

Abstract
Myotonic dystrophy type 1 (DM1) is an inherited multisystem neuromuscular disorder caused by a CTG trinucleotide repeat expansion in the DMPK gene. Recent evidence documents that DM1 patients have an increased risk of certain cancers, but whether skin cancer risks are elevated is unclear. Using the U.K. Clinical Practice Research Datalink (CPRD), we identified 1,061 DM1 patients and 15,119 DM1-free individuals matched on gender, birth year (±2 years), attending practice, and registration year (±1 year). We calculated the hazard ratios (HRs) and 95% confidence intervals (CIs) for the association of DM1 diagnosis with skin cancer risk using Cox proportional hazards models, for all skin cancers combined and by histological subtype. Follow-up started at the latest of the age at practice registration, DM1 diagnosis/control selection or January 1st 1988, and ended at the earliest of the age at first skin cancer diagnosis, death, transfer out of the practice, last date of data collection or the end of the CPRD record (October 31, 2016). During a median follow-up of 3.6 years, 35 DM1 patients and 108 matched DM1-free individuals developed a skin cancer. DM1 patients had an increased risk of skin cancer overall (HR=5.44, 95% CI=3.33–8.89, p<.0001), and basal cell carcinoma (BCC) (HR=5.78, 95% CI=3.36–9.92, p<.0001). Risks did not differ by gender, or age at DM1 diagnosis (P-heterogeneity>0.5). Our data confirm suggested associations between DM1 and skin neoplasms with the highest risk seen for BCC. Patients are advised to minimize ultraviolet light exposure and seek medical advice for suspicious lesions.
Keywords: Myotonic dystrophy, cancer, skin, basal cell carcinoma, melanoma, non-melanoma
INTRODUCTION
Myotonic dystrophy type 1 (dystrophia myotonica, DM1, also called Steinert’s disease) is an autosomal dominant multisystem disorder caused by unstable (CTG)n trinucleotide repeat expansion in the 3′ noncoding region of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19q13.31–3. The prevalence of DM1 ranges from 0.5/100,000 in Taiwan4 to 1/550 in Northestern Quebec5; the estimate for Europe ranges from 6.8/100,000 to 36.2/100,0006–8. Myotonia and muscle weakness are the main clinical presentations of DM1. Other prevalent manifestations include posterior subcapsular cataracts, cardiac conduction abnormalities, central nervous system dysfunctions, and endocrine abnormalities9. Recently, large epidemiological studies indicated that cancer is part of the DM phenotype10–12, but lacked the information required to adequately assess the risk of skin cancers.
Case reports and small case series have suggested a possible link between DM and both pilomatricoma -a rare, benign, calcifying cutaneous tumor arising from the hair matrix,13 and basal cell carcinoma (BCC)13–15. Large DM registry studies from the U.S., Italy, and the U.K. have shown that cancers of the skin are the most common cancer in DM1 patients (n=32/781, 6/255, and 4/231, respectively)16–18; however, comparisons of risk with that in the general population were not available. Two small studies from Italy comparing dermatological findings of DM1 patients with controls showed significantly higher frequencies of pilomatricoma19, dysplastic nevi19, 20, and melanoma19 in DM1 patients, but found contradictory results with BCC19, 20. Similar results for pilomatricoma were reported in a single Spanish center study using patient medical records21. No significant BCC prevalence difference between DM1 patients and controls was noted; however, DM1 patients developed BCC at a younger age than controls. In the current study, we used computerized primary care physician records from the U.K. to evaluate the risk of skin cancers (overall and by subtype) in a large cohort of patients with DM1 compared with age and sex-matched DM1-free controls.
METHODS
Data sources
The U.K. Clinical Practice Research Datalink (CPRD) is one of the world’s largest anonymized longitudinal databases of electronic primary care medical records, derived from more than 4 million active patients and 650 general care practices around the U.K.22 CPRD started in June 1987, first known as Value Added Medical Products (VAMP) Research Databank, but earlier data are available. The database includes demographic information, clinical diagnoses, test results, immunization and referral records, selected lifestyle factors, and prescription records. Clinical diagnoses are recorded using Read codes, a unique clinical terms coding system used in the U.K. National Health Service (https://data.gov.uk/dataset/uk-read-code). All patients in CPRD are linkable to practice level Indices of Multiple Deprivation (IMD, a proxy measure for socioeconomic measure), and approximately 57% of the participating CPRD practices in the U.K. and 75% of CPRD practices located in England are linkable to the Hospital Episode Statistics (HES) inpatient records database from April 1997 to February 201623.
Patients attending CPRD participating practices were found to be representative of the U.K. population with regard to age and sex22.
Study population
From the October 2016 CPRD data release, we identified all patients with a DM1 diagnosis (n=1,061) using Read codes F392011: Steinert’s disease, and F392000: Dystrophia myotonica (Steinert’s disease). For each DM1 patient, we randomly selected up to 20 individuals from the pool of DM1-free individuals registered in the same practice and who were alive at the index date (defined as the date at 1st DM1 record for patients diagnosed after their date of practice registration, or the date of practice registration, if diagnosed prior to enrollment). DM1 patients and DM1-free individuals were additionally matched on gender, year of birth (± 2 years), and practice registration year (± 1 year); the total number of DM1-free individuals was 15,119. Figure 1 provides a flow chart of participant selection.
