintaining an equivalent elimination rate to plasma after 4 h (Fig. 4C).
Fig. 4.
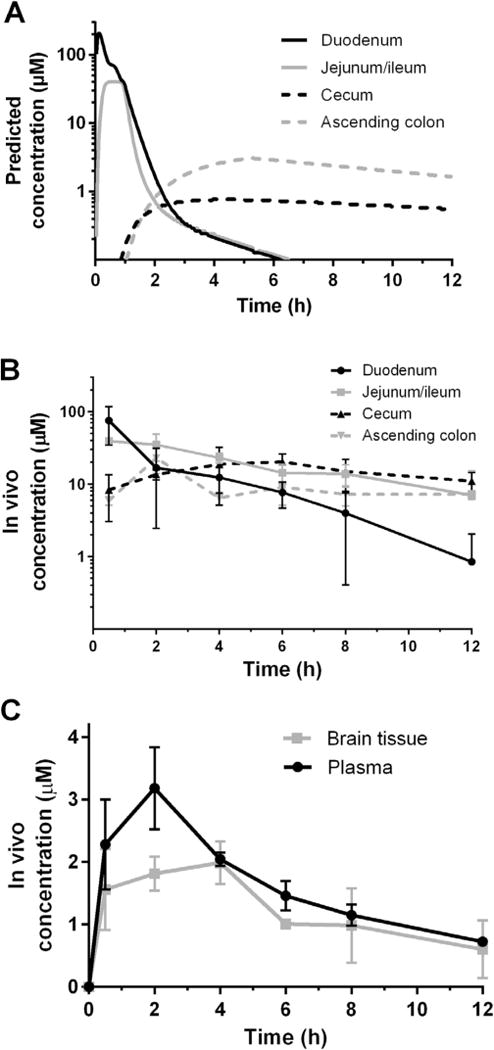
Bumped kinase inhibitor (BKI) 1369 concentrations in mouse gastrointestinal tissues, plasma and brain tissue after a single oral dose of BKI 1369 at 30 mg/kg of body weight. (A) Gastroplus predicted concentrations of BKI 1369 in gastrointestinal tract tissues. (B) In vivo concentrations of gastrointestinal tract tissues. (C) In vivo brain tissue concentrations and predicted and in vivo plasma concentrations on BKI 1369.
3.7. Metabolite identification, formation and excretion
Plasma samples taken from adult BALB/c mice dosed PO with 60 mg/kg of BKI 1369 were analysed on LC-MS/MS, yielding the positive identification of two major metabolites in vivo (Fig. 5). The first metabolite (metabolite 1) was formed by a demethylation of the methyl-piperidine-methyl group, resulting in an identical structure to the previously investigated BKI 1318 (Hulverson et al., 2017). This compound was observed to be active in vitro against C. parvum (EC50 = 3.2 μM), but lacked efficacy when orally dosed in vivo in mice. The second metabolite (metabolite 2) was formed by a conversion of the quinoline ethyl ether to a 2-oxy-quinoline. This metabolite was inactive in vitro against Nluc expressing C. parvum, with an EC50 of >20 μM. Metabolite 2 was also tested for in vitro cytotoxicity against mammalian CRL-8155 and HepG2 cells and showed no toxicity up to 80 μM. Similarly, metabolite 1 (BKI 1318) has shown no toxicity against these cell lines (Hulverson et al., 2017).
Fig. 5.
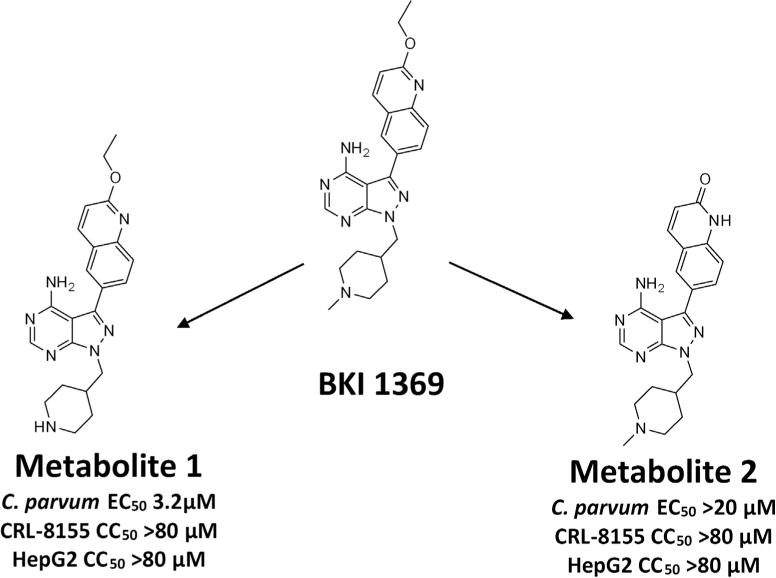
Metabolite identification. Adult BALB/c mice orally dosed with bumped kinase inhibitor (BKI) 1369 (60 mg/kg of body weight) yielded two major metabolites based on LC-MS/MS analysis of plasma samples. They were a product of the demethylation of the BKI 1369 methyl-piperidine-methyl group (metabolite 1) or a conversion of the quinoline ethyl ether to a 2-oxy-quinoline (metabolite 2). Each of the metabolites appears to be non-toxic to human cells.
