

Abstract
The mitochondrial carrier system (MCS) transports small molecules between mitochondria and the cytoplasm. It is integral to the core mitochondrial function to regulate cellular chemistry by metabolism. The mammalian MCS comprises the transporters of the 53 member canonical SLC25A family and a lesser number of identified non-canonical transporters. The recent discovery and investigations of the mitochondrial pyruvate carrier (MPC) illustrate the diverse effects a single mitochondrial carrier may exert on cellular function. However, the transport selectivities of many carriers remain unknown, and most have not been functionally investigated in mammalian cells. The mechanisms coordinating their function as a unified system remain undefined. Increased accessibility to molecular-genetic and metabolomic technologies now greatly enables investigation of the MCS. Continued investigation of the MCS may reveal how mitochondria encode complex regulatory information within chemical thermodynamic gradients. This understanding may enable precision modulation of cellular chemistry to counteract the dysmetabolism inherent in disease.
Keywords: Mitochondria, carrier, transporter, systems biology, redox cycles, MPC
The Mitochondrial Carrier System
The mitochondrial carrier system (MCS) (“see glossary”) transports small molecules between mitochondria and the cytoplasm, thereby regulating the function of both compartments [1–3]. In mammals, the MCS comprises the transporters of the 53 member canonical SLC25A family and a lesser number of identified non-canonical transporters. Mitochondrial carriers transport an array of substrates, including citric acid cycle intermediates, amino acids, nucleotides, vitamins, metals, and minerals. This diversity in transport activities confers extraordinary capacity for complex, precisely tuned regulation of cellular chemistry. Higher-order processes obligately routed through the MCS include carbon fuel oxidation, the urea cycle, redox cycles, and synthesis of sugars, lipids, pyrimidine nucleotides, and heme.
The first discovered mitochondrial carriers were identified as transport activities that could be purified from mitochondria, and several were discovered in the pre-molecular era. These include the mitochondrial ADP/ATP carrier (ANT1; SLC25A4) [4], uncoupling protein (UCP1; UCP; SLC25A7) [5], phosphate carrier (PiC; SLC25A3) [6], oxoglutarate carrier (OGC; SLC25A11) [7], citrate carrier (CIC; SLC25A1) [8], and carnitine/acylcarnitine carrier (CACT; SLC25A20) [9]. Mutations in 14 SLC25A family members including ANT1 [10], PiC [11], CIC [12, 13], and CACT [14, 15] are now known to cause human disease [3]. Many rigorously performed biochemical studies have provided a wealth of information on mitochondrial carrier substrate selectivity [16]. However, the substrates transported by many mitochondrial carriers remain undefined, most have never been functionally investigated in living mammalian cells, and the mechanisms coordinating their function as a unified system are not understood.
The recent discovery and investigations of the mitochondrial pyruvate carrier (MPC) illustrate several critical functional principles of the larger MCS [17–23]. They demonstrate the pleiotropy inherent in the function of a single mitochondrial carrier and the systems-level flexibility of the MCS to adapt to disruption. Molecular genetic technologies available when the MPC was identified have allowed rapid extension to functional metabolomic investigations in cell culture and mouse models. They now readily permit similar studies on carriers discovered decades earlier. The intent of this review is to highlight recent findings on the MPC to illustrate emerging concepts underlying the systems-level function of the MCS. This is significant because understanding the systems-level function of the MCS may provide new opportunities for therapeutic metabolic reprogramming. For detailed coverage of the history of mitochondrial carrier research, the reader is referred to several excellent review articles [1–3, 24, 25].
The Mitochondrial Pyruvate Carrier
The mitochondrial pyruvate carrier (MPC) transports pyruvate from the cytosol to the mitochondrial matrix, thereby linking glycolysis with mitochondrial metabolism. The MPC was first identified as a proton-linked transport activity in purified rat mitochondria in 1971 [26]. This characterization was followed by the identification of specific inhibitors [27], reconstitution of the MPC activity into artificial liposomes [28], and several attempts at purification and identification [29–32]. A candidate gene for the MPC was identified in S cerevisiae [33], but was subsequently shown to be an NAD+ transporter [34]. In 2003, MPC deficiency was implicated in the early death of a human patient [35]. Finally, in 2012, two groups simultaneously identified the Brp44 and Brp44L paralogs as encoding the MPC, which were renamed as MPC1 and MPC2, respectively [17, 19]. This discovery included identification of the causative mutation in the human case of MPC deficiency, which changed a conserved arginine to tryptophan [17].
The molecular identification of the MPC revealed it to be a non-canonical mitochondrial carrier. Canonical mitochondrial carrier SLC25A family members harbor a tripartite structure with three tandem repeats, and functional transporters are formed by homodimerization [3, 36]. In contrast, the MPC1 and MPC2 genes encode 12 and 14 kD proteins, respectively, that associate in an unknown heteromeric stoichiometry to form the MPC. MPC1 and MPC2 show partial similarity to the bacterial semi-SWEET sugar transports [37], which may reflect a shared heritage in microbial carbohydrate metabolism.
The discovery of the MPC has enabled molecular-genetic studies that illuminate the numerous cellular processes regulated by mitochondrial pyruvate uptake. As will be discussed in more detail in the sections that follow, overexpression, knockdown, and conditional alleles have been utilized to understand the role of the MPC in diverse systems including cancer and mouse models of diabetes [18, 20, 21, 38–43]. Nonetheless, reports on selective deletion in adult tissues are currently limited to pancreatic β-cells [44] and the liver [18, 20], which will be highlighted as examples of the role of the MPC in energy sensing and regulation of mitochondrial substrate selection. For broad and detailed coverage of the MPC the reader may refer to several recent review articles [39, 45–49].
The MPC regulates cell fate
The MPC regulates cell fate during development and in cancer. Similar to the case of human MPC deficiency [35], whole-body MPC deficiency in mice results in embryonic lethality [50–52]. Lethality occurs in part because of energetic insufficiency in the central nervous system and may result from metabolic vulnerabilities during development. This may not apply to the adult brain, as recent studies found that chemical inhibition of MPC activity in the brain attenuated neurodegeneration [53] and excitatory toxicity [54]. Another investigation found that a hypomorphic Mpc1 allele results in perinatal lethality, and that Mpc1 and Mpc2 mRNA levels in the brain, heart, and liver increase from late gestation through the early postnatal period [51]. These findings are consistent with an increased dependence upon mitochondrial pyruvate metabolism during later development and terminal differentiation. Thus, changes in MPC expression during development may help tune relative glycolytic verses oxidative glucose utilization, depending on oxygen availability and specific biosynthetic demands (Figure 1A).
Figure 1. The mitochondria pyruvate carrier regulates cellular decisions.