Figure 1.
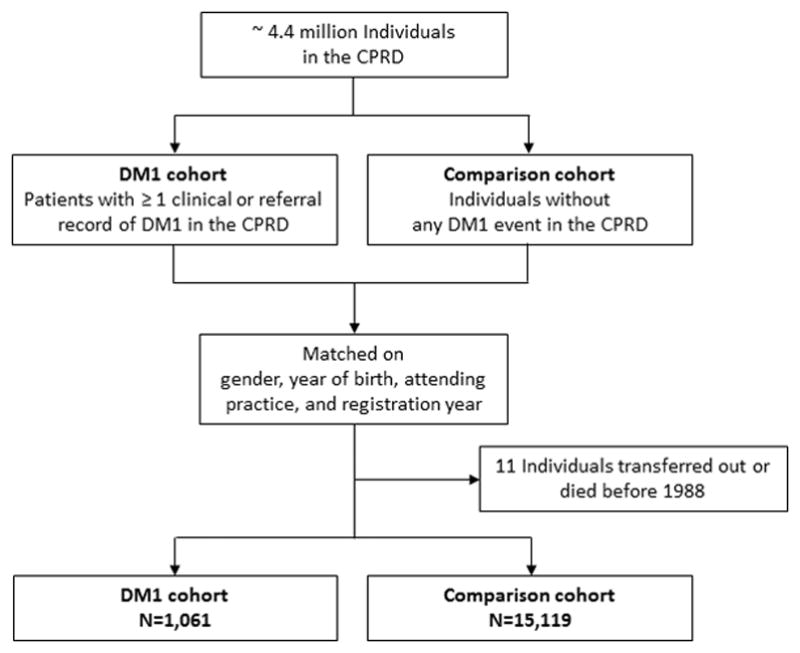
Flow chart of study participants
The study protocol was reviewed and approved by the CPRD Independent Scientific Advisory Committee (ISAC; protocol 16_005RA2R). Our use of CPRD database was approved by the National Institutes of Health Office of Human Subject Research Protection.
Skin cancer outcomes
The outcome of interest was the first skin cancer (all types combined and stratified by histological subtypes) occurring during follow-up. In the main analysis, we used Read codes (available in Supplemental Table 1) to identify skin cancers from primary care physicians’ records. Primary care physicians in the UK are the center of health care delivery; they treat, refer, and follow-up, therefore their records capture patient information through the health care continuum. In a sensitivity analysis, we used ICD-10 codes C43 (malignant melanoma of skin), and C44 (other malignant neoplasms of skin) to identify skin cancers from hospital records using HES database. This analysis included the subset of patients who are linkable to HES.
Statistical analysis
We used Cox proportional hazard regression models to calculate hazard ratios (HRs) and 95% confidence intervals (CIs) for the association of having DM1 with the first diagnosis of skin cancer occurring during follow-up, overall and by subtype. Skin cancer risk in DM1 patients was compared with that in matched DM1-free subjects. The proportional hazards assumption was evaluated using Schoenfeld residuals, and no significant violation was observed.
We used age as the time scale for all analyses. Follow-up started at the latest of age at index date, or January 1st 1988 (after the start of CPRD database). For the sensitivity analysis using the HES database, we started follow-up at the latest of age at index date or April 1st 1997 (after the start of HES data linkage). Late entry into the cohort was accounted for in PROC PHREG procedure, SAS 9.324. Follow-up ended at the earliest of age at 1st record of any skin cancer, death, transfer out of the practice, last data collection, or end of database record (for CPRD: October 31, 2016, for HES: February 29, 2016). For subtype analysis, skin cancer diagnosis other than the subtype of interest were treated as censored (i.e. follow-up ended at 1st skin cancer of any type).
The matched design of DM1 and DM1-free subjects was accommodated by stratifying the baseline hazard function on the matching ID. The models were additionally adjusted for yearly average number of clinic visits, calculated as the total number of clinical events (maximum one per day) after start of follow-up until 1 year prior to the skin cancer or censor date, divided by the number of years of follow-up.
The analysis was further stratified by gender (male, female), patient registration year at the clinic (<1991, 1991–2000, >2000), age at DM1 diagnosis (<30, ≥30), and geographical region of the practice (north, central, south). We tested the difference of the magnitude of the associations between DM1 and skin cancer across categories using a Wald test, computed as the difference of the estimates squared divided by the sum of the variances, as the estimates for different strata are independent.