Plasma samples for all time points from the previous 30 mg/kg dose experiment, where GI tissues and brains were collected (Fig. 4), were then analysed by LC-MS/MS to quantify concentrations of BKI 1369, metabolite 1 and metabolite 2 (Fig. 6). Metabolite 1 was observed to rise to approximately 25% of the AUC of BKI 1369. The observed in vivo elimination rate of metabolite 1 appeared to be identical to BKI 1369, suggesting that its elimination was formation rate limited. Metabolite 2 maintained a consistent concentration of approximately 0.5 μM for 12 h post dose, even as parent BKI 1369 and metabolite 1 were eliminated.
Fig. 6.
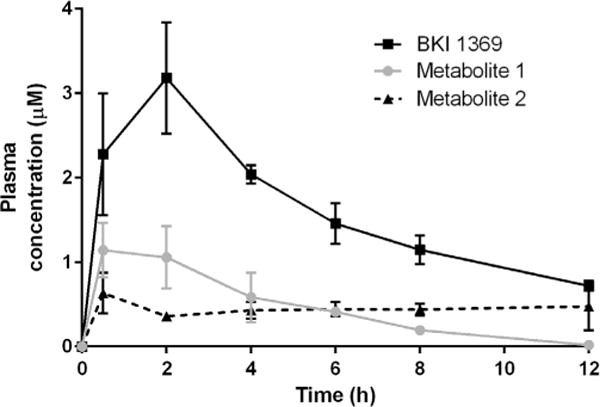
Bumped kinase inhibitor 1369 and metabolite concentrations in mouse plasma following a single dose (30 mg/kg of body weight) in 3% ethanol/7% Tween 80/90% saline.
BKI 1369 concentrations in plasma, urine and faeces were analysed from samples taken at intervals over a 48 h period after a single 60 mg/kg oral dose (Fig. 7). Concentrations and the rate of elimination in plasma (Fig. 7A) were observed to be similar for BKI 1369 and metabolite 2 to those seen after the 30 mg/kg dose (Fig. 6), with exposure increasing linearly for the increased dose. Metabolite 1 concentrations and the rate of elimination in plasma were approximately equivalent for both the 30 and 60 mg/kg doses (Figs. 6 and 7A), further supporting the observation that its formation rate is limited. Excretion of BKI 1369 in urine (Fig. 7B) showed an observed renal clearance rate of 67.2 μL/min, in agreement with the predicted renal clearance in female mice of 88 μL/min (fractional unbound * glomerular filtration rate) (Qi et al., 2004). Excretion in the faeces (Fig. 7C) demonstrated that exposure within the GI tract is sustained longer than systemic exposure. Concentrations of BKI 1369 rose to a maximum in faeces between 16 and 24 h, and remained at detectable levels through 48 h.
Fig. 7.
