

Abstract
Following fasting, satiety is accompanied by neuronal activation in brain areas including the central amygdalar nucleus (CEA). Since CEA is known to inhibit food intake, we hypothesized that CEA contributes to the termination of meal during refeeding. To better understand the organization of this satiety-related circuit, the interconnections of the CEA with refeeding-activated neuronal groups were elucidated using retrograde (Cholera Toxin-β subunit, CTB) and anterograde (Phaseolus Vulgaris Leucoagglutinin, PHA-L) tracers in male rats. C-Fos-immunoreactivity was used as marker of neuronal activation. The refeeding-activated input of the CEA primarily originated from the paraventricular thalamic, parasubthalamic and parabrachial nuclei. Few CTB-c-Fos double-labeled neurons were detected in the prefrontal cortex, lateral hypothalamic area, nucleus of the solitary tract (NTS) and the bed nuclei of the stria terminalis (BNST). Only few refeeding-activated proopiomelanocortin-producing neurons of the arcuate nucleus projected to the CEA. Anterograde tract tracing revealed a high density of PHAL-labeled axons contacted with refeeding-activated neurons in the BNST, lateral hypothalamic area, parasubthalamic, paraventricular thalamic and parabrachial nuclei and NTS; a low density of labeled axons was found in the paraventricular hypothalamic nucleus. Chemogenetic activation of the medial CEA (CEAm) inhibited food intake during the first hour of refeeding, while activation of lateral CEA had no effect. These data demonstrate the existence of reciprocal connections between the CEA and distinct refeeding-activated hypothalamic, thalamic and brainstem nuclei, suggesting the importance of short feedback loops in the regulation of satiety and importance of the CEAm in the regulation of food intake during refeeding.
Keywords: satiety, anterograde tract tracing, retrograde tract tracing, c-Fos, connectivity map, parabrachial nucleus, parasubthalamic nucleus
Introduction
Obesity arises from a chronic imbalance of energy intake and energy expenditure that can be caused by a delayed satiety response during food intake (French et al. 1993; Geliebter et al. 1988). Although the nucleus of the solitary tract (NTS) is known as a primary integration site for central and peripheral satiety signals involved in the determination of meal size (Grill and Hayes 2009; Gibson et al. 2010), interaction of the NTS and forebrain structures, like the hypothalamic arcuate nucleus (ARC), is necessary for the integration of adiposity and satiety signals (Johnstone et al. 2006; Woods and D’Alessio 2008; Fekete et al. 2012). During refeeding after two days of fasting, animals terminate their food intake after two hours (Ji and Friedman 1999; Sato et al. 2005; Barrachina et al. 1997), accompanied by a well defined pattern of neuronal activation (Zseli et al. 2016; Singru et al. 2007; Timofeeva et al. 2002; Wu et al. 2014). Therefore, this fasting-refeeding paradigm can be used as a model to facilitate the elucidation of the neuronal networks that determine meal size and terminate food intake.
The central amygdalar nucleus (CEA) is well known to regulate food intake and integrate homeostatic and reward related signals. Lesions of this nucleus result in increased food intake and promote body weight gain (Rollins and King 2000). In addition, the CEA is critical for the inhibition of food intake by learned aversive cues (Petrovich et al. 2009). These data suggest that the CEA is an important anorexic center in the brain. In accordance with these data, the refeeding of fasted animals results in marked neuronal activation in the CEA (Timofeeva et al. 2002; Wu et al. 2014). Since optogenetic activation of CEA results in inhibition of food intake (Carter et al. 2013), we hypothesize that activation of CEA neurons during refeeding contributes to the development of satiety. To test this hypothesis, we determined whether early, chemogenetic activation of subnuclei of the CEA reduces food intake during refeeding, and to better understand the integration of the CEA into the network of satiety-related neuronal groups, we used retrograde and anterograde tract tracing to map the origins of the refeeding-related inputs and the refeeding-related targets of this nucleus. Since our laboratories previously demonstrated that during the early phase of refeeding, activation of the anorexigenic, proopiomelanocortin (POMC) neurons in the ARC has a critical role in the determination of meal size (Singru et al. 2007), we also determined whether refeeding-activated POMC neurons in the ARC project to the CEA.
Materials and methods
Animals
Adult, male, Wistar rats (280–300g body weight) were maintained in a light-and temperature-controlled environment (lights on 0600–1800h; 22°C) with free access to food and water.
Stereotaxic injection of hSyn-hM3D(Gq)-mCherry AAV
The hSyn-hM3D(Gq)-mCherry AAV virus (University of North Carolina Vectore Core, Chapel Hill, NC) was injected into the CEA of 24 Wistar rats. The surgeries were performed in a biosafety level 2 (BSL-2) virus injection facility. The rats were anesthetized and their head positioned in a stereotaxic apparatus with the Bregma and Lambda in the horizontal plane. Through a burr hole in the skull, a glass pipette (20-μm outer tip diameter) connected to a Nanoject II/Nanoliter 2000 microinjector (Drummond Scientific Co. or WPI Inc.) was lowered into the brain at stereotaxic coordinates corresponding to the medial part of CEA (CEAm) (anteroposterior: −2.4 mm, mediolateral: −3.9 mm, dorsoventral: −8.2 mm), or to the laterocapsular part of CEA (CEAlc) (anteroposterior: −2.4 mm, mediolateral: −4.45 mm, dorsoventral: −8.25 mm) based on the atlas of Paxinos and Watson (Paxinos and Watson 1998). The virus containing solution (80–100nl; 4.5×1012 virus/ml) was injected unilaterally into the CEA with 5nl/sec speed. Five minutes after the injection, the pipette was removed slowly, the scalp was sutured and the rats were housed in BSL-2 quarantine for 2 weeks before experimentation.
Design of experiments using chemogenetic activation of CEA
Experimental animals were transferred to the biosafety level 1 (BSL-1) room 2d before the beginning of the experiment to habituate them to the metabolic cages of the TSE Phenomaster system (TSE Systems GmbH, Bad Homburg, Germany). Each cage had a calibrated food sensor that recorded consumed food to a sensitivity of 0.01g. Food spillage was minimized by a catch tray. One hour before the experiment, the body composition of the rats was determined using an EchoMRI 700 Body Composition Analyser (EchoMRI LLC, Houston TX).
Half of the rats were injected ip with clozapine-N-oxide (CNO; RD-4936/50, Tocris Bioscience (Bristol, UK)) dissolved in saline (3mg/kg BW) and the other half of the animals were treated with saline alone after 40h fasting. Fifteen min after the injection, rats received food, and the food intake was monitored during the 20 hours refeeding period. One week later, the experiment was repeated as follows: rats that had been injected with CNO in the first experiment received saline injection, and the saline treated rats received CNO injection after 40h fasting. The food intake was recorded at intervals of 1 min, and the cumulative food intake was calculated from these data. The food intake of CEAm and CEAlc injected rats was compared after saline or CNO injection. Data were analyzed by repeated measures ANOVA followed by Tukey HSD test.
One week later, after the 40h fasting period, the first injection protocol was repeated with the difference that the animals did not receive food. Two hours after the treatment, the animals were deeply anesthetized with ketamine-xylazine and perfused transcardially with 20 ml 0.01M PBS; pH 7.4, followed by 150 ml of 4% paraformaldehyde in 0.1M PB, pH 7.4. The brains were rapidly removed and cryoprotected by immersion in 30% sucrose in PBS overnight. Coronal, 25μm thick sections of the forebrain and brainstem were cut with a freezing microtome (Leica Microsystems, Wetzlar, Germany). Series Series of sections, obtained at 100 μm intervals, were collected into antifreeze solution (30% ethylene glycol; 25% glycerol; 0.05M PB) and stored at −20°C until their use for immunohistochemistry.
Meal pattern analysis
Meal pattern analysis was carried out in order to investigate feeding motivation of the saline-and CNO-treated rats during refeeding. To analyze the meal pattern, the food and water intake data each minute were exported from the TSE PhenoMaster software. Data from the first and second hours of refeeding were calculated separately. The resulted data series were divided into meal intervals, when food and/or drink consumption occurred with less than a 6 minute break, as well as intermeal intervals longer than 5 minutes between two meal intervals.
The average and total duration of meal and intermeal intervals, the amount of food consumed during a meal interval and the number of meal and intermeal intervals were calculated in the first and second hour after the onset of refeeding.
Localization of the virus injection sites
The virus injection site was detected based on the fluorescence of hM3D(Gq)-mCherry fusion protein. Sections were mounted onto glass slides and coverslipped with DAPI-containing Vectashield mounting medium. Only animals with virus injection site confined within the CEA were included in the studies.
Mapping of the refeeding-activated areas projecting to the CEA
The retrograde tracer cholera toxin β subunit (CTB; List Biological Laboratories, Campbell, CA) was injected by iontophoresis into the region of CEA of 23 animals. Rats were anesthetized i.p. with ketamine-xylazine (ketamine: 50mg/kg; xylazine: 10mg/kg body weight) and their head positioned in a stereotaxic apparatus with the Bregma and Lambda in the horizontal plane. Through a burr hole in the skull, a glass micropipette (17.5μm outer tip diameter) filled with 0.5% CTB in 0.01M PB at pH 8.0 was lowered into the brain at stereotaxic coordinates corresponding to the CEA described above. The CTB was deposited for 10min (5μA positive current, pulsed on —off at 7s intervals) using a constant-current source (Stoelting, Wood Dale, IL). 7–10 day after tracer deposition, rats were fasted for 48h and then given free access to food for 2h. Immediately after refeeding, the animals were perfused with fixative and sections were prepared as described above.