To assess the robustness of our findings, we conducted several sensitivity analyses. First, we repeated the analysis restricted to DM1 patients (and their matched controls) who had their 1st DM1 event recorded at the current clinic after the start of CPRD (on or after January 1st 1988; N=538 and 6,849 for DM1 patients and DM1-free subjects, respectively). This would restrict the analysis to patients with prospectively recorded diagnoses and thus bring greater certainty about the exact date of first diagnosis of DM1 and skin cancer. Second, we repeated the analysis including only patients with first DM1 record after the date CPRD identified the practice recording to be “up-to-standard”22 (N=403 and 4,849 for DM1 patients and DM1-free subjects, respectively). Again, this would ensure better data quality. Third, we excluded individuals with skin cancer records before the start of study follow-up (N=21 DM1, and 73 DM1-free). Fourth, we restricted the analysis to a subset of patients who were linked to the HES database (N=573 and 7,614 for DM1 patients and DM1-free subjects with unique HES ID, respectively), in which skin cancer outcomes were identified only from the hospital records. Lastly, we restricted the analysis to DM1 patients with unique HES ID who had DM diagnosis records in both HES (ICD-10 code G71.1) and CPRD (N=374) and their 5,435 DM1-free matched subjects.
All p values were two-sided with statistical significance defined as p<0.05. All analyses were conducted using SAS version 9.3 (SAS Institute, Cary, NC) and R version 3.3.3.
RESULTS
Characteristics of DM1 patients and matched DM1-free subjects
DM1 patients were first diagnosed between 1944 and 2016, at a mean age of 32.7 years (SD=18.6). The mean age at the start of follow-up was 38.1 years (SD=17.0) for DM1 patients and 35.6 years (SD=16.6) for the matched DM1-free subjects. The median follow-up time was 5.4 years for DM1 patients and 3.5 years for the DM1-free individuals. Approximately 51% of both cohorts were female, and 80% were from England. More clinic visits were noted for the DM1 patients than the DM1-free cohort (mean number of annual visits=10.4 (SD=12.2) and 5.0 (SD=8.4), respectively). The characteristics of DM1 patients and matched DM1-free subjects are presented in Table 1.
Table 1.
Study cohort characteristics
| Characteristics | DM1 patients (N=1,061) | DM1-free subjects (N=15,119) |
|---|---|---|
|
| ||
| N (%) | N (%) | |
| Follow-up (person-years) | 8054.3 | 82400.4 |
| Age at start of follow-up, year | ||
| ≤20 | 155 (14.6%) | 2446 (16.2%) |
| >20, ≤30 | 207 (19.5%) | 3609 (23.9%) |
| >30, ≤40 | 210 (19.8%) | 3068 (20.3%) |
| >40, ≤50 | 206 (19.4%) | 2729 (18.0%) |
| >50 | 283 (26.7%) | 3267 (21.6%) |
| Age at 1st DM diagnosis in CPRD, year | ||
| ≤15 | 191 (18.0%) | |
| >15, ≤30 | 286 (27.0%) | |
| >30, ≤45 | 285 (26.9%) | |
| >45, ≤60 | 225 (21.2%) | |
| >60 | 74 (7.0%) | |
| Gender | ||
| Male | 520 (49.0%) | 7350 (48.6%) |
| Female | 541 (51.0%) | 7769 (51.4%) |
| Year at registration to the practice | ||
| Before 1991 | 336 (31.7%) | 4239 (28.0%) |
| 1991–2000 | 293 (27.6%) | 4689 (31.0%) |
| After 2000 | 432 (40.7%) | 6191 (40.9%) |
| Practice located in England | 844 (79.5%) | 12346 (81.7%) |
| UK Region of included practices1 | ||
| North | 370 (34.9%) | 5079 (33.6%) |
| Central | 340 (32.0%) | 4701 (31.1%) |
| South | 351 (33.1%) | 5339 (35.3%) |
| Socioeconomic status2 based on practice location, quintile | ||
| 1 (Most affluent) | 158 (14.9%) | 2206 (14.6%) |
| 2 | 181 (17.1%) | 2467 (16.3%) |
| 3 | 196 (18.5%) | 2795 (18.5%) |
| 4 | 221 (20.8%) | 3175 (21.0%) |
| 5 | 305 (28.7%) | 4476 (29.6%) |
| Average Annual number of practice visit3 | ||
| 0–1 | 187 (17.6%) | 6049 (40.0%) |
| >1, ≤5 | 224 (21.1%) | 4295 (28.4%) |
| >5 | 650 (61.3%) | 4775 (31.6%) |
North: North West England, Yorkshire & The Humbler, Northern Ireland, North East England, and Scotland
Central: East of England, Wales, West Midlands, and East Midlands
South: South West England, South East Coast, South Central England and London
Using practice level Indices of Multiple Deprivation data
Average number of clinic visit/year after the start date until 1 year prior to the end of follow-up
The association between DM1 and skin cancer risk
During 90,455 person-years of follow-up, 35 DM1 and 108 matched DM1-free subjects developed skin cancer, corresponding to crude incidence rates of 434.6 and 131.1 per 100,000 person-years among DM1 and DM1-free subjects, respectively. The mean (SD) age at 1st skin cancer diagnosis during follow-up in DM1 patients was 57.3 years (11.0), versus 63.3 years (13.0) in the DM1-free subjects.
