

Summary
The Interleukin-1 (IL-1) family cytokines are cytosolic proteins that exhibit inflammatory activity upon release into the extracellular space. These factors are released following various cell death processes, with pyroptosis being a common mechanism. Recently, it was recognized that phagocytes can achieve a state of hyperactivation, which is defined by their ability to secrete IL-1 while retaining viability, yet it is unclear how IL-1 can be secreted from living cells. Herein, we report that the pyroptosis regulator gasdermin D (GSDMD) was necessary for IL-1β secretion from living macrophages that have been exposed to inflammasome activators, such as bacteria and their products or host-derived oxidized lipids. Cell- and liposome-based assays demonstrated that GSDMD pores were required for IL-1β transport across an intact lipid bilayer. These findings identify a non-pyroptotic function for GSDMD, and raise the possibility that GSDMD pores represent conduits for the secretion of cytosolic cytokines under conditions of cell hyperactivation.
eTOC
Inflammasomes elicit pyroptosis or cell hyperactivation, with the latter defined as living cells that release IL-1. Evavold et al report that the pore-forming protein gasdermin D regulates IL-1 release from hyperactive macrophages. Cell- and liposome-based assays revealed that gasdermin D pores permit IL-1 passage across intact lipid bilayers.
INTRODUCTION
Interleukin-1 (IL-1) family cytokines induce inflammatory responses in numerous tissues of the body. These pyrogens are produced as cytosolic factors that lack an N-terminal secretion signal, and are therefore not released from cells via the conventional secretory pathway (Garlanda et al., 2013). Whereas the inflammatory functions of extracellular IL-1 are well-defined, the mechanisms by which these cytokines are released from cells remain elusive.
Central to the function of IL-1 are inflammasomes (Martinon et al., 2002), which are supramolecular organizing centers (SMOCs) that assemble in the cytosol in response to infection, ionic imbalance, and mitochondrial dysfunction (Latz et al., 2013; Kagan et al., 2014; Martinon et al., 2009). Inflammasomes consist of a sensor protein, an adaptor protein, and an inflammatory caspase effector protein (e.g. caspase-1). Caspase-1 is capable of cleaving IL-1 family cytokines that are translated in a pro-form, such as IL-1β and IL-18 (Cerretti et al., 1992; Garlanda et al., 2013). Cleavage of these factors is necessary for inflammatory activity.
Caspase-1 (and caspase-11) also cleave the cytosolic protein gasdermin D (GSDMD) (Kayagaki et al., 2015; Shi et al., 2015). Upon cleavage, the N-terminal fragment of GSDMD oligomerizes into ring-shaped structures in membranes (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). GSDMD rings form a pore in the plasma membrane that ultimately cause cell lysis. This cell death process (pyroptosis) is a highly inflammatory event, and provides a mechanism of IL-1 release (Kayagaki et al., 2015; Shi et al., 2015).
Pyroptosis is not the only means by which IL-1 is released from cells. For example, a set of oxidized lipids (oxPAPC) derived from dead mammalian cells induces inflammasome-dependent release of IL-1, but not cell death (Zanoni et al., 2016). The N-acetyl glucosamine (NAG) fragment of bacterial peptidoglycan (PGN) induces inflammasome-mediated IL-1 release from living macrophages (Wolf et al., 2016). Bacterial lipopolysaccharides (LPS) induce inflammasome-mediated release of IL-1 from living human monocytes (Gaidt et al., 2016), and living neutrophils also release IL-1 (Chen et al., 2014).
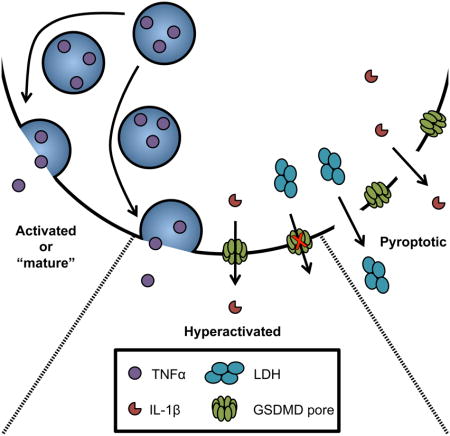
Viable cells that release inflammatory cytokines via the secretory pathway are known as “activated” or “mature” cells (Mellman et al., 1998), whereas dead cells that release IL-1 are considered “pyroptotic” (Kovacs and Miao, 2017). Viable cells that release IL-1 along with other cytokines exist in a state that differs from their traditionally activated or pyroptotic counterparts. This distinct activation state has been dubbed “hyperactive” (Zanoni et al., 2016). The recognition that phagocytes can release IL-1 while maintaining viability reignites the question of how IL-1 can be released from these cells. IL-1 family cytokines have a diameter of 4.5 nanometers (van Oostrum et al., 1991), which is theoretically narrow enough to pass through the GSDMD pore (inner diameter of 10–15 nanometer) (Liu et al., 2016; Sborgi et al., 2016).
Herein, we report that GSDMD is required for IL-1 release under conditions of macrophage hyperactivation, and under pyroptosis-inducing conditions where plasma membrane rupture is experimentally prevented. Using reconstituted 293T cells and liposome-based analyses, we provide evidence that GSDMD pores may serve as conduits for the transport of IL-1 family cytokines across intact lipid bilayers.
RESULTS
GSDMD regulates inflammasome-induced pore formation and IL-1 release from intact cells
Pyroptotic cells are commonly defined as necrotic cells with inflammasome-dependent plasma membrane rupture. This loss of membrane integrity results in the release of cytosolic lactate dehydrogenase (LDH) into the extracellular space, and the staining of intracellular nucleic acids by the membrane impermeable dye propidium iodide (PI) (Davis et al., 2011; Latz et al., 2013). As PI can pass through pores that form in intact cells (Fink and Cookson, 2006), PI staining alone cannot unequivocally identify lysed cells. We consider pyroptotic events those that result in LDH release and PI staining.
To determine if GSDMD influences IL-1 release from intact cells, we utilized the osmoprotectant glycine to prevent membrane rupture and pyroptosis-associated cytolysis (Fink and Cookson, 2006). Glycine was used to prevent membrane rupture downstream of the normally pyroptotic NLRP3 inflammasome activator nigericin after LPS priming (Latz et al., 2013). Using immortalized bone marrow derived macrophages (iBMDMs), LPS treatment alone (or nigericin alone) did not induce LDH release into the extracellular media (Fig. 1A). In contrast, LDH release was observed when LPS-primed cells were exposed to nigericin (Fig. 1A). Accompanying LDH release was the release of IL-1β (Fig. 1B), and the cell population exhibited signs of pore formation, as assessed by real time monitoring of PI staining (Fig. 1C). Glycine buffering of the extracellular media almost completely prevented LDH release (Fig. 1A), but pore formation and IL-1β release were largely unaffected (Fig. 1B, C). These findings support the idea that PI staining identifies intact cells that contain pores and cells whose plasma membranes have ruptured. LDH release, in contrast, is specifically indicative of pyroptosis.
Figure 1. GSDMD is required for IL-1β release when lysis is prevented.

(A, F) WT and Gsdmd−/− iBMDMs were primed with LPS for 3 hours (or not), and then treated nigericin for 2 hours (A) or Flatox (PA+LFn-Fla) for 2 hours (F). Stimulations contained 0 mM Glycine or 5 mM Glycine. LDH present in the extracellular media was then quantified.