Evaluation of the CTB injection sites
The core of the CTB injection sites were assessed by double-labeling of the sections for CTB and c-Fos. The early gene, c-Fos, was used to mark the CEA, since neurons of CEA are activated by refeeding, while the surrounding regions do not show c-Fos-immunoreactivity in refed animals. Sections pre-treated, as described, above were incubated in a mixture of primary antisera: goat anti-CTB (1:5000, List Biological Laboratories, Campbell, CA) and rabbit antic-Fos (1:1500, Oncogene, MA, USA) for 48h. After washing in PBS, sections were incubated in biotinylated donkey anti-sheep IgG (Jackson ImmunoResearch, West Grove, PA) at 1:500 for 2h at room temperature, and after further rinsing in PBS, incubated in ABC (1:1000) for 1h. Following rinses in PBS, sections were subjected to biotinylated tyramide amplification. After further washes, the sections were incubated in the cocktail of Fluorescein DTAF-conjugated Streptavidin (1:250) and Alexa555-conjugated donkey anti-rabbit IgG (1:500, Invitrogen, Carlsbad, CA, USA) for 2h, mounted onto glass slides and coverslipped with Vectashield mounting medium.
Immunocytochemical detection of the origins of the refeeding-activated inputs of the CEA
Double-labeling, light microscopic immunocytochemistry was performed to map the location of refeeding-activated neurons projecting to the CEA. Following pretreatment as described above, the sections were incubated in rabbit antiserum against c-Fos (1:10,000). After washing in PBS, sections were incubated in biotinylated donkey anti-rabbit IgG (1:500, Jackson Laboratories) for 2h, and after further rinsing in PBS, incubated in ABC (1:1000) for 1h. The tissue-bound peroxidase activity was visualized by NiDAB developer (0.05% diaminobenzidine and 0.15% nickel ammonium sulfate with 0.005% H2O2 in 0.05 M Tris buffer at pH 7.6) followed by silver-gold intensification (Liposits et al. 1984). In the second step, the sections were incubated in goat anti-CTB (1:5000) serum for two days at 4°C, followed by biotinylated donkey anti-sheep IgG (1:500) for 2h and ABC (1:1000) for 1h. The CTB immunoreactive (IR) sites were visualized with brown DAB developer (0.05% diaminobenzidine with 0.005% H2O2 in 0.05 M Tris buffer). The sections were mounted onto glass slides and coverslipped with DPX mounting medium (Fluka Chemie AG, Buchs, Switzerland).
Involvement of refeeding-activated POMC neurons of the ARC in the innervation of the CEA
To determine whether refeeding-activated POMC neurons in the ARC participate in the innervation of CEA, triple-labeling immunofluorescence was performed for CTB, c-Fos and POMC. Following pre-treatment, sections of CTB-injected animals were incubated in a mixture of the following primary antisera: goat anti-CTB (1:10,000) and rabbit anti-c-Fos (1:1500) sera. After washing in PBS, the sections were incubated in biotinylated donkey anti-sheep IgG at 1:500 for 2h at room temperature. Following ABC-BT amplification steps as described above, CTB immunoreactivity was visualized with DTAF-conjugated Streptavidin (1:250), which was applied simultaneously for 2h with Alexa555-conjugated donkey anti-rabbit IgG (1:500) to detect c-Fos immunoreactivity. After further washes, sections were immersed in rabbit anti-POMC serum (1:2000, Phoenix Pharmaceuticals Inc., Belmont, CA, USA) for 48h at 4°C, washed in PBS, incubated in Cy5-conjugated donkey anti-rabbit IgG (1:100, Jackson), and then mounted onto glass slides and coverslipped with Vectashield mounting medium.
Involvement of POMC neurons of the ARC in the innervation of refeeding-activated neurons in the parasubthalamic nucleus
To determine whether neurons of the parasubthalamic nucleus relay refeeding-related inputs to the CEA originating from POMC neurons in the ARC, triple-labeling immunofluorescence was performed for CTB, c-Fos and POMC as described above.
Identification of refeeding-activated neuronal populations contacted by axons originating from the CEA
The anterograde tracer, Phaseolus vulgaris leuco-agglutinin (PHA-L; Vector, 2.5% PHA-L in 0.01M phosphate buffer, PB at pH 8.0), was injected by iontophoresis over 20min into the region of CEA in 11 animals. Stereotaxic surgery was performed using methods similar to that used for the retrograde tract tracing experiments.
Evaluation of PHA-L injection sites
Double-labeling, fluorescent immunohistochemistry for PHA-L and c-Fos was performed to reveal the location and extent of injection sites using refeeding-induced c-Fos activation as a marker for the CEA. Following standard pre-treatment as described above, sections were incubated in a mixture of primary antisera: goat anti-PHA-L serum (1:1000, Vector Laboratories) and rabbit anti-c-Fos serum (1:1500), diluted in PBS containing 2% normal horse serum and 0.2% sodium azide (antibody diluent) for 1 day at room temperature. After washing in PBS, sections were incubated in biotinylated donkey anti-sheep IgG (1:500) for 2h at room temperature. After further rinsing in PBS, the sections were incubated in ABC (1:1000) for 1h, and then subjected BT amplification as described above. After further washes, the sections were incubated in a cocktail of Fluorescein DTAF-conjugated Streptavidin (1:250) and Alexa555-conjugated donkey anti-rabbit IgG (1:500) for 2h, mounted onto glass slides and coverslipped with Vectashield mounting medium.
Mapping refeeding-activated neuronal groups targeted by the CEA
Double-labeling, light microscopic immunocytochemistry was performed to map areas where PHA-L-IR fibers contact refeeding-activated neurons. Following pretreatment as described above, the sections were incubated in goat anti-PHA-L (1:5000) and in rabbit anti-c-Fos (1:2000) antisera. After washing in PBS, sections were incubated in biotinylated donkey anti-sheep IgG (1:500) for 2h at room temperature, and then in ABC (1:1000) for 1h. Following rinses in PBS, the tissue-bound peroxidase activity was visualized by silver-gold intensified NiDAB developer. In the second step, the sections were incubated in biotinylated donkey anti-rabbit IgG (1:500) for 2h and ABC (1:1000) for 1h. The c-Fos-IR nuclei were visualized with DAB developer, and the sections were mounted onto glass slides and coverslipped with DPX.
Determine whether the axons of CEA neurons establish contact with refeeding-activated neurons
To demonstrate that PHA-L fibers form appositions on c-Fos positive neurons in the target areas of the CEA, triple-immunofluorescent labeling was carried out. Neuronal cytoplasm was immunolabeled using antiserum against the neuronal RNA-binding protein, HuCD, and following standard pre-treatment as described above for double labeling, sections were incubated in a mixture of the following primary antisera: goat anti-PHA-L (1:5000), rabbit antic-Fos (1:2000) and mouse anti-HuCD (1:500, Molecular Probes, Eugene, OR, USA). After incubation in biotinylated donkey anti-sheep IgG (1:500, 2h), the sections were rinsed in PBS followed by the ABC-BT amplification steps. PHA-L immunoreactivity was detected with Alexa555-conjugated Streptavidin and the sections were than incubated in the mixture of Cy5-conjugated anti-mouse IgG (1:100, Jackson) and FITC-conjugated anti-rabbit IgG (1:250, Jackson) for 2h, mounted onto glass slides and coverslipped with Vectashield mounting medium.
Identify brain areas directly activated by the chemogenetic stimulation of CEA neurons
To discover brain areas activated by the stimulation of the CEA, triple immunofluorescent labeling was carried out on sections obtained from the chemogenetic experiment as described above to detect c-Fos expressing neuronal groups directly contacted by axons of CEA neurons. Following standard pre-treatment as described above, sections were incubated in a mixture of the following primary antisera: rabbit anti-c-Fos (1:10000) and mouse anti-HuCD (1:500). After incubation in biotinylated donkey anti-rabbit IgG (1:500, 2h), the sections were rinsed in PBS followed by the ABC-BT amplification steps. C-Fos immunoreactivity was detected with DTAF-conjugated Streptavidin (1:250) and the sections were then incubated in Dylight 649-conjugated anti-mouse IgG (1:100, Jackson). They were then incubated in rabbit anti-red fluorescent protein (RFP) antibody (1:3000, Rockland Immunochemicals Inc. Limerick, PA) for 48h. After rinsing in PBS, immunoreaction was visualized with Alexa555-conjugated donkey anti-rabbit IgG (1:500) for 2h, mounted onto glass slides and coverslipped with Vectashield mounting medium. In this experiment, c-Fos-immunoreactivity labeled the activated neurons, HuCD-immunoreactivity labeled the cytoplasm of neurons, while the RFP-immunoreactivity labeled the axons of the CEA neurons. The HuCD labeling of neuronal cytoplasm enabled us to examine the RFP-IR contacts on the surface of c-Fos-IR neurons.
The number of the c-Fos-IR cell nuclei in the different brain regions was counted with ImageJ software (http://rsbweb.nih.gov/ij/) using automatic particle analysis. The color images were converted to black and white images. The monochrome images were thresholded. The threshold range was set to differentiate the c-Fos containing nuclei apart from the background. The association of CEA axons and the activated neurons were studied in the activated regions.
Image analysis
Double-labeling immunofluorescence for the determination of the injection sites was examined with a Zeiss AxioImager M1 epifluorescent microscope (Carl Zeiss AG, Göttingen, Germany). Sections were examined under fluorescent illumination through Zeiss Filter Set 23: excitation 475–495 and 540–552 nm, beam splitter 500 and 560 nm, emission 515–530 and 580–630 nm. Images were captured with a Zeiss AxioImager M1 microscope using an AxioCam MRc 5 digital camera (Zeiss) and AxioVision 4.6 software (Zeiss).
Double immunolabeled sections for light microscopic analysis of PHA-L/c-Fos CTB/c-Fos neurons were examined with the same Zeiss AxioImager M1 epifluorescent microscope and images were captured with the same AxioCam MRc 5 digital camera and AxioVision Rel. 4.6 software. After composite images were taken of the entire section containing either CTB-c-Fos double-labeled neurons or c-Fos-IR neurons surrounded with PHA-L fibers, schematic drawings were made using Inkscape software. Labeled regions were identified using the rat brain atlas (Swanson 2000) and also Nissl-stained sections from the corresponding rostro-caudal level.