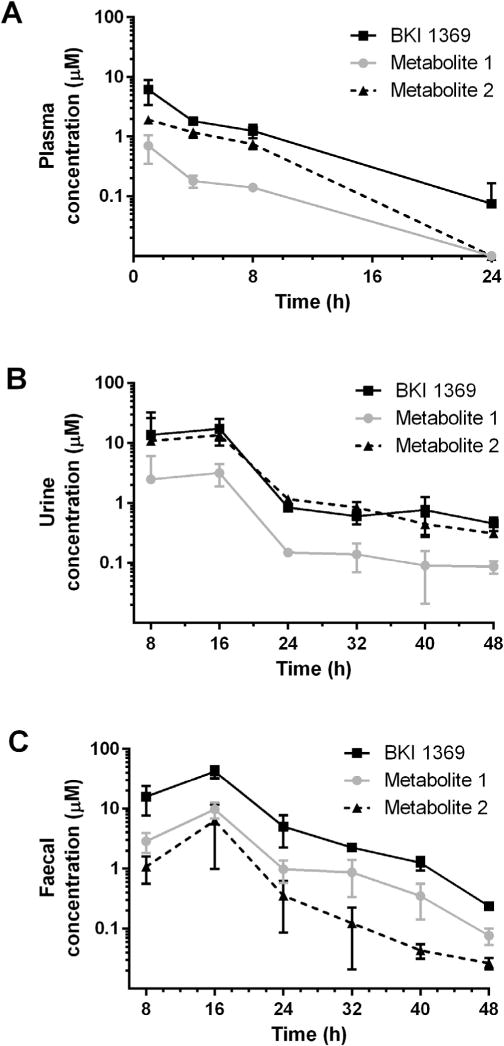
Bumped kinase inhibitor 1369, metabolite 1 and metabolite 2 concentrations in mice in plasma, urine and faeces over 24–48 h after a single oral dose (60 mg/kg of body weight) in 0.2 mL of 3% ethanol/7% Tween 80/90% saline. (A) Plasma concentrations. (B) Urine concentrations. (C) Faecal concentrations.
3.8. Additional toxicity and cardiotoxicity screening
BKI 1369 was tested for additional toxicity signals including the in vitro micronucleus and S9-activated 24-well Ames screening assay for genotoxicity (Table 5). The Ames 24-well screen result was negative, but the in vitro micronucleus test was positive. The high frequency of false positives for in vitro micronucleus assays (Magkoufopoulou et al., 2011; Fowler et al., 2012) indicates the need to further explore this compound in vivo for micronucleus formation within bone marrow or blood erythrocyte cells in rats. A kinome bioprofile of BKI 1369 was constructed using 81 human kinases for potential off-target toxicity. The kinome profile yielded only three hits at 10 μM, with IC50s of 0.036 μM for PRKCN, 0.775 μM for RIPK2, and 1.08 μM for PI3K (p110delta/p85alpha). The remaining 78 kinases profiled in this assay all resulted in an IC50 of >10 μM. The results for BKI 1369 in the 81 kinase panel are consistent with a kinobead lysate profiling assay, where only PRKCN1/2/3 show sub-micromolar competition of the >200 kinases assayed (Golkowski et al., 2017).
Table 5.
Genotoxicity screening using the 24-well Ames and in vitro micronucleus assays, and cardiotoxicity screening using the Qpatch assay for ether-à-go-go channel activity and the QTiSA-HT assay for contractility, effective refractory period, and threshold excitability of bumped kinase inhibitor 1369.
| 24-well Ames assay | In vitro micronucleus | Qpatch hERG | Change in contractility from buffer (%)
|
Change in ERP from buffer (ms)
|
Change in threshold excitability from buffer (V/cm)
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) |
IC50 (μM) |
MSC (μM) |
30 μM (% change) |
IC50 (μM) |
MSC (μM) |
30 μM (% change) |
IC50 (μM) |
MSC (μM) |
30 μM (% change) |
||
| Negative | Positive | 1.51 | 20 | 4.17 | −57.4 | >30 | 6.68 | 42.3 | >30 | 9.06 | 0.7 |
MSC, minimum significant concentration.
BKI 1369 previously showed a result of 10 μM in the in vitro thallium flux assay for hERG activity (Hulverson et al., 2017). This demonstrated the need for additional testing for cardiotoxicity liabilities, so BKI 1369 was assayed in the QPatch assay for hERG channel activity that could lead to QT interval elongation and the QTiSA-HT assay for repolarization (QT), inotropicity (i), and excitability caused by sodium channel blockade in a high throughput (HT) screening assay (SA) (Table 5). In the QTiSA-HT assays, minimum significant concentrations (MSCs) of 4.17 μM for contractility, 6.68 μM for change in effective refractory period (ERP), and 9.06 μM for change in threshold excitability were obtained (Table 5). The QPatch IC50 result of 1.51 μM was the lowest value and is of concern. For comparison of safety parameters with minimum efficacious dose, we used mouse efficacy peak plasma concentrations, as only in mice have we performed a complete dose response for efficacy. At the minimum dose for complete efficacy in mice, 30 mg/kg, the peak plasma levels were 1.6 ± 0.2 μM. Thus, each of the QTiSA MSC levels are below the plasma exposure, but the Q-Patch level is approximately the same as the peak plasma exposure. However, given the 77% human plasma protein binding of BKI 1369, the fractional unbound peak concentration during effective therapy would remain below 1 μM, suggesting that a safety window exists. These studies highlight the need for further in vivo cardiotoxicity screening of BKI 1369 to better determine its therapeutic window.