(B, G) WT and Gsdmd−/− iBMDMs were primed with LPS for 3 hours (or not), and then treated with nigericin for 2 hours or Flatox (2 μg/ml PA and 0.5 μg/ml LFn-Fla) for 2 hours. Stimulations contained 0 mM Glycine or 5 mM Glycine. IL-1β release was monitored by ELISA.
(C, H) WT and Gsdmd−/− iBMDMs were primed with LPS for 3 hours (or not), and then treated with nigericin for 2 hours or Flatox (PA+LFn-Fla) for 2 hours. Stimulations contained 0 mM Glycine or 5 mM Glycine. PI (5 μM) was added to assay membrane permeability over time.
(D, E) Immunoblot analysis of cell-associated (D) or extracellular (E) IL-1β in Gsdmd−/− iBMDMs after 3 hours of LPS priming and 30 min of nigericin treatment. Stimulations contained 0 mM Glycine or 5 mM Glycine.
(I, J) Immunoblot analysis of cell-associated cleaved (I) or extracellular (J) IL-1β in Gsdmd−/− iBMDMs after 3 hours of LPS priming and 2 hours of Flatox treatment at 2 μg/ml PA and 0.5 μg/ml LFn-Fla. Data with error bars are represented as mean ± SEM. Each panel is a representative experiment of at least 3 repeats. **** p<0.0001 as determined by two-way ANOVA with Tukey’s multicomparison correction
See also Figure S1.
To determine if GSDMD regulates nigericin-induced pore formation and IL-1β release, we used Cas9-based reverse genetics to inactivate Gsdmd in iBMDM. Specifically, we used two gRNAs to generate Gsdmd−/− iBMDMs. These gRNAs were validated for Gsdmd ablation in iBMDMs or mice (Shi et al., 2015). This approach resulted in the elimination of the GSDMD protein from two independent clonal iBMDM populations (Fig. S1A). Gsdmd−/− cells displayed all expected phenotypes (Shi et al., 2015), in that they could not release LDH, could not form pores, and could not release IL-1β upon LPS and nigericin treatment (Fig. 1A–C, and S1B–D). LPS-induced tumor necrosis alpha (TNF-α) secretion was largely unaffected by GSDMD deficiency (Fig. S1E). Gsdmd−/− cells also retained the ability to form active inflammasomes after LPS priming with subsequent nigericin treatment, as evidenced by the presence of cleaved IL-1β in Gsdmd−/− lysates (Fig. 1D). The ability of nigericin to induce LDH, IL-1β release and pore formation in LPS-primed cells was sensitive to high concentrations of extracellular potassium (Fig. S1F–H), as expected (Perregaux and Gabel, 1994). These observations are consistent with the role of GSDMD as a pyroptosis effector that acts downstream of inflammasome assembly.
In the presence or absence of glycine, Gsdmd−/− cells were unable to form pores or release IL-1β (Fig. 1B, C). Immunoblot analysis verified these results, as the cleaved fragment of IL-1β was present in the extracellular media of nigericin-treated wild type (WT) cells that were primed with LPS, in the presence or absence of glycine (Fig. 1E). In contrast, Gsdmd−/− cells were unable to release cleaved IL-1β (Fig. 1E), even though this cleaved fragment was detected in cell lysates (Fig. 1D). These data indicate that IL-1β can be released from intact cells, and that GSDMD is required for this process.
To complement this analysis, we utilized Flatox as an activator of the NAIP-NLRC4 inflammasome. Flatox is an anthrax toxin derivative that consists of a Lethal Factor-flagellin fusion protein (LFn-Fla) and Protective Antigen (PA) (von Moltke et al., 2012). When PA and LFn-Fla are combined Flatox is formed, which delivers flagellin into the cytosol where it activates pyroptosis (von Moltke et al., 2012). Flatox-treated cells induced LDH release from iBMDM (Fig. 1F), but IL-1β release was only observed if cells were primed with LPS (Fig. 1G). LFn-Fla treatments in the absence of PA led to no LDH release (Fig. 1F). Glycine buffering of the extracellular media almost completely prevented LDH release induced by Flatox (Fig. 1F), but did not prevent pore formation or IL-1β release (Fig. 1G, H). Under all conditions examined, Gsdmd−/− cells were unable to form pores, release IL-1β or LDH (Fig. 1F–H). Immunoblot analysis demonstrated that GSDMD was required for IL-1β release, but not processing in the cytosol (Fig. 1I). No cleaved IL-1β was detected in the extracellular media of Gsdmd−/− cells (Fig. 1J). GSDMD was not required for LPS-induced TNF-α secretion (Fig. S1I). These data indicate that GSDMD is required for pore formation and IL-1β release in response to activators of distinct inflammasomes, even when lysis is prevented.
GSDMD is required for IL-1 release from hyperactive macrophages that were stimulated by bacteria and their products
Naturally, pyroptosis and IL-1β release can be uncoupled under conditions of cell hyperactivation, where phagocytes release IL-1β while maintaining viability. To determine if GSDMD regulates IL-1β release from living cells, we utilized S. aureus mutants that lack the gene encoding O-acetyltransferase A (OatA). This strain induces NLRP3-dependent IL-1β release from living macrophages (Shimada et al., 2010; Wolf et al., 2016). Consistent with this idea, we found that OatA-deficient S. aureus did not induce LDH release from iBMDMs (Fig. 2A), but did promote pore formation and IL-1β release (Fig. 2B, C). The extent of pore formation observed during bacterial infection was less than that observed for pyroptotic stimuli (compare PI staining in Fig. 2C to similar experiments in Fig. 1). This difference in the extent of pore formation may explain the ability of these infected macrophages to achieve a state of hyperactivation, as opposed to pyroptosis.
Figure 2. GSDMD regulates IL-1β release in response to bacteria and oxidized lipids that hyperactivate macrophages.

(A) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then treated with SA113 ΔoatA bacteria at an MOI of 10 and 30 for 12 hours. LDH present in the extracellular media was then quantified.
(B) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then treated with SA113 ΔoatA bacteria at an MOI of 10 and 30 for 12 hours. IL-1β release was monitored by ELISA.
(C) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then treated with SA113 ΔoatA bacteria at an MOI of 10 and 30 for 12 hours with PI (5 μM) in the culture media during infection. Pore formation was assessed after 12 hours by quantifying PI fluorescence intensity.
(D) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then infected with SA113 ΔoatA for 12 hours at an MOI 30 for 12 hours. IL-1β release was monitored by ELISA.
(E) Immunoblot analysis of cleaved IL-1β in the extracellular media of WT and Gsdmd−/− iBMDMs after 4 hours of LPS priming or unprimed, and then infected with SA113 ΔoatA for 12 hours at an MOI of 10 and 30.
(F) Immunoblot analysis of cleaved IL-1β in lysates of WT and Gsdmd−/− iBMDMs after 4 hours of LPS priming or unprimed, and then infected with SA113 ΔoatA for 12 hours at an MOI of 10 and 30.
(G) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then treated with PGN for 6 hours. LDH present in the extracellular media was then quantified. (H) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then treated with PGN for 6 hours. IL-1β release was monitored by ELISA.
(I) Immunoblot analysis of cleaved IL-1β in the extracellular media of WT and Gsdmd−/− iBMDMs after 4 hours of LPS priming or unprimed, and then treated with PGN for 6 hours.
(J) Immunoblot analysis of cleaved IL-1β in lysates of Gsdmd−/− iBMDMs after 4 hours of LPS priming or unprimed, and then treated with PGN for 6 hours.
(K) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then treated with PGPC or POVPC for 6 hours. LDH present in the extracellular media was then quantified.