The triple-labeled fluorescent sections were analyzed using a Radiance 2100 confocal microscope (Bio-Rad Laboratories, Hemel Hempstead, UK). Confocal images were taken using line by line sequential scanning with laser excitation lines and filters: 488nm for DTAF or FITC 543nm for Alexa555 637nm for CY5 and dichroic/emission filters 560nm/500–530nm for DTAF or FITC, 650nm/570–620nm for Alexa555 and 660nm long-pass filter for CY5. All confocal images processed for analysis were collected using a x20 oil immersion objective for the c-Fos/CTB/POMC immunostaining and 40x objective for the c-Fos/PHA-L/HuCD immunolabeled sections, z-step 1.8 or 0.8μm, respectively; and image size 512×512 pixels.
Analyses of c-Fos-IR neurons that were either retrogradely labeled or contacted by PHAL-IR axons were restricted to areas that have been shown to be activated by refeeding in our earlier studies (Zseli et al. 2016).
Controls for the influence of CTB injection on the c-Fos immunoreactivity
In our previous experiments, we have shown that the deposition of CTB into the parabrachial nucleus did not influence the pattern of c-Fos immunoreactivity in refed animals, indicating that tracer does not interfere with neuronal activation (Zseli et al. 2016).
Antibody Characterization
The antibodies used are summarized in Table 1. According to descriptions of the manufacturers, the PHA L antiserum was produced by hyperimmunization of goat with purified PHA-L, and specific antibodies to PHA-L were isolated by affinity chromatography on PHAL-agarose columns, whereas the CTB antiserum forms an immunoprecipitin band against a 0.5 mg/ml solution of CTB. Because PHA-L and CTB are not present in the brain enogenously, the specificities of PHA-L and CTB antisera were verified by the lack of any labeling in brain sections from animals that were not injected with PHA-L and CTB.
Table 1.
| Name of the antibody | Structure of the immunogen | Manufacturing | Dilution |
|---|---|---|---|
| c-Fos | Synthetic peptide (SGFNADYEASSS RC) corresponding to amino acids 4–17 of human c-Fos | Merck Millipore, rabbit, polyclonal, #cat. PC38 RRID AB_213663 | 1:10000 |
| CTB | Purified CTB isolated from Vibrio cholerae | List Biological Laboratories, goat, polyclonal, #cat. 703 RRID AB_2314252 | 1:5000 |
| PHAL | Purified PHAL | Vector Laboratories, goat, polyclonal, #cat. AS-2224 RRID AB_2315141 | 1:5000 |
| POMC | Amino acids of 27–52 of porcine POMC precursor | Phoenix Pharmaceuticals, rabbit, polyclonal, #cat. H-029-30 RRID AB_2307442 | 1:2000 |
| HuCD | Recognizes human HuC/HuD neuronal protein and the expressed product of the ELAVL3 gene | Molecular Probes, mouse, monoclonal, #cat. A-21271 RRID AB_221448 | 1:500 |
| CRH | human/rat (h/r)CRF peptide (SEEPPI SLDLTFHLLREVL EMARAEQLAQQA HSNRKLMEII-NH2), | Peninsula Laboratories International, Inc, rabbit, polyclonal, #cat. T-4037 RRID AB_2314240 | 1:3000 |
| RFP | The immunogen is a Red Fluorescent Protein (RFP) fusion protein corresponding to the full length amino acid sequence (234aa) derived from the mushroom polyp coral Discosoma. | Rockland Immunochemicals Inc, rabbit, polyclonal, #cat. 600-401-379 RRID AB_2335885 | 1:3000 |
The anti-c-Fos antibody was raised against the N-terminal part (residues 4–17) of c-Fos protein that is conserved among human, mouse, and rat. This antibody recognized a 55-kDa c-Fos on Western blot, which corresponds to the expected molecular weight of c-Fos (manufacturer’s technical information). Additionally, a commercially available goat anti-c-Fos antibody raised against a peptide containing residues 3–16 of the amino terminus of human c-Fos (Santa Cruz Biotechnology, Santa Cruz, CA; product No. sc-52-G, lot No. 02506) in stress tissues resulted in a staining pattern identical to that produced by the rabbit anti-c-Fos antibody used in this study (Reznikov et al. 2008; Stratford and Finger 2011). Preabsorbing the serum with c-Fos protein (Elmquist et al. 1996) or the synthetic peptide immunogen (Serrats and Sawchenko 2006) eliminated staining in the rat brain. This antiserum also co-localized with c-fos mRNA in rat brain (Serrats and Sawchenko 2006).
The anti-POMC serum (Phoenix Pharmaceuticals, Inc., Belmont, CA; catalog No. H-029-30, lot R163-5) was raised in a rabbit against the porcine POMC precursor (AA 27–52). The specificity of this antiserum was confirmed by dual immunofluorescent labeling of rat and mouse hypothalamic sections using a sheep α-MSH antiserum (Wittmann et al. 2013).
Neuronal protein HuC/HuD (HuC/D) monoclonal (clone 16A11) antibody labeled neuronal cell nuclei and perikarya, recognizing an epitope within the carboxy-terminal domain of HuD (Marusich et al. 1994). Staining pattern of cellular morphology and distribution is the same as previously described (Lin et al. 2002; Murphy et al. 2007). The staining pattern of biotin-conjugated HuC/D monoclonal antibody is identical to that of the unconjugated antibody.
According to the manufacturer, the anti-RFP antibody was prepared from monospecific antiserum by immunoaffinity chromatography using Red Fluorescent Protein (Discosoma) coupled to agarose beads followed by solid phase adsorption(s) to remove any unwanted reactivities. Assay by immunoelectrophoresis resulted in a single precipitin arc against anti-rabbit serum and purified and partially purified Red Fluorescent Protein (Discosoma). No reaction was observed against human, mouse or rat serum proteins. ELISA was used to confirm specificity at less than 0.1% of target signal. Immunoreaction was not detected in brains if fluorescent protein was not expressed.
Results
Localization of the hSyn-hM3D(Gq)-mCherry AAV injection sites
To selectively activate neurons located in the CEA, hSyn-hM3D(Gq)-mCherry AAV was used to express the hM3Dq receptor-mCherry fusion protein in this nucleus. This receptor has a high affinity for the pharmacologically inert ligand CNO, but is not activated by endogenous neurotransmitters (Alexander et al. 2009).
The virus injection site in 10 cases was localized within the borders of the CEA. In 6 animals the center of the injection site was in the CEAm, and did not spread into the CEAlc. In the additional 4 cases, the virus spread only the CEAlc, without reaching the medial subnucleus (Fig. 1). Within the CEA, the CNO induced activation resulted in c-Fos expression only in the region covered by the injection site. In brains where the injection site was confined to the CEAm, the CNO treatment did not result in c-Fos activation in the CEAlc, and in brains with CEAlc injections, the neurons of CEAm were not activated by CNO (Figure 2).
Figure 1.

Localization of hSyn-hM3D(Gq)-mCherry AAV injection sites. The animals shown on (A-F) were used to study the effects of CEAm activation, while animals shown in (G–J) were used in experiments studying the effects of the activation of CEAlc. Scale bar = 500μm
Figure 2.

Effect of chemogenetic activation of CEA subnuclei on c-Fos expression in this nucleus. Saline treatment did not cause neuronal activation in the CEA of rats injected with hM3D(Gq)-mCherry expressing AAV in the CEAm (A) or CEAlc (C). C-Fos-immunoreactivity is labeled with green color, while the red fluorescence of the mCherry labels the injection site. CNO treatment induced marked neuronal activation in the CEAm, but did not influence c-Fos expression in the CEAlc in the CEAm injected animals (B). Conversely, CNO treatment induced marked neuronal activation in the CEAlc, but did not influence c-Fos expression in the CEAm in the CEAlc injected animals (D). Scale bar =500μm
Effect of chemogenetic activation of CEA subnuclei on food intake during refeeding
Activation of the CEAm neurons by ip CNO administration resulted in a significant decrease in food intake during the first 60 min. of refeeding. Following CNO treatment, animals ate 15.3±1.5mg/g lean body weight whereas after saline injection, they ate 23.3±2.1mg/g lean body weight (p=0.004). After CNO treatment, the rats had shorter meal intervals, than after saline treatment (CNO vs. saline (min): 34.83±4.56 vs. 53.67±3.29; P=0.01), and CNO treatment significantly decreased meal size compared to saline treatment (CNO vs. saline (g): 4.21±0.86 and 7.55±0.75; P=0.01).
In the second hour of refeeding, the CNO injected animals consumed significantly more food (10.5 ±2.1mg/g lean body weight) than after saline injection (2.5±1.1mg/g lean body weight) (p=0.006). Following CNO treatment, the rats demonstrated a tendency for longer meal intervals in the second hour (P=0.05), ate more during a meal interval (CNO vs. saline (g): 2.90±0.72 vs. 0.79±0.34; P=0.02), and their average intermeal intervals were shorter (CNO vs. saline (min): 14.08±6.13 vs. 45.92±6.61; P=0.03) than after saline treatment. There were also significant differences between the total length of the meal and intermeal intervals following CNO or saline treatment (CNO vs. saline cumulative length of meals (min): 40.17±7.60 vs. 11.33±4.35 P=0.01; CNO vs. saline cumulative length of intermeal intervals (min): 19.83±7.60 vs. 48.67±4.35; P=0.01).
When the parameters of the meal pattern were compared between the first and second hours, there was no significant difference after CNO treatment. In contrast, the average meal length and size and the total meal length were markedly decreased during the second hour when the animals received saline.
Cumulative food intake during the first two hours of refeeding did not differ significantly between the saline-and CNO-treated animals.