To assess the cardiotoxicity of BKI 1369 in rats, three rats were dosed with IV infusions of 3, 10 and 30 mg/kg. Plasma samples were taken and rats were observed for effects on mean arterial pressure (MAP), heart rate (HR), and left ventricular contractility (dT/dP@50) (Table 6). No significant effects, considered to be a change of >15%, were observed on MAP at any of these dose concentrations and significant effects were only seen in HR and dT/dP@50 at 30 mg/kg, which reached a Cmax of 5.0 ±0.5 μM in plasma. This demonstrates that a window of safety still exists from the minimum efficacious exposure in mice of 1.6 ± 0.2 μM, but cardiotoxicity is the toxicity parameter that must be probed in further animal models and in human testing if BKI 1369 is advanced to a clinical candidate.
Table 6.
Rat cardiotoxicity screening of bumped kinase inhibitor 1369.
| IV infusion dose (mg/kg) |
Maximum plasma concentration ± S.D. (μM) |
MAP (% change) |
HR (% change) |
dP/dt@50 (% change) |
|---|---|---|---|---|
| 3 | 0.73 ± 0.7 | −1 | −4 | 3 |
| 10 | 2.62 ± 0.3 | −7 | −10 | −3 |
| 30 | 5.00 ± 0.5 | −12 | −21 | −23 |
A change of ± >15% was considered significant.
MAP, mean arterial pressure; HR, heart rate; dT/dP@50, left ventricular contractility.
3.9. Efficacy in a calf clinical model
An oral dose of 5 mg/kg twice daily (BID) for 5 days (days 2–6 p. i.) was administered to neonatal calves infected with C. parvum. No toxicity was observed with BKI 1369 therapy. Calves treated with BKI 1369 showed significant improvement compared with controls for daily clinical health score on days 5–7 (P < 0.05), total daily faecal volumes on days 4–8 (P < 0.05), and faecal consistency scores on days 4–8 (P < 0.05) (Fig. 8A–C). Diarrhoea became less severe in treated calves within 24–72 h of the start of treatment, resolved by days 6–7, and did not return. Additionally, urine output and net weight gain were significantly higher for treated calves compared with controls (Table 7), supporting the observation that treated calves were less dehydrated and had a shorter duration of diarrhoea. The mean amount of C. parvum DNA shed daily by treated calves was significantly lower than controls (P < 0.05) for days 3–8 (Fig. 8D). The mean total numbers of oocysts shed in faeces, as measured by C. parvum DNA, over the duration of observation were measured at 3.1 × 109 for the treated calves and 8.9 × 1010 for the controls, representing an approximately 30-fold reduction in parasite excretion by calves undergoing BKI 1369 treatment. Faecal and plasma samples were analysed to determine BKI 1369 concentrations in the treated group (Table 7). Plasma concentrations of BKI 1369 at 12 h after dosing reached 5.3 ± 2.4 μM. Metabolites 1 and 2 were also observed in calf plasma (Table 7), but proportionate to one another, each appeared to be formed and eliminated at much different rates than in mice. Faecal concentrations of BKI 1369 were observed throughout the dosing period, reaching a high of 9.4 ± 6.1 μM and remaining at detectable levels for 48 to 96 h after the final dose.
Fig. 8.
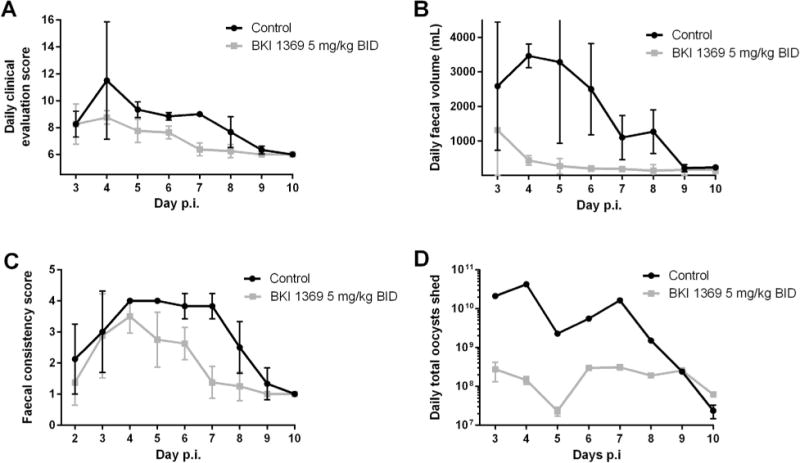
Efficacy of bumped kinase inhibitor 1369 in the Cryptosporidium parvum-infected neonatal calf clinical model. Calves were dosed twice daily (BID) orally (5 mg/kg of body weight) on days 2–6 p.i. (A) Daily clinical scores. (B) Daily faecal volumes. (C) Daily faecal consistency scores. (D) Daily total oocysts shed.