(L) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then treated with PGPC or POVPC for 6 hours. IL-1β release was monitored by ELISA.
(M) WT and Gsdmd−/− iBMDMs were primed with LPS for 4 hours (or not), and then treated with PGPC or POVPC for 6 hours with PI (5 μM) in the culture media. Pore formation was assessed after 6 hours by quantifying PI fluorescence intensity.
(N) Immunoblot analysis of cleaved IL-1β of cell culture supernatants from WT and Gsdmd−/− iBMDMs after 4 hours of LPS priming (or not), and then challenged with PGPC or POVPC for 6 hours.
(O) Immunoblot analysis of cell-associated cleaved IL-1β in Gsdmd−/− iBMDMs after 4 hours of LPS priming or unprimed, and then challenged with PGPC or POVPC for 6 hours.
Data with error bars are represented as mean ± SEM. Each panel is a representative experiment of at least 3 repeats.
See also Figure S2.
In contrast to WT cells, OatA-deficient S. aureus could not induce pore formation, IL-1β or IL-1α release in Gsdmd−/− cells (Fig. 2B–D). A second clonal population of Gsdmd−/− macrophages yielded similar results (Fig. S2A, B). Immunoblot analysis demonstrated that GSDMD was necessary for the release of cleaved IL-1β during infection but was not necessary for IL-1β processing within cells (Fig. 2E, F).
To determine if PGN or NAG induce GSDMD-dependent IL-1β release, we treated cells with PGN or transfected NAG into the cytosol. PGN treatment or NAG transfection did not induce LDH release (Fig. 2G, S2C), but did induce IL-1β release (Fig. 2H, S2D). Under these conditions, Gsdmd−/− cells released no IL-1β (Fig. 2H, S2D). Although Gsdmd−/− cells did not release cleaved IL-1β (Fig. 2I, S2E), lysates from Gsdmd−/− cells contained cleaved IL-1β (Fig. 2J, S2F). Thus, the ability of several microbial hyperactivating stimuli to induce IL-1 release is dependent on GSDMD.
GSDMD controls IL-1 release from macrophages stimulated with oxPAPC components
In addition to microbial products, self-derived damage associated molecular patterns (DAMPs) induce macrophage hyperactivation. For example, isolated lipid components from oxPAPC induce NLRP3-dependent IL-1 release from living macrophages (Zanoni et al., 2017). These lipids are PGPC (1-palmitoyl-2-glutaryl-sn-glycero-3-phosphocholine) and POVPC (1-palmitoyl-2-(5′-oxo-valeroyl)-sn-glycero-3-phosphocholine). We treated LPS-primed macrophages with PGPC or POVPC, and observed pore formation and IL-1β release, but no release of LDH (Fig. 2K, L, M). No IL-1β was released from Gsdmd−/− cells, and these cells were also defective for pore formation in response to PGPC or POVPC (Fig. 2L, M). A second clonal population of Gsdmd−/− cells yielded similar results (Fig. S2G, H). In addition, no cleaved IL-1β was detected in the extracellular media of Gsdmd−/− cells (Fig. 2N), whereas cleaved IL-1β was detected in the Gsdmd−/− lysates (Fig. 2O). oxPAPC component lipids were similar to S. aureus in their ability to elicit IL-1β release by a process that is largely insensitive to high concentrations of extracellular potassium (Fig. S2I–L). oxPAPC components and S. aureus therefore induce a mechanism of NLRP3 activation that is distinct from that induced by traditional pyroptotic stimulators of NLRP3, in that high extracellular potassium prevents the activity of the latter (S1F–H). These data indicate that several DAMPs that hyperactivate macrophages promote the GSDMD-dependent release of IL-1.
Multiple stimuli that hyperactivate macrophages promote inflammasome assembly within living cells
If hyperactive phagocytes truly represent viable cells that release IL-1, then we should be able to visualize inflammasomes within living cells. To test this hypothesis, we used primary macrophages from mice containing a transgene for the inflammasome adaptor ASC, which was fused to the fluorescent protein citrine (Tzeng et al., 2016). The ASC-citrine protein allowed us to identify cells that contained ASC “specks”—which are recognized as inflammasomes (Stutz et al., 2013). LPS-primed primary macrophages from these mice released IL-1β upon treatment with nigericin, ATP, PGN, OatA-deficient S. aureus, PGPC or POVPC (Fig. S3A–D). LPS-primed cells stimulated with nigericin and ATP appeared swollen and rounded (Fig. S3E), which are characteristic features of pyroptosis (Fink and Cookson, 2006). LPS treatment alone or treatment of LPS-primed macrophages with OatA-deficient S. aureus, PGN, PGPC or POVPC did not induce cell rounding or swelling, and the cells remained adherent to the plate (Fig. S3E). Thus, macrophages derived from ASC-citrine expressing mice behaved similarly to other macrophages used in this study and elsewhere (Wolf et al., 2016; Zanoni et al., 2016).
Cells were stimulated with LPS alone, or were primed with LPS and treated with pyroptotic or hyperactivating stimuli. 16–22 hours later, live cell confocal microscopy was used to identify macrophages containing ASC specks. ASC speck containing cells were monitored for the ability to maintain mitochondrial membrane potential by staining with MitoTracker Red CMXROS. Alternatively, ASC-citrine macrophages were treated with fluorescent zymosan, which allowed us to monitor phagocytic activity. LPS treatment alone did not induce inflammasome assembly, as revealed by a lack of ASC specks in these cells (Fig. 3A and Video S1). These cells exhibited MitoTracker Red CMXROS staining (Fig 3A, B) and readily ingested zymosan (Fig. 3C, and Video S1). The opposite finding was made upon inspection of LPS-primed cells that were treated with nigericin or ATP. Under these conditions, ASC specks were formed, yet cells containing specks did not stain for MitoTracker Red CMXROS and were unable to undergo phagocytosis (Fig. 3A–C and Video S2). The absence of any cellular activities within ATP- or nigericin-treated macrophages supports the idea that these stimuli induce pyroptosis.
Figure 3. Hyperactive stimuli induce inflammasome assembly within living macrophages.

(A) Confocal imaging in the red and green channels of live cell ASC-citrine expressing BMDMs primed with LPS for 3 hours, with second stimulations of nigericin, 5 mM ATP, 50 μg/ml PGN, MOI 30 of SA113 ΔoatA, POVPC, or PGPC for 16–22 hours. Red signal corresponds to MitoTracker Red CMXRos. Green signal corresponds to ASC-citrine fusion protein diffusely cytosolic or oligomerized into inflammasome specks.
(B) Enumeration of cells with visible ASC specks that also contain polarized mitochondria as a sign of viability. (n > 25 speck containing cells per condition)
(C) Enumeration of cells with visible ASC specks that also phagocytose zymosan particles as a sign of viability. (n > 28 speck containing cells per condition)
See also Figure S3
LPS-primed cells that were stimulated with PGN, OatA-deficient S. aureus, PGPC or POVPC induced the formation of ASC specks (Fig. 3A). The vast majority of ASC speck-containing cells treated with these stimuli retained mitochondrial and phagocytic activity (Fig. 3A–C and Videos S3–S4). We noted that a small proportion of cells appeared dead after treatment with hyperactive stimuli. Of these stimuli, OatA-deficient S. aureus induced the least amount of cell death (Fig. 3B, C), but induced the highest amount of IL-1β release (Fig. S3B–D). In contrast, the most toxic hyperactivating stimulus (PGN) (Fig. 3B, C), induced the lowest amount of IL-1β release (Fig. S3B–D). This anti-correlation between cell death and IL-1β release that is associated with hyperactivating stimuli supports the idea that these inflammasome activators induce IL-1 release from living cells that contain ASC specks.