In multivariable analysis, DM1 patients had a statistically significantly increased risk of all skin cancers combined compared with their matched DM1-free subjects (HR=5.44, 95% CI=3.33–8.89, p<0.0001). The risk was highest for BCC (HR=5.78, 95% CI=3.36–9.92, p<0.0001). Although not statistically significant, DM1 patients had an approximately two-fold increase in melanoma risk (HR=2.40, 95% CI=0.56–10.31, p=0.24). No squamous cell carcinomas (SCCs) were reported in DM1 patients. Similar results were observed when excluding patients with skin cancer diagnoses within the first 6 months of the start of follow-up (potential prevalent cancer cases)(Table 2).
Table 2.
Risk of skin cancers, overall and by histological subtype in patients with DM1.
| Outcome | All Patients (N=16,180) | Excluding individuals with skin cancer event occurred within 6 months of start of follow-up (N=16,171) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| N event (crude incident rate1) | HR2 (95% CI) | P | N event (crude incident rate1) | HR2 (95% CI) | p | |
| 1st skin cancer3 | ||||||
| DM1-free subjects4 | 108 (131.07) | 1.00 (reference) | 100(121.36) | 1.00 (reference) | ||
| DM1 patients | 35 (434.55) | 5.44 (3.33–8.89) | <.0001 | 34 (422.14) | 5.81 (3.52–9.59) | <.0001 |
| Melanoma | ||||||
| DM1-free subjects | 20 (24.27) | 1.00 (reference) | 20 (24.27) | 1.00 (reference) | ||
| DM1 patients | <5 (37.25) | 2.40 (0.56–10.31) | 0.24 | <5 (37.25) | 2.40 (0.56–10.31) | 0.24 |
| Basal cell carcinoma | ||||||
| DM1-free subjects | 80 (97.09) | 1.00 (reference) | 73 (88.59) | 1.00 (reference) | ||
| DM1 patients | 30 (372.47) | 5.78 (3.36–9.92) | <.0001 | 29 (360.06) | 6.48 (3.71–11.34) | <.0001 |
per 100,000 person-years.
Models were stratified on matched set, and adjusted for average number of clinic visit/year after the start date until 1 year prior to the end of follow-up.
In addition to the melanoma and BCC cases in the table, there were 6 SCC (all in DM1-free subjects), and <5 skin cancer, not otherwise specified in all patients; person-year of follow-up=82400 for DM1-free subjects, and 8054 for DM1 patients in analysis of all patients, and 82399 and 8054, respectively in analysis excluding cases within first 6 months of follow-up
DM1-free individuals were matched to DM1 patients on gender, birth year (±2 years), attending practice, and registration year (±1 year).
In stratified analyses, there was no evidence of differences in the magnitude of risk of overall skin cancer or BCC by gender (p-heterogeneity=0.99 and 0.56, respectively) or geographical region (p-heterogeneity=0.98 and 0.88, respectively). We also found no evidence of heterogeneity in the risk of all skin cancer combined and BCC by registration year to the practice (p-heterogeneity=0.91 and 0.72, respectively), or age at DM1 diagnosis (p-heterogeneity=0.40 and 0.50, respectively) (Table 3).
Table 3.
The association between skin cancer and DM1 stratified by selected characteristics.
| Characteristics | 1st skin cancer (all subtypes)1 | Basal cell carcinoma | ||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| DM1 patients | DM1-free subjects2 | HR3 (95% CI) | P | DM1 patients | DM1-free subjects2 | HR3 (95% CI) | p | |
|
|
|
|||||||
| N event (crude incident rate4) | N event (crude incident rate4) | |||||||
| Gender | ||||||||
| Male | 18 (451.96) | 63 (151.81) | 5.54 (2.86–10.73) | <.0001 | 15 (376.63) | 46 (110.84) | 5.09 (2.45–10.54) | <.0001 |
| Female | 17 (417.53) | 45 (110.02) | 5.51 (2.64–11.49) | <.0001 | 15 (368.41) | 34 (83.13) | 7.01 (3.10–15.90) | <.0001 |
| Pheterogeneity | 0.99 | 0.56 | ||||||
| Registration year | ||||||||
| Before 1991 | 18 (471.86) | 55 (145.05) | 5.06 (2.63–9.74) | <.0001 | 16 (419.43) | 41 (108.13) | 5.94 (2.93–12.05) | <.0001 |
| 1991–2000 | 9 (374.18) | 29 (103.08) | 6.39 (2.21–18.54) | <.001 | 8 (332.61) | 21 (74.64) | 7.43 (2.19–25.27) | <.01 |
| After 2000 | 8 (436.12) | 24 (146.80) | 4.68 (1.58–13.87) | 0.01 | 6 (327.09) | 18 (110.10) | 3.75 (1.12–12.57) | 0.03 |
| Pheterogeneity | 0.91 | 0.72 | ||||||
| Age at 1st DM diagnosis | ||||||||
| < 30 | 5 (122.06) | 12 (29.95) | 3.02 (0.68–13.39) | 0.15 | <5 (97.64) | 8 (19.97) | 3.22 (0.51–20.46) | 0.22 |
| ≥ 30 | 30 (758.00) | 96 (226.77) | 5.94 (3.53–9.98) | <.0001 | 26 (656.94) | 72 (170.08) | 6.25 (3.55–11.01) | <.0001 |
| Pheterogeneity | 0.40 | 0.50 | ||||||
| Region5 | ||||||||
| North | 12 (398.23) | 43 (145.66) | 5.18 (2.21–12.12) | <.001 | 11 (365.04) | 35 (118.56) | 6.65 (2.68–16.55) | <.0001 |
| Central | 11 (434.89) | 32 (132.46) | 5.49 (2.36–12.75) | <.0001 | 8 (316.28) | 22 (91.07) | 4.77 (1.80–12.63) | <.01 |
| South | 12 (477.80) | 33 (114.89) | 5.81 (2.39–14.11) | <.001 | 11 (437.98) | 23 (80.08) | 5.98 (2.33–15.40) | <.001 |
| Pheterogeneity | 0.98 | 0.88 | ||||||
Includes melanoma, basal cell carcinoma, squamous cell carcinoma of skin and other skin cancer, not otherwise specified.