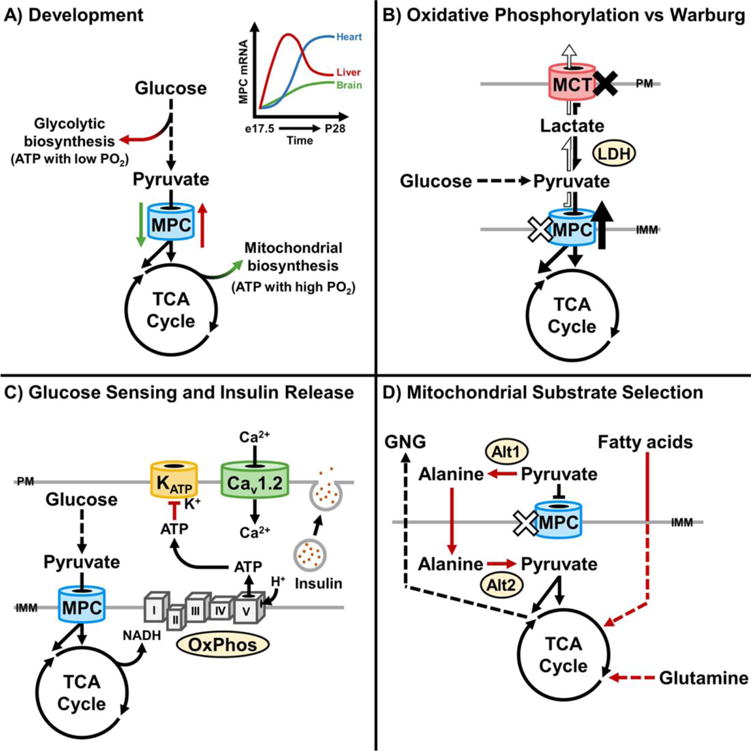
A) MPC expression changes during differentiation and development. Lower expression may facilitate glycolysis-dependent ATP production and biosynthesis when PO2 is low. Higher expression may facilitate oxidative phosphorylation and TCA cycle-dependent biosynthesis during terminal differentiation, when PO2 is increased. The upper right panel is a qualitative representation of relative changes in mouse MPC mRNA abundance from embryonic day 17.5 (e17.5) to 28 days postnatal (P28), based on quantitative changes in Mpc1 and Mpc2 mRNA abundance previously reported [51]. B) MPC activity promotes pyruvate entry into the TCA cycle and oxidative phosphorylation. Decreased MPC activity promotes the Warburg Effect, where glycolysis and lactate production are favored over oxidative phosphorylation [21, 38–43]. Inhibition of MCTs impairs cellular lactate export, increases MPC activity, and promotes pyruvate oxidation [38]. C) The MPC facilitates glucose sensing and insulin release in pancreatic β-cells [44, 52, 55]. Increased glucose levels drive MPC-dependent pyruvate oxidation. Increased pyruvate oxidation increases ATP levels. This leads to closure of ATP-gated potassium channels (KATP) and membrane depolarization. Depolarization increases calcium uptake by the voltage-gated calcium channels, thereby stimulating exocytosis of insulin granules. D) Disruption of MPC activity promotes pyruvate-alanine cycling, fatty acid oxidation, and glutaminolysis [18, 20, 22, 23, 51]. These mechanisms sustain TCA cycle flux. In hepatocytes, they also support gluconeogenesis [18, 20].
In contrast, loss of MPC activity and impaired carbohydrate oxidation may contribute to cellular transformation in cancer, where the MPC functions as a repressor of the Warburg Effect [21, 38–43]. Loss of MPC activity is associated with cancer cell stemness, and genetically increasing MPC activity decreases growth of HCT15 and HT29 colon cancer cells in spheroids and xenografts, but not in adherent culture as a monolayer [21]. Thus, in these cases, loss of MPC activity may contribute to escape from anchorage-dependent growth and tumorigenesis. Interestingly, another report shows that impairing cellular lactate export by disruption of plasma membrane monocarboxylate transporter (MCT) activity increases MPC activity [38]. This may signify the presence of an MCT-MPC axis that is dysregulated in cancer (Figure 1B). The precise mechanisms by which MPC activity regulates development and stemness remain to be discovered, but may involve links among mitochondrial substrate selection, energy sensing, and genetic control.
The MPC functions in energy sensing
The role of the MPC in energy sensing is illustrated in pancreatic β-cells, where it regulates insulin secretion (Figure 1C) [44, 52, 55]. Rises in blood glucose lead to increased β-cell glucose uptake and glycolysis. Increased glycolysis results in increased mitochondrial pyruvate uptake through the MPC, which stimulates pyruvate oxidation and ATP production. Increases in ATP close sulfonylurea sensitive KATP channels, leading to membrane depolarization, Ca2+ influx, and release of insulin granules. Disruption of MPC activity by chemical inhibition or RNAi in isolated rat islets and INS-1 cells decreases oxygen consumption, ATP levels, and glucose-stimulated insulin release [55]. In vivo, mice expressing a whole-body hypomorphic Mpc2 allele show impaired glucose-stimulated insulin secretion [52]. Mice with β-cell-restricted Mpc2 deletion are also glucose intolerant due to impaired insulin secretion [44]. Isolated islets from these mice display decreased respiration on glucose, KATP channel hyperactivity, and impaired insulin release. The same study shows congruent results in Drosophila melanogaster, where deletion of dMPC1 in insulin-producing cells leads to chronic hyperglycemia. This raises the possibility that the MPC and other components of the MCS exert specific functions in energy sensing and signaling, depending on cell type and nutritional context.
The MPC regulates broad mitochondrial substrate selection
Several studies in mouse and cell culture models illustrate the role the MPC fulfills in determining mitochondrial substrate selection (Figure 1D). Patient fibroblasts deficient in MPC activity display normal capacity for glutamine oxidation, consistent with a selective defect in pyruvate oxidation [17]. In cancer cells and cultured myoblasts, use of metabolic tracers revealed that disruption of MPC activity by chemical inhibition or RNAi increases glutamine oxidation [22, 23]. Increased glutamine-driven lipogenesis [23] and fatty acid oxidation [22] were also observed. In both cases, utilization of alternative substrates sustains TCA cycle flux when pyruvate utilization is decreased. These adaptive themes extend to mouse liver in vivo. Selective disruption of the Mpc1 or Mpc2 genes in hepatocytes decreases liver glutamine concentrations, increases fatty acid oxidation, and increases ketone levels as a secondary marker of fatty acid oxidation [18, 20]. Thus, the MPC regulates mitochondrial substrate selection by two mechanisms, directly by gating carbohydrate oxidation, and indirectly when changes in activity stimulate compensatory changes in amino acid and lipid metabolism.
These studies also illustrate a role for the MPC as a conduit for mitochondria-dependent biosynthesis. Numerous biosynthetic pathways flow through the TCA cycle and are dependent upon mitochondrial pyruvate import and thus MPC activity. Consistent with this role, 13C isotope-tracing demonstrate that disruption of the MPC decreases pyruvate-driven gluconeogenesis in vivo in the liver and ex vivo in primary hepatocytes [18, 20]. Furthermore, these studies revealed that MPC disruption decreases flux of pyruvate into citrate, which is exported from mitochondria as a precursor for cholesterol and fatty acid synthesis. In accord, serum cholesterol and fasting serum triglyceride levels were decreased in MPC liver-specific knockout mice [18]. These findings await more detailed investigation by quantitative tracing of pyruvate into the sterol and fatty acid synthesis pathways, with and without MPC disruption.