IL-1β release occurs upon GSDMD pore formation in the plasma membrane of intact cells
To determine if GSDMD pores facilitate the secretion of proteins from living cells, we used 293T cells as a model. We created cDNAs encoding the N- and C-terminal fragments of GSDMD (NT-GSDMD-EGFP and CT-GSDMD-EGFP), and full-length GSDMD (GSDMD-EGFP), each containing an EGFP tag. Plasmids encoding each of these GSDMD alleles were electroporated into 293T cells that were stably producing a fluorescent IL-1β-tdTomato fusion protein. Rupture of the plasma membrane during our analysis was prevented by performing all experiments in the presence of glycine.
Forward and side scatter analysis by flow cytometry focused attention on intact cells (Fig. S4A). At 6 hours post-electroporation, all Gsdmd alleles were expressed detectably, except the GSDMD N-terminus (Fig. 4A). Thus, we included in our analysis a hypomorphic GSDMD N-terminus that contains an I105N mutation (Kayagaki et al., 2015). This mutation reduces the efficiency of pore formation and renders this protein more detectable within cells than the WT GSDMD N-terminus (Aglietti et al., 2016) and (Fig. 4A).
Figure 4. Cleaved GSDMD acts as a conduit for the release of cytosolic or encapsulated IL-1 family members.

(A) EGFP signal was monitored by flow cytometry in 293T cells stably expressing IL-1β-tdTomato after electroporation with GSDMD alleles tagged with EGFP.
(B) 7-AAD signal was monitored by flow cytometry in 293T cells stably expressing IL-1β-tdTomato after electroporation with GSDMD alleles tagged with EGFP.
(C) tdTomato signal was monitored by flow cytometry in 293T cells stably expressing IL-1β-tdTomato after electroporation with GSDMD alleles tagged with EGFP.
(D) Representative histogram of tdTomato fluorescence overlays for 0 and 7 hours post electroporation with GSDMD alleles tagged with EGFP.
(E–G) EM micrographs of PC:PS unextruded liposomes treated with recombinant GSDMD alone (E) caspase-11 alone (F) or with combined treatment (G). Scale bar equals 50nm
(H) Immunoblot analysis of IL-1β present within liposomes (pellet), or supernatants after ultracentrifugation of liposomes that were untreated, treated with caspase-11, or treated with caspase-11 and GSDMD for 30 and 120 minutes.
(I) Densitometry quantification of western blot band density for IL-1β release from liposomes.
(J) Immunoblot analysis of IL-18 present within liposomes (pellet), or supernatants after ultracentrifugation of liposomes that were untreated, treated with caspase-11, or treated with caspase-11 and GSDMD for 30 and 120 minutes.
(K) Densitometry quantification of western band density for IL-18 release from liposomes.
Data with error bars are represented as mean ± SEM. Each panel is a representative experiment of at least 3 repeats.
See also Figure S4
To determine which GSDMD variant induced pore formation, cells were stained with the membrane impermeable DNA dye 7-AAD. Two functionally distinct groups of GSDMD variants were identified. One group exhibited low intensity 7-AAD staining, and consisted of full-length GSDMD, its C-terminal fragment, and full length GSDMD I105N (Fig. 4B, S4B). The second group exhibited high intensity 7-AAD staining, and consisted of the N-terminal pore-forming fragments of WT and I105N GSDMD (Fig. 4B, S4B). To determine if pore forming activity correlated with IL-1β release, IL-1β-tdTomato fluorescence intensity was monitored. We observed a transition from a single tdTomato positive population at 0 hours to positive and negative populations at 7 hours post-electroporation (Fig. 4C, D). The pore forming N-terminal WT and I105N GSDMD fragments exhibited similar and substantial decrease in red fluorescence over time (Fig. 4C, D). In contrast, the full-length GSDMD, its C-terminal fragment, and full length GSDMD I105N exhibited minimal loss of red fluorescence over time (Fig. 4C, D). The pore forming activity of GSDMD therefore correlates with the ability of cells to release IL-1β. These studies suggest that the GSDMD pore is sufficient to mediate IL-1 release from intact cells.
IL-1 family cytokines pass through GSDMD pores that form in intact liposomes
The correlation between pore formation and IL-1β release in intact cells suggests that GSDMD pores serve as a conduit for IL-1β secretion. To test this possibility, a reductionist approach was taken to examine the release of IL-1β from the lumen of liposomes. We generated liposomes that consist of 1-palmitoyl- 2-oleoyl-sn-glycero-3-phosphocholine (PC) and 1,2-dioleoyl-sn-glycero-3-(phospho-l-serine) (PS). Electron microscopy of liposomes treated with GSDMD or caspase-11 did not reveal evidence of pore formation (Fig. 4E, 4F). In contrast, GSDMD and caspase-11 co-treatment generated liposomes that contained pores (Fig. 4G), as expected (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). To determine if GSDMD pores stimulated membrane rupture, liposomes were generated in the presence of fluorescent dextrans of variable sizes. No dextran release was observed when liposomes were treated with GSDMD or caspase-11, but dextran release was observed upon GSDMD and caspase-11 co-treatment (Fig. S4C). Dextrans with a diameter smaller than the GSDMD pore passed into the extra-liposomal space (4kDa and 20Kda with hydrodynamic radii of less than 4 nm) (Armstrong et al., 2004) (Fig. S4C). By contrast, dextrans with hydrodynamic radii of 27 nm (2,000 kDa) were largely retained in the liposome lumen (Fig. S4C). We also examined the release of enzymatically active tetrameric LDH from the liposome lumen, which has a diameter of 10nm (Kovacs and Miao, 2017). LDH release was minimal under conditions of GSDMD and caspase-11 co-treatment (Fig. S4D). These data indicate that GSDMD generates pores within intact liposomes.
Similar studies were performed to determine if GSDMD pores facilitate IL-1β release from the liposome lumen. Treatment with GSDMD or caspase-11 did not release IL-1β (Fig. 4H, I). In contrast, treatment of liposomes with GSDMD and caspase-11 induced the release of ~80% of the total IL-1β that was present in the lumen (Fig. 4H, I). Similar results were obtained when we examined IL-18 release (Fig. 4J, K). These findings are similar to what we observe under conditions of cell hyperactivation, where IL-1 family cytokines, but not LDH, are released from intact cells. These collective results raise the possibility that GSDMD, in addition to promoting pyroptosis, serves as a direct conduit for the release of IL-1 family cytokines.
DISCUSSION
Based on data presented in this study, we propose that GSDMD has distinct functions in the context of two different cell fate decisions. One function is to execute the process of cell death, which occurs when the cell fate decision of pyroptosis is made. This activity of GSDMD results in the indirect release of IL-1 after membrane disruption. The pyroptotic cell fate decision offers the benefit of a massive inflammatory response that can be induced locally, at the site of infection. However, the cost of pyroptosis is that the dead cell can no longer participate in any immunomodulatory activities. These costs and benefits are somewhat balanced by the second cell fate decision that we now consider to be mediated by GSDMD—cell hyperactivation. The benefit of this cell fate decision is that the phagocyte will have added IL-1 family cytokines to the repertoire of secreted factors, and that these cells may continue to influence immunomodulatory events. Indeed, conditions inducing dendritic cell hyperactivation lead to stronger antigen-specific T cell responses in mice than those that induce traditional dendritic cell activation states (Zanoni et al., 2016). The cost of cell hyperactivation however, may be that a more modest abundance of IL-1 in the infected tissue is achieved, as compared to a pyroptotic cell fate decision.