Table 7.
Neonatal calf clinical model for efficacy of bumped kinase inhibitor 1369 against Cryptosporidium parvum.
| Experimental groups | Total urine output (L) |
Net weight gain (% change) |
BKI 1369 plasma conc. at 12 h after 2nd dose (Avg ± S.D.) (μM) |
Metabolite 1 plasma conc. at 12 h after 2nd dose (Avg ± S.D.) (μM) |
Metabolite 2 plasma conc. at 12 h after 2nd dose (Avg ± S.D.) (μM) |
Highest observed BKI 1369 conc. in faeces (Avg ± S.D.) (μM) |
|---|---|---|---|---|---|---|
| Control (n = 3) | 30.2 | −0.06 | NA | NA | NA | NA |
| Treated (n = 4) | 53.7 | +4.7 | 5.3 ± 2.4 | 0.2 ± 0.1 | 8.8 ± 3.5 | 9.4 ± 6.1 |
Calves were treated twice daily with oral doses (5 mg/kg of body weight) of bumped kinase inhibitor 1369 for 5 days.
Conc., concentration; NA, not applicable; Avg, average.
4. Discussion
The prevalence, mortality and morbidity of cryptosporidiosis in malnourished children under 2 years of age speaks to the need for a new efficacious drug for Cryptosporidium therapy. Yet the vulnerability of the malnourished and <2 year olds target population necessitates that any new therapies will satisfy a strict set of safety requirements. Despite their excellent efficacies in mice, the failure of BKIs 1534 and 1649 in the mouse pregnancy safety tests eliminates both from further consideration for treatment of this target group, as safety in pregnancy is part of the paediatric safety package required by the FDA. The additional toxicities observed during multiple dosing, through interference with peristalsis for BKI 1534 and accumulation at extremely high systemic concentrations for BKI 1649, also limit their potential as reasonably safe therapeutic options for treatment in any other age group for humans or for veterinary use. Conversely, safety of BKI 1369 in the mouse pregnancy model, its lack of toxicity until multiple doses at 300 mg/kg (>10 times its efficacious dose for mice), and its relatively low systemic exposure, keep it in place as a viable candidate for human and animal use. Cardiotoxicity, discussed below, remains the toxicity parameter to monitor for BKI 1369.
BKI 1369 has consistently shown relatively low systemic exposures. Even at a high dose of 150 mg/kg daily in mice, plasma levels only reached an average of 3.9 ± 1.5 μM (Table 1). This could prove to be a valuable asset for BKI 1369 as a potential therapy, as low systemic exposure will most likely alleviate many of its potential toxicity liabilities (Charmot, 2012). With therapeutic doses only reaching plasma concentrations of 5 μM or less in mice, rats and calves, CNS toxicity is less likely, as brain tissue concentrations of BKI 1369 remain approximately equal to its plasma concentrations and is eliminated from the brain at the same rate as plasma. Its high plasma protein binding in cattle and humans could further mitigate toxicity risks in these important species, as fractional unbound drug levels will be below concentrations of concern. Conversion of BKI 1369 to non-toxic metabolites which are similarly eliminated and never reach high concentrations in plasma is also beneficial in terms of toxicity.
Low systemic exposures will prove to be especially important in terms of cardiotoxicity liabilities of BKI 1369. The minimum significant concentration of 4.17 μM for contractility seen in the QTiSA-HT assay matches the results of the in vivo rat cardiovascular screen, where plasma levels had to rise to 5.0 ± 0.5 μM with the highest dose before causing a significant change in contractility or HR. This threshold is not reached at therapeutic level treatments in mice, suggesting that there is still an opportunity to develop a sufficiently safe treatment regimen for both animals and humans. Potential cardiotoxicity, as demonstrated in the Q-Patch, Q-TiSA, and rat cardiovascular assays, is the parameter that will have to be monitored if BKI 1369 is advanced as a clinical candidate. It will be worth exploring BKI 1369 in additional in vivo cardiovascular screens, and particularly in dogs, where hERG inhibition may demonstrate prolonged QT intervals with greater sensitivity than in rats.