Redundant Mechanisms of Mitochondrial Pyruvate Utilization
Studies on the liver MPC also reveal how pyruvate-alanine cycling sustains mitochondrial pyruvate utilization when the MPC is disrupted [18, 20] (Figure 1D). In this case, cytosolic pyruvate is transaminated to alanine by the cytosolic alanine transaminase, Alt1. Alanine is then imported into the mitochondrial matrix and deaminated back to pyruvate by the mitochondrial alanine transaminase, Alt2. Importantly, while glutamine and fatty acids may function as alternative substrates when MPC activity is lost, pyruvate-alanine cycling functions as a mechanism for MPC-independent mitochondrial pyruvate utilization. This bypass was also observed in fibroblasts, indicating that it is not limited to hepatocytes [51].
In some cell systems, pyruvate may also bypass the MPC by pyruvate-lactate cycling through separate cytosolic and mitochondrial matrix lactate dehydrogenase activities, similar to pyruvate-alanine cycling through Alt1 and Alt2. The existence of an intracellular mitochondrial lactate-shuttle in normal differentiated tissue has been debated extensively in the literature [56, 57], with evidence for direct mitochondrial lactate metabolism in some cancer cells [58, 59]. Recently, novel double stable-isotope tracing approaches were utilized to understand the observation that lactate is preferred over glucose as a substrate for lipogenesis in cultured HeLa and H460 cells [60]. Direct mitochondrial import of lactate was proposed as a contributing mechanism. Retention of the 2-2H label on 13C, 2H-lactate in isolated mitochondria indicated that lactate was directly imported into mitochondria, rather than converted to pyruvate prior to import and re-converted to lactate during isolation. The double-labeling technique employed in this study may be generally useful for detecting mitochondrial import when the identity of the transporter in unknown, as it is for a mitochondrial lactate carrier. Overall, these and other experiments produced results consistent with oxidation of lactate to pyruvate by a mitochondria-localized lactate dehydrogenase B and subsequent flux through pyruvate dehydrogenase towards lipogenesis [60].
There and back again: From the MPC to the larger mitochondrial carrier system (MCS)
Although by gene family the MPC is a non-canonical mitochondrial carrier, its proton-linked symport mode of transport activity is utilized by several canonical SLC25A family members [2]. Furthermore, the metabolic adaptations evoked by MPC disruption demonstrate that it is functionally connected with the larger MCS. This section will highlight fundamental systems-level concepts of MCS function. Recent findings on the MPC will be utilized to illustrate several critical functional principles of the larger MCS.
The functional redundancy of the MCS is manifest at three levels. The first is at the individual carrier level, because of overlapping substrate selectivities. As an example, the aspartate-glutamate carrier 1 (AGC1; SCL25A12), aspartate-glutamate carrier 2 (AGC2; SLC25A13), glutamate carrier 1 (GC1; SLC25A22), and glutamate carrier 2 (GC2; SLC25A18) all import glutamate. AGC1 and AGC2 are calcium-stimulated exchangers that import glutamate while exporting aspartate [61]. This exchange activity is a key step in the malate-aspartate shuttle [62, 63]. As examples of physiologic importance, AGC1 is critical for calcium-stimulated increases in neuronal respiration [64] and for neuronal export of aspartate to glia, which supports brain glutamate and glutamine synthesis and thus GABAergic signaling [65]. In contrast to AGC1 and AGC2, GC1 and GC2 co-import glutamate with a proton (H+), but with different kinetics and tissue distributions [66]. Interestingly, deficiency in either AGC1 or GC1, which predominate in the brain, results in hypomyelination and infantile epilepsy [67, 68]. Thus, in cells that express all four of these carriers, there are at least four distinct portals for glutamate import (Figure 2A). But, importantly, because of differences in co-transported substrates and mechanisms of regulation, functional redundancy is partial rather than complete and in some systems insufficient to prevent disease. Comparisons among other groups of mitochondrial carriers illustrate similar themes.
Figure 2. The mitochondrial carrier system enables redundant substrate utilization.
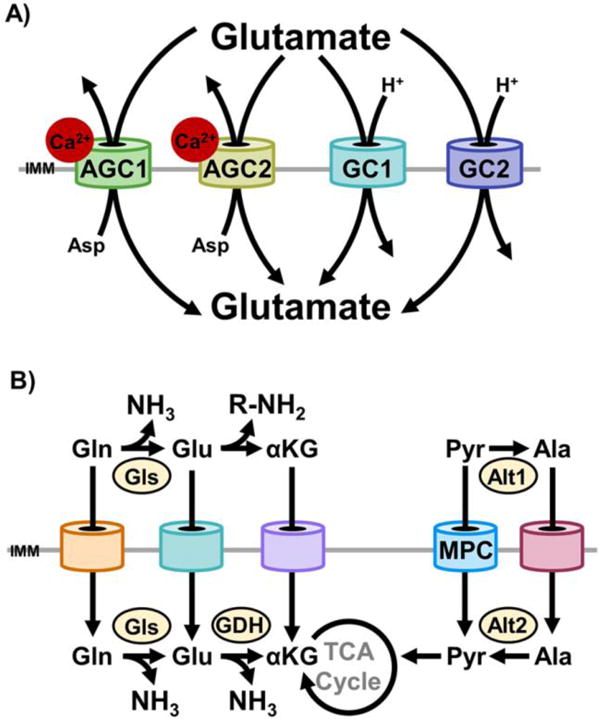
A)AGC1, AGC2, glutamate carrier 1 (GC1), and glutamate carrier 2 (GC2) each transport glutamate into mitochondria. Import through AGC1/2 is in exchange for aspartate export [61]. Import through GC1/2 is by proton (H+) symport [66]. In cells that express each of these carriers, there are at least four distinct portals for glutamate import. Because of differences in co-transported substrates, mechanisms of regulation, and kinetics, functional redundancy is partial rather than absolute. Thus, each mode of glutamate import exerts unique effects on cellular function. B) The presence of distinct mitochondrial carriers for immediate chemical neighbors endows the mitochondrial carrier system with systems-level redundancy. Two examples are illustrated here. In the first, mitochondrial import of glutamine, glutamate, and α-ketoglutarate may all be utilized to anaplerotically replenish the TCA cycle with α-ketoglutarate. However, whether glutamine and glutamate are deaminated in the cytosol or mitochondria changes intracellular nitrogen partitioning. In the second, the MPC and the currently unidentified mitochondrial alanine carrier are functionally redundant. In coordination with the cytosolic (Alt1) and mitochondrial (Alt2) alanine transaminase, either carrier may be utilized to generate mitochondrial pyruvate or alanine. This is exemplified by the pyruvate-alanine cycling employed to bypass disrupted MPC activity [18, 20, 51].