Central to our working hypothesis is that hyperactive cells are alive. In this study, we provided additional data to support this claim, as live cell microscopy revealed hyperactive cells containing inflammasomes, and most of these cells maintained mitochondrial and phagocytic activity. This ability to maintain biological activities after inflammasome assembly is a hallmark of cell viability, and supports our conclusion that hyperactivating stimuli induce IL-1 release from living cells.
The presence of a small percent of dead cells that have been exposed to hyperactivating stimuli is worth discussing. These dead cells likely contribute to some of the IL-1 release observed under hyperactivating conditions, as these cells contain inflammasomes. However, three lines of evidence invalidate the suggestion that these dead cells are the sole source of IL-1 released into the extracellular media. First, a comparison of several hyperactivating stimuli revealed an anti-correlation between amount of IL-1β release and the extent of cell death. If dead cells were solely responsible for IL-1 release, then a direct correlation should be observed. Second, NLRP3 stimuli that promoted pyroptosis were sensitive to high extracellular potassium concentrations, whereas NLRP3 stimuli that promoted cell hyperactivation were largely insensitive to similar treatments. This finding is inconsistent with the idea that hyperactivating stimuli are merely weak activators of pyroptosis. Third, if we were to propose that dead cells were the sole source of extracellular IL-1, then we must also propose that the vast majority of living hyperactive cells that contain inflammasomes are unable to process IL-1 and GSDMD. Based on the compendium of evidence supporting inflammasomes as the sites of IL-1 and GSDMD processing, this possibility is unlikely. We do, however, recognize the limitations of our assays, in that we have demonstrated a genetic requirement of GSDMD for IL-1 release from macrophages, but have used model 293T cells and liposomes to monitor protein secretion. Unimpeachable evidence supporting our conclusions awaits the development of assays that monitor viability, inflammasome assembly and IL-1 release simultaneously from single cells. Despite this caveat, the simplest interpretation of our data is that GSDMD pores represent a mechanism of IL-1 secretion from hyperactive cells. Validation of this hypothesis would provide a mandate to better understand the conventional and unconventional mechanisms of cytokine release in the innate immune system.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jonathan C. Kagan (jonathan.kagan@childrens.harvard.edu).
Experimental Model and Subject Details
Cell lines, Transfection, and Retroviral Transduction
Immortalized bone marrow derived macrophages (iBMDMs) were cultured in DMEM containing 10% FBS, Penicillin and Streptomycin (Pen+Strep), and supplements of L-glutamine and sodium pyruvate. This media is referred to below as complete DMEM. Cells were washed in PBS pH 7.4 containing 2 mM EDTA to detach cells for passage. Cells were passaged 1:10 every 3 days. HEK 293T cells were cultured in complete DMEM. Cells were washed in PBS pH 7.4 then lifted from culture flasks with 0.25% Trypsin. Trypsin was deactivated by addition of serum containing media. The IL-1β-tdTomato fusion construct was created by PCR amplification of the DNA sequence corresponding to the amino acids of cleaved murine IL-1β with the addition of a starting methionine and terminal BglII and SalI restriction enzyme cut sites. The amplicon and vector ptdTomato-N1 were digested with BglII and SalI enzymes per the manufacturer’s instructions. The amplicon and vector were gel extracted and ligated together with T7 ligase. GsdmD-EGFP, NT-GsdmD-EGFP, and CT-GsdmD-EGFP were amplified from mouse GsdmD cDNA and cloned in the same manner into the fusion vector pEGFP-N1. When used to make stably expressing cell lines, fusion constructs were subcloned into the MSCV-IRES-hCD2 retroviral vector by digestion with the restriction enzymes BglII and NotI, gel purified, and ligated with T7 ligase. For generating stable cell lines with transgenes, retrovirus particles were produced by transfecting 293T cells with plasmids pCL-Eco, pCMV-VSV-G, and MSCV-IRES-hCD2 containing the gene of interest. For lentiviral mediated CRISPR/cas9 editing of cells, lentiviral particles were produced by transfecting 293T cells with plasmids psPAX2, pCMV-VSV-G, and lentiCRISPRv2. Plasmids were transfected into HEK 293T cells in 10 cm dishes at a confluency of 50–70% with polyethylenimine (PEI) at a DNA to PEI ratio of 1:3. Media was changed 12 hours post DNA transfection and viral supernatants were collected 24 hours post media change. Viral supernatants were spun at 400 x g to remove cellular debris then passed through a 0.45 μm PVDF filter via syringe. To generate an IL-1β-tdTomato stably transduced cell line, viral supernatants were placed directly on growing 293T cells for 24 hours. These cell lines were sorted based on double positivity of hCD2 surface expression and intrinsic fluorescence from the IL-1β-tdTomato protein.
Generation of Gsdmd-deficient iBMDMs
The Gsdmd-targeting lentivirus was packaged using lentiCRISPRv2 system. In brief, iBMDMs were plated into a 6-well tissue culture plate and were transduced the next day with the lentiviral particle expressing Cas9 and one of two Gsdmd-specific guide RNAs (CAGCATCCTGGCATTCCGAG, AAAGTCTCTGATGTCGTCGA). At 48 hours-post transduction, fresh media containing 20 μg/ml puromycin was added to select for cells transduced with the Gsdmd-targeting lentivirus. The puromycin resistant cells were further subjected to single-cell cloning by limited dilution. After culturing in puromycin-containing media for an additional 2–3 weeks, single colonies were picked, expanded, and then analyzed for GSDMD protein by western analysis. iBMDMs expressing the lentiCRISPRv2 vector with a GFP targeting sequence were used as WT controls.
Differentiation of ASC-citrine bone marrow derived macrophages
L929 fibroblast cells producing M-CSF were cultured in complete DMEM. Supernatants from L929 fibroblasts were cleared of cellular debris by spinning at 400 x g for 5 minutes. Pooled supernatants from several culture flasks were combined and passed through a 0.22 μm filter. M-CSF conditioned supernatants were aliquoted and frozen at −20 C. Leg bones of transgenic ASC-citrine mice were removed from dead mice. Cleaned bones were cut with scissors and flushed with sterile PBS pH 7.4 via syringe. Bone marrow suspension was passed through a 70 μm cell strainer to exclude clumps. Cells were plated at 10E6 bone marrow cells per untreated 10 cm dish in macrophage differentiation media consisting of complete DMEM with 30% L-929 M-CSF conditioned media. Plates were fed with 5 ml of additional macrophage differentiation media on day 3 of differentiation. Cell culture media was removed on day 7 and day 8 cultures. Cells were detached from non-treated plates with cold PBS pH 7.4 containing 2 mM EDTA. Cells were then resuspended in media and plated on 96 well plates for IL-1β release assays and ibidi disposable live cell imaging dishes for imaging studies.
Immortalization protocol for bone marrow derived macrophages
Primary BMDMs for immortalization were cultured in complete RPMI with 15% FBS, 30% L929 conditioned supernatant and Pen+Strep. Conditioned supernatant collected from the CREJ2 cell line carrying the J2 retrovirus was used to immortalize primary BMDMs. In brief, differentiated primary BMDMs (day 7) were further incubated with 50% J2 conditioned supernatant and 50% L929 conditioned supernatant for 7 days, with one new batch of mixed J2 supernatant and L929 supernatant added at day 3. Transduced BMDMs were then cultured in complete DMEM plus 30% L929 supernatant until 90% confluent. Cells were then passed into new medium containing 25% L929 supernatant. Following this trend, the L929 supernatant concentration in complete DMEM was decreased by 5% during each passage. The immortalization process was completed when the BMDMs grew normally in complete DMEM in the absence of L929 supernatant.