Despite its low systemic exposure, the compound is relatively stable and remains present in the system for longer than 12 h after a therapeutic dose. Its prolonged exposure in the faeces of over 40 h post dose is of obvious benefit due to localisation of Cryptosporidium in the gut, but its sustained levels in plasma and urine over 24–48 h indicate that much of the compound is absorbed and metabolized. There are some benefits to this systemic exposure, as immunocompromised patients, especially HIV-positive individuals, may require exposures outside of the GI tract for therapy to be successful (Hunter and Nichols, 2002). Further studies on exposure to BKI 1369 in bile duct cannulated rats would be warranted since the biliary tract is a possible repository of infection in immunocompromised patients (Verdon et al., 1998). Inhibition of additional CYP enzymes by BKI 1369 should also be explored to avoid drug-to-drug interactions or effects on treatments for certain coinfections. This is especially important as the prevalence of cryptosporidiosis in Africa and other tropical or resource limited areas (Kotloff et al., 2013) increases the likelihood of co-treatments for HIV, malaria or other infectious diseases.
BKI 1369 clearly has potential advantages over the current clinical treatment, nitazoxanide. BKI 1369 is able to clear Cryptosporidium infection in immunocompromised mice, which nitazoxanide is unable to do (Theodos et al., 1998). This would be an obvious advantage if this result were to translate to humans or other animal species, as nitazoxanide has also been ineffective at treating Cryptosporidium in HIV-positive and other immune compromised patients (Amadi et al., 2009) and calves (Schnyder et al., 2009). It is also worth noting that BKI 1369 is more potent in vitro than nitazoxanide against C. hominis and three different strains of C. parvum (Table 4). BKI 1369 activity against C. hominis, the dominant species in humans, is especially significant from a public health perspective. Further testing against additional C. hominis strains would be ideal to confirm broad efficacy. However, strains adapted for stable laboratory propagation are very limited at this time.
The successful result in the neonatal calf clinical model is one of the strongest arguments for advancing BKI 1369 as a human and veterinary candidate. Cryptosporidium parvum DNA excretion was reduced approximately 30-fold and dropped significantly after a single dose, remaining well below the control counts for the duration of dosing. However, C. parvum DNA was still detectable in the stool of treated calves, even after therapy with BKI 1369. Whether this represents intact, infective oocysts requires further experimentation to attempt to transmit the infection to susceptible animals. Other BKIs have previously improved clinical scores and reduced oocyst counts and diarrhoea severity significantly in calves, but failed to fully alleviate diarrhoea or other clinical signs (Schaefer et al., 2016). BKI 1369 significantly improved all clinical signs including diarrhoea and associated dehydration, leading to improved weight gain and absence of significant signs of illness after only 5 days of treatment. Thus, we conclude that BKI 1369 remains a viable preclinical candidate for human and animal studies.
Supplementary Material
Acknowledgments
The authors thank Geno De Hostos and Robert Choy from PATH Drug Solutions (San Francisco, CA, USA) for their consultation in the drug development pathway. Research studies reported in this publication were supported by National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH), USA, under award numbers R01AI089441, R01AI111341 and R01HD080670. The work was also supported by award number 2014-06183 from the United States Department of Agriculture National Institute of Food and Agriculture.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ijpara.2017.08.006.