A second level of MCS redundancy occurs when different carriers transport readily interconvertible chemical neighbors (Figure 2B). This is exemplified by the MPC bypasses outlined earlier in this review. Although the identity of the mitochondrial alanine carrier is currently unknown, it is functionally redundant with the MPC because of cytoplasmic and mitochondrial pyruvate-alanine interconversions. This principle is also illustrated by a recent report of an α-ketoglutarate bypass of the CIC in H460 non-small lung cancer cells [69]. Mitochondria support lipogenesis by importing pyruvate derived from glucose, and converting it to citrate. Citrate is then exported by the CIC and converted to acetyl-CoA by ATP-dependent citrate lyase as the starting molecule for acyl-CoA chain elongation. Yet, similar to the effects of MPC disruption on gluconeogenesis, disruption of the CIC impaired, but did not block, lipogenesis from glucose [69]. 13C tracer metabolomic experiments showed that mitochondrial α-ketoglutarate, produced from citrate was exported to the cytoplasm, reductively carboxylated to isocitrate, isomerized to citrate, and utilized for lipogenesis [69]. Thus, mitochondrial α-ketoglutarate transport capacity provides a level of functional redundancy for CIC activity. The α-ketoglutarate transport activity was not specified, but could originate from the oxodicarboxylate carrier (ODC, SLC25A21) [70] and the OGC [71]. These carriers exchange α-ketoglutarate with oxoadipate and malate, respectively.
A third level of MCS redundancy occurs at the systems-level. Because of myriad potential metabolite interconversions within the cytosol and mitochondrial matrix, diverse patterns of mitochondrial transport may be utilized to support the same higher-order process. This is exemplified by the adaptive metabolism evoked by disruption of hepatic MPC activity. As discussed in a preceding section, TCA cycle flux and gluconeogenesis are sustained by both pyruvate-alanine cycling and increased glutaminolysis [18, 20]. Yet, adaptive changes in mitochondrial transport are more extensive. These changes were accompanied by increased urea cycle activity, presumably in coordination with ornithine carrier 1 (ORC1; SLC25A15) activity. ORC1 exchanges ornithine and citrulline and is critical for urea cycle function. ORC1 deficiency results in Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome [72], which was not observed in mice lacking MPC activity in the liver. Thus, adaptation for disruption of MPC activity is achieved through complex changes in flux through multiple mitochondrial carriers.
This principle also extends to the adaptive metabolism highlighted above when CIC activity is lost. Increased reductive carboxylation of α-ketoglutarate partially sustained lipogenesis, but numerous metabolite concentrations and patterns of flux were altered [69]. Critically, CIC knockout cells remained competent for the numerous biosynthetic processes required for the higher-order process of cell division. Thus, the MCS provides cells with redundant systems-level mechanisms for maintaining essential homeostatic processes.
Substrate exchange cycles routed through the MCS regulate mitochondrial and cytoplasmic redox balance
The systems-level redundancy of the MCS is employed to selectively regulate mitochondrial and cytoplasmic redox balance by substrate exchange cycles. Classically, this is illustrated by the malate-aspartate shuttle, which transfers NADH equivalents from the cytoplasm to the mitochondria, but also may run in the reverse direction. A series of cyclical mitochondrial and cytosolic interconversions are linked by the activities of the OGC and AGC1/2. Malate import in exchange for α-ketoglutarate export by the OGC is coordinated with glutamate import in exchange for aspartate export by the AGC. In net, this cycle transfer electrons from cytoplasmic NADH to NAD+ in the mitochondrial matrix (Figure 3A). Importantly, this does not change carbon and nitrogen balance between mitochondria and the cytoplasm. Thus, in the absence of other energetic limitations, high cytosolic levels of NADH in respiring cells may drive the malate-aspartate shuttle indefinitely.
Figure 3. The mitochondrial carriers coordinate redox cycles.
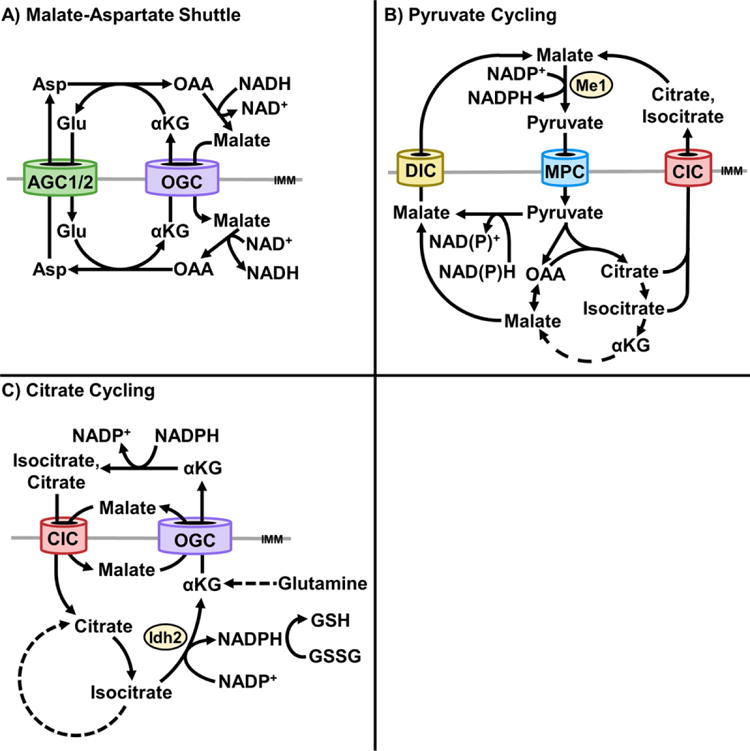
A) The aspartate-glutamate carrier (AGC) and oxoglutarate (α-ketoglutarate) carrier (OGC) coordinate the malate-aspartate shuttle. This cycle transfers NADH equivalents from the cytosol to the mitochondrial matrix. Cytosolic oxaloacetate is reduced to malate, coupled to oxidation of NAD+ to NADH, by cytosolic malate dehydrogenase. Malate is imported into mitochondria and then re-oxidized to oxaloacetate, coupled to reduction of NAD+ to NADH, by mitochondrial malate dehydrogenase. Malate import in exchange for α-ketoglutarate export, by the OGC, is coordinated with glutamate import in exchange for aspartate export, by the AGC. B) Pyruvate cycling through the mitochondrial pyruvate carrier (MPC) and dicarboxylate carrier (DIC) [75] or Citrate Carrier (CIC) [77] regenerates cytosolic NADPH. Pyruvate imported through the CIC may be converted to malate or citrate, exported, and re-converted to pyruvate by malic enzyme 1 (ME1), coupled with reduction of NADP+ to NADPH. C) A glutaminolysis-dependent, citrate-α-ketoglutarate cycle transfers NADPH equivalents from the cytosol to mitochondria [79]. Cytosolic α-ketoglutarate is reductively carboxylated to isocitrate, coupled to NADPH oxidation to NADP+, and isomerized to citrate. Citrate is imported into mitochondria and oxidized to α-ketoglutarate by isocitrate dehydrogenase 2 (IDH2), coupled to reduction of NADP+ to NADPH. NADPH may be utilized to reduce glutathione (GSSG to GSH). In cancer cells, α-ketoglutarate may be re-exported to the cytosol to supply lipogenesis [69]. A potential alternative fate of exported α-ketoglutarate is reconversion to citrate, to promote sustained α-ketoglutarate-citrate cycling as an NADPH shuttle.