In animals with hM3D(Gq)-mCherry expression in the CEAlc, CNO treatment did not cause significant alterations in food intake: CNO vs. saline in the first hour (g/lean body weight): 20.9±1.2 vs. 24.7±0.7; p= 0.24; CNO vs. saline in the second hour (g/lean body weight): 3.4±2.2 vs. 2.3±0.8; p= 0.96). The data are summarized in Figure 3 and Table 2–4.
Figure 3.
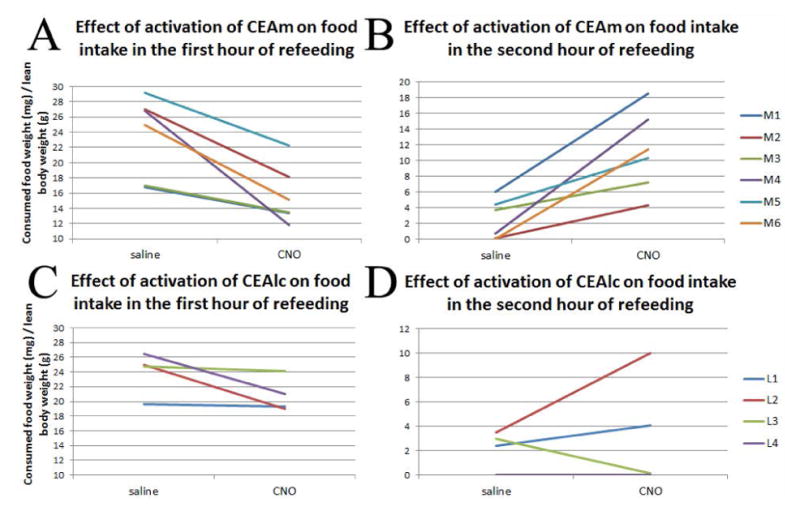
Effect of chemogenetic activation of the CEA subnuclei on food intake during refeeding. CNO-induced activation of the neurons of CEAm significantly decreased food intake during the first hour of refeeding compared to food intake of the same animal after saline injection (A; p=0.007). The color lines label the differences of food intake of the same animal after saline and CNO injection. In the second hour, the animals ate significantly more food when the CEAm was activated by CNO (B; p=0.006). Chemogenetic activation of CEAlc did not cause a significant change of food intake. First hour food intake is shown in (C) (p= 0.13). Second hour food intake is shown in (D) (p= 0.55). M1–6= rats with CEAm injection; L1–4= rats with CEAlc injection.
Table 2.
summarizes the effects of the chemogenetic activation of the CEAm and the CEAlc on the food intake in the first and the second hour of refeeding. Data are presented as means±SEM.
| Consumed food (mg/g lean bodyweight) | Consumed food (mg/g bodyweight) | |||||||
|---|---|---|---|---|---|---|---|---|
| First hour | Second hour | First hour | Second hour | |||||
| saline | CNO | Saline | CNO | saline | CNO | Saline | CNO | |
| CEAm activation | 23.61±2.19 | 15.69±1.58* | 2.48±1.03 | 11.14±2.10* | 20.32±14.63 | 13.52±10.51* | 2.17±0.90 | 9.60±1.82* |
| CEAlc activation | 24.00±1.50 | 20.86±1.16 | 2.23±0.77 | 3.54±2.35 | 20.75±1.49 | 18.00±1.00 | 1.92±0.66 | 3.03±2.01 |
significantly different from the saline treated animals P<0.01
Table 4.
Comparison the first and second hour meal pattern of refed rats with hM3D(Gq)-mCherry expression in the CEAm
| Saline | CNO | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| First hour | Second hour | T-test for dependent samples p value |
First hour | Second hour | T-test for dependent samples p value |
|||||
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |||
| Avarage Meal length (min) | 53,67 | 3,29 | 11,33 | 4,35 | 0,001 | 34,83 | 4,56 | 35,75 | 9,10 | 0,940 |
| Avarage food intake/meal (g) | 7,55 | 0,75 | 0,79 | 0,34 | 0,001 | 4,21 | 0,86 | 2,90 | 0,68 | 0,945 |
| Avarage intermeal inteval length (min) | 6,33 | 3,29 | 45,92 | 6,61 | 0,006 | 16,67 | 4,26 | 14,08 | 5,49 | 0,658 |
| Total length of meal intervals (min) | 53,67 | 3,29 | 11,33 | 4,35 | 0,002 | 43,33 | 4,26 | 40,17 | 7,16 | 0,680 |
| Total length of intermeal intervals (min) | 6,33 | 3,29 | 48,67 | 4,35 | 0,002 | 16,67 | 4,26 | 19,83 | 7,16 | 0,680 |
| Number of meal intervals | 1,00 | 0,00 | 0,67 | 0,21 | 0,170 | 1,33 | 0,21 | 1,33 | 0,21 | 1,000 |
| Number of intermeal intervals | 0,50 | 0,22 | 1,17 | 0,17 | 0,025 | 1,00 | 0,00 | 1,00 | 0,37 | 1,000 |
Localization of the CTB/PHA-L injection sites
In five, CTB-injected and three, PHA-L-injected animals, the injection site was centered in the CEA. In these cases, the injection site filled the CEA and did not spread to the surrounding regions. Only these brains were included in the analysis. In all studied brains, the patterns of CTB-IR neurons and of PHA-L fibers were similar. Images of representative injection sites and mapping of the injection sites to the planes of the Paxinos and Watson rat brain atlas (Paxinos and Watson 1998) are shown in Fig. 4 and 5.
Figure 4.

Representative images of CTB and PHA-L injection sites in the CEA. A: Core of CTB injection site deposited into the CEA. B: Core of PHA-L injection site centered in the CEA. Double-labeling immunohistochemistry for CTB (green; A1, A2), PHA-L (green; B1,B2) and c-Fos (red; A3, B3) was performed to reveal the injection sites. Areas adjacent to the CEA are not activated by refeeding. st= stria terminalis Scale bars: 500μm.
Figure 5.

Schematic representation of the core of CTB and PHAL injection sites within the CEA in 3 or 4 anteroposterior levels, respectively. The injection sites were mapped to planes of the Paxinos rat brain atlas (Paxinos and Watson 1998). Case numbers and the pattern demonstrating the boundaries of each injection core are indicated above the images.
Origin of the refeeding-activated inputs of the CEA
After iontophoretic injection of CTB into the CEA, the majority of the double-labeled neurons containing both c-Fos and CTB (Fig. 5.) were detected ipsilateral to the injection site. A large number of double-labeled cells were observed in the paraventricular thalamic nucleus (Fig. 6N), parasubthalamic nucleus (Fig. 6N, Q), parabrachial nucleus (Fig. 6O, S) and in the ventrolateral division of the periaqueductal gray (Fig. 6M, P). Few CTB-c-Fos double-labeled neurons were detected in the prelimbic (Fig. 6. A, B), agranular insular (Fig. 6. C, D), visceral (Fig. 6C), piriform (Fig. 6J), primary (Fig. 6C) and supplementary somatosensory area of cortex (Fig. 6C, E), bed nuclei of the stria terminalis (Fig. 6F, G), arcuate hypothalamic nucleus (Fig. 6N, R) perifornical nucleus (Fig. 6H, I), lateral hypothalamic area, (Fig. 6K), zona incerta and NTS (Fig. 6L).
Figure 6.


Schematic drawings illustrate the brain regions where the refeeding-activated (c-Fos-positive, black) neurons that projects to the CEA (CTB-immunoreactive perikarya, brown) are located. These double-labeled neurons are present in the prelimbic area (PrL; A, B), agranular insular area, posterior part (AIp; C, D), visceral area (VISC; D), supplementary somatosensory area (SSs; D, F), primary somatosensory cortex, barrel field (SSp-bfd; D), bed nuclei of the stria terminalis (BST; E, G), perifornical nucleus (PeF; H, I), piriform area (PIR; J), lateral hypothalamic area (LH; K), nucleus of the solitary tract (NTS; L), ventrolateral division of the periaqueductal gray (PAGvl; M, P), paraventricular thalamic nucleus (PVT; N), parasubthalamic nucleus (PSTN; N, Q), arcuate hypothalamic nucleus (ARC; N, R), PBN (O, S). Representative images illustrate the distribution of the double labeled neurons in these areas. Arrows indicate examples of double-labeled neurons. Scale bar = 500μm in the schematic drawing and 20μm in the photomicrograph.
Involvement of anorexigenic ARC POMC neurons in the innervation of the CEA and refeeding-activated neurons of the parasubthalamic nucleus that project to the CEA
After iontophoretic injection of CTB into the CEA, confocal microscopic analysis of sections immunolabeled for c-Fos, CTB and POMC revealed that only few (1–3/section) refeeding-activated POMC-IR neurons were found to project to the CEA (Fig. 7A). In contrast, sections containing the parasubthalamic nucleus revealed that in this nucleus, POMC-IR fibers frequently formed appositions on refeeding-activated neurons projecting to the CEA (Fig. 7B).
Figure 7.

Connections between the POMC neurons of the arcuate hypothalamic nucleus and the CEA. Confocal microscopic image (A) of triple-immunolabeled section indicates the presence of CTB (green) in a refeeding-activated (c-Fos-positive, red), POMC-producing (blue) perikarion in the arcuate hypothalamic nucleus of brain previously injected with CTB into the CEA. Arrow points to a triple-labeled neuron. The image was made by projecting 7 consecutive 1.8 μm thick confocal optical slices into one plane. To facilitate identification of triple-labeled cells, CTB, c-Fos and POMC-labeling of the same cells are shown in separate images in the insets. Note that the vast majority of the c-Fos-containing POMC neurons do not contain CTB-immunoreactivity.