DM1 patients and DM1-free subjects were matched on gender, birth year (±2 years), attending practice, and registration year (±1 year).
Models were stratified on matched set, and adjusted for average number of clinic visit/year after the start date to 1 year prior to the end of follow-up.
per 100,000 person-years.
North: North West England, Yorkshire & The Humbler, Northern Ireland, North East England, and Scotland
Central: East of England, Wales, West Midlands, and East Midlands
South: South West England, South East Coast, South Central England and London
Results from sensitivity analyses were consistent with those of the main analysis. Specifically, similar results were observed from models restricted to: 1) patients diagnosed with DM1 after the start of CPRD (HR=5.61, 95% CI=3.01–10.45 for all skin cancers combined, and HR=6.19, 95% CI=3.18–12.08 for BCC), 2) patients diagnosed with DM1 after the clinic “up-to-standard” date (HR=5.08, 95% CI=2.45–10.53 for skin cancer combined, and HR=5.84, 95% CI=2.71–12.57 for BCC only). In analysis restricted to patients with no prior history of skin cancer, the observed risk estimates slightly attenuated for both all skin cancer combined (HR=4.87, 95% CI=2.85–8.29), and for BCC (HR=4.86, 95% CI=2.68–8.82).
In subgroup analysis using HES database (573 DM1 and 7,614 DM1-free), having DM1 was associated with an approximately four-fold excess in the risk of non-melanoma skin cancers (NMSC) (HR=3.78, 95% CI=1.44–9.90, p=0.01). These data also suggested a possible risk for melanoma skin cancer, however not statistically significant (HR=3.38, 95% CI=0.25–46.17, p=0.36).
When restricting the analysis to patients with DM codes in both HES and CPRD (N=374) and their matched DM1-free cohort (N=5,435), DM1 patients showed an approximately seven-fold excess in the risk of all skin cancer combined (HR=7.41, 95% CI=3.31–16.59) and that of BCC (HR=6.71, 95% CI=2.86–15.76). The risk estimates were attenuated when the analyses were repeated in DM1 cases whose diagnosis were identified from one source (CPRD only) (for all skin cancer combined: HR=4.54, 95% CI=2.33–8.85, for BCC: HR=5.34, 95% CI=2.51–11.39).
DISCUSSION
In this large cohort of 1,061 patients with DM1 and 15,119 DM1-free matched individuals, we used electronic primary care health records to quanify the risk of skin cancer in those patients. We showed that DM1 patients are at a particularly high risk for basal cell carcinoma, and possibly melanoma, but no evidence of an excess risk of squamous cell carcinoma.
DM1 patients in this study had a 6-fold increase in the risk of BCC compared with matched DM1-free individuals. On the contrary, none of the DM1 patients had records of squamous cell carcinoma compared with 6 cases in the DM1-free individuals, suggesting that DM1 patients may be at a lower risk of cutaneous SCC. Because NMSC, particularly BCC is generally underreported in cancer registries25, 26, adequate comparative studies were not available with the exception of data from Denmark which suggested an excess risk of NMSC in DM patients (standardized incidence ratio (SIR)=2.08; 95% CI=1.2–3.4)10. Results related to risk of SCC need to be interpreted with caution since a validation study of primary care recording for cutaneous SCC in the UK has shown that physicians tend to use non-organ specific codes for recording cutaneous SCC (53%)27. Here, we used skin-specific SCC codes to ensure organ specificity, therefore it is possible that SCC cases were underascertained. Yet, concerns related to possible differential misclassification bias are lessened since both DM1 patients and DM1-free subjects were selected from the same practice and therefore similar reporting patterns are expected. Our estimated risk for melanoma in this study agrees with that previously reported in other DM population- and clinic-based studies (SIR=2.3 in Scandinavian patients10, 1.7 in patients from the Basque, Spain12, and 2.05 in a US cohort11); none of these risk estimates reached statistical significance.