The MCS supports reducing equivalent exchange through additional cycles involving pyruvate, citrate, and malate. Pyruvate cycling is a process whereby pyruvate is transported into mitochondria, converted to malate, citrate, or isocitrate, exported to the cytosol, and converted back to pyruvate by malic enzyme 1 (ME1) [73, 74] (Figure 3B). This results in NADPH production that may support cytosolic biosynthesis, glutathione reduction, and glucose-stimulated insulin secretion from pancreatic β-cells. Pyruvate cycling in β-cells has been demonstrated to utilize the activities of the dicarboxylate carrier (DIC; SLC25A10) [75], OGC [76], and CIC [77]. The effect of MPC disruption on glucose-stimulated insulin secretion may also be partially attributable to impaired pyruvate cycling [44, 52, 55, 78]. Of note, in contrast to the malate-aspartate shuttle, pyruvate cycling normally transfers reducing equivalents from the mitochondrial matrix to the cytosol.
Recent findings in H460 cancer cells demonstrate an additional role for the CIC in regulation of redox balance. A CIC-dependent, α-ketoglutarate-citrate cycle was observed to function as a mechanism for transporting cytosolic NADPH equivalents into mitochondria. It was found to be important for ROS neutralization, which is required for escape from anchorage-dependent growth [79]. In this cycle, glutamine-derived α-ketoglutarate is reductively carboxylated in the cytosol by isocitrate dehydrogenase 1 (IDH1) to produce isocitrate. Isocitrate is isomerized to citrate, which is imported into mitochondria by the CIC. Mitochondrial citrate is then isomerized back to isocitrate, which is oxidized to α-ketoglutarate by isocitrate dehydrogenase 2 (IDH2). The IDH2 reaction also reduces NADP+ to NADPH, which is utilized to reduce glutathione and augment mitochondrial ROS defense (Figure 3C). Re-export of α-ketoglutarate may then be utilized for lipogenesis, as outlined earlier in this review [69]. This raises the possibility that citrate and α-ketoglutarate may be perpetually cycled to regenerate mitochondrial NADPH. Like pyruvate cycling, a citrate-α-ketoglutarate cycle may utilize the CIC and OGC.
Mitochondrial substrate selection regulates nitrogen partitioning
The systems-level redundancy of the MCS is also employed to regulate nitrogen partitioning between mitochondria and the cytoplasm. As addressed earlier, disruption of MPC activity in the liver stimulates increased pyruvate-alanine cycling and glutaminolysis to sustain TCA cycle flux [18, 20]. Whether pyruvate, pyruvate-alanine cycling, or glutaminolysis feeds the TCA cycle results in distinct effects on cellular nitrogen balance. Because pyruvate lacks nitrogen, its oxidation is nitrogen neutral. Similarly, pyruvate-alanine cycling is nitrogen neutral at the cellular level because of reciprocal transamination events in the cytosol and mitochondria. In contrast, net oxidation of glutamine and other amino acids is ammoniagenic and therefore not nitrogen neutral. Thus, the relative balance between MPC and mitochondrial amino acid carrier activities regulates cellular nitrogen balance.
The MCS also regulates nitrogen partitioning by importing amino acids before deamination or as α-ketoglutarate after deamination. For example, flux of glutamine into the TCA cycle requires deamidation to glutamate, followed by deamination to α-ketoglutarate. Each of these steps may occur in the cytoplasm or the mitochondria. The MCS activities for import of glutamine, glutamate, or α-ketoglutarate may each function as a step in glutamine oxidation (also Figure 2B). However, which of these transport activities is utilized is important because it determines whether nitrogen is removed in the cytoplasm or the mitochondrial matrix. Depending on physiological context and cell type, this is important for channeling nitrogen into biosynthetic pathways or waste disposal within the mitochondria or cytoplasm. In the liver, mitochondrial glutamine and glutamate import may be favored when rates of amino acid oxidation are high to promote efficient channeling of ammonia into the urea cycle. In this case, carbamoyl phosphate synthetase 1 (CPS1), the rate-limiting enzyme of the urea cycle that resides in the mitochondrial matrix, may readily scavenge ammonia generated by matrix deamination events [80]. Of note, this is consistent with the increased amino acid catabolism and urea cycle activity observed when MPC activity is disrupted [18, 20]. Thus, regulation of nitrogen partitioning by the MCS is thematically similar to regulation of redox state. Different patterns of amino acid carrier activity may support equivalent rates of carbon fuel oxidation while selectively regulating nitrogen partitioning.
The MCS as a master hub of cellular communication
The diverse, complex patterns of small molecule transport orchestrated by the MCS endow mitochondria with vast capacity to function as a cellular information processing hub. It is well appreciated that the reticular structure and distribution of mitochondria throughout the cell enable efficient distribution of ATP. This morphological utility extends to the function of mitochondria to efficiently sense and modulate overall cellular chemistry. Allostery is a critical, rapid mode of metabolic regulation. Numerous mitochondrial enzymes are regulated by allostery, with many more likely to be discovered [81]. Because of the numerous small molecules transported by the MCS, it links mitochondrial allosteric sensory capacity with the cytosolic chemical environment. Thus, the MCS enables mitochondria to function as a sensory net that rapidly responds to homeostatically counter disturbances in cellular chemistry.
Chemical homeostatic responses mediated through the MCS also extend to regulation of specific interfaces between cellular metabolism and signaling. As examples, the MCS may modulate AMPK activity by changes in rate of ATP production, cytosolic and mitochondrial sirtuin activity by operation of specific substrate shuttles that impinge on redox balance, and calcium signaling by changing rates of calcium import and export. Furthermore, the many contacts between mitochondria, ER, and other organelles may enable mitochondria to sense and respond to changes in chemistry in other organelles [82]. Thus, mitochondria are distributed throughout the cell, possess exquisite capacity for chemical sensing, and utilize numerous patterns of small molecule exchange via the MCS. Therefore, the MCS confers mitochondria with the capacity to mediate temporally and spatially tuned intracellular communication.
Concluding Remarks and Future Perspectives
Several critical challenges must be solved to achieve a mechanistic understanding of MCS function (see Outstanding Questions). First, the in vivo transport selectivities of each carrier must be defined. 24 of the 53 mammalian SLC25A members lack empirically defined substrate selectivities [3]. Furthermore, the molecular identities for key mitochondrial transport activities remain unknown, including glutamine, alanine, and lactate transport. As the discoveries of the MPC [17, 19] and the mitochondrial calcium uniporter (MCU) [83, 84] these may extend to non-canonical mitochondrial transporters. Second, the specific mechanisms regulating the function of each individual mitochondrial carrier must be defined. Similar to other mitochondrial proteins, these may include allosteric regulation, post-translational regulation by acylation and other modifications, transcriptional regulation, and regulated interactions with adaptor proteins [85–89]. Third, the mechanisms coordinating the activities of individual mitochondrial carriers to function as a unified system must be determined. As discussed, several investigations have observed increases in glutaminolysis when MPC activity is disrupted [18, 20, 22, 23]. This is partially dependent upon increased glutamate dehydrogenase activity, but the mechanisms coordinating changes in mitochondrial transport activities remain undefined [23]. Fourth, and perhaps most challenging, the discreet regulatory effects of different patterns of mitochondrial small molecule transport must be deciphered. This will require determining how patterns of transport that are partially functionally redundant at the systems-level are selectively utilized to exert differential effects on cellular chemistry. This effort will be enhanced by the recent advent of CRISPR/Cas9 systems for gene disruption, which greatly enables the synthetic lethality approaches that will be required to understand the functional connections within the MCS. Over time, this understanding may enable precise modulation of cellular chemistry to counteract the dysmetabolism inherent in disease.