Confocal image of a triple-immunolabeled section (B) shows POMC-IR (blue) boutons on refeeding-activated neurons (c-Fos-IR, red) in the parasubthalamic nucleus (PSTN) that projects to the CEA (CTB-IR, green). Arrowheads point to POMC-IR varicosities on the surface of double-labeled, c-Fos-and POMC-IR neurons. The cells in squares are shown in higher magnification in the insets. B3 shows the c-Fos-IR nucleus of the neuron illustrated in B2. The POMC-IR varicosities contacting the perikaryon and the dendrite of this double-labeled neuron are pointed by arrows. B3 also illustrates a refeeding-activated and CEA-projecting neuron contacted by a POMC-IR varicosity (arrow). Note that primary antibodies against c-Fos and POMC were produced in rabbit, thus immunolabeling was performed sequentially, which prevented cross-reaction in the cytoplasm, but not in the cell nuclei. Scale bars: 20μm.
Innervation of refeeding-activated neuronal populations by CEA neurons
Numerous PHA-L-IR fibers were detected in several refeeding-activated brain regions (Fig. 8A–G) on the ipsilateral side of tracer injection including the bed nuclei of the stria terminalis, lateral hypothalamic area, posterior magnocellular part of the hypothalamic paraventricular nucleus, parasubthalamic nucleus, paraventricular thalamic nucleus, periaqueductal gray, NTS, and parabrachial nucleus. Only few PHA-L fibers were identified in the medial parvicellular part of the hypothalamic paraventricular nucleus. Confocal microscopic analysis of sections immunolabeled for PHA-L/c-Fos and a neuronal marker, HuCD, revealed that PHA-L-containing axons contact refeeding-activated neurons in all of these areas (Fig. 8A–G). Connectivity of the CEA with refeeding-activated brain areas is summarized in Fig. 9.
Figure 8.


Schematic drawings and representative images illustrate the distribution of PHA-L-immunoreactive axons in refeeding-activated areas: the bed nuclei of the stria terminalis (BST; A1, A2), paraventricular hypothalamic nucleus, medial parvicellular part (PVHmp; B1, B2), paraventricular hypothalamic nucleus, posterior magnocellular part (PVHpm; B1, B3), lateral hypothalamus (LHA; C1, C2), periaqueductal gray (PAG; D1, D2), parasubthalamic nucleus (PSTN; E1, E2), paraventricular thalamic nucleus (PVT; E1, E3), parabrachial nucleus (PB; F1, F2), nucleus tractus solitarius (NTS; G1, G2),. The
 indicates a high density of PHA-L fibers, whereas
indicates a high density of PHA-L fibers, whereas
 indicates a low density of PHA-L fibers. Confocal images illustrate PHA-L (red) fibers among refeeding-activated neurons. Arrowheads point to PHA-L-immunoreactive (red) contacts on c-Fos (green)/HuCD (blue) double-labeled neurons. Higher magnification of framed areas is shown in the insets. Scale bars: 500μm in the schematic drawings and 20μm in the confocal images.
indicates a low density of PHA-L fibers. Confocal images illustrate PHA-L (red) fibers among refeeding-activated neurons. Arrowheads point to PHA-L-immunoreactive (red) contacts on c-Fos (green)/HuCD (blue) double-labeled neurons. Higher magnification of framed areas is shown in the insets. Scale bars: 500μm in the schematic drawings and 20μm in the confocal images.
Figure 9.
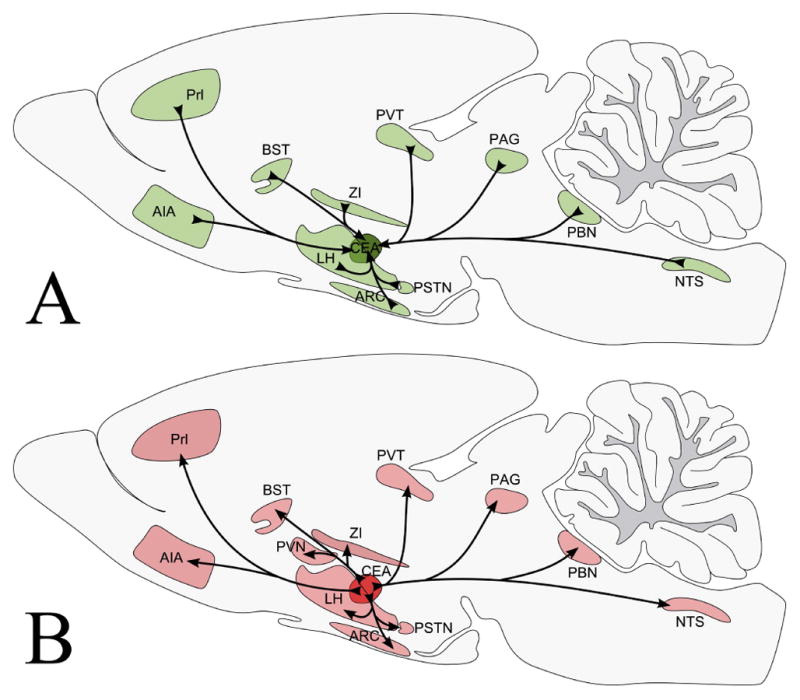
Schematic illustration of the afferent (A) and efferent (B) connections of the CEA with refeeding-activated brain areas.
Brain areas directly activated by stimulated CEA neurons
To understand which brain areas may be directly activated by CEA neurons during refeeding, refeeding-activated brain areas were identified where chemogenetic activation of CEA subnuclei induced c-Fos expression, and the activated neurons are contacted by the axons of CEA neurons. The chemogenetic activation of CEAm neurons induced significant increase of c-Fos-IR nuclei in the BNST, parasubthalamic nucleus and parabrachial nucleus compared to virus-injected, saline-treated animals (Fig. 10). Axons of CEAm neurons contacted c-Fos-IR neurons in all of these areas at the ipsilateral site (Fig. 11). The activation of CEAlc did not influence the activation of BNST and parasubthalamic nucleus, but it resulted in significant increase of neuronal activation in the parabrachial nucleus. The increase in the number of c-Fos-IR nuclei was less than that induced by the activation of CEAm.
Figure 10.

Graphs summarizing the effects of the chemogenetic activation of CEAlc and CEAm on the number of c-Fos-IR nuclei in brain regions shown to respond to refeeding by neuronal activation. Control animals (ctr) were also injected with hSyn-hM3D(Gq)-mCherry AAV into the CEAlc or CEAm, but received saline treatment instead of CNO. In the BNST and PSTN, the activation of CEAlc did not influence the number of c-Fos-IR neurons, but activation of CEAm caused a marked increase of the number of c-Fos-IR neurons. In the parabrachial nucleus (PBN), both the activation of the CEAlc and the CEAm induced significant increase of the number of c-Fos-IR neurons, however, the effect of the CEAm was more pronounced. (*=p<0.05, **=p<0.01, ***=p<0.001)
Figure 11.

Confocal images of triple-immunolabeled sections show mCherry-containing axons (red) that label the axons of CEA origin; and activated (c-Fos-positive, green), HuCD-producing neurons (blue) in saline (A, D, G)-and CNO (B,E,H)-treated rats in the following areas: BNST (A–C), PSTN (D–F), PBN (G–I). Arrowheads show the activated neurons that receive inputs from mCherry positive fibers. Cells in the white boxes in (C, F and I) are shown in higher magnification in insets. ac= anterior commissure, cp= cerebral peduncle; STh= subthalamic nucleus; scp= superior cerebellar peduncle; Scale bar: 50μm in A,B,D,E,G,H 10μm on C, F, I.
Discussion
To better understand satiety-related neuronal networks in the brain, we used a fasting-refeeding paradigm as a satiety model, and have begun to build a connectivity map of the refeeding-activated neuronal groups. As a first step, we mapped the refeeding-activated neuronal groups in the rat brain with a particular focus on connectivity of the parabrachial nucleus (Zseli et al. 2016). In the current manuscript, we focus on the role of CEA in the regulation of food intake during refeeding, and describe its connections with the other refeeding-activated neuronal populations using classic anterograde and retrograde tract-tracing methods to reveal the sources of the refeeding-related inputs of these neurons and the regions where they send refeeding-related information.
The functional role of the CEA in the regulation of food intake has been demonstrated using optogenetic tools (Carter et al. 2013), but only the role of the PBN CGRP neuron-PBNlc pathway was investigated (Carter et al. 2013). However, our earlier tract tracing studies (Zseli et al. 2016) and the observation that refeeding-induced activation of neurons was also observed in the CEAm, suggested that a PBNm-CEAm pathway is also involved in the regulation of food intake during refeeding. To test this possibility, we studied whether activation of CEA subnuclei alter the food intake during refeeding.
Chemogenetic activation caused neuronal activation only in the virus infected cells within the CEA. This specific activation pattern allowed the examination of the effects of CEAm and CEAlc activations separately. During refeeding, the activation of CEAlc neurons had no significant effect on food intake. In contrast, activation of CEAm resulted in a marked decrease of food intake during the first hour of refeeding. However, the second hour of food intake of the CEAm-activated rats was greater than after saline treatment, such that at the end of the two hour refeeding interval, total food consumption was similar for both CNO and saline treatments. To better understand the CEAm-activation-induced changes in feeding behavior, we studied the effect of CNO treatment on the meal pattern of the rats with hM3D(Gq)-mCherry expression in the CEAm. CNO treatment reduced the average meal length and size during the first hour, but had a tendency to increase the average meal length and increase meal size and the total length of time spent with feeding during the second hour when the parameters were compared to saline treatment.
We observed that the rats were already satiated in the second hour of refeeding when they received saline, as indicated by the marked decrease in their meal length and meal size in the second hour compared to the first hour. In contrast, the rats had very similar food intake during the first and second hour when they were treated with CNO. This observation suggests that CEAm-activated rats eat less vigorously and need more time to reach satiety than saline controls, but the overall amount of food causing satiety was not affected by the treatment. Further studies will be necessary to determine whether CEAm activation influences the motivation to eat, or it decreases meal size by stimulating the effects of meal termination signals.