Method Details
Ligand and chemical reconstitution
Lipopolysaccharide (LPS) from E. coli, serotype 0111:B4 was bought in a ready to use format at a stock concentration of 1 mg/ml from Enzo. LPS was used in assays at a concentration of 1 μg/ml. Nigericin was purchased from Invivogen and suspended in sterile ethanol to a stock concentration of 6.7 mM. Nigericin was used at a concentration of 10 μM. Peptidoglycan (PGN) from S. aureas was supplied from Sigma and resuspended in sterile PBS pH 7.4 at a stock concentration of 2 mg/ml. Ligand was vortexed and well mixed prior to dilution and addition to cells as PGN is poorly soluble. N-acetyl glucosamine (NAG) was resuspended at a concentration of 1 M in sterile, freshly opened optiMEM transfection media and complexed with lipofectamine 2000 for transfection into cells. Glycine was purchased from Sigma and prepared as a stock solution of 500 mM in PBS pH 7.4, sterile filtered, and kept at 4C for no longer than 2 months. KCl was purchased from Amresco and stock solution was prepared in complete DMEM at a concentration of 2M. PA and LFn-Fla protein preparations were kind gifts from Russell Vance (UC Berkeley). Propidium iodide (PI) was purchased from Sigma in a ready to use stock solution at a concentration of 1 mg/ml. 7-AAD was purchased from BioLegend as a ready to use solution and used per manufacture instructions. Recombinant mouse IL-1β and human IL-18 were supplied by BioLegend at a concentration of 0.2 mg/ml in the absence of a carrier protein. L-LDH isolated from rabbit muscle was supplied by Sigma in a ready to use solution at a concentration of 10 mg/ml. Fluorescent FITC conjugated dextrans of various sizes (4 kDa, 20 kDa, and 2000 kDa) were supplied by Sigma and reconstituted in liposome buffer at a concentration of 50 mg/ml. Cell mask deep red plasma membrane dye for live cell imaging was purchased from ThermoFisher in a ready to use format. Red fluorescent zymosan was purchased from ThermoFisher and reconstituted in sterile PBS pH 7.4 through vortex and sonication per the manufacturer’s directions. MitoTracker Red CMXRos was purchased from ThermoFisher and used within the range suggested by the manufacturer’s protocol. PS and PC lipids were supplied by Avanti polar lipids in chloroform. POVPC and PGPC were supplied by Cayman Chemical. Commercially available POVPC and PGPC were supplied as a solution in ethanol in clear glass vials. To remove the organic solvent, a gentle nitrogen gas stream was used to evaporate the ethanol, leaving only the lipids in the glass vials. Pre-warmed serum-free media was then immediately added to the dried lipids so that the final concentrations of POVPC and PGPC were at 1 mg/ml. Reconstituted lipids were incubated at 37 C for 5 mins and were vortexed vigorously for 10s before adding to cells. Cells were stimulated with 100 μg/ml of resuspended POVPC or PGPC.
LDH assay, ELISAs, and antibody usage
Cell culture supernatants were cleared of cells by spinning 96 well plates at 400 x g for 5 minutes. Supernatants were transferred to round bottom, non-treated 96 well plates and spun again at 400 x g for 5 minutes. Supernatants were once more transferred to a new 96 well plate for use or storage at −20C. Supernatants were assayed for LDH release freshly after stimulation time courses per the manufacturer’s protocol from the Pierce LDH cytotoxicity colorimetric assay kit. Measurements for absorbance readings were performed on a Tecan plate reader at wavelengths of 490 nm and 680 nm. Mouse IL-1β, IL-1α, and TNFα were quantitatively measured from cell-free culture supernatants using the eBioscience Ready-SET-Go! (now ThermoFisher) ELISA kits according to the manufacturer’s protocol. Supernatants for immunoprecipitation (IP) were isolated from 6 well cultures. Supernatants were cleared of cells through transfer to 5 ml FACS tubes with tightly sealing caps and spun at 400 x g for 5 minutes. Supernatants were transferred to a new FACS tubes for IP reaction. IL-1β immunoprecipitation used biotinylated polyclonal goat anti-IL-1β antibody from R&D Systems at 0.5 μg of antibody in conjunction with 20 μl of washed neutravidin agarose beads from ThermoFisher per IP sample. These reactions occurred at 4C overnight on a wheel rotator. IL-1β from supernatant IPs was blotted with rabbit polyclonal anti-IL-1β antibody from Genetex at a dilution of 1:1000. Cell associated IL-1β in cell lysates was blotted with unconjugated goat anti-IL-1β antibody from R&D Systems at a dilution of 1:500. Cell associated β-actin was assayed for loading control through either mouse monoclonal anti-β-actin from Sigma or rabbit polyclonal anti-β-actin from Cell Signaling at a dilution of 1:5000. Liposome and supernatant associated IL-1β was detected with rabbit polyclonal anti-IL-1β antibody from Genetex at a dilution of 1:1000. Liposome and supernatant associated human IL-18 was detected with mouse monoclonal anti-IL-18 from R&D Systems at a dilution of 1:1000.
Real-time cell permeability plate reader assay
1E5 C9-WT and Gsdmd−/− iBMDMs were plated in 200 μl per well of a tissue culture treated, black 96 well plate with flat optical transparent bottoms from Costar. After at least 6 hours, the media was changed with warmed media containing no LPS or 1 μg/ml LPS. After 3 hours of priming with LPS, media was removed and replaced with secondary stimuli in warmed plate reader media (HBSS + 20 mM HEPES + 10% FBS). Wells had 100 μl 2X PI (10 μM) solution added to them either with no glycine or 2X glycine (10 mM). Lysed positive controls had 100 μl of 2X Triton X (1%) solution made in warmed plate reader media. Untreated wells had 100 μl of warm plate reader media added to them. Nigericin treated cells had 100 μl of 2X Nigericin (20 μM) solution added to their respective wells. PA treated cells had 100 μl of 2X PA (4 μg/ml) added to their respective wells. LFn-Fla treated cells had 100 μl of 2X LFn-Fla (1.0 μg/ml) added to their respective wells. Flatox (PA + LFn-Fla) treated cells has 100 μl of 2X Flatox (4 μg/ml PA + 1.0 μg/ml LFn-Fla) added to their respective wells. Cells were allowed to equilibrate with PI for 10 minutes in a 37 C incubator. The plate was spun at 400xg for 5 minutes to ensure all cells were at the bottom of the well prior to adding the plate to the plate reader. A Tecan plate reader was used for real time fluorescence monitoring. The program settings for kinetic PI inclusion assay were bottom reading of fluorescence with an excitation wavelength of 530 nm with a bandwidth of 9 nm and emission wavelength of 617 nm with a bandwidth of 20 nm. Ideal gain was calculated from one of the positive lysed replicate wells to make the signal from that well be 70% of max detectable signal. The number of flashes was set to 25 with an integration time of 20 μs with no lag time or settle time. The machine was set to maintain 37 C for the period of the assay.