Outstanding Questions Box.
What are the molecular identities of each mitochondrial carrier? What are the substrates transported by each?
What are the mechanisms regulating the function of each mitochondrial carrier?
What are the mechanisms coordinating the regulation of individual mitochondrial carriers, to function together as a system?
How are different patterns of mitochondrial carrier activity utilized to regulate cellular programming, by precisely modulating cellular chemistry and energetics?
How does the function of the mitochondrial carrier system change in different disease states? How can the activity of the mitochondrial carrier system be targeted during disease, to correct specific defects in chemical homeostasis, or to elicit protective, hormetic responses?
Trends Box.
Increased accessibility of metabolomics and molecular genetic technologies now greatly enables functional investigation of the mitochondrial carrier system.
The mitochondrial pyruvate carrier regulates multiple dimensions of cellular biology including differentiation state, nutrient sensing, and mitochondrial substrate selection.
The mitochondrial carrier system is functionally redundant at the individual carrier- and systems-levels.
The mitochondrial carrier system regulates cellular chemistry, including relative mitochondrial-cytosolic redox and nitrogen balances.
Acknowledgments
Research in the author’s laboratory is funded by NIH R01 DK104998. The author thanks Dr. Adam Rauckhorst, Dr. Ryan Sheldon, Dr. Lawrence Gray, and Maria Noterman for contributing to figure construction and providing helpful feedback on a draft manuscript.
Glossary
- Aspartate-glutamate carrier 1 and 2 (AGC1: SLC25A12; AGC2: SLC25A13)
Exchange glutamate and aspartate across the mitochondrial inner membrane. Glutamate import and aspartate export is the primary direction of exchange. AGC1 and ACG2 are expressed in several tissues, and their activities are stimulated by calcium. However, AGC2 is expressed in the liver whereas AGC1 is not. The AGCs help facilitate the aspartate-malate shuttle, which transfers NADH-equivalents from the cytosol to the mitochondria
- Alanine transaminase 1 and 2 (ALT1, ALT2)
Cytosolic and mitochondrial matrix enzymes that catalyze the transfer of an amino group from glutamate to pyruvate, thereby producing alanine and α-ketoglutarate. They also readily catalyze the reverse reaction. ALT1 is localized in the cytosol, whereas ALT2 is localized in the mitochondrial matrix
- Citrate carrier (CIC: SLC25A1)
Exchanges citrate and malate across the mitochondrial inner membrane. CIC activity contributes to lipogenesis and facilitates the transfer of NADPH equivalents into mitochondria under conditions of high glutaminolysis
- Dicarboxylate carrier (DIC: SLC25A10)
Exchanges malate and phosphate across the mitochondrial inner membrane. Its activity may contribute to multiple cycles for transferring reducing equivalents between the cytosol and mitochondria
- Malic enzyme 1 (ME1)
A cytosolic enzyme that decarboxylates malate to pyruvate, coupled to reduction of NADP+ to NADPH. Its activity is important for pyruvate cycling, because it may regenerate pyruvate from malate exported from mitochondria, which was produced from pyruvate imported into mitochondria from the cytosol
- Mitochondrial Carrier
A mitochondrial inner membrane protein that transports small molecules between the cytosol and mitochondria, thereby regulating the function of both compartments. The mitochondrial carriers include the transporters of the canonical SL25A family. They also include non-canonical transporters like the mitochondrial pyruvate carrier and mitochondrial calcium uniporter
- Mitochondrial carrier system (MCS)
The working system of mitochondrial carriers, where activities of individual carries are coordinated to function as a unified system for balanced, dynamic regulation of cellular metabolism and chemical homeostasis
- Mitochondrial pyruvate carrier (MPC)
A non-canonical mitochondrial carrier that symports pyruvate with a proton (H+) across the mitochondrial inner membrane. It comprises two obligate subunits, encoded by the MPC1 and MPC2 genes
- Monocarboxylate transporter (MCT)
Transports the monocarboxylates pyruvate and lactate across the plasma membrane, in symport with a proton (H+). Several isoforms exist, with different tissue distributions
- Oxoglutarate carrier (OGC: SLC25A11)
Exchanges α-ketoglutarate (oxoglutarate) and malate across the mitochondrial inner membrane. In coordination with other mitochondrial carriers, its activity may contribute to multiple cycles for transferring reducing equivalents between the cytosol and mitochondria, including the malate-aspartate shuttle
- Pyruvate-alanine cycling
Functions as a mechanism for bypassing the MPC to transfer pyruvate from the cytosol into mitochondria. In this case, cytoplasmic pyruvate is transaminated to alanine by ALT1, imported into mitochondria, and then deaminated back to pyruvate by ALT2
- SLC25A mitochondrial carrier family
The canonical family of mitochondrial carriers. There are 53 family members in mammalian systems. They are characterized by a tripartite structure with 3 tandem repeats. Each of these repeats contains the SLC25A signature motif and hydrophobic α-helices connected by a hydrophilic matrix loop. The N- and C-termini reside on the cytosolic side of the mitochondrial inner membrane. They catalyze the transport of diverse molecules including citric acid cycle intermediates, amino acids, nucleotides, vitamins, metals, and minerals
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gutierrez-Aguilar M, Baines CP. Physiological and pathological roles of mitochondrial SLC25 carriers. Biochem J. 2013;454(3):371–86. doi: 10.1042/BJ20121753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med. 2013;34(2–3):465–84. doi: 10.1016/j.mam.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Palmieri F, Monne M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim Biophys Acta. 2016;1863(10):2362–78. doi: 10.1016/j.bbamcr.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Aquila H, et al. Complete amino acid sequence of the ADP/ATP carrier from beef heart mitochondria. Hoppe Seylers Z Physiol Chem. 1982;363(3):345–9. [PubMed] [Google Scholar]
- 5.Aquila H, et al. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane. Embo j. 1985;4(9):2369–76. doi: 10.1002/j.1460-2075.1985.tb03941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Runswick MJ, et al. Sequence of the bovine mitochondrial phosphate carrier protein: structural relationship to ADP/ATP translocase and the brown fat mitochondria uncoupling protein. Embo j. 1987;6(5):1367–73. doi: 10.1002/j.1460-2075.1987.tb02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Runswick MJ, et al. Sequence of the bovine 2-oxoglutarate/malate carrier protein: structural relationship to other mitochondrial transport proteins. Biochemistry. 1990;29(50):11033–40. doi: 10.1021/bi00502a004. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan RS, et al. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J Biol Chem. 1993;268(18):13682–90. [PubMed] [Google Scholar]