References
- Amadi B, Mwiya M, Musuku J, Watuka A, Sianongo S, Ayoub A, Kelly P. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial. Lancet. 2002;360:1375–1380. doi: 10.1016/S0140-6736(02)11401-2. [DOI] [PubMed] [Google Scholar]
- Amadi B, Mwiya M, Sianongo S, Payne L, Watuka A, Katubulushi M, Kelly P. High dose prolonged treatment with nitazoxanide is not effective for cryptosporidiosis in HIV positive Zambian children: a randomised controlled trial. BMC Infect Dis. 2009;9:195. doi: 10.1186/1471-2334-9-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SLM, Choi R, Hulverson MA, Schaefer DA, Vinayak S, Vidadala RSR, McCloskey MC, Whitman GR, Huang W, Barrett LK, Ojo KK, Fan E, Maly DJ, Riggs MW, Striepen B, Van Voorhis WC. Necessity of bumped kinase inhibitor gastrointestinal exposurein treating Cryptosporidium infection. J Infect Dis. 2017;216:53–63. doi: 10.1093/infdis/jix247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfor PN, Gintant GA, Lipari JM, Zocharski PD. A novel intravenous vehicle for preclinical cardiovascular screening of small molecule drug candidates in rat. J Pharmacol Toxicol Methods. 2016;82:62–67. doi: 10.1016/j.vascn.2016.07.002. [DOI] [PubMed] [Google Scholar]
- Charmot D. Non-systemic drugs: a critical review. Curr Pharm Des. 2012;18:1434–1445. doi: 10.2174/138161212799504858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danker T, Moller C. Early identification of hERG liability in drug discovery programs by automated patch clamp. Front Pharmacol. 2014;5:203. doi: 10.3389/fphar.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler P, Smith K, Young J, Jeffrey L, Kirkland D, Pfuhler S, Carmichael P. Reduction of misleading (“false”) positive results in mammalian cell genotoxicity assays. I. Choice of cell type. Mutat Res. 2012;742:11–25. doi: 10.1016/j.mrgentox.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Goedken ER, Argiriadi MA, Banach DL, Fiamengo BA, Foley SE, Frank KE, George JS, Harris CM, Hobson AD, Ihle DC, Marcotte D, Merta PJ, Michalak ME, Murdock SE, Tomlinson MJ, Voss JW. Tricyclic covalent inhibitors selectively target Jak3 through an active site thiol. J Biol Chem. 2015;290:4573–4589. doi: 10.1074/jbc.M114.595181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkowski M, Vidadala RS, Lombard CK, Suh HW, Maly DJ, Ong SE. Kinobead and single-shot LC-MS profiling identifies selective PKD inhibitors. J Proteome Res. 2017;16:1216–1227. doi: 10.1021/acs.jproteome.6b00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulverson MA, Vinayak S, Choi R, Schaefer DA, Castellanos-Gonzalez A, Vidadala RS, Brooks CF, Herbert GT, Betzer DP, Whitman GR, Sparks HN, Arnold SL, Rivas KL, Barrett LK, White AC, Jr, Dustin JM, Riggs MW, Striepen B, Van Voorhis WC, Ojo KK. Bumped-kinase inhibitors for therapy of cryptosporidiosis. J Infect Dis. 2017 doi: 10.1093/infdis/jix120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter PR, Nichols G. Epidemiology and clinical features of Cryptosporidium infection in immunocompromised patients. Clin Microbiol Rev. 2002;15:145–154. doi: 10.1128/CMR.15.1.145-154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acácio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- Love MS, Beasley FC, Jumani RS, Wright TM, Chatterjee AK, Huston CD, Schultz PG, McNamara CW. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl Trop Dis. 2017;11:e0005373. doi: 10.1371/journal.pntd.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magkoufopoulou C, Claessen SM, Jennen DG, Kleinjans JC, van Delft JH. Comparison of phenotypic and transcriptomic effects of false-positive genotoxins, true genotoxins and non-genotoxins using HepG2 cells. Mutagenesis. 2011;26:593–604. doi: 10.1093/mutage/ger021. [DOI] [PubMed] [Google Scholar]
- Murphy RC, Ojo KK, Larson ET, Castellanos-Gonzalez A, Perera BG, Keyloun KR, Kim JE, Bhandari JG, Muller NR, Verlinde CL, White AC, Merritt EA, Van Voorhis WC, Maly DJ. Discovery of Potent and Selective Inhibitors of Calcium-Dependent Protein Kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med Chem Lett. 2010;1:331–335. doi: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolette J, Diehl M, Sonders P, Bryce S, Blomme E. In vitro micronucleus screening of pharmaceutical candidates by flow cytometry in Chinese hamster V79 cells. Environ Mol Mutagen. 2011;52:355–362. doi: 10.1002/em.20631. [DOI] [PubMed] [Google Scholar]