Bacterial infections with SA113 ΔoatA
WT and ΔoatA SA113 were generous gifts from the lab of David Underhill (Cedars Sinai), and were first identified by Friedrich Götz and colleagues. Glycerol bacterial stocks of SA113 ΔoatA were initially streaked to colonies on TSA agar plates containing sheep blood from ThermoFisher. Single colonies were used for individual experiments. Colonies were picked and grown in Todd-Hewitt broth from Sigma supplemented with Kanamycin selection at 50 μg/ml in a volume of 5 ml for 18–24 hours at 37 C while shaking. Cultures were washed 3 times in sterile PBS pH 7.4. Indicated MOI of bacteria were prepared in warm DMEM with 10% FBS, L-glutamine and sodium pyruvate supplementation without antibiotics. Cell culture media was replaced with media of indicated MOI and plates were spun at 400 x g for 5 minutes to synchronize bacterial uptake by macrophages. Macrophages were either unprimed or primed with 1 μg/ml of LPS for 3–4 hours prior to infection. After 1–2 hours of infection in antibiotic free media, media was replaced with warm DMEM with 10% FBS, L-glutamine and sodium pyruvate supplementation with Pen+Strep antibiotics and supplemented with gentamycin to kill extracellular bacteria. For experiments examining PI staining, PI was included in the culture media to monitor transient pore formation during the 12 hour course of infection. 12 hours post initial infection, LDH was measured on cell-free supernatants from 96 well plates that had been plated with 1E5 macrophages 6 hours prior to priming. Additional supernatants were used for ELISAs. Western analysis of cleaved IL-1β in the supernatant and lysate of macrophages was from 1E6 macrophages plated in 6 well plates 6 hours prior to priming.
Live-cell imaging of cells with inflammasome specks
Leg bones from ASC-citrine transgenic mice expressing ubiquitous Cre on a WT ASC background were generous gifts from the lab of Douglas Golenbock (UMASS). ASC-citrine bone marrow cells were differentiated with 30% L929 supernatants containing M-CSF for 7 days in 10 ml of supplemented media on 10 cm untreated suspension cell culture dishes to prevent full adherence. On day 3, 5 ml of supplemented media containing 30% L929 supernatants were added to dishes to feed the cells. On day 7 and 8 cells were used for experiments by gentle lifting with PBS with 2 mM EDTA, cell counting with hemocytometer, and plating 2.5E5 macrophages onto ibidi glass bottom disposable 35 mm, high live cell imaging plates. Plated cells were primed with 1 μg/ml of LPS for 3 hours then stimulated with second stimuli as described elsewhere. Stimuli were left overnight and imaging was conducted the following morning. Bright field images were taken prior to confocal imaging to note general morphology of cells. For mitochondrial potential experiments, MitoTracker Red CMXRos was used at a dilution of 1:7000 and stained for 30 minutes prior to imaging. Cell mask deep red was used to stain plasma membrane at a dilution of 1:1000 for 5–10 minutes prior to staining. Cells were washed in warm DMEM to remove excess stains. Warm Fluorbrite colorless DMEM was used for live cell imaging supplemented with 20 mM HEPES. For experiments involving phagocytosis of red fluorescent zymosan, zymosan was added at an MOI of 5 to cells prior to imaging. The imaging chamber was kept at 37 C. Images were obtained with a 40X oil immersion lens on a Zeiss confocal microscope. ASC-citrine signal was obtained with the green channel, MitoTracker Red CMXRos or red fluorescent zymosan in the red channel, and cell mask deep red in the infrared channel. Cells were identified as having visual ASC specks by the operator, and positions were logged. Automatic recall positions were visited over the course of 30 minutes for phagocytosis imaging assays to collect time-lapse movies. Videos were assembled side by side with indicator arrows added to identify specks using the video editing software iMovie.
Neon electroporation
Electroporation was conducted using the Neon transfection machine, pipette, tips, and associated commercial buffers from ThermoFisher. For experiments involving electroporation of plasmid DNA into cells, 6E6 cells were suspended in 120 ul of R buffer from the Neon transfection kit. DNA was added to the cellular suspension at a concentration of 1.8 μg/1E6 cells. Cells were drawn into the neon transfection pipette and electroporated with the parameters of Voltage=1150, pulse width=20ms, and pulse number=1.
Flow cytometric analysis of electroporated cells
IL-1β-tdTomato stable HEK 293T cells were lifted with trypsin. Cells were counted and electroporated with GSDMD-EGFP fusion constructs as described above. For FACS experiments, ~6E5 electroporated cells were pipetted into FACS tubes in 0.5 ml of complete DMEM containing 5 mM glycine. When time points were taken, tubes were placed on ice and 7-AAD was added to assay for membrane permeability. Cellular debris from electroporation was excluded with FSC and SSC gating. EGFP, tdTomato, and 7-AAD fluorescence signals were recorded for cells at each time point on a FACS Canto II machine and analyzed with FlowJo v10.3.0.
Protein expression and purification
Full-length human gasdermin D (GSDMD) was cloned into pDB.His.MBP vector with a TEV cleavable N-teminal His6.MBP tag using NdeI and Xho I restriction sites. To obtain full length GSDMD, E. coli BL21 (DE3) cells harbouring pDB-His6-MBP-GSDMD were grown in LB medium supplemented with 50 μg/ml kanamycin. Protein expression was induced overnight at 18 ° C with 0.5 mM isopropyl-B-D-thiogalactopyranos ide (IPTG) after OD600 nm reached 0.8. Cells were harvested and resuspended in a lysis buffer containing 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 20 mM imidazole and 5 mM 2-mercaptoethanol, and homogenized by ultrasonication. The cell lysate was clarified by centrifugation at 18,000 rpm at 4 °C for 1 hours. The supernatant containing the target protein was incubated with Ni-NTA resin (Qiagen) that was pre-equilibrated with the lysis buffer for 30 min at 4 °C. After incubation, the resin-supernatant mixture was poured into a column and the resin was washed with the lysis buffer. The proteins were eluted by the lysis buffer supplemented with 500 mM imidazole. The His6-MBP-tagged protein was further purified by HiTrap Q ion-exchange and Superdex G200 gel-filtration chromatography (GE Healthcare Life Sciences). The MBP tag was removed by overnight TEV protease digestion at 16 °C. The cleaved protein was purified using HiTrap Q ion-exchange and Superdex 200 gel-filtration column.