- 9.Indiveri C, et al. The mitochondrial carnitine carrier protein: cDNA cloning, primary structure and comparison with other mitochondrial transport proteins. Biochem J. 1997;321(Pt 3):713–9. doi: 10.1042/bj3210713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaukonen J, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289(5480):782–5. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 11.Mayr JA, et al. Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am J Hum Genet. 2007;80(3):478–84. doi: 10.1086/511788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nota B, et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am J Hum Genet. 2013;92(4):627–31. doi: 10.1016/j.ajhg.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seifert EL, et al. The mitochondrial phosphate carrier: Role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem Biophys Res Commun. 2015;464(2):369–75. doi: 10.1016/j.bbrc.2015.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huizing M, et al. Cloning of the human carnitine-acylcarnitine carrier cDNA and identification of the molecular defect in a patient. Am J Hum Genet. 1997;61(6):1239–45. doi: 10.1086/301628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Indiveri C, et al. The mitochondrial carnitine/acylcarnitine carrier: function, structure and physiopathology. Mol Aspects Med. 2011;32(4–6):223–33. doi: 10.1016/j.mam.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Monne M, et al. The substrate specificity of mitochondrial carriers: mutagenesis revisited. Mol Membr Biol. 2013;30(2):149–59. doi: 10.3109/09687688.2012.737936. [DOI] [PubMed] [Google Scholar]
- 17.Bricker DK, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337(6090):96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray LR, et al. Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis. Cell Metab. 2015;22(4):669–81. doi: 10.1016/j.cmet.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herzig S, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337(6090):93–6. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- 20.McCommis KS, et al. Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell Metab. 2015;22(4):682–94. doi: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schell JC, et al. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol Cell. 2014;56(3):400–13. doi: 10.1016/j.molcel.2014.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vacanti NM, et al. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol Cell. 2014;56(3):425–35. doi: 10.1016/j.molcel.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang C, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56(3):414–24. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monne M, et al. Mitochondrial transporters for ornithine and related amino acids: a review. Amino Acids. 2015;47(9):1763–77. doi: 10.1007/s00726-015-1990-5. [DOI] [PubMed] [Google Scholar]
- 25.Palmieri F. Mitochondrial transporters of the SLC25 family and associated diseases: a review. J Inherit Metab Dis. 2014;37(4):565–75. doi: 10.1007/s10545-014-9708-5. [DOI] [PubMed] [Google Scholar]
- 26.Papa S, et al. The transport of pyruvate in rat liver mitochondria. FEBS Lett. 1971;12(5):285–288. doi: 10.1016/0014-5793(71)80200-4. [DOI] [PubMed] [Google Scholar]
- 27.Halestrap AP, Denton RM. Specific inhibition of pyruvate transport in rat liver mitochondria and human erythrocytes by alpha-cyano-4-hydroxycinnamate. Biochem J. 1974;138(2):313–6. doi: 10.1042/bj1380313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nalecz MJ, et al. Extraction, partial purification and functional reconstitution of two mitochondrial carriers transporting keto acids: 2-oxoglutarate and pyruvate. FEBS Lett. 1986;196(2):331–6. doi: 10.1016/0014-5793(86)80273-3. [DOI] [PubMed] [Google Scholar]
- 29.Thomas AP, Halestrap AP. Identification of the protein responsible for pyruvate transport into rat liver and heart mitochondria by specific labelling with [3H]N-phenylmaleimide. Biochem J. 1981;196(2):471–9. doi: 10.1042/bj1960471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolli R, et al. Monocarboxylate and alpha-ketoglutarate carriers from bovine heart mitochondria. Purification by affinity chromatography on immobilized 2-cyano-4-hydroxycinnamate. J Biol Chem. 1989;264(30):18024–30. [PubMed] [Google Scholar]
- 31.Capuano F, et al. The monocarboxylate carrier from rat liver mitochondria. Purification and kinetic characterization in a reconstituted system. FEBS Lett. 1990;261(1):39–42. doi: 10.1016/0014-5793(90)80631-r. [DOI] [PubMed] [Google Scholar]
- 32.Nalecz MJ, et al. Purification and functional characterisation of the pyruvate (monocarboxylate) carrier from baker’s yeast mitochondria (Saccharomyces cerevisiae) Biochim Biophys Acta. 1991;1079(1):87–95. doi: 10.1016/0167-4838(91)90028-x. [DOI] [PubMed] [Google Scholar]
- 33.Hildyard JC, Halestrap AP. Identification of the mitochondrial pyruvate carrier in Saccharomyces cerevisiae. Biochem J. 2003;374(Pt 3):607–11. doi: 10.1042/BJ20030995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Todisco S, et al. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J Biol Chem. 2006;281(3):1524–31. doi: 10.1074/jbc.M510425200. [DOI] [PubMed] [Google Scholar]
- 35.Brivet M, et al. Impaired mitochondrial pyruvate importation in a patient and a fetus at risk. Mol Genet Metab. 2003;78(3):186–92. doi: 10.1016/s1096-7192(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 36.Kunji ER, et al. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim Biophys Acta. 2016;1863(10):2379–93. doi: 10.1016/j.bbamcr.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Xu Y, et al. Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature. 2014;515(7527):448–52. doi: 10.1038/nature13670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Compan V, et al. Monitoring Mitochondrial Pyruvate Carrier Activity in Real Time Using a BRET-Based Biosensor: Investigation of the Warburg Effect. Mol Cell. 2015;59(3):491–501. doi: 10.1016/j.molcel.2015.06.035. [DOI] [PubMed] [Google Scholar]
- 39.Rauckhorst AJ, Taylor EB. Mitochondrial pyruvate carrier function and cancer metabolism. Curr Opin Genet Dev. 2016;38:102–109. doi: 10.1016/j.gde.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, et al. Mitochondrial pyruvate carrier function is negatively linked to Warburg phenotype in vitro and malignant features in esophageal squamous cell carcinomas. Oncotarget. 2016 doi: 10.18632/oncotarget.13717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, et al. MPC1 and MPC2 expressions are associated with favorable clinical outcomes in prostate cancer. BMC Cancer. 2016;16(1):894. doi: 10.1186/s12885-016-2941-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong Y, et al. Application of mitochondrial pyruvate carrier blocker UK5099 creates metabolic reprogram and greater stem-like properties in LnCap prostate cancer cells in vitro. Oncotarget. 2015;6(35):37758–69. doi: 10.18632/oncotarget.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olson KA, et al. Pyruvate and Metabolic Flexibility: Illuminating a Path Toward Selective Cancer Therapies. Trends Biochem Sci. 2016;41(3):219–30. doi: 10.1016/j.tibs.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCommis KS, et al. An ancestral role for the mitochondrial pyruvate carrier in glucose-stimulated insulin secretion. Mol Metab. 2016;5(8):602–14. doi: 10.1016/j.molmet.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schell JC, Rutter J. The long and winding road to the mitochondrial pyruvate carrier. Cancer Metab. 2013;1(1):6. doi: 10.1186/2049-3002-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gray LR, et al. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014;71(14):2577–604. doi: 10.1007/s00018-013-1539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanderperre B, et al. Mitochondrial pyruvate import and its effects on homeostasis. Curr Opin Cell Biol. 2015;33:35–41. doi: 10.1016/j.ceb.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 48.McCommis KS, Finck BN. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J. 2015;466(3):443–54. doi: 10.1042/BJ20141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bender T, Martinou JC. The mitochondrial pyruvate carrier in health and disease: To carry or not to carry? Biochim Biophys Acta. 2016;1863(10):2436–42. doi: 10.1016/j.bbamcr.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 50.Vanderperre B, et al. Embryonic Lethality of Mitochondrial Pyruvate Carrier 1 Deficient Mouse Can Be Rescued by a Ketogenic Diet. PLoS Genet. 2016;12(5):e1006056. doi: 10.1371/journal.pgen.1006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bowman CE, et al. Requirement for the Mitochondrial Pyruvate Carrier in Mammalian Development Revealed by a Hypomorphic Allelic Series. Mol Cell Biol. 2016;36(15):2089–104. doi: 10.1128/MCB.00166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vigueira PA, et al. Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep. 2014;7(6):2042–53. doi: 10.1016/j.celrep.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghosh A, et al. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci Transl Med. 2016;8(368):368ra174. doi: 10.1126/scitranslmed.aag2210. [DOI] [PubMed] [Google Scholar]