The molecular mechanism underlying skin tumorigenesis in DM1 patients is still unknown, but several mechanisms have been hypothesized, including aberrant β-catenin accumulation via the Wnt/β-catenin signaling pathway13, and depletion/malfunction of the RNA binding protein-muscleblind like splicing regulator 1(MBLN-1)14. A recent study suggested a role for Vitamin D homeostasis in DM skin abnormalities including dysplastic nevi; an inverse correlation between Vitamin D level and the presence of dysplastic nevi was observed20.
Our study showed no gender differences in the relative risks of BCC (HR=5.09 vs 7.01, in men and women), or melanoma (3.27 vs 1.97, in men and women) in DM1 patients. This finding is similar to those previously reported in DM1 patients from Sweden and Denmark for cancers other than that of the reproductive organs10. Yet, this contrasts skin cancer statistics from the U.K. general population, in which men are at higher risk of BCC28, and women are at higher risk of melanoma29. Other known skin cancer risk factors include older age, fair skin color, light eye color and a tendency to burn on sun exposure30–33. In our study, DM1 patients appeared to develop melanoma skin cancer at a relatively early age. All melanoma cases among DM1 patients were diagnosed at <65 years of age (median age=43.8 years) versus 70% in the controls. In the U.K., about 50% of melanoma cases are diagnosed among people aged ≥65 years34. The age difference at skin cancer diagnosis was less clear in BCC, in which DM1 patients were diagnosed at a slightly younger age than DM1-free controls (58.5 vs. 62.3). It is possible that the early age at skin cancer diagnosis in DM1 patients represents ascertainment bias due to the frequent and close medical surveillance they experience in the course of their care for a serious, multisystem disorder.
Our data showed no significant association between age at DM1 diagnosis (an indicator of disease severity) and skin cancer risk. This is similar to our previous finding in DM-related brain cancer, in which no association between risk and age at DM diagnosis was observed35. Similarily, tumor development in DM patients did not correlate with the size of nucleotide repeat measured in patient blood (another proxy of disease severity) in several studies12, 16, 17, 19.
The strengths of the current study include its relatively large sample size, longitudinal design that ensured the identification of incident cancer cases, and the use of matched comparison cohort design. The ascertainment of cancer diagnosis from clinical records minimized the possible recall bias often associated with survey studies. The use of data from the primary health care setting allowed the inclusion of the full spectrum of DM1 cases, minimizing selection bias associated with identifying patients only from hospital records or tertiary care centers. Additionally, and in contrast to most cancer registries, the CPRD data captured NMSC.
Several limitations existed including our inability to directly adjust for the known skin cancer risk factors such as sun exposure and cutaneous phenotype, which are not uniformly captured with UK primary care records. Suggestive associations between risk of skin malignancies and pigmentation phenotype or reaction to sun exposure have been observed in a previous DM study36. Given the close medical attention DM1 patients recieve, it is likely that our observed association is affected by detection bias. To minimize this possibility, we adjusted our models for the yearly average number of doctor visits. Additionally, it is possible that DM1 diagnosis in CPRD may not be accurately recorded. We investigated DM1 diagnosis validity in 516 DM1 patients with any HES record, and found that 374 of them had DM1 records in both sources. The stronger associations we observed when restricting the analysis to those with DM1 diagnosis in both sources suggest that our results are valid and that possible bias that may be associated with DM1 misclassification is pulling the results toward the null.
In conclusion, our study showed that patients with DM1 are at increased risk of basal cell carcinoma and possibly melanoma. It is important that DM1 patients adhere to sun protective behaviors, minimizing exposure to ultraviolet light, and to seek medical advice if suspicious skin lesions appeared. Molecular studies aiming at elucidating the biological pathways involved in DM1 skin carcinogenesis are warranted, since it may provide novel insights into our understanding of DM-related carcinogenesis, in general.
Supplementary Material
Novelty and Impact.
Patients with myotonic dystrophy type 1 (DM1), an inherited tri-nucleotide repeat disorder, are at high risk of certain cancers. However, risk of skin cancers in those patients was not comprehensively evaluated. Using data from the UK Clinical Practice Datalink, we showed that DM1 patients are at high risk of basal cell carcinoma (HR=5.8, p<0.0001), and possibly melanoma (HR=2.4, p=0.24). The findings provide evidence that skin cancer is part of the DM phenotype.
Acknowledgments
The authors would like to thank Ms. Julie Buckland, Ms. Emily Carver and Mr. David Ruggieri of Information Management Services, Inc. Rockville, MD for their valuable contribution in study data management.
The study is funded by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, USA.
Abbreviations
- BCC
Basal cell carcinoma
- CPRD
Clinical Practice Research Datalink
- DM1
Myotonic dystrophy type 1
- DMPK
Dystrophia myotonica protein kinase
- HES
Hospital Episode Statistics
- HR
Hazard ratio
- IMD
Indices of Multiple Deprivation
- MBLN-1
Muscleblind like splicing regulator 1
- NMSC
Non-melanoma skin cancers
- SCC
Squamous cell carcinoma
- SD
Standard deviation
- SIR
Standardized incidence ratio
- 95% CI
95% confidence interval
Footnotes
CONFLICT OF INTEREST
Dr. Wilhelmine Meeraus is currently employed as a respiratory epidemiologist by GlaxoSmithKline, and holds GlaxoSmithKline shares. Dr. Meeraus was employed by CPRD when the study was designed and conducted. There is no other conflict of interest to disclose.