Liposome encapsulation, pore-dependent release assay, and electron microscopy
Phosphocholine (PC):phosphatidylserine (PS) liposomes were created by mixing PC and PS dissolved in chloroform in a glass test tube at a mass ratio of 3:2. Chloroform was evaporated by gentle application of a stream of inert nitrogen gas over the lipid solution in a chemical fume hood. Dried lipid film was resuspended in buffer containing 25 mM Tris-HCl (pH 8.0) and 150 mM NaCl to make a final concentration of lipid in solution of 5 μg/μl. 5 μg of recombinant protein IL-1β or IL-18 was incubated in 500 μl of reconstituted buffer/lipid solution. 10 μg of L-LDH isolated from rabbit muscle was incubated in 500 μl of reconstituted buffer/lipid solution. To increase reconstitution of dried lipid film with recombinant proteins and buffer, test tubes containing lipid, buffer, and recombinant proteins were covered in parafilm and incubated at 37 C for 10–15 minutes. The combined solution was then vortexed continuously for 5 minutes at room temperature to encapsulate recombinant proteins into large liposomes. The liposome solution was spun at 100,000 x g for 15 minutes to pellet liposomes containing the recombinant protein. Supernatant containing the free protein was removed. The liposome pellet was gently washed 3 times with liposome buffer, resuspended in buffer, and then spun again at 100,000 x g for 15 minutes to pellet liposomes containing recombinant protein. The liposome pellet was gently washed again 3 times with liposome buffer and resuspended in a final volume of 1200 μl for protein release experiments. 170 μl of the liposome solution were aliquoted into 7 ultracentrifuge tubes. One tube corresponding to the maximal pellet signal and minimal supernatant signal was spun at 100,000 x g for 30 minutes. The pellet was resuspended in the same volume of fresh liposome buffer as the harvested supernatant. 7.5 μg of recombinant caspase-11 was added to liposome solution aliquots for indicated time points. 22.5 μg of recombinant GSDMD was added to 200 μl of liposome solutions for indicated time points. Caspase-11 was thawed prior to experiments and left in the fridge for 24 hours to ensure optimal activity. GSDMD was thawed directly prior to experiments on the bench top at room temperature. For IL-1β or IL-18 experiments, 5X SDS buffer was added to resuspended pellet or harvested supernatants for subsequent western blotting. For LDH experiments, resuspended pellet was lysed with 10X lysis buffer from the Pierce LDH colorimetric quantification kit and liposome buffer to make a final volume equivalent to harvested supernatants. 50 μl of lysed pellet and supernatants were incubated with 50 μl of colormetric LDH substrate from the Pierce LDH quantification kit for 15–30 minutes to assay LDH activity. Absorbance was read at 490 nm and background at 680 nm to quantify LDH release from liposome experiments. Absorbance values were normalized using the mean of 3 technical replicates of lysed pellet set to 100% activity. All liposome release assays were repeated at least 3 times, and 4 batches of purified recombinant proteins were used with slight variation in potency noted between batches.
Fluorescent dextrans of differing molecular weights were resuspended in liposomes buffer containing 25 mM Tris-HCl (pH 8.0) and 150 mM NaCl to a concentration of 50 mg/ml. Liposomes were prepared by drying and resuspension in diluted dextran solutions at a final concentration of 25 mg/ml of dextrans with 5 mg/ml of total 3:2 PC:PS lipid. Maximum fluorescence was set to the lysed pellet of untreated equivalent aliquoted resuspended liposomes loaded with fluorescent dextrans where each MW of dextran was compared to its own lysed sample to account for loading and labeling variability between the different sizes.
For electron microscopy of PC:PS liposomes in the presence of recombinant proteins, copper grids coated with a layer of thin carbon were rendered hydrophilic immediately before use by glow-discharge in air with 25 mA current for 45 seconds. Liposome solutions were loaded onto the grids, air-dried for ~1 min and blotted, leaving a thin layer of sample on the grid surface. The grids were floated on a drop of staining solution containing 1.0% uranyl formate for 60 seconds. After air-drying, the grids were examined by the Tecnai G2 Spirit BioTWIN electron microscope.
Quantification and Statistical Analysis
Statistical significance for experiments with more than two groups was tested with two-way ANOVA with Tukey multiple comparison test correction. Adjusted p values were calculated with Prism 7.0 from GraphPad and the designation of **** in the figures corresponds to a p ≤ 0.0001. Data presented is representative of at least 3 independent repeats unless otherwise designated. Data with error bars are represented as mean ± SEM.
Supplementary Material
Related to Figure 3. Analysis of phagocytosis in LPS stimulated ASC-citrine BMDMs.
Related to Figure 3. Analysis of phagocytosis in pyroptotic ASC-citrine BMDMs.
Related to Figure 3. Analysis of phagocytosis in hyperactive ASC-citrine BMDMs.
Related to Figure 3. Analysis of phagocytosis in hyperactive ASC-citrine BMDMs.
Highlights.
Multiple microbial and self-derived stimuli induce IL-1 release from living macrophages
Inflammasomes can be detected within cells that display multiple signs of viability
Living macrophages require gasdermin D to induce pore formation and IL-1 release
Gasdermin D pores facilitate the release of IL-1 from liposomes and intact cells
Acknowledgments
We thank Douglas Golenbock, Russell Vance and David Underhill for providing essential reagents. This work was supported by NIH grants AI093589 and AI116550 to J.K., and NIH grants HD087988 and Al124491 to H.W. J.K. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. C.E. is supported by the Harvard Herchel Smith and Landry Fellowships. Y.T. is a postdoctoral fellow of the Jane Coffin Childs Fund (the Merck Fellow).
Footnotes
Author Contributions
C.E. and Y.T. performed experiments. J.R., S.X., and H.W. provided reagents and performed electron microscopy. J.K. supervised all research. All authors discussed results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JK, Wenby RB, Meiselman HJ, Fisher TC. The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys J. 2004;87:4259–4270. doi: 10.1529/biophysj.104.047746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera A, Herbert S, Jakob A, Vollmer W, Götz F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Molecular Microbiology. 2005;55:778–87. doi: 10.1111/j.1365-2958.2004.04446.x. [DOI] [PubMed] [Google Scholar]
- Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, March CJ, Kronheim SR, Druck T, Cannizzaro LA, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science. 1992;256:97–100. doi: 10.1126/science.1373520. [DOI] [PubMed] [Google Scholar]
- Chen KW, Groß CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, Schroder K. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Reports. 2014;8(2):570–582. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annual review of immunology. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cellular microbiology. 2006;8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T, Hornung V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44:833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nature reviews. Immunology. 2014 doi: 10.1038/nri3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017 Sep;27(9):673–684. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nature reviews Immunology. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annual review of immunology. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Mellman I, Turley SJ, Steinman RM. Antigen processing for amateurs and professionals. Trends in cell biology. 1998;8:231–237. doi: 10.1016/s0962-8924(98)01276-8. [DOI] [PubMed] [Google Scholar]
- Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Muller DJ, Broz P, Hiller S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. The EMBO journal. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Gotz F, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell host & microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. Journal of Biological Chemistry. 1994;269(21):15195–15203. [PubMed] [Google Scholar]
- Stutz A, Horvath GL, Monks BG, Latz E. ASC speck formation as a readout for inflammasome activation. Methods Mol Biol. 2013;1040:91–101. doi: 10.1007/978-1-62703-523-1_8. [DOI] [PubMed] [Google Scholar]
- Tzeng TC, Schattgen S, Monks B, Wang D, Cerny A, Latz E, Fitzgerald K, Golenbock DT. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks during In Vivo Infection. Cell Rep. 2016 Jul 12;16(2):571–82. doi: 10.1016/j.celrep.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oostrum J, Priestle JP, Grütter MG, Schmitz A. The structure of murine interleukin-1 beta at 2.8 A resolution. J Struct Biol. 1991 Oct;107(2):189–95. doi: 10.1016/1047-8477(91)90021-n. [DOI] [PubMed] [Google Scholar]
- von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, Vance RE. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu NI, Coggeshall KM, Arditi M, Underhill DM. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell. 2016;166:624–636. doi: 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, Kagan JC. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352:1232–1236. doi: 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Springstead JR, Kagan JC. By Capturing Inflammatory Lipids Released from Dying Cells, the Receptor CD14 Induces Inflammasome-Dependent Phagocyte Hyperactivation. Immunity. 2017;47:697–709. doi: 10.1016/j.immuni.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figure 3. Analysis of phagocytosis in LPS stimulated ASC-citrine BMDMs.
Related to Figure 3. Analysis of phagocytosis in pyroptotic ASC-citrine BMDMs.
Related to Figure 3. Analysis of phagocytosis in hyperactive ASC-citrine BMDMs.
Related to Figure 3. Analysis of phagocytosis in hyperactive ASC-citrine BMDMs.