- Ojo KK, Eastman RT, Vidadala R, Zhang Z, Rivas KL, Choi R, Lutz JD, Reid MC, Fox AM, Hulverson MA, Kennedy M, Isoherranen N, Kim LM, Comess KM, Kempf DJ, Verlinde CL, Su XZ, Kappe SH, Maly DJ, Fan E, Van Voorhis WC. A Specific Inhibitor of PfCDPK4 Blocks Malaria Transmission: Chemical-genetic Validation. J Infect Dis. 2014;209:275–284. doi: 10.1093/infdis/jit522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo KK, Pfander C, Mueller NR, Burstroem C, Larson ET, Bryan CM, Fox AM, Reid MC, Johnson SM, Murphy RC, Kennedy M, Mann H, Leibly DJ, Hewitt SN, Verlinde CL, Kappe S, Merritt EA, Maly DJ, Billker O, Van Voorhis WC. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J Clin Investig. 2012;122:2301–2305. doi: 10.1172/JCI61822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platts-Mills JA, Babji S, Bodhidatta L, Gratz J, Haque R, Havt A, McCormick BJ, McGrath M, Olortegui MP, Samie A, Shakoor S, Mondal D, Lima IF, Hariraju D, Rayamajhi BB, Qureshi S, Kabir F, Yori PP, Mufamadi B, Amour C, Carreon JD, Richard SA, Lang D, Bessong P, Mduma E, Ahmed T, Lima AA, Mason CJ, Zaidi AK, Bhutta ZA, Kosek M, Guerrant RL, Gottlieb M, Miller M, Kang G, Houpt ER, Investigators, M.-E.N. Pathogen-specific burdens of community diarrhoea in developing countries: a multisite birth cohort study (MAL-ED) Lancet Glob Health. 2015;3:e564–575. doi: 10.1016/S2214-109X(15)00151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Whitt I, Mehta A, Jin J, Zhao M, Harris RC, Fogo AB, Breyer MD. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol Renal Physiol. 2004;286:F590–596. doi: 10.1152/ajprenal.00324.2003. [DOI] [PubMed] [Google Scholar]
- Riggs MW, McGuire TC, Mason PH, Perryman LE. Neutralization-sensitive epitopes are exposed on the surface of infectious Cryptosporidium parvum sporozoites. J Immunol. 1989;143:1340–1345. [PubMed] [Google Scholar]
- Riggs MW, Perryman LE. Infectivity and neutralization of Cryptosporidium parvum sporozoites. Infect Immun. 1987;55:2081–2087. doi: 10.1128/iai.55.9.2081-2087.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer DA, Betzer DP, Smith KD, Millman ZG, Michalski HC, Menchaca SE, Zambriski JA, Ojo KK, Hulverson MA, Arnold SL, Rivas KL, Vidadala RS, Huang W, Barrett LK, Maly DJ, Fan E, Van Voorhis WC, Riggs MW. Novel Bumped Kinase Inhibitors Are Safe and Effective Therapeutics in the Calf Clinical Model for Cryptosporidiosis. J Infect Dis. 2016;214:1856–1864. doi: 10.1093/infdis/jiw488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnyder M, Kohler L, Hemphill A, Deplazes P. Prophylactic and therapeutic efficacy of nitazoxanide against Cryptosporidium parvum in experimentally challenged neonatal calves. Vet Parasitol. 2009;160:149–154. doi: 10.1016/j.vetpar.2008.10.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatipaka HB, Gillespie JR, Chatterjee AK, Norcross NR, Hulverson MA, Ranade RM, Nagendar P, Creason SA, McQueen J, Duster NA, Nagle A, Supek F, Molteni V, Wenzler T, Brun R, Glynne R, Buckner FS, Gelb MH. Substituted 2-phenylimidazopyridines: a new class of drug leads for human African trypanosomiasis. J Med Chem. 2014;57:828–835. doi: 10.1021/jm401178t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodos CM, Griffiths JK, D’Onfro J, Fairfield A, Tzipori S. Efficacy of nitazoxanide against Cryptosporidium parvum in cell culture and in animal models. Antimicrob Agents Chemother. 1998;42:1959–1965. doi: 10.1128/aac.42.8.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdon R, Polianski J, Grodet A, Garry L, Carbon C. Cryptosporidium parvum biliary tract infection in adult immunocompetent and immunosuppressed mice. J Med Microbiol. 1998;47:71–77. doi: 10.1099/00222615-47-1-71. [DOI] [PubMed] [Google Scholar]
- Vidadala RS, Rivas KL, Ojo KK, Hulverson MA, Zambriski JA, Bruzual I, Schultz TL, Huang W, Zhang Z, Scheele S, DeRocher AE, Choi R, Barrett LK, Siddaramaiah LK, Hol WG, Fan E, Merritt EA, Parsons M, Freiberg G, Marsh K, Kempf DJ, Carruthers VB, Isoherranen N, Doggett JS, Van Voorhis WC, Maly DJ. Development of an Orally Available and Central Nervous System (CNS) Penetrant Toxoplasma gondii Calcium-Dependent Protein Kinase 1 (TgCDPK1) Inhibitor with Minimal Human Ether-a-go-go-Related Gene (hERG) Activity for the Treatment of Toxoplasmosis. J Med Chem. 2016;59:6531–6546. doi: 10.1021/acs.jmedchem.6b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, Cipriano MJ, Striepen B. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature. 2015;523:477–480. doi: 10.1038/nature14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.