Disclaimer: The results were presented at the 2017 International Conference on Pharmacoepidemiology & Therapeutic Risk Management.
References
- 1.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 2.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–5. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 3.Cho DH, Tapscott SJ. Myotonic dystrophy: Emerging mechanisms for DM1 and DM2. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2007;1772:195–204. doi: 10.1016/j.bbadis.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Hsiao KM, Chen SS, Li SY, Chiang SY, Lin HM, Pan H, Huang CC, Kuo HC, Jou SB, Su CC, Ro LS, Liu CS, et al. Epidemiological and genetic studies of myotonic dystrophy type 1 in Taiwan. Neuroepidemiology. 2003;22:283–9. doi: 10.1159/000071191. [DOI] [PubMed] [Google Scholar]
- 5.Yotova V, Labuda D, Zietkiewicz E, Gehl D, Lovell A, Lefebvre JF, Bourgeois S, Lemieux-Blanchard E, Labuda M, Vezina H, Houde L, Tremblay M, et al. Anatomy of a founder effect: myotonic dystrophy in Northeastern Quebec. Hum Genet. 2005;117:177–87. doi: 10.1007/s00439-005-1298-8. [DOI] [PubMed] [Google Scholar]
- 6.Lefter S, Hardiman O, Ryan AM. A population-based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology. 2017;88:304–13. doi: 10.1212/WNL.0000000000003504. [DOI] [PubMed] [Google Scholar]
- 7.Suominen T, Bachinski LL, Auvinen S, Hackman P, Baggerly KA, Angelini C, Peltonen L, Krahe R, Udd B. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet. 2011;19:776–82. doi: 10.1038/ejhg.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindberg C, Bjerkne F. Prevalence of myotonic dystrophy type 1 in adults in western Sweden. Neuromuscul Disord. 2017;27:159–62. doi: 10.1016/j.nmd.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Meola G, Cardani R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2015;1852:594–606. doi: 10.1016/j.bbadis.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 10.Gadalla SM, Lund M, Pfeiffer RM, et al. Cancer risk among patients with myotonic muscular dystrophy. JAMA. 2011;306:2480–6. doi: 10.1001/jama.2011.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Win AK, Perattur PG, Pulido JS, Pulido CM, Lindor NM. Increased Cancer Risks in Myotonic Dystrophy. Mayo Clin Proc. 2012;87:130–5. doi: 10.1016/j.mayocp.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernández-Torrón R, García-Puga M, Emparanza J-I, Maneiro M, Cobo A-M, Poza J-J, Espinal J-B, Zulaica M, Ruiz I, Martorell L, Otaegui D, Matheu A, et al. Cancer risk in DM1 is sex-related and linked to miRNA-200/141 downregulation. Neurology. 2016;87:1250–7. doi: 10.1212/WNL.0000000000003124. [DOI] [PubMed] [Google Scholar]
- 13.Mueller CM, Hilbert JE, Martens W, Thornton CA, Moxley RT, 3rd, Greene MH. Hypothesis: neoplasms in myotonic dystrophy. Cancer Causes Control. 2009;20:2009–20. doi: 10.1007/s10552-009-9395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zemtsov A. Association between basal, squamous cell carcinomas, dysplastic nevi and myotonic muscular dystrophy indicates an important role of RNA-binding proteins in development of human skin cancer. Arch Dermatol Res. 2010;302:169–70. doi: 10.1007/s00403-009-0997-8. [DOI] [PubMed] [Google Scholar]
- 15.Goolamali SI, Edmonds EVJ, Francis N, Bunker CB. Myotonic dystrophy and basal cell carcinomas: coincidence or true association? Clin Exp Dermatol. 2009;34:e370-e. doi: 10.1111/j.1365-2230.2009.03321.x. [DOI] [PubMed] [Google Scholar]
- 16.Das M, Moxley RT, III, Hilbert JE, Martens WB, Letren L, Greene MH, Gadalla SM. Correlates of tumor development in patients with myotonic dystrophy. J Neurol. 2012;259:2161–6. doi: 10.1007/s00415-012-6476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianchi ML, Leoncini E, Masciullo M, Modoni A, Gadalla SM, Massa R, Rastelli E, Terracciano C, Antonini G, Bucci E, Petrucci A, Costanzi S, et al. Increased risk of tumor in DM1 is not related to exposure to common lifestyle risk factors. J Neurol. 2016;263:492–8. doi: 10.1007/s00415-015-8006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alsaggaf R, Wang Y, Marini-Bettolo C, Wood L, Nikolenko N, Lochmuller H, Greene MH, Gadalla SM. Benign and malignant tumors in the UK myotonic dystrophy patient registry. Muscle Nerve. 2017 doi: 10.1002/mus.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zampetti A, Silvestri G, Manco S, Khamis K, Masciullo M, Bianchi MLE, Damiani A, Santoro M, Linder D, Bewley A, Feliciani C. Dysplastic nevi, cutaneous melanoma, and other skin neoplasms in patients with myotonic dystrophy type 1: A cross-sectional study. J Am Acad Dermatol. 2015;72:85–91. doi: 10.1016/j.jaad.2014.09.038. [DOI] [PubMed] [Google Scholar]