To determine the source of the refeeding related inputs of CEA neurons, retrograde tracing was performed. It is well recognized that satiety-related information primarily reaches the brain through two, main brains areas, the NTS and the ARC (Schwartz 2000). These areas contain large number of activated neurons during refeeding (Singru et al. 2007; Timofeeva et al. 2002). The NTS is the primary neuroanatomical site receiving meal-related, visceral, afferent information from the gastrointestinal tract required for negative feedback control of food intake (Schwartz et al. 2000) via afferent fibers of the vagus nerve. These fibers are activated by mechanoreceptors and chemoreceptors in the esophagus, stomach and the small intestine, and their stimulation results in dose-dependent reductions in subsequent food intake and meal size (Peters et al. 2006; Schwartz 2000). The NTS integrates these signals with humoral signals released by enteroendocrine cells in the stomach, pancreas, and small intestine in response to food intake (Grill and Hayes 2009), and by glucose (Balfour and Trapp 2007; Ritter et al. 2000) and amino acids (Blouet and Schwartz 2012).
One might speculate from the literature that the NTS is directly involved in refeeding-induced activation of the CEA. However, this does not appear to be the case, as only few refeeding-activated neurons of the NTS were observed to project to the CEA in our study. In contrast, a major refeeding-related input to the CEA was found to originate from the parabrachial nucleus. As we have previously demonstrated (Zseli et al. 2016) that refeeding-activated neurons in the NTS densely innervate parabrachial neurons, a NTS-parabrachial-CEA pathway is suggested that may have a major role in refeeding-induced activation of CEA neurons. A similar anorexigenic NTS-parabrachial-CEA pathway mediating toxin-induced anorexia was described by Carter et al. (Carter et al. 2013) involving CGRP neurons located in the outer external lateral subdivision of the parabrachial nucleus of mice. Refeeding-activation of the CEA, however, also involves neurons in the medial parabrachial nucleus, suggesting that the toxin-induced anorexia and refeeding-induced satiety may be mediated by separate groups of parabrachial neurons and involve different pathways.
Similar to the NTS, the ARC also serves as an important metabolic sensor area in the brain (Schwartz et al. 2000), responding to humoral signals such as leptin, insulin, ghrelin and metabolites including glucose, amino acids and fatty acids (Schwartz et al. 2000). ARC neurons sense and integrate these signals and transmit the processed information to second order feeding-related brain centers (Schwartz et al. 2000). One group of feeding-related neurons in the ARC are those that express POMC (Schwartz et al. 2000). Alpha-melanocyte-stimulating hormone (α-MSH) cleaved from the POMC prohormone has a potent effect to suppress food intake and increase energy expenditure by binding to MC4 melanocortin receptors (Schwartz et al. 2000). Deletion of POMC or MC4-R produces an obesity syndrome (Huszar et al. 1997; Ste Marie et al. 2000), substantiating the role of α-MSH in the regulation of feeding and energy homeostasis. Refeeding activates POMC neurons independently of the vagus nerve and the NTS, likely through direct effects of circulating hormones and metabolites (Fekete et al. 2012). Although melanocortin receptors are expressed in a number of distinct brain areas that could mediate the effects of POMC on energy balance (Kishi et al. 2003), data in the literature suggest that the hypothalamic paraventricular nucleus is the major center for the action of the melanocortin signaling system on food intake (Wirth et al. 2001; Balthasar et al. 2005). In accordance, we observed only few refeeding-activated POMC neurons projecting to the CEA. In contrast, a major refeeding-activated input to the CEA originating from the parasubthalamic nucleus was observed. The existence of parasubthalamic nucleus-CEA projection has been already shown by tract tracing studies (Goto and Swanson 2004; Chometton et al. 2016). Currently, little is known about the role of this nucleus in the regulation of energy homeostasis. Our data demonstrate that the CEA-projecting refeeding-activated neurons of the parasubthalamic nucleus are densely innervated by POMC-IR axons, suggesting that the parasubthalamic nucleus mediates satiety effects of refeeding-activated POMC neurons toward the CEA. In addition, we have observed that the refeeding-activated neurons of the parasubthalamic nucleus also receives a dense input from the parabrachial nucleus, indicating that the parasubthalamic nucleus may integrate refeeding-related inputs from both the hypothalamus and the brainstem and then sends this information to the CEA. This hypothesis is supported by findings of Chometton et al (Chometton et al. 2016) showing that CEA projecting parasubthalamic neurons receive CGRP-IR innervation from the parabrachial nucleus. These authors (Chometton et al. 2016) also suggest that the parasubthalamic nucleus may be involved in the processing of the hedonic value of food.
The paraventricular thalamic nucleus is also a component of the neural circuitry controlling feeding behavior and was found to be a major source of the refeeding-activated inputs of the CEA. The paraventricular thalamic nucleus projects to and receives inputs from a number of other feeding-related sites such as the parabrachial nucleus, NTS and lateral hypothalamic area (Moga et al. 1995; Chen and Su 1990; Cornwall and Phillipson 1988). Many of these connections contain feeding-related peptides, such as glucagon-like peptide 1 (Llewellyn-Smith et al. 2011), cocaine and amphetamine regulated transcript (Kirouac et al. 2006), cholecystokinin (Otake 2005) and orexin (Kirouac et al. 2005), all which have been shown to influence food intake. Lesions or inhibition of paraventricular thalamic neurons have been reported to increase food intake and weight gain in rats (Bhatnagar and Dallman 1999; Stratford and Wirtshafter 2013), supporting its role in the termination of food intake.
A less dense refeeding-related input to the CEA was found to originate from parts of the prefrontal cortex. This observation is of interest as the prefrontal cortex has been implicated in goal-directed actions, including food reward and memories required for obtaining food. Specifically, the prelimbic area is heavily involved in instrumental learning and performance (Baldwin et al. 2000; Baldwin et al. 2002; Balleine and Dickinson 1998), and the agranular insular cortical area in working memory for food reward value (DeCoteau et al. 1997), as anticipatory discriminability is impaired in rats with agranular insular area lesions (Kesner and Gilbert 2007). We also observed refeeding–activated, retrogradely-labeled neurons in the supplementary somatosensory area and in the barrel field of the primary somatosensory cortex. However, projection of these areas to the CEA was not described by Bienkowski and Rinaman (Bienkowski and Rinaman 2013) and therefore, further studies are needed to clarify the importance of these refeeding-activated neuronal groups in the innervation of the CEA.
To elucidate the neuronal pathways mediating the effects of refeeding-activated CEA neurons on food intake, the efferent connections of the CEA with refeeding-activated neuronal groups were also studied. These experiments demonstrated that refeeding-activated CEA neurons have bidirectional connections with the parasubthalamic nucleus, parabrachial nucleus, bed nuclei of the stria terminalis and the paraventricular thalamic nucleus. Since these areas appear to mediate the effects of peripheral refeeding-related signals to the CEA, projections from the CEA to these cell populations may serve as a short feedback loop that facilitates or restrains refeeding-activated neuronal inputs to the CEA. Although the CEA projects to the NTS, it does not receive major, refeeding-related, direct inputs from the NTS. Nevertheless, the NTS may still be capable of relaying satiety information to the CEA indirectly via the NTS-parabrachial nucleus-CEA pathway. Since the CEA-NTS pathway is primarily GABA-ergic (Serrats and Sawchenko 2006), it is likely that the CEA projection to the NTS decreases the sensitivity of the NTS neurons to the peripheral signals rather than facilitate the satiety response.
Another refeeding related region of the brain demonstrating reciprocal connections with the CEA is the midbrain periaqueductal gray. Reciprocal connections between these two regions were previously recognized by Rizvi et al (Rizvi et al. 1991). This region is of interest because similar to the CEA, the periaqueductal gray is also involved in emotional and limbic behaviors and cardiovascular responses, hence, it is suggested to be an important structure mediating functions elicited from CEA (Rizvi et al. 1991). Support for this region in feeding responses was recently reported by Stachniak et al (Stachniak et al. 2014), showing increased food intake after chemogenetic inhibition of the descending glutamatergic input to the ventrolateral portion of the periaqueductal gray. Refeeding, however, appears to differentially regulate two, separate populations of cells in the ventrolateral periaqueductal gray including a cell group in the lateral part of this region that is inhibited by refeeding, and a medial group that is activated by refeeding (Zseli et al. 2016). Thus, the ventrolateral periaqueductal gray may contain both orexigenic and anorexigenic neurons. Further studies are needed to characterize these cells, support the existence of orexigenic cells in this brain region and determine whether the refeeding-inhibited cells also receive input from the CEA. Given that the CEA has major, GABAergic, efferent projections (Serrats and Sawchenko 2006), the possibility that some ventrolateral periaqueductal neurons may be inhibited is quite possible. In fact, there may be other refeeding-related targets of CEA that show inhibition during refeeding and would not be detected by c-Fos immunocytochemistry. Thus, it is likely that CEA may have an even wider refeeding-related output than recognized by these studies.
The CEA also has a major unilateral projection to the lateral hypothalamic area. This hypothalamic region is an integrative site for signals underlying the motivation to eat that includes physiological signals from the body and external signals from the environment contributing to feeding, reward, and motivation (Elmquist et al. 1999; Swanson 2000; Wise 1974; Morton et al. 2006; Kelley et al. 2005). Results to date suggest that glutamate in the lateral hypothalamic area may have a major role in the generation of feeding, whereas GABA induces satiety (Stanley et al. 2011; Delgado and Anand 1953; Turenius et al. 2009). Since the CEA contains both glutamatergic and GABA-ergic neurons (Sah et al. 2003), further experiments are needed to reveal the classical neurotransmitter content of the CEA-lateral hypothalamic area pathway and understand its role in the development of refeeding-induced satiety. Since neurons in the lateral hypothalamus have an important role in food reward, the CEA-lateral hypothalamic area pathway may also modulate the reward value of food that facilitates termination of food intake.
The involvement of CEA in the direct activation of neurons in the BNST, parasubthalamic and parabrachial nuclei is further supported by the chemogenetic experiment showing that specific activation of CEAm neurons directly induces neuronal activation in these brain regions. Since CEA neurons contact activated neurons in more brain areas during refeeding than after chemogenetic activation of CEA, we hypothesize that in some brain regions, like in the refeeding-activated neurons in the NTS, periaqueductal gray and lateral hypothalamic area, GABA-ergic CEA neurons may inhibit the refeeding-activated neuronal populations.
In summary, the present tract-tracing experiments reveal that the CEA is highly interconnected with refeeding-activated brain areas, in particular the parabrachial nucleus, parasubthalamic nucleus, paraventricular thalamic nucleus and the BNST. Information from the NTS, however, would appear to be primarily transmitted to the CEA from the parabrachial nucleus and from the ARC POMC neurons through the parasubthalamic nucleus. These data support the utility of the fasting-refeeding model as a way to identify brain centers regulating the food intake including novel loci, and facilitate understanding of how they communicate with each other to regulate feeding behavior.
Table 3.
Effect of CNO treatment ont he meal pattern of refed rats with hM3D(Gq)-mCherry expression in the CEAm
| First hour | Second hour | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Saline | CNO | T-test for dependent samples p value |
Saline | CNO | T-test for dependent samples p value |
|||||
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |||
| Avarage Meal length (min) | 53.67 | 3.29 | 34.83 | 4.56 | 0.01 | 11.33 | 4.35 | 35.75 | 9.10 | 0.05 |
| Avarage food intake/meal (g) | 7.55 | 0.75 | 4.21 | 0.86 | 0.01 | 0.79 | 0.34 | 2.90 | 0.68 | 0.02 |
| Avarage intermeal inteval length (min) | 6.33 | 3.29 | 16.67 | 4.26 | 0.13 | 45.92 | 6.61 | 14.08 | 5.49 | 0.03 |
| Total length of meal intervals (min) | 53.67 | 3.29 | 43.33 | 4.26 | 0.13 | 11.33 | 4.35 | 40.17 | 7.16 | 0.01 |
| Total length of intermeal intervals (min) | 6.33 | 3.29 | 16.67 | 4.26 | 0.13 | 48.67 | 4.35 | 19.83 | 7.16 | 0.01 |
| Number of meal intervals | 1.00 | 0.00 | 1.33 | 0.21 | 0.17 | 0.67 | 0.21 | 1.33 | 0.21 | 0.10 |
| Number of intermeal intervals | 0.50 | 0.22 | 1.00 | 0.00 | 0.08 | 1.17 | 0.17 | 1.00 | 0.37 | 0.70 |
Acknowledgments
The authors are grateful for Vera Maruzs and Ágnes Simon for the expert technical assistant. This work was supported by Grants from the Hungarian Science Foundation (OTKA K109710), Hungarian National Brain Research Program, Lendület Award of the Hungarian Academy of Sciences, the Dr. Gerald J. and Dorothy R. Friedman New York Foundation for Medical Research and the NIDDK HD087958 grant.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
Role of authors:
Györgyi Zséli: Anterograde tract tracing, comparing c-Fos immunoreactivity in fasted and refed animals in order to choose the areas of interest, DREADD virus injection, c-Fos analysis of DREADD virus injected animals, immunohistochemistry, image preparation and analysis.
Barbara Vida: Retrograde tract tracing, DREADD virus injection, immunohistochemistry, image analysis.
Anett Szilvásy-Szabó: analyses of feeding behavior.
Mónika Tóth: analyses of feeding behavior.
Ronald M. Lechan: data interpretation, manuscript preparation
Csaba Fekete: Experimental design, data acquisition, drafting, revising and interpretation of the work, takes primary responsibility.
Ethical approval: All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were reviewed and approved by the Animal Welfare Committee of the Institute of Experimental Medicine, Hungarian Academy of Sciences, Budapest, Hungary.
References
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, McNamara JO, Roth BL. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63(1):27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AE, Holahan MR, Sadeghian K, Kelley AE. N-methyl-D-aspartate receptor-dependent plasticity within a distributed corticostriatal network mediates appetitive instrumental learning. Behavioral neuroscience. 2000;114(1):84–98. doi: 10.1037//0735-7044.114.1.84. [DOI] [PubMed] [Google Scholar]
- Baldwin AE, Sadeghian K, Kelley AE. Appetitive instrumental learning requires coincident activation of NMDA and dopamine D1 receptors within the medial prefrontal cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22(3):1063–1071. doi: 10.1523/JNEUROSCI.22-03-01063.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balfour RH, Trapp S. Ionic currents underlying the response of rat dorsal vagal neurones to hypoglycaemia and chemical anoxia. The Journal of physiology. 2007;579(Pt 3):691–702. doi: 10.1113/jphysiol.2006.126094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine BW, Dickinson A. Goal-directed instrumental action: contingency and incentive learning and their cortical substrates. Neuropharmacology. 1998;37(4–5):407–419. doi: 10.1016/s0028-3908(98)00033-1. [DOI] [PubMed] [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Barrachina MD, Martinez V, Wang L, Wei JY, Tache Y. Synergistic interaction between leptin and cholecystokinin to reduce short-term food intake in lean mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(19):10455–10460. doi: 10.1073/pnas.94.19.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, Dallman MF. The paraventricular nucleus of the thalamus alters rhythms in core temperature and energy balance in a state-dependent manner. Brain research. 1999;851(1–2):66–75. doi: 10.1016/s0006-8993(99)02108-3. [DOI] [PubMed] [Google Scholar]
- Bienkowski MS, Rinaman L. Common and distinct neural inputs to the medial central nucleus of the amygdala and anterior ventrolateral bed nucleus of stria terminalis in rats. Brain structure & function. 2013;218(1):187–208. doi: 10.1007/s00429-012-0393-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouet C, Schwartz GJ. Brainstem nutrient sensing in the nucleus of the solitary tract inhibits feeding. Cell metabolism. 2012;16(5):579–587. doi: 10.1016/j.cmet.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ME, Soden ME, Zweifel LS, Palmiter RD. Genetic identification of a neural circuit that suppresses appetite. Nature. 2013;503(7474):111–114. doi: 10.1038/nature12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Su HS. Afferent connections of the thalamic paraventricular and parataenial nuclei in the rat--a retrograde tracing study with iontophoretic application of Fluoro-Gold. Brain research. 1990;522(1):1–6. doi: 10.1016/0006-8993(90)91570-7. [DOI] [PubMed] [Google Scholar]
- Chometton S, Pedron S, Peterschmitt Y, Van Waes V, Fellmann D, Risold PY. A premammillary lateral hypothalamic nuclear complex responds to hedonic but not aversive tastes in the male rat. Brain structure & function. 2016;221(4):2183–2208. doi: 10.1007/s00429-015-1038-3. [DOI] [PubMed] [Google Scholar]
- Cornwall J, Phillipson OT. Afferent projections to the dorsal thalamus of the rat as shown by retrograde lectin transport. II. The midline nuclei. Brain research bulletin. 1988;21(2):147–161. doi: 10.1016/0361-9230(88)90227-4. [DOI] [PubMed] [Google Scholar]
- DeCoteau WE, Kesner RP, Williams JM. Short-term memory for food reward magnitude: the role of the prefrontal cortex. Behavioural brain research. 1997;88(2):239–249. doi: 10.1016/s0166-4328(97)00044-2. [DOI] [PubMed] [Google Scholar]
- Delgado JM, Anand BK. Increase of food intake induced by electrical stimulation of the lateral hypothalamus. The American journal of physiology. 1953;172(1):162–168. doi: 10.1152/ajplegacy.1952.172.1.162. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22(2):221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Scammell TE, Jacobson CD, Saper CB. Distribution of Fos-like immunoreactivity in the rat brain following intravenous lipopolysaccharide administration. The Journal of comparative neurology. 1996;371(1):85–103. doi: 10.1002/(SICI)1096-9861(19960715)371:1<85::AID-CNE5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Fekete C, Zseli G, Singru PS, Kadar A, Wittmann G, Fuzesi T, El-Bermani W, Lechan RM. Activation of anorexigenic pro-opiomelanocortin neurones during refeeding is independent of vagal and brainstem inputs. Journal of neuroendocrinology. 2012;24(11):1423–1431. doi: 10.1111/j.1365-2826.2012.02354.x. [DOI] [PubMed] [Google Scholar]
- French SJ, Murray B, Rumsey RD, Sepple CP, Read NW. Preliminary studies on the gastrointestinal responses to fatty meals in obese people. International journal of obesity and related metabolic disorders: journal of the International Association for the Study of Obesity. 1993;17(5):295–300. [PubMed] [Google Scholar]
- Geliebter A, Westreich S, Gage D. Gastric distention by balloon and test-meal intake in obese and lean subjects. The American journal of clinical nutrition. 1988;48(3):592–594. doi: 10.1093/ajcn/48.3.592. [DOI] [PubMed] [Google Scholar]
- Gibson CD, Carnell S, Ochner CN, Geliebter A. Neuroimaging, gut peptides and obesity: novel studies of the neurobiology of appetite. Journal of neuroendocrinology. 2010;22(8):833–845. doi: 10.1111/j.1365-2826.2010.02025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M, Swanson LW. Axonal projections from the parasubthalamic nucleus. The Journal of comparative neurology. 2004;469(4):581–607. doi: 10.1002/cne.11036. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Hayes MR. The nucleus tractus solitarius: a portal for visceral afferent signal processing, energy status assessment and integration of their combined effects on food intake. International journal of obesity. 2009;33(Suppl 1):S11–15. doi: 10.1038/ijo.2009.10. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Ji H, Friedman MI. Compensatory hyperphagia after fasting tracks recovery of liver energy status. Physiology & behavior. 1999;68(1–2):181–186. doi: 10.1016/s0031-9384(99)00173-0. [DOI] [PubMed] [Google Scholar]
- Johnstone LE, Fong TM, Leng G. Neuronal activation in the hypothalamus and brainstem during feeding in rats. Cell metabolism. 2006;4(4):313–321. doi: 10.1016/j.cmet.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Kelley AE, Baldo BA, Pratt WE, Will MJ. Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward. Physiology & behavior. 2005;86(5):773–795. doi: 10.1016/j.physbeh.2005.08.066. [DOI] [PubMed] [Google Scholar]
- Kesner RP, Gilbert PE. The role of the agranular insular cortex in anticipation of reward contrast. Neurobiology of learning and memory. 2007;88(1):82–86. doi: 10.1016/j.nlm.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac GJ, Parsons MP, Li S. Orexin (hypocretin) innervation of the paraventricular nucleus of the thalamus. Brain research. 2005;1059(2):179–188. doi: 10.1016/j.brainres.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Kirouac GJ, Parsons MP, Li S. Innervation of the paraventricular nucleus of the thalamus from cocaine-and amphetamine-regulated transcript (CART) containing neurons of the hypothalamus. The Journal of comparative neurology. 2006;497(2):155–165. doi: 10.1002/cne.20971. [DOI] [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. The Journal of comparative neurology. 2003;457(3):213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- Lin Z, Gao N, Hu HZ, Liu S, Gao C, Kim G, Ren J, Xia Y, Peck OC, Wood JD. Immunoreactivity of Hu proteins facilitates identification of myenteric neurones in guinea-pig small intestine. Neurogastroenterology and motility: the official journal of the European Gastrointestinal Motility Society. 2002;14(2):197–204. doi: 10.1046/j.1365-2982.2002.00317.x. [DOI] [PubMed] [Google Scholar]
- Liposits Z, Setalo G, Flerko B. Application of the silver-gold intensified 3,3′-diaminobenzidine chromogen to the light and electron microscopic detection of the luteinizing hormone-releasing hormone system of the rat brain. Neuroscience. 1984;13(2):513–525. doi: 10.1016/0306-4522(84)90245-8. [DOI] [PubMed] [Google Scholar]
- Llewellyn-Smith IJ, Reimann F, Gribble FM, Trapp S. Preproglucagon neurons project widely to autonomic control areas in the mouse brain. Neuroscience. 2011;180:111–121. doi: 10.1016/j.neuroscience.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusich MF, Furneaux HM, Henion PD, Weston JA. Hu neuronal proteins are expressed in proliferating neurogenic cells. Journal of neurobiology. 1994;25(2):143–155. doi: 10.1002/neu.480250206. [DOI] [PubMed] [Google Scholar]
- Moga MM, Weis RP, Moore RY. Efferent projections of the paraventricular thalamic nucleus in the rat. The Journal of comparative neurology. 1995;359(2):221–238. doi: 10.1002/cne.903590204. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Murphy EM, Defontgalland D, Costa M, Brookes SJ, Wattchow DA. Quantification of subclasses of human colonic myenteric neurons by immunoreactivity to Hu, choline acetyltransferase and nitric oxide synthase. Neurogastroenterology and motility: the official journal of the European Gastrointestinal Motility Society. 2007;19(2):126–134. doi: 10.1111/j.1365-2982.2006.00843.x. [DOI] [PubMed] [Google Scholar]
- Otake K. Cholecystokinin and substance P immunoreactive projections to the paraventricular thalamic nucleus in the rat. Neuroscience research. 2005;51(4):383–394. doi: 10.1016/j.neures.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego, CA: 1998. [Google Scholar]
- Peters JH, Ritter RC, Simasko SM. Leptin and CCK selectively activate vagal afferent neurons innervating the stomach and duodenum. American journal of physiology Regulatory, integrative and comparative physiology. 2006;290(6):R1544–1549. doi: 10.1152/ajpregu.00811.2005. [DOI] [PubMed] [Google Scholar]
- Petrovich GD, Ross CA, Mody P, Holland PC, Gallagher M. Central, but not basolateral, amygdala is critical for control of feeding by aversive learned cues. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(48):15205–15212. doi: 10.1523/JNEUROSCI.3656-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikov LR, Reagan LP, Fadel JR. Activation of phenotypically distinct neuronal subpopulations in the anterior subdivision of the rat basolateral amygdala following acute and repeated stress. The Journal of comparative neurology. 2008;508(3):458–472. doi: 10.1002/cne.21687. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT, Zhang Y. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain research. 2000;856(1–2):37–47. doi: 10.1016/s0006-8993(99)02327-6. [DOI] [PubMed] [Google Scholar]
- Rizvi TA, Ennis M, Behbehani MM, Shipley MT. Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: topography and reciprocity. The Journal of comparative neurology. 1991;303(1):121–131. doi: 10.1002/cne.903030111. [DOI] [PubMed] [Google Scholar]
- Rollins BL, King BM. Amygdala-lesion obesity: what is the role of the various amygdaloid nuclei? American journal of physiology Regulatory, integrative and comparative physiology. 2000;279(4):R1348–1356. doi: 10.1152/ajpregu.2000.279.4.R1348. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiological reviews. 2003;83(3):803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Sato I, Arima H, Ozaki N, Watanabe M, Goto M, Hayashi M, Banno R, Nagasaki H, Oiso Y. Insulin inhibits neuropeptide Y gene expression in the arcuate nucleus through GABAergic systems. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25(38):8657–8664. doi: 10.1523/JNEUROSCI.2739-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GJ. The role of gastrointestinal vagal afferents in the control of food intake: current prospects. Nutrition. 2000;16(10):866–873. doi: 10.1016/s0899-9007(00)00464-0. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404(6778):661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Serrats J, Sawchenko PE. CNS activational responses to staphylococcal enterotoxin B: T-lymphocyte-dependent immune challenge effects on stress-related circuitry. The Journal of comparative neurology. 2006;495(2):236–254. doi: 10.1002/cne.20872. [DOI] [PubMed] [Google Scholar]
- Singru PS, Sanchez E, Fekete C, Lechan RM. Importance of melanocortin signaling in refeeding-induced neuronal activation and satiety. Endocrinology. 2007;148(2):638–646. doi: 10.1210/en.2006-1233. [DOI] [PubMed] [Google Scholar]
- Stachniak TJ, Ghosh A, Sternson SM. Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus-->midbrain pathway for feeding behavior. Neuron. 2014;82(4):797–808. doi: 10.1016/j.neuron.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley BG, Urstadt KR, Charles JR, Kee T. Glutamate and GABA in lateral hypothalamic mechanisms controlling food intake. Physiology & behavior. 2011;104(1):40–46. doi: 10.1016/j.physbeh.2011.04.046. [DOI] [PubMed] [Google Scholar]
- Ste Marie L, Miura GI, Marsh DJ, Yagaloff K, Palmiter RD. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(22):12339–12344. doi: 10.1073/pnas.220409497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford JM, Finger TE. Central representation of postingestive chemosensory cues in mice that lack the ability to taste. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31(25):9101–9110. doi: 10.1523/jneurosci.0404-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford TR, Wirtshafter D. Injections of muscimol into the paraventricular thalamic nucleus, but not mediodorsal thalamic nuclei, induce feeding in rats. Brain research. 2013;1490:128–133. doi: 10.1016/j.brainres.2012.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW. Cerebral hemisphere regulation of motivated behavior. Brain research. 2000;886(1–2):113–164. doi: 10.1016/s0006-8993(00)02905-x. [DOI] [PubMed] [Google Scholar]
- Timofeeva E, Picard F, Duclos M, Deshaies Y, Richard D. Neuronal activation and corticotropin-releasing hormone expression in the brain of obese (fa/fa) and lean (fa/?) Zucker rats in response to refeeding. The European journal of neuroscience. 2002;15(6):1013–1029. doi: 10.1046/j.1460-9568.2002.01942.x. [DOI] [PubMed] [Google Scholar]
- Turenius CI, Htut MM, Prodon DA, Ebersole PL, Ngo PT, Lara RN, Wilczynski JL, Stanley BG. GABA(A) receptors in the lateral hypothalamus as mediators of satiety and body weight regulation. Brain research. 2009;1262:16–24. doi: 10.1016/j.brainres.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Wirth MM, Olszewski PK, Yu C, Levine AS, Giraudo SQ. Paraventricular hypothalamic alpha-melanocyte-stimulating hormone and MTII reduce feeding without causing aversive effects. Peptides. 2001;22(1):129–134. doi: 10.1016/s0196-9781(00)00367-3. [DOI] [PubMed] [Google Scholar]
- Wise RA. Lateral hypothalamic electrical stimulation: does it make animals ‘hungry’? Brain research. 1974;67(2):187–209. doi: 10.1016/0006-8993(74)90272-8. [DOI] [PubMed] [Google Scholar]
- Wittmann G, Hrabovszky E, Lechan RM. Distinct glutamatergic and GABAergic subsets of hypothalamic pro-opiomelanocortin neurons revealed by in situ hybridization in male rats and mice. The Journal of comparative neurology. 2013;521(14):3287–3302. doi: 10.1002/cne.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, D’Alessio DA. Central control of body weight and appetite. The Journal of clinical endocrinology and metabolism. 2008;93(11 Suppl 1):S37–50. doi: 10.1210/jc.2008-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Lemus MB, Stark R, Bayliss JA, Reichenbach A, Lockie SH, Andrews ZB. The temporal pattern of cfos activation in hypothalamic, cortical, and brainstem nuclei in response to fasting and refeeding in male mice. Endocrinology. 2014;155(3):840–853. doi: 10.1210/en.2013-1831. [DOI] [PubMed] [Google Scholar]
- Zseli G, Vida B, Martinez A, Lechan RM, Khan AM, Fekete C. Elucidation of the anatomy of a satiety network: Focus on connectivity of the parabrachial nucleus in the adult rat. The Journal of comparative neurology. 2016;524(14):2803–2827. doi: 10.1002/cne.23992. [DOI] [PMC free article] [PubMed] [Google Scholar]