- 54.Divakaruni AS, et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J Cell Biol. 2017 doi: 10.1083/jcb.201612067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patterson JN, et al. Mitochondrial metabolism of pyruvate is essential for regulating glucose-stimulated insulin secretion. J Biol Chem. 2014;289(19):13335–46. doi: 10.1074/jbc.M113.521666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brooks GA. Cell-cell and intracellular lactate shuttles. J Physiol. 2009;587(Pt 23):5591–600. doi: 10.1113/jphysiol.2009.178350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gladden LB. Current trends in lactate metabolism: introduction. Med Sci Sports Exerc. 2008;40(3):475–6. doi: 10.1249/MSS.0b013e31816154c9. [DOI] [PubMed] [Google Scholar]
- 58.Passarella S, et al. The mitochondrial L-lactate dehydrogenase affair. Front Neurosci. 2014;8:407. doi: 10.3389/fnins.2014.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pizzuto R, et al. l-Lactate metabolism in HEP G2 cell mitochondria due to the l-lactate dehydrogenase determines the occurrence of the lactate/pyruvate shuttle and the appearance of oxaloacetate, malate and citrate outside mitochondria. Biochim Biophys Acta. 2012;1817(9):1679–90. doi: 10.1016/j.bbabio.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 60.Chen YJ, et al. Lactate metabolism is associated with mammalian mitochondria. Nat Chem Biol. 2016;12(11):937–943. doi: 10.1038/nchembio.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amoedo ND, et al. AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim Biophys Acta. 2016;1863(10):2394–412. doi: 10.1016/j.bbamcr.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 62.Pardo B, et al. Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem. 2006;281(2):1039–47. doi: 10.1074/jbc.M507270200. [DOI] [PubMed] [Google Scholar]
- 63.Marmol P, et al. Requirement for aralar and its Ca2+-binding sites in Ca2+ signal transduction in mitochondria from INS-1 clonal beta-cells. J Biol Chem. 2009;284(1):515–24. doi: 10.1074/jbc.M806729200. [DOI] [PubMed] [Google Scholar]
- 64.Llorente-Folch I, et al. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci. 2013;33(35):13957–71. 13971a. doi: 10.1523/JNEUROSCI.0929-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pardo B, et al. Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J Cereb Blood Flow Metab. 2011;31(1):90–101. doi: 10.1038/jcbfm.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fiermonte G, et al. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2002;277(22):19289–94. doi: 10.1074/jbc.M201572200. [DOI] [PubMed] [Google Scholar]
- 67.Wibom R, et al. AGC1 deficiency associated with global cerebral hypomyelination. N Engl J Med. 2009;361(5):489–95. doi: 10.1056/NEJMoa0900591. [DOI] [PubMed] [Google Scholar]
- 68.Molinari F, et al. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am J Hum Genet. 2005;76(2):334–9. doi: 10.1086/427564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang L, et al. Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein. Metab Eng. 2016 doi: 10.1016/j.ymben.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fiermonte G, et al. Identification of the human mitochondrial oxodicarboxylate carrier. Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J Biol Chem. 2001;276(11):8225–30. doi: 10.1074/jbc.M009607200. [DOI] [PubMed] [Google Scholar]
- 71.Iacobazzi V, et al. Sequences of the human and bovine genes for the mitochondrial 2-oxoglutarate carrier. DNA Seq. 1992;3(2):79–88. doi: 10.3109/10425179209034000. [DOI] [PubMed] [Google Scholar]
- 72.Camacho JA, et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet. 1999;22(2):151–8. doi: 10.1038/9658. [DOI] [PubMed] [Google Scholar]
- 73.Jensen MV, et al. Metabolic cycling in control of glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab. 2008;295(6):E1287–97. doi: 10.1152/ajpendo.90604.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MacDonald MJ, et al. Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am J Physiol Endocrinol Metab. 2005;288(1):E1-15. doi: 10.1152/ajpendo.00218.2004. [DOI] [PubMed] [Google Scholar]
- 75.Huypens P, et al. The dicarboxylate carrier plays a role in mitochondrial malate transport and in the regulation of glucose-stimulated insulin secretion from rat pancreatic beta cells. Diabetologia. 2011;54(1):135–45. doi: 10.1007/s00125-010-1923-5. [DOI] [PubMed] [Google Scholar]
- 76.Odegaard ML, et al. The mitochondrial 2-oxoglutarate carrier is part of a metabolic pathway that mediates glucose- and glutamine-stimulated insulin secretion. J Biol Chem. 2010;285(22):16530–7. doi: 10.1074/jbc.M109.092593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Joseph JW, et al. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J Biol Chem. 2006;281(47):35624–32. doi: 10.1074/jbc.M602606200. [DOI] [PubMed] [Google Scholar]
- 78.Fisher-Wellman KH, et al. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem J. 2015;467(2):271–80. doi: 10.1042/BJ20141447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang L, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532(7598):255–8. doi: 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martinez AI, et al. Genetic, structural and biochemical basis of carbamoyl phosphate synthetase 1 deficiency. Mol Genet Metab. 2010;101(4):311–23. doi: 10.1016/j.ymgme.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 81.Orsak T, et al. Revealing the allosterome: systematic identification of metabolite-protein interactions. Biochemistry. 2012;51(1):225–32. doi: 10.1021/bi201313s. [DOI] [PubMed] [Google Scholar]
- 82.Murley A, Nunnari J. The Emerging Network of Mitochondria-Organelle Contacts. Mol Cell. 2016;61(5):648–53. doi: 10.1016/j.molcel.2016.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baughman JM, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–5. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chaudhuri D, et al. MCU encodes the pore conducting mitochondrial calcium currents. Elife. 2013;2:e00704. doi: 10.7554/eLife.00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Floyd BJ, et al. Mitochondrial Protein Interaction Mapping Identifies Regulators of Respiratory Chain Function. Mol Cell. 2016;63(4):621–32. doi: 10.1016/j.molcel.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hirschey MD, Zhao Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol Cell Proteomics. 2015;14(9):2308–15. doi: 10.1074/mcp.R114.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell. 2014;54(1):5–16. doi: 10.1016/j.molcel.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Still AJ, et al. Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. J Biol Chem. 2013;288(36):26209–19. doi: 10.1074/jbc.M113.483396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lytovchenko O, Kunji ER. Expression and putative role of mitochondrial transport proteins in cancer. Biochim Biophys Acta. 2017 doi: 10.1016/j.bbabio.2017.03.006. [DOI] [PubMed] [Google Scholar]