- 20.Campione E, Botta A, Di Prete M, Rastelli E, Gibellini M, Petrucci A, Bernardini S, Novelli G, Bianchi L, Orlandi A, Massa R, Terracciano C. Cutaneous features of myotonic dystrophy types 1 and 2: Implication of premature aging and vitamin D homeostasis. Neuromuscul Disord. 2017;27:163–9. doi: 10.1016/j.nmd.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Marcoval J, Olive M, Bonfill-Orti M, Martinez-Molina L, Talavera-Belmonte A. Cutaneous Neoplasms in Myotonic Dystrophy Type 1. Dermatology. 2017 doi: 10.1159/000456074. [DOI] [PubMed] [Google Scholar]
- 22.Herrett E, Gallagher AM, Bhaskaran K, Forbes H, Mathur R, van Staa T, Smeeth L. Data Resource Profile: Clinical Practice Research Datalink (CPRD) Int J Epidemiol. 2015;44:827–36. doi: 10.1093/ije/dyv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medicines and Healthcare products Regulatory Agency. CPRD Linked Data. https://www.cprd.com/dataAccess/linkeddata.asp.
- 24.Allison PD. Survival Analysis Using SAS: A Practical Guide. 2. SAS Institute; 2010. [Google Scholar]
- 25.Goodwin RG, Holme SA, Roberts DL. Variations in registration of skin cancer in the United Kingdom. Clin Exp Dermatol. 2004;29:328–30. doi: 10.1111/j.1365-2230.2004.01523.x. [DOI] [PubMed] [Google Scholar]
- 26.McLoone NM, Middleton RJ, Gavin AT, Walsh M, Dolan OM. Audit of basal cell carcinoma: registration practice. Br J Dermatol. 2003;148:371. doi: 10.1046/j.1365-2133.2003.05097_7.x. [DOI] [PubMed] [Google Scholar]
- 27.Chiesa Fuxench ZC, Troxel AB, Gelfand JM. Validity of diagnostic codes for identifying cutaneous squamous cell carcinoma in The Health Improvement Network. Br J Dermatol. 2016 doi: 10.1111/bjd.14957. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.National Cancer Intelligence Network. Non-melanoma skin cancer in England, Scotland, Northern Ireland, and Ireland, NCIN Data Briefing. 2013. [Google Scholar]
- 29.Arnold M, Holterhues C, Hollestein LM, Coebergh JW, Nijsten T, Pukkala E, Holleczek B, Tryggvadottir L, Comber H, Bento MJ, Diba Ch S, Micallef R, et al. Trends in incidence and predictions of cutaneous melanoma across Europe up to 2015. J Eur Acad Dermatol Venereol. 2014;28:1170–8. doi: 10.1111/jdv.12236. [DOI] [PubMed] [Google Scholar]
- 30.Khalesi M, Whiteman DC, Tran B, Kimlin MG, Olsen CM, Neale RE. A meta-analysis of pigmentary characteristics, sun sensitivity, freckling and melanocytic nevi and risk of basal cell carcinoma of the skin. Cancer Epidemiol. 2013;37:534–43. doi: 10.1016/j.canep.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Gandini S, Sera F, Cattaruzza MS, Pasquini P, Picconi O, Boyle P, Melchi CF. Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure. Eur J Cancer. 2005;41:45–60. doi: 10.1016/j.ejca.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Gandini S, Sera F, Cattaruzza MS, Pasquini P, Zanetti R, Masini C, Boyle P, Melchi CF. Meta-analysis of risk factors for cutaneous melanoma: III. Family history, actinic damage and phenotypic factors. Eur J Cancer. 2005;41:2040–59. doi: 10.1016/j.ejca.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 33.Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. Journal of Photochemistry and Photobiology B: Biology. 2001;63:8–18. doi: 10.1016/s1011-1344(01)00198-1. [DOI] [PubMed] [Google Scholar]
- 34.Cancer Research UK. Skin cancer incidence statistics. http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/skin-cancer/incidence#ref-1.
- 35.Gadalla SM, Pfeiffer RM, Kristinsson SY, Bjorkholm M, Landgren O, Greene MH. Brain tumors in patients with myotonic dystrophy: a population-based study. Eur J Neurol. 2016;23:542–7. doi: 10.1111/ene.12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gadalla SM, Hilbert JE, Martens WB, Givens S, Moxley RT, 3rd, Greene MH. Pigmentation phenotype, photosensitivity and skin neoplasms in patients with myotonic dystrophy. Eur J Neurol. 2017;24:713–8. doi: 10.1111/ene.